

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 10/104/25. The contractual start date was in July 2012. The draft report began editorial review in November 2017 and was accepted for publication in July 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Alexander Szubert reports grants from National Institute for Health Research (NIHR) and the Medical Research Council (MRC) during the conduct of the study. Robert Tilley reports personal fees from NIHR Clinical Research Network outside the submitted work. Peter Wilson reports personal fees from 3M Advisory Panel, Roche Drug Safety Monitoring Board and Merck Sharp & Dohme Corp. (MSD; Hoddesdon, UK) outside the submitted work. Achyut Guleri reports receiving fees from Novartis as a member of advisory boards and speaker panels, and consultancy fees from Astellas (Chertsey, UK), AstraZeneca (Cambridge, UK), MSD and Schering-Plough (Hoddesdon, UK); he also received support to attend scientific conferences, including accommodation and travel payments from Becton Dickinson (Winnersh Triangle, UK), Carefusion UK (Winnersh Triangle, UK), Janssen-Cilag (High Wycombe, UK) and MSD. Paul R Chadwick reports non-financial support from Novartis (Frimley, UK), and grants and personal fees from NIHR outside the submitted work. Bernadette Young reports grants from the Wellcome Trust outside the submitted work. M Estee Török reports grants from Academy of Medical Sciences/The Health Foundation, grants from Medical Research Council (MRC), grants from NIHR Cambridge Biomedical Research Centre, grants from MRC/Department of Biotechnology Partnership Grant, and personal fees from Oxford University Press outside the submitted work. Martin J Llewelyn reports personal fees from Pfizer (Walton Oaks, UK) outside the submitted work and is a member of the panel that develops the European Society of Clinical Microbiology and Infectious Diseases/Infectious Diseases Society of America clinical practice guideline on Staphylococcus aureus bacteraemia.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Thwaites et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
This chapter includes material that has been adapted from the trial protocol published in Thwaites et al. 1 © Thwaites et al. ; licensee BioMed Central Ltd. 2012. This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution and reproduction in any medium, provided the original work is properly cited.
Background
Staphylococcus aureus bacteraemia is one of the most common and serious bacterial infections worldwide. There were over 12,000 cases of S. aureus bacteraemia in the UK in 2016/17, and around 25% of patients died. 2,3 Current treatment guidelines recommend that S. aureus bacteraemia should be treated with ≥ 14 days of an intravenous (i.v.) beta-lactam antibiotic, or a glycopeptide if the bacterium is meticillin resistant. Combination antimicrobial therapy is generally not recommended, except in severe meticillin-resistant Staphylococcus aureus (MRSA) infections (e.g. endocarditis) or in the presence of prosthetic joint infections. 4–7 Most of the recommendations are based on uncontrolled observational studies and clinical experience, and views of how to manage S. aureus bacteraemia differ widely. 8,9
How might adjunctive rifampicin improve outcomes from S. aureus bacteraemia?
Three properties make rifampicin an attractive, if unproven, antibiotic for S. aureus bacteraemia treatment. First, it has good oral bioavailability. 10 Second, it penetrates cells, tissues and biofilms better than beta-lactam and glycopeptide antibiotics (the current mainstays of S. aureus bacteraemia treatment) and, therefore, in combination with these agents, may resolve serious S. aureus infections faster and more effectively. 11 Third, it is cheap: a daily 600 mg dose costs £0.73 by mouth and £7.67 intravenously. 12
The best clinical predictor of complications and death from S. aureus bacteraemia is the persistence of bacteria in blood 48–96 hours after the start of active antimicrobial therapy. 13–15 Persistent bacteraemia (> 48 hours) occurs in around 40% of patients, despite prompt removal of any infected foci and effective antimicrobial therapy,13,14 and increases the patient’s risk of metastatic complications and death nearly fivefold. 13 Why S. aureus persists in blood despite treatment with antibiotics with good in vitro activity is uncertain, but is probably explained by the failure of currently recommended first-line antibiotics (beta-lactams and glycopeptides) to kill the bacteria associated with pus (dead or dying neutrophils), viable cells or biofilms. The well-documented survival of S. aureus within each of these ecological niches may lead to persistent bacterial seeding of the bloodstream and recurrent–recalcitrant infection. In addition, it has been proposed that bloodstream neutrophils may act as ‘Trojan horses’ for S. aureus dissemination, providing bacteria with further protection from first-line antibiotics with poor intracellular activity, such as the recommended beta-lactams and glycopeptides. 16
Rifampicin, clindamycin, the tetracyclines and the fluoroquinolones are all concentrated within cells, but, with the exception of rifampicin, their activity is reduced in the acidic environments found within intracellular phagolysosomes. 17,18 Rifampicin has been repeatedly shown to be highly effective against S. aureus within cells18,19 and against bacteria associated with biofilms and prostheses. 11,20 Beta-lactams and glycopeptides do not pass easily into eukaryotic cells or biofilms, and do not kill S. aureus associated with these niches as effectively as free, extracellular bacteria. 21,22 Data from animal models of severe S. aureus infections have generally shown rifampicin-containing antibiotic combinations to be superior with respect to reduced bacteria counts, sterilisation and cure rates, independent of the model used. 11 Yet, despite the breadth of these experimental findings, the potential advantages of adjunctive rifampicin for the treatment of severe S. aureus infections in humans remain theoretical. There are insufficient data from only 246 patients randomised between rifampicin- and non-rifampicin-containing regimens in controlled trials to confirm or refute a beneficial effect.
What are the potential problems of using adjunctive rifampicin for S. aureus bacteraemia?
There are three important potential problems with using rifampicin for the treatment of S. aureus bacteraemia: the development of rifampicin-resistant bacteria, interactions with other drugs and hepatic toxicity. Resistance can be acquired rapidly when rifampicin is used alone in treatment, resulting from mutations in the drug binding site [the β-subunit of the bacterial deoxyribonucleic acid (DNA)-dependent ribonucleic acid (RNA) polymerase]. Interactions with other drugs are mediated by the ability of rifampicin to increase its metabolism through the potent induction of the hepatic cytochrome P450 system. Lastly, rifampicin can cause hepatic toxicity, although the enormous worldwide experience of using rifampicin for the prevention and 6-month treatment of tuberculosis confirms that the drug is extremely well-tolerated and causes clinically significant hepatitis in < 1% of patients. 23
The frequency with which rifampicin resistance develops during the combination therapy of S. aureus bacteraemia and the factors associated with its development are difficult to assess from the published literature. New resistance was not reported in any of the 433 patients treated with adjunctive rifampicin in three non-randomised clinical studies of S. aureus bacteraemia and other serious S. aureus infections,24–26 giving an observed incidence of 0% with upper 97.5% confidence limit of 0.8%. However, other clinical series have reported the emergence of rifampicin resistance in 20–40% of patients after a median of 9–12 days of treatment (range 5–58 days). 27–29 One of these studies, a retrospective description of 42 rifampicin-treated patients with native valve S. aureus endocarditis, reported that those who developed resistance (21%) were more likely to have prolonged bacteraemia than a selected control group who were not given rifampicin, although the control group had a significantly less severe form of the disease at the start of treatment. 27 The investigators also reported that rifampicin had clinically important interactions with other drugs in 52% of patients, but a high proportion of patients were coinfected with human immunodeficiency virus (HIV) (18%) and/or hepatitis C (48%) and required methadone (which interacts with rifampicin) for opiate addiction (57%). This population were also at a high risk of having rifampicin-related hepatic toxicity, but hepatic dysfunction occurred in only nine patients; all patients were infected with hepatitis C and had abnormal liver function tests before starting rifampicin.
In summary, there are insufficient clinical data to determine the true incidence of rifampicin resistance, drug interactions and hepatic toxicity. Only one large, randomised controlled trial (RCT) will provide these data and allow the potential risks of adjunctive rifampicin to be properly balanced against the potential benefits.
Adjunctive rifampicin for S. aureus bacteraemia: current clinical evidence, guidelines and practice
Four RCTs, involving 246 patients in total, have examined the effectiveness of adjunctive rifampicin for serious S. aureus infections, including patients with bacteraemia. 30–33 The first two trials, published > 25 years ago, enrolled adults with any serious S. aureus infection, of whom 47 out of 121 (39%) were bacteraemic at randomisation. 30,31 The third trial enrolled 42 adults, all of whom had S. aureus bacteraemia and endocarditis,32 and the fourth enrolled 83 adults who were admitted to intensive care with MRSA pneumonia, but only 9 out of 83 (11%) were bacteraemic. 33 A stratified meta-analysis of the results from these trials was performed; subgroup analysis of bacteraemic adults was possible for all but the fourth trial, which did not provide sufficient data. Overall, adjunctive rifampicin reduced infection-related deaths by 55% (p = 0.02) and bacteriological failure by 58% (p = 0.004), with similar (54%, 77%) but non-significant (p = 0.22, p = 0.17) reductions in the bacteraemic subgroup (n = 89).
The daily dose of rifampicin in these studies varied from 600 mg to 1200 mg. Significant drug interactions were not reported in any of the studies, and details concerning hepatic toxicity were not provided in the first three trials. 30–32 The most recent trial reported that 6 out of 41 (15%) patients treated with rifampicin developed hyperbilirubinaemia (vs. one control patient) but the impact on treatment was not described. 33 This trial was also the only one to report rifampicin resistance developing on treatment: new resistance was found in 14 out of 41 (34%) rifampicin-treated patients, although it did not appear to have a significant impact on clinical cure rates. 33
There are limited data from uncontrolled, observational studies supporting the use of adjunctive rifampicin, although, given the potential for confounding by indication, their results must be interpreted cautiously. A prospective study of 381 adults with S. aureus bacteraemia found that the mortality of those with severe disease was halved in those who received adjunctive rifampicin (mortality 38% vs. 17%; p < 0.001), without an increased incidence of rifampicin resistance. 25 A retrospective analysis of patients with staphylococcal sternal wound infections, 35% of whom had S. aureus bacteraemia, reported that adjunctive rifampicin was independently associated with a reduced risk of treatment failure [hazard ratio (HR) 0.26, 95% confidence interval (CI) 0.10 to 0.64; p = 0.004]. 26 A recent observational study of 964 patients with S. aureus bacteraemia reported that 512 (53%) patients received combination therapy and the majority (301/512, 59%) received rifampicin. 34 Combination therapy was not associated with reduced mortality in all patients, but was associated with reduced deaths and infection-related complications in those suffering from device-related infections.
The current observational study found that 17% of NHS patients with S. aureus bacteraemia were treated with rifampicin, but with large variations in use across the six centres (range 1–75% of patients). 35 Rifampicin was used to treat 21% of MRSA bacteraemia and 15% of meticillin-susceptible bacteraemia, and was not reserved for severe, complex disease as the guidelines suggest;4–7 13% of uncomplicated i.v. catheter-related bacteraemia were treated with rifampicin. However, rifampicin was given more often to patients with MRSA bacteraemia resulting from foci other than i.v. catheters – although even in this indication only 24% received it. An unadjusted comparison of inpatient mortality showed that 23% of patients not treated with rifampicin died compared with 13% given rifampicin (p = 0.03). The impact on survival appeared to be more marked in those with a non-removable focus of infection (whose inpatient mortality was higher), although there was no statistical evidence supporting smaller relative effects of adjunctive rifampicin in those with removable foci (p = 0.39).
Rationale
The results of the meta-analysis, together with data from observational studies indicate, that adjunctive rifampicin may have a surprising and substantial impact on survival from S. aureus bacteraemia. They do not, however, constitute evidence of sufficient rigour to influence current treatment guidelines, clinical practice, or indeed the equipoise of clinicians recruiting patients into the proposed trial – even clinicians in centres using rifampicin in a greater proportion of patients have indicated their willingness to randomise, as they recognise the lack of evidence supporting their practice. In particular, although statistically significant, the results from the trial meta-analysis are not convincing, as they are based on a small number of patients in a small number of trials over a wide period of time. In addition, the potential negative impact of rifampicin toxicity, interactions and resistance cannot reliably be assessed in these studies. Current guidelines recommend adjunctive rifampicin only for the treatment of severe MRSA infections, specifically endocarditis, bone and joint infections, and infections involving prostheses (category II evidence). 5,7 But with weak support for these recommendations it is unsurprising that few physicians follow them in practice. The ARREST (Adjunctive Rifampicin to Reduce Early mortality from STaphylococcus aureus bacteraemia) trial was designed to provide a definitive answer to the role of adjuvant rifampicin therapy in the treatment of S. aureus.
Objectives
The hypothesis addressed by the ARREST trial is that adjunctive rifampicin will enhance the killing of S. aureus early in the course of antibiotic treatment, sterilise infected foci and blood faster, and thereby reduce the risk of dissemination, metastatic infection and death. Therefore, the primary objective of the trial was to investigate the impact of adjunctive rifampicin on bacteriologically confirmed failure/recurrence or death through 12 weeks from randomisation. Secondary objectives included evaluating the impact of rifampicin on all-cause mortality up to 14 days from randomisation, or clinically defined failure/recurrence or death, toxicity [serious and grade 3/4 adverse events (AEs), any modification of treatment as a result of drug interactions], emergence of resistance and duration of bacteraemia, and assessing the cost-effectiveness of adjunctive rifampicin for S. aureus bacteraemia in the NHS.
Substudies
There were three ancillary studies to the main trial. First, with assistance from the trial public and patient representative, Jennifer Bostock, the process of obtaining consent to enter the trial was examined. Patients/legal representatives who did not consent to participation in the trial were offered the opportunity to complete a questionnaire exploring reasons for this; participants/legal representatives at one trial centre who did consent were offered the opportunity to be interviewed by the ARREST trial patient and public representative to explore their experiences of trial participation.
Samples were collected for two further ancillary studies for which funding will be sought separately. Participants enrolled at Guy’s and St Thomas’ NHS Foundation Trust, Cambridge University Hospitals NHS Trust, Oxford University Hospitals NHS Trust, The Royal Liverpool and Broadgreen University Hospitals NHS Trust, and Brighton and Sussex University Hospitals NHS Trust were approached for additional consent for a pharmacokinetic/pharmacodynamic (PK/PD) substudy – a population PK/PD study of rifampicin, flucloxacillin and vancomycin for the treatment of S. aureus. The aim of the substudy is to determine the pharmacological parameters of rifampicin that best predict treatment success and provide a rational basis from which optimal dose, frequency and route of administration can be modelled statistically and/or explored in future studies.
All participants were also approached for additional consent for the host DNA/RNA substudy to investigate the influence of host and bacterial genetics on disease severity and outcome from S. aureus. The aim is to identify host and bacterial genetic factors that influence disease severity (e.g. the development of metastatic complications) and poor outcomes from S. aureus bacteraemia.
The samples for the PK/PD and DNA/RNA substudies have been archived at the King’s College London Biobank until funding has been secured.
Chapter 2 Methods
This chapter includes material that has been adapted from the trial protocol published in Thwaites et al. 1 © Thwaites et al. ; licensee BioMed Central Ltd. 2012. This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution and reproduction in any medium, provided the original work is properly cited.
Trial setting
Patients were recruited from 29 large UK NHS Hospital Trusts:
-
Guy’s and St Thomas’ NHS Foundation Trust
-
Oxford University Hospitals NHS Trust
-
University College London (UCL) Hospitals NHS Foundation Trust
-
Royal Free London NHS Foundation Trust
-
King’s College Hospital NHS Foundation Trust
-
Brighton and Sussex University Hospitals NHS Trust
-
The Royal Liverpool and Broadgreen University Hospitals NHS Trust
-
Sheffield Teaching Hospitals NHS Foundation Trust
-
Cambridge University Hospitals NHS Foundation Trust
-
Royal United Hospital Bath NHS Trust
-
Royal Devon and Exeter NHS Foundation Trust
-
Plymouth Hospitals NHS Trust
-
Hull and East Yorkshire Hospitals NHS Trust
-
South Tees Hospitals NHS Foundation Trust
-
Heart of England NHS Foundation Trust
-
St George’s Healthcare NHS Trust
-
Portsmouth Hospitals NHS Trust
-
University Hospital Southampton NHS Foundation Trust
-
Blackpool Teaching Hospitals NHS Foundation Trust
-
The Leeds Teaching Hospital NHS Trust
-
Aintree University Hospital NHS Foundation Trust
-
Bradford Teaching Hospitals NHS Foundation Trust
-
County Durham and Darlington NHS Foundation Trust
-
Dartford and Gravesham NHS Trust
-
North Cumbria University Hospitals
-
University Hospitals of Leicester NHS Trust
-
Wirral University Teaching Hospital NHS Foundation Trust
-
The Newcastle Upon Tyne Hospitals NHS Foundation Trust
-
Salford Royal NHS Foundation Trust.
The main criteria for selecting participating hospitals were that they had an existing S. aureus bacteraemia ward consultation service and sufficient numbers of S. aureus bacteraemias to be able to recruit patients (potential to recruit a minimum of one patient per month), as well as the necessary research infrastructure to conduct the trial.
The overall trial design is summarised in Figure 1.
FIGURE 1.
Trial schema. a, Incapacitated adults would be eligible, provided that they had an appropriate legal representative.
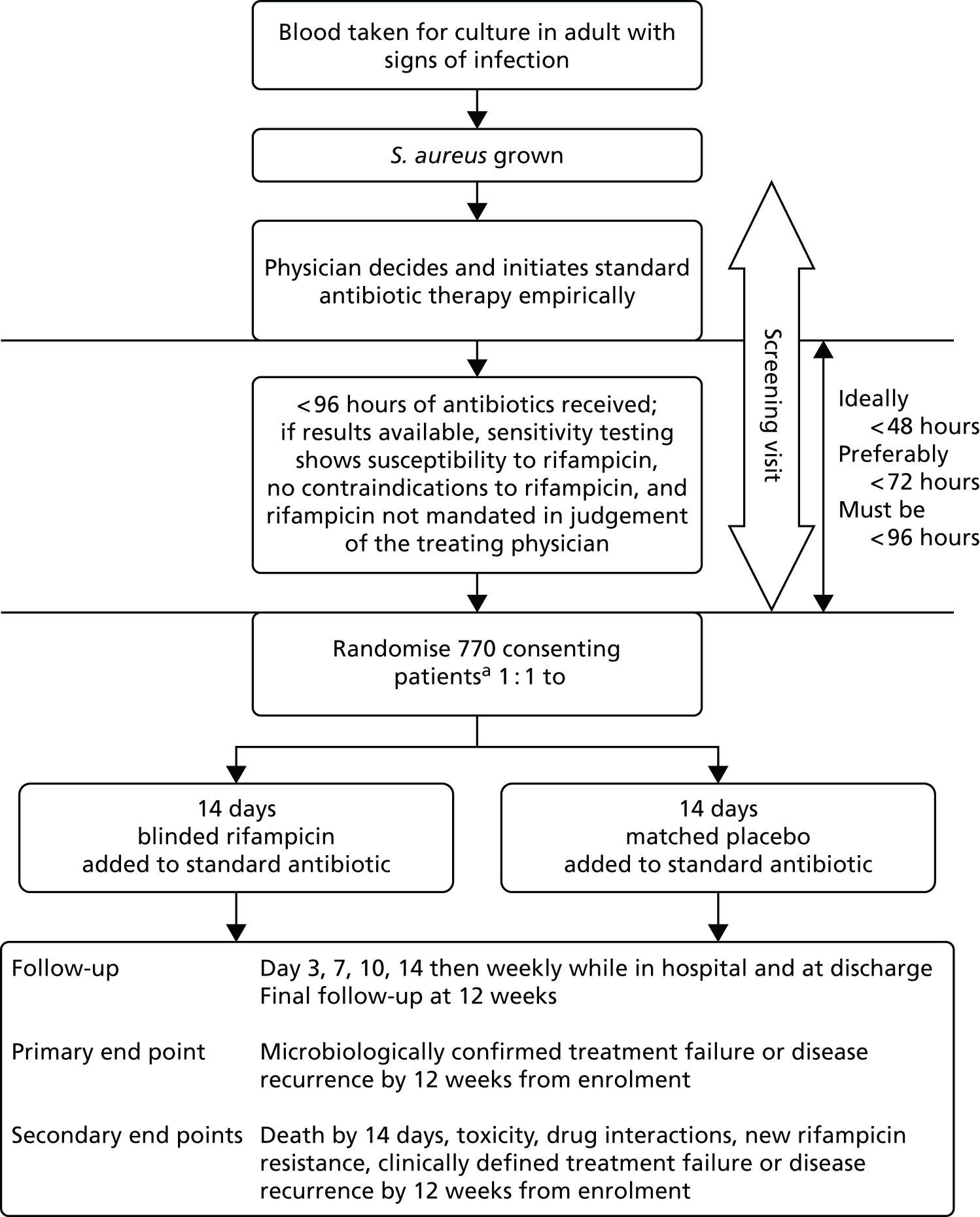
Patient selection
As S. aureus bacteraemia is a serious infection for which the standard treatment requires i.v. antibiotics, all eligible patients were hospital inpatients at the time of recruitment. Patients were identified via the clinical microbiology laboratory and the infectious diseases/microbiology consult service at each centre. When possible, patients were screened for eligibility on the day that their blood cultures were flagged positive with S. aureus. Written informed consent was obtained from patients. Incapacitated adults were eligible provided that they had an appropriate legal representative to provide consent. The principal investigator (PI) or another experienced independent physician was required to follow the Mental Capacity Act 200536 to formally assess the capacity of the individual to make an informed decision to participate in the trial. If incapacity was confirmed, then written informed consent was sought from either a personal (e.g. a relative) or a nominated (e.g. consultant intensivist caring for the patient, but not involved in the trial) legal representative.
Inclusion criteria
The trial enrolled adults aged ≥ 18 years who had S. aureus (meticillin susceptible or resistant) grown from at least one blood culture, who had received < 96 hours of active antibiotic therapy for the current infection (not including rifampicin, and excluding any stat doses), and the patient or legal representative had provided written informed consent for participation in the trial.
Although the formal inclusion criteria stated that patients must have received < 96 hours of active antibiotic therapy for the current infection, the best clinical predictor of complications and death from S. aureus bacteraemia is the persistence of bacteria in blood 48–96 hours after the start of active antimicrobial therapy. 13–15 Therefore, patients were included in the trial as soon after initiation of active antibiotic therapy as possible, within 48 hours whenever possible and ideally within 72 hours.
Exclusion criteria
Patients were excluded from the trial if they had an infection not caused by S. aureus alone in the opinion of the infection specialist (e.g. S. aureus was considered a blood culture contaminant, or polymicrobial culture with another organism was likely to be contributing clinically to the current infection); if sensitivity results were already available and demonstrated rifampicin-resistant S. aureus [defined by British Society for Antimicrobial Chemotherapy in vitro disc susceptibility testing or by Vitek® (bioMérieux, Marcy l’Etoile, France) testing]; if the infection specialist, in consultation with the treating physician, considered rifampicin to be contraindicated for any reason; if the infection specialist, in consultation with the treating physician, considered rifampicin treatment to be mandatory for any reason; if the infection specialist suspected active infection with Mycobacterium tuberculosis; or if the patient had been previously been randomised in the ARREST trial for a prior episode of S. aureus bacteraemia.
As the underlying hypothesis was that rifampicin may improve outcomes by increasing the rate of early bacterial killing, results of in vitro sensitivity testing were not required before randomisation, as it was important to initiate rifampicin as soon as S. aureus was identified. This also ensures that results are generalisable to empiric treatment of S. aureus bacteraemia in the future. However, if for any reason in vitro susceptibility results were already available at the point when randomisation would be considered, and demonstrated rifampicin resistance, then the patient was not eligible.
Randomisation
Eligibility was confirmed by the ARREST trial site investigators (PI, co-PI or research nurse) via the online ARREST trial database, and patients were randomised into two parallel groups in a 1 : 1 ratio, to standard i.v. antibiotic therapy plus 14 days of placebo, or standard i.v. antibiotic therapy plus 14 days of rifampicin. The choice and duration of the standard antibiotic therapy was left to the attending physician. Randomisation was stratified by clinical centre, as blinded drug (in fully made-up and labelled treatment packs) was pre-shipped to local pharmacies. A computer-generated sequential randomisation list using variably sized permuted blocks was prepared by the trial statistician and incorporated securely into the online trial database. The list was concealed until allocation, after eligibility was confirmed by researchers at the local hospitals, who then performed the randomisation. A 24-hour web-based randomisation service was provided via the online ARREST trial database.
Trial intervention
Rifampicin/placebo was given by oral or i.v. route, depending on the attending physician’s preference and the patient’s status. Providing that a patient could swallow safely, the preference was to use rifampicin orally. Intravenous administration was permitted for patients who were not able to swallow or absorb tablets. Rifampicin is a well-established, widely used drug, and was not used outside its licensed indication during the course of the trial.
The oral investigational medicinal product (IMP) was prepared by a Clinical Trials Supplier (Sharp Clinical Services, Crickhowell, UK). It was supplied as 300 mg of rifampicin capsules (Sanofi-Aventis, Guildford, UK) [summary of product characteristics (SPC); www.medicines.org.uk/emc/medicine/21223/SPC/Rifadin±300mg±Capsules/ (accessed 4 June 2018)] or placebo oral 300-mg capsules containing cellulose. The rifampicin capsules were over-encapsulated so that they were identical in appearance to the placebo capsules. The capsules were supplied to trial centres as individual participant-blinded treatment packs so that they were dosed and dispensed in the same way.
The i.v. IMP was provided via standard hospital stock and consisted of either rifampicin for i.v. infusion [600 mg of rifampicin for i.v. injection (Sanofi-Aventis) SPC www.medicines.org.uk/emc/medicine/6435 (accessed 4 June 2018)] or standard saline as the placebo. Participants receiving i.v. infusions in the intensive care unit (ICU) could have their infusion volume altered in accordance with standard local practice and the SPC. The trial pharmacist at each hospital had access to a copy of the randomised allocations for each ARREST trial number for their centre in order to prescribe i.v. rifampicin if required.
Dose
The dose of rifampicin/placebo was prescribed on the basis of the patient’s weight:
-
those < 60 kg received 600 mg every 24 hours
-
those ≥ 60 kg received 900 mg every 24 hours.
Oral doses could be given once or twice daily depending on clinician and patient preference, and subgroup analysis was prespecified according to initial oral dosing frequency (elicited at randomisation). If taken twice daily, 900 mg daily (three capsules) was taken as unequal divided doses (600 mg in the morning and 300 mg in the afternoon). As rifampicin can also be taken once daily, this provided adequate exposure.
Where i.v. was prescribed, it was administered to the patient over 2–3 hours.
Blinding and masking
Rifampicin for i.v. infusion is supplied as a vial of red powder that requires reconstitution with 10 ml of water for infusion with saline. The resulting fluid for i.v. infusion is orange. It was impossible to safely and reliably produce a red-powder placebo that produced an identical orange infusion. Therefore, the ward nurse making up the i.v. drug for the infusion was not blind to the treatment, nor was the hospital pharmacist dispensing either rifampicin or saline for i.v. administration. The ward nurses were instructed not to divulge the colour of the drug to the physicians caring for the patient. In addition, the infusion was covered by an opaque bag to disguise the treatment. As far as possible, the trial physicians, research nurses and other physicians caring for the patient remained blinded, as were all trial and data management staff except for statisticians.
Rifampicin can turn urine (and tears/sweat) reddish-orange. It is impossible to safely replicate this effect with a placebo; therefore, urine discolouration was a potential source of unblinding, particularly for the participant. There is, however, considerable inter- and intra-individual variability in the effect of rifampicin on urine colour. In addition, the opportunity for physicians to examine the urine at the bedside occurred only in participants with urinary catheters. Catheters were not required by all participants and were removed at the earliest opportunity. The opportunity for physicians to inspect urine was also limited by ensuring that the catheter bags were emptied regularly and urine was not allowed to accumulate in large volumes. The success of blinding was assessed at the final 12-week visit, when physicians and participants were asked which treatment they believed they had received.
Dose modifications, interruptions and discontinuations
Toxicity was managed in both randomised groups according to standard clinical practice. In some situations, changes in the patient’s condition meant that the dose of rifampicin needed to be reduced or stopped altogether. Whenever possible, this was done without unblinding. Unblinding was performed only when knowledge of the allocated treatment had a direct bearing on clinical management. Patients were not put at any additional risk by trial randomisation, as any patient who developed a suspected adverse reaction to a study drug was managed as if they were receiving rifampicin, and the study drug was discontinued.
The most important rifampicin toxicity is liver impairment, although serious hepatic toxicity is rare (< 1% of patients). The study drug (rifampicin/placebo) was withdrawn without unblinding if significant liver toxicity was observed [blood aspartate aminotransferase (AST)/alanine transaminase (ALT) of > 5× upper limit of normal (ULN)] without other probable causes, and was withdrawn for grade 4 liver toxicity (blood AST/ALT of > 10× ULN) regardless of probable cause. The dose of the study drug was reduced if less severe liver dysfunction occurred according to the judgement of the treating physician. Other medications (including other antibiotics) were continued at the discretion of the treating physician. Rifampicin-related hepatic toxicity requires no specific treatment other than its withdrawal and, therefore, knowledge of whether the patient was receiving rifampicin or placebo was not mandated for patient management.
Rifampicin has a number of other uncommon side effects, which include anorexia, nausea, vomiting and diarrhoea, headache and drowsiness, haemolytic anaemia, thrombocytopenic purpura, disseminated intravascular coagulation and leucopenia, flushing, urticaria and rashes, and a flu-like syndrome with fever (although this is usually associated with administration twice or three times per week).
Rifampicin/placebo was discontinued before 14 days in two specific situations:
-
When other antibiotics being used to treat S. aureus bacteraemia were stopped before 14 days after randomisation. This was to prevent rifampicin being given as monotherapy, which could theoretically increase the risk of resistance.
-
When results from S. aureus susceptibility testing became available after the patient had been randomised and initiated on rifampicin/placebo and indicated resistance to rifampicin. This was to prevent any toxicity from an additional but ineffective drug being used. Primary rifampicin resistance was expected in < 1% enrolled patients based on observational study data. 35
Other antibiotics
Infection specialist consultation, with advice on management to non-specialists caring for the trial participants, followed normal clinical practice in all sites. Attending physicians could change ‘backbone’ antibiotics on the basis of clinical need and infection specialist advice and use open-label rifampicin after 14 days. When judged clinically necessary, the attending physicians could stop the blinded trial drug before 14 days to use open-label rifampicin, with participants continuing follow-up ‘off study drug, on study’.
Assessments and follow-up
Trial assessment schedule
All participants were followed by the centre trial teams for 12 weeks for evaluation of all-cause mortality, morbidity and toxicity. To assess the outcome measures, patients were visited on the ward by the centre PI, one of their clinical team (e.g. a specialist registrar) or a research nurse. The schedule for timing, frequency and method of collection of all study data is summarised subsequently. Assessments were performed as close as possible to the required time point.
Screening and randomisation visits
Patients were identified through the clinical microbiology laboratory and the infectious diseases/microbiology consult service of each centre. All the trial centres ran a clinical consult service for all cases of S. aureus bacteraemia and identified such patients as soon as their blood cultures become positive. The screening visit took place as soon as possible after a potential patient had been identified by the microbiology laboratory. The trial’s central hypothesis was that early intervention with rifampicin enhances bacterial killing and improves clinical outcome. Therefore, it was essential that patients were randomised as early as possible in their treatment and by the limit defined by the inclusion criteria of < 96 hours of active antibiotic therapy for the current infection. For this reason, patient consent to recruitment was requested within 2 hours of the screening assessment whenever possible, and ideally within 4 hours.
Written informed consent to enter into the trial and be randomised was obtained from patients or a person with responsibility (a legal representative, such as a legal authority).
After consent was obtained from the patient or their legal representative, clinical information including medical history and examination as well as weight were recorded. C-reactive protein (CRP) and liver function tests are routine investigations for patients with suspected S. aureus bacteraemia and results were also recorded.
Randomisation took place as soon as possible after eligibility was confirmed and consent was signed.
Follow-up
At each main clinical assessment (days 0, 3, 7, 10, 14, weekly until discharge, week 12 final visit), the following was undertaken:
-
Assessment of new or ongoing foci of infection together with arrangements to identify, remove or drain the foci if necessary.
-
Assessment of clinical treatment response, including whether or not the patient was febrile (> 37.5 °C) in the previous 24 hours.
-
All grade 3 or 4 AEs, all serious adverse events (SAEs) and all AEs of any grade leading to modification of rifampicin/placebo dose or its interruption/early discontinuation were recorded. With the exception of events leading to modification/interruption/discontinuation of the study drug, the severity and likely relationship of these AEs to rifampicin/placebo was documented by a physician. Any drug interactions leading to dose modification of any drug (including concomitant medications) were also recorded.
-
Assessment of adherence to rifampicin/placebo (missed pills).
-
Assessment of resource utilisation (medications, procedures, laboratory tests and other relevant resource use categories).
Blood cultures were repeated on days 0, 3 and 7 to assess duration of bacteraemia in all patients as persistent bacteraemia is strongly predictive of worse outcomes. Blood cultures could be taken at any other time points necessary for clinical management, but were additionally taken if potential treatment failure was suspected (e.g. in patients who still had a positive blood culture on day 7 and in whom transoesophageal echocardiography was being considered) or when S. aureus bacteraemia recurrence was suspected. CRP levels were measured on days 0, 3, 10 and 14 to assess treatment response. Levels of ALT, bilirubin and alkaline phosphatase were assessed on days 3 and 10 to evaluate liver toxicity. Full blood count was measured at baseline in all patients as total white blood cell count/total neutrophils may be important baseline prognostic determinants. The ethylenediaminetetraacetic acid plasma (2.5 ml of blood) and PAXgene blood RNA tube (2.5 ml of blood) were taken from patients on day 0 and stored for later DNA/RNA extraction when consent had been provided for this. If a patient had already been discharged from hospital before day 7, 10, or 14, these additional investigations requiring a blood draw (culture, CRP, ALT, alkaline phosphatase, bilirubin, serum storage) were not required, so patients were not asked to attend ARREST trial-specific outpatient appointments on these days, but returned at 12 weeks only.
The EuroQol-5 Dimensions (EQ-5D) for quality-of-life assessment was administered on days 0, 7, 14 and at the final visit.
Those patients discharged before 12 weeks were managed and followed up through each centre’s infectious diseases outpatient clinic. Final follow-up at 12 weeks was either by a ward visit (if the patient was still admitted to hospital) or by a clinic visit with interview and clinical assessment. In the event that the patient was unable to attend clinic, the follow-up visit could take place over the telephone. If failure or S. aureus bacteraemia recurrence was suspected then repeat blood cultures were performed together with a clinical assessment and EQ-5D.
The trial end was defined as the final 12-week visit of the last patient to be randomised. At the end of the trial, the vital status of all participants was ascertained from electronic NHS records, and consent was sought for this.
Procedures for assessing efficacy
The primary outcome was:
-
time to death or bacteriologically confirmed failure/disease recurrence up to 12 weeks from randomisation.
This outcome measure was assessed by visiting the patient on days 3, 7, 10 and 14, and weekly thereafter until discharge from hospital, and the final clinical assessment 12 weeks after recruitment [either by a ward visit (if the patient was still admitted to hospital) or by a clinic visit or telephone call]. Consent to contact the patient’s general practitioner (GP) was also obtained.
The definition of bacteriologically confirmed failure was:
-
symptoms and signs of infection ongoing for > 14 days from randomisation
-
the isolation of same strain of S. aureus (confirmed by genotyping) from either blood or another sterile site (e.g. joint fluid, pus from tissue) indicating blood-borne dissemination of the bacteria.
The definition of bacteriologically confirmed disease recurrence was:
-
the isolation of the same strain of S. aureus from a sterile site after > 7 days of apparent clinical improvement.
As defined, failure reflected both the speed of killing of S. aureus and sterilisation of infected foci/blood, and both failure and recurrence reflected the risk of dissemination and metastatic infection. Outcome measures included S. aureus infection of sterile sites other than just blood, because such disseminated infection can be the consequence of failure to treat initial infections adequately. Asymptomatic bacteraemia without any sign or symptom of infection was not considered failure. Additional blood cultures were requested as soon as the PI/study physician suspected failure or recurrence. All bacterial isolates (initial and all subsequent) from patients randomised in the trial were originally intended to be genotyped by multilocus sequence and spa-typing and tested for susceptibility to rifampicin.
A substantial proportion of bacteriological failure/recurrences did not have both baseline and failure/recurrence isolates stored [17 (61%) of 28 failures/recurrences where S. aureus was isolated from a sterile site]. In order to avoid excluding a substantial proportion of potential primary end points, the statistical analysis plan specified that the primary analysis would include all bacteriologically confirmed failures and recurrences (i.e. without restricting to the same strain).
In the 11 pairs of baseline and failure/recurrence isolates that were stored, same strain was defined by whole-genome sequencing using Illumina technology (San Diego, CA, USA) on the basis of 40 single nucleotide variants between baseline and failure/recurrence isolates. All failure/recurrence isolates were within 12 single nucleotide variants of the baseline isolate {median 1 [interquartile range (IQR) 1–6; range 0–12] single nucleotide variant}.
The secondary efficacy outcome measures were:
-
time to all-cause mortality up to 14 days
-
time to clinically defined failure/recurrence/death by 12 weeks
-
duration of bacteraemia
-
AEs (grade 3/4 AEs, SAEs, AEs of any grade leading to modification of rifampicin/placebo dose or interruption/early discontinuation) (all AEs reported, primary comparisons based on time to first event)
-
the proportion modifying any treatment (including concomitant medications) as a result of drug interactions
-
the proportion developing rifampicin-resistant S. aureus
-
cost-effectiveness of rifampicin.
Mortality was reported on the ARREST trial database on a SAE electronic case report form (eCRF). Clinically defined failure/recurrence was assessed clinically in the same manner as bacteriologically confirmed failure or recurrence; however, microbiological confirmation was not required (e.g. patients who failed clinically but for whom blood cultures were not taken). Clinically defined failure/recurrence was primarily determined by radiological evidence for an ongoing or new active infection focus by 12 weeks and the requirement for ongoing or new antibiotic therapy.
The PIs were required to report all potential failures/recurrences and these were adjudicated as trial end points by an independent end-point committee. The blinded independent review committee consisted of two infectious disease physicians with experience in acute/general medicine (Professor Tim Peto, Oxford University Hospitals NHS Foundation Trust, Oxford, UK; Dr Graham Cooke, Imperial College Healthcare NHS Trust, London, UK; see Acknowledgements). Potential failures/recurrences were also identified through questions regarding signs and symptoms of ongoing or new S. aureus infections on routine case report forms, and S. aureus isolated from any microbiological specimen. For all such potential failures/recurrences a structured clinical narrative was completed by the site physician and approved by the site PI. All reported failures, recurrences and deaths were then adjudicated using standardised proformas by the committee without knowledge of randomised allocation.
Blood cultures were taken on days 3 and 7 following randomisation to assess duration of bacteraemia. Sensitivity to rifampicin was repeated on the day 3 and 7 blood cultures and in all subsequent S. aureus isolates grown at scheduled time points or at failure/recurrence in order to assess the secondary end point: development of rifampicin-resistant S. aureus.
The levels of CRP were measured longitudinally as a continuous measure of response to infection.
Procedures for assessing safety
Hepatitis is the most important side effect of rifampicin. Liver function tests were performed twice while on rifampicin/placebo (days 3 and 10) to assess laboratory safety parameters. Additional safety blood tests or investigations were performed to investigate symptoms or monitor emergent laboratory test abnormalities as clinically indicated.
Grade 3 and 4 AEs and SAEs were elicited at the regular clinical assessments, through consultation with the patient, their medical team or their medical records. All such AEs were reported on eCRFs, together with AEs of any grade leading to modification of rifampicin/placebo dose or its interruption/early discontinuation. All AEs (clinical and laboratory) were graded using the Common Toxicity Criteria grading scale v3.0. The SAEs were defined following the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use37 as events that led to death, were life-threatening, caused or prolonged hospitalisation (excluding elective procedures), caused permanent disability, or were other important medical conditions or with a real, not hypothetical risk of one of the previous categories. The SAEs were reported to the Medical Research Council (MRC) Clinical Trial Unit (CTU) at UCL according to standard timelines. All SAEs were reported on study eCRFs, unless they were specifically related to the S. aureus bacteraemia episode for which the patient was originally admitted (in which case they were reported as infection-related events). The protocol specifically exempted events related to S. aureus bacteraemia from AE reporting, unless the event was fatal, to avoid double-counting. The severity and likely relationship of any AEs to rifampicin/placebo were documented by a physician. All reported AEs were coded centrally at the MRC CTU at UCL using the Medical Dictionary for Regulatory Activities (MedDRA).
All modifications to rifampicin/placebo dose or administration were recorded, as were all significant drug interactions requiring modification of study and non-study medication.
Procedures for assessing health-related costs of S. aureus and quality of life
Health-care-related costs of S. aureus bacteraemia in the NHS and the evaluation of health-related quality of life (HRQoL) were evaluated using the EQ-5D. These assessments were used further to inform the cost-effectiveness of adjunctive rifampicin and relevant antibiotic regimens for S. aureus bacteraemia (see Chapter 5). Information on health-care-related costs of patients in the trial was collected, starting from when the first positive blood culture was taken and continuing for the duration of follow-up. Information on hospitalisation costs (including procedures, laboratory tests and concomitant medications) was collected at the regular clinical assessments, and data on other health-care resource utilisation (post-discharge outpatient visits, medications and procedures) were collected at the 12-week visit.
Within-trial assessments of HRQoL (using the EQ-5D) were also used in the economic analysis. The EQ-5D scores were used to weight lifetime lived by its quality; the EQ-5D tariff developed for the UK was used to derive the scores from the participants’ responses to the EQ-5D descriptive system. The cost-effectiveness analysis thus used quality-adjusted life-years (QALYs) as the outcome measure.
Sample size
The trial was originally designed with two coprimary outcomes: all-cause mortality by 14 days and bacteriological failure/recurrence or death by 12 weeks. Assuming 80% power, a two-sided alpha of 0.025 (to adjust for multiple testing given two coprimary outcomes) and a 10% loss to follow-up by 12 weeks, 920 participants were needed to detect a 30% relative reduction in bacteriological failure/death from 35% to 25%, an absolute difference of 10% corresponding to an number needed to treat (NNT) of 10 participants. Assuming 80% power, a two-sided alpha of 0.025 and a lower 4% loss to follow-up by 14 days (as most participants remained in hospital over this time scale), 940 participants were needed to detect a 45% relative reduction in mortality from 16% to 9%, an absolute 7% difference and a NNT of 14 participants. The total sample size was originally therefore 940 participants.
Recruitment to the trial was slower than anticipated. To facilitate successful completion of the trial and at the request of the trial funder, after 3 years of recruitment 14-day mortality was moved from a coprimary to a secondary outcome. Therefore, 12-week bacteriological failure/recurrence or death became the sole primary outcome with consequent decrease in sample size [as a result of the increase in the two-sided alpha (type I error) from 0.025 (two coprimary outcomes) to 0.05 (one primary outcome)]. With 12-week bacteriological failure/recurrence or death as the sole primary outcome, the total sample size became 770 participants (alpha = 0.05, other assumptions as above).
The protocol and statistical analysis plan specified that the primary outcome (bacteriologically confirmed failure/recurrence or death) would be analysed using time-to-event methods as described in Statistical methods. The sample size calculation treated this outcome as binary, in order to produce a conservative estimate of sample size given uncertainties in the underlying assumptions, and because all patients were to be followed for a fixed 12-week period (i.e. no additional power was gained from longer follow-up in some patients).
Statistical methods
Randomised groups were compared following the principle of intention to treat, including all follow-up regardless of changes to treatment. The statistical analysis plan prespecified that any patient who was randomised in error (defined as realising that the patient should not have been randomised before taking blinded study drug and not ever taking study drug and, hence, not followed up) would be excluded. The blinding means that there was no possibility that knowledge of randomised allocation affected this judgement about what was an error. Any participants who were randomised in good faith (i.e. not by mistake) but never took study drug were included in all analyses.
The time-to-event analyses measured time from randomisation. Analyses of clinical outcomes censored at the earliest of 12 weeks from randomisation and the last clinical information. Analyses of mortality censored at the earliest of the time scale being considered (2 weeks, 12 weeks) or last vital status information (including that ascertained at trial closure through the NHS records).
The primary analyses were unstratified because the randomisation stratification factor (centre) was expected to have some small strata and participants in these strata might then not contribute to comparisons. Results from secondary stratified analyses (stratified log-rank test and stratified Cox regression) were very similar (data not shown). Lost to follow-up was defined as not having been assessed in person or by telephone at the 12-week final visit (within a window of 1 week before to 8 weeks after week 12) by a trial clinician and not having information on whether or not signs/symptoms of S. aureus were present (e.g. from the patient’s GP).
Primary analysis of the primary end point included all randomised participants other than those considered randomised in error (following the statistical analysis plan): secondary analysis of the primary end point was to exclude those (expected < 1%) who were subsequently identified as having had a rifampicin-resistant S. aureus bacteraemia on susceptibility testing. As no patients were identified after randomisation as having had a rifampicin-resistant S. aureus bacteraemia at enrolment, this analysis was identical to the primary analysis. In the statistical analysis plan (but not the protocol), a per-protocol analysis was also specified for the primary end point, including all participants in the primary intention-to-treat analysis who received active/placebo for ≥ 80% of days from the start of trial drug to earliest of 14 days subsequently/death/discontinuation of active antibiotics (not including trial drug).
Safety analyses included all data between randomisation and 12 weeks post randomisation (inclusive). Non-fatal events related to S. aureus bacteraemia were not considered AEs/SAEs in the protocol.
When composite outcomes did not include all-cause mortality as part of the composite, competing risks analysis methods were used. Analogous to a Kaplan–Meier estimate, competing risk methods use cumulative incidence functions to estimate the probability of the event. The effect of randomised group on the subdistribution hazard that corresponds to this cumulative incidence function was estimated. Stratification is not possible with the estimating equation approach used to estimate these subdistribution hazards and so these analyses were conducted unstratified.
The CRP and liver function test results were compared between randomised groups over time using generalised estimating equations (normal distribution, independent correlation structure) with randomised group, adjusting for the stratification factor, baseline values and scheduled visit week as categorical independent variables and interaction between baseline values and scheduled visit week. The closest measurement to each scheduled visit date within equally spaced windows was used as the measurement at each scheduled visit. The midpoint between two scheduled assessment days was taken as belonging to the latter window (e.g. a measurement taken on day 12, midpoint between day 10 and day 14, would be considered as day 14 in the analysis). When there were two values within one of these equally spaced windows, but both equidistant from the nominal assessment day, the later value was used. Analyses were based on observed data. To account for CRP values above the limit of quantification in one centre (i.e. CRP levels only reported as > 156 mg/l if above this threshold), mean CRP level was estimated using normal interval regression. For analyses of change from baseline, these values were assumed equal to the limit of quantification.
For blood cultures, baseline (used to define baseline resistance/susceptibility) was defined as the closest up to and including day 0, and up to 1 day post randomisation providing this was on or before date of start of the trial drug. Cultures prior to randomisation were used in preference to cultures the same number of days after randomisation, but on or before the date of start of trial drug. As eligibility was based on the screening of positive blood cultures, and because the intention was to characterise persisting bacteraemia, baseline bacteraemia included cultures on day 1 when a culture on the day of or on the day prior to randomisation was not available. For duration of bacteraemia, baseline was defined as the closest up to and including day 0 within the preceding day, and up to 1 day post randomisation.
For laboratory measurements (e.g. CRP levels), baseline was defined as the closest up to and including day 0 within the preceding 4 days, and up to 1 day post randomisation providing this was on or before date of start of trial drug. Measurements prior to randomisation were used in preference to measurements the same number of days after randomisation, but on or before the date of start of trial drug.
A deep infection focus was defined as infection of implanted vascular device, native/prosthetic heart value, native/prosthetic bone/joint, or deep tissue infection/abscess (including vertebral bone/disc or other bone infection, epidural or intraspinal empyema, infected intravascular thrombus, brain infection).
Information on all antibiotics received through 12 weeks was collected, but not according to specific indication. Primary antibiotic treatment, and its duration, was therefore defined by complete cessation of all antibiotics for 2 days, with the exception of vancomycin when intermittent dosing up to 1 week was allowed. The cessation of vancomycin was defined by adding the number of days between the last two doses to the date of the final dose.
Subgroup analyses
Subgroup analyses were conducted to assess consistency of effects across different participant characteristics. The primary method of assessing subgroup effects was an interaction test within a Cox proportional hazards regression. For the continuous factors, both categorisation and natural cubic splines were used [five knots at the 10th, 25th, 50th, 75th and 90th centiles; four knots at the 10th, 33rd, 67th and 90th centiles for Charlson Comorbidity Index score (as 10th and 25th centiles identical)] to test for interactions. Subgroup analyses were conducted unstratified to avoid losing information from small strata with no events in one randomised group. No formal adjustment for multiple testing was made for subgroup analyses.
Twelve subgroup analyses were prespecified in the protocol for the primary end point, namely time from initiation of antibiotics to initiation of randomised treatment, time from randomisation to initiation of randomised treatment, initial oral randomised treatment frequency (once vs. twice daily), initial treatment with oral trial drug only or regimen containing i.v. trial drug, class of primary antibiotic treatment, other antibiotic adjuncts (e.g. gentamicin), MRSA/meticillin-sensitive Staphylococcus aureus (MSSA), i.v. catheter-associated infection/other, deep focus/no deep focus, endocarditis/no endocarditis, age, and CRP levels (terciles).
The statistical analysis plan included six additional subgroup analyses, but prioritised the subgroup analyses into three priority groups as follows (with all analyses in each group being given equal priority) (* = in protocol).
1. *Time from initiation of first active antibiotic treatment to initiation of randomised treatment (0–24, > 24–48, > 48–72, > 72 hours).
1. *Class of initial antibiotic treatment, and according to individual drugs when these are used by > 10% of the trial population.
1. *MRSA/MSSA.
1. *i.v. catheter (central/peripheral venous line)/implanted vascular device-associated infection versus other (based on portal of entry).
1. *Deep focus (implanted vascular device, native/prosthetic heart valve, native/prosthetic joint, deep tissue infection/abscess)/no deep focus (based on foci of infection).
1. *Endocarditis (main focus/foci of infection at time first positive blood culture taken = native heart valve/prosthetic heart valve)/no endocarditis.
1. *Foci of infection known/not known.
1. *Age (terciles).
2. *Initial oral randomised treatment frequency (once vs. twice daily).
2. *Initial treatment with oral trial drug only or regimen containing i.v. trial drug.
2. *Whether or not gentamicin was administered between first positive blood culture and 48 hours post randomisation, regardless of activity.
2. Whether or not any active antibiotic other than that first administered (excluding trial drug), trial drug and gentamicin was administered between first positive blood culture and 48 hours post randomisation (yes vs. no).
2. *Baseline CRP (terciles).
2. Charlson Comorbidity Index score (0, 1–2, 3–4, ≥ 5).
3. Time from randomisation to initiation of randomised treatment (0–4, > 4–12, > 12–24, > 24 hours).
3. Community, health-care associated and nosocomial acquisition.
3. Calendar year of randomisation.
3. Baseline neutrophils (terciles).
Additional exploratory subgroups defined by initial total daily dose (600 vs. 900 mg) were also considered, as well as whether or not the patient was bacteraemic at randomisation, leading to 20 subgroups in total.
Data collection and handling
Data were entered by staff at each NHS trust hospital on to eCRFs on the online ARREST trial database. Staff with data entry responsibilities were required to complete database training before they were granted access to the database. Data were exported into Stata® (v15.1) (StataCorp LLC, College Station, TX, USA) for analysis.
Interim analyses
The trial was reviewed by the ARREST trial’s Data Monitoring Committee (DMC). It met four times in strict confidence over the course of the trial: 14 November 2013, 31 October 2014, 26 May 2015 and 24 February 2016. DMC recommendations were communicated through a letter to the Trial Steering Committee (TSC) following each meeting.
Clinical site monitoring
Trial monitoring was carried out in accordance with the protocol. Trial centres agreed to provide access to source data and consent was gained from patients for direct access to patient notes. All centres that had a minimum of four patients who had completed follow-up (week 12 visit or death) were monitored on site at least once during the trial. The following data were validated from source documents:
-
eligibility and signed consent
-
trial drug and antibiotic management
-
safety events
-
any data concerns raised by central monitoring.
Patient and public involvement
The ARREST trial was developed with the Healthcare Associated Infection Service Users Research Forum (SURF) [www.hcaisurf.org (accessed 4 June 2018)], in particular, Jennifer Bostock, who was the patient and public involvement (PPI) representative on the ARREST Trial Steering Committee. Jennifer Bostock advised on the inclusion of incapacitated adults, the application of the Mental Capacity Act 200536 and the information provided to patients. SURF is no longer active, but Ms Bostock is helping to disseminate the trial results beyond the academic and health-care professional community to other patient groups that she works with, including MRSA Action UK.
In particular, given recruitment challenges, Ms Bostock developed and led the substudy investigating patients’ and carers’ reasons for, and for not, participating in the trial. This is reported in full in Chapter 4.
Protocol changes
The trial was approved by the London (Westminster) Research Ethics Committee (reference number 12/LO/0637). See Appendix 1 for changes to the protocol.
Chapter 3 Results
Participant flow diagram
Between 10 December 2012 and 25 October 2016, 770 participants from 29 UK hospital groups were randomised to add placebo (n = 396) or rifampicin (n = 374) to their ‘backbone’ antibiotic treatment (Figure 2). A total of 2896 participants were screened for entry to the trial. The most common reason for not randomising a potentially eligible participant was that they had already received > 96 hours of antibiotics (n = 664). In 364 cases, the participant was not willing. Rifampicin was considered mandatory in 232 cases. Known rifampicin resistance occurred in only 19 cases; however, 139 cases were not eligible because of pre-existing liver disease raising concerns about rifampicin treatment and 167 cases because of predicted drug interactions.
FIGURE 2.
Participant flow diagram. a, Reasons are not mutually exclusive and, therefore, total is more than the number of participants not randomised. b, Seven participants with predicted drug interaction, two misdiagnosed (S. aureus not grown from blood but only other samples), rifampicin considered mandatory in one, a clinician considered one participant should not have been randomised because of acute kidney injury, a clinician considered one participant should not have been randomised as they were in another study (not of an IMP). c, Final 12-week visit could occur any time from 11 weeks onwards in accordance with the protocol. Consent withdrawals not included in these numbers. d, Time-to-event analyses included all time at risk from randomisation to the earliest of the event or last clinical follow-up if the event had not occurred. © The Author(s). Published by Elsevier Ltd. This is an open access article published under the CC BY 4.0 licence.
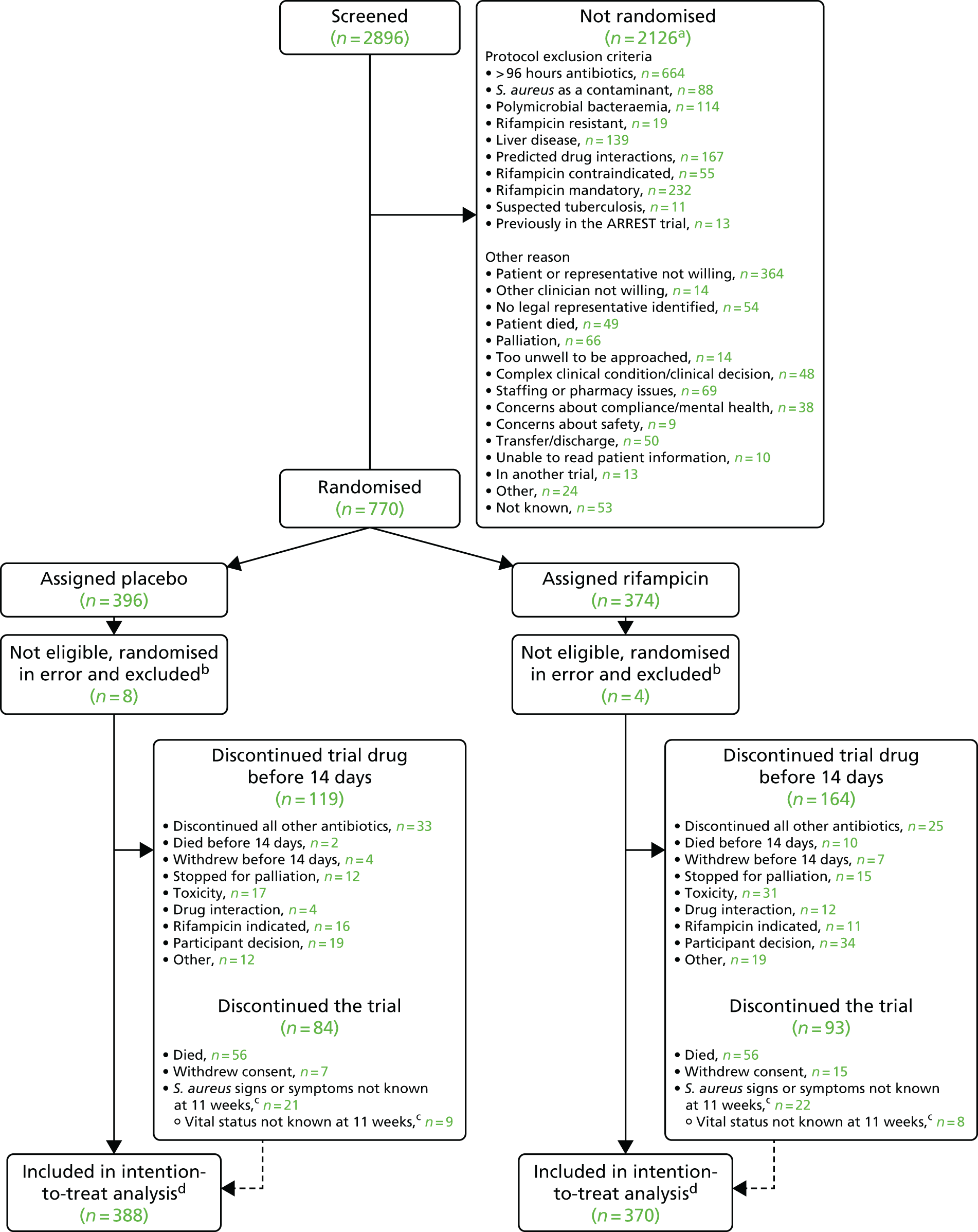
A total of 12 participants (placebo, n = 8; rifampicin, n = 4) were randomised in error (the participant should not have been randomised and never received trial drug) and were excluded following the statistical analysis plan. Of these 12 participants, seven participants had predicted drug interactions, two were misdiagnosed (S. aureus was not grown from blood), rifampicin was considered mandatory in one patient, a clinician considered that one participant should not have been randomised owing to acute kidney injury, and a clinician considered that one participant should not have been randomised as they were in another study (not of an IMP, allowed according to the protocol).
Thus, 758 (placebo, n = 388; rifampicin, n = 370) participants were included in the analyses. The median number of patients recruited per centre was 11 (IQR 4–30 patients, range 1–163 patients). A total of 415 (54.7%) participants were recruited from five centres (Oxford, n = 163; Guy’s and St Thomas’, n = 99; Liverpool, n = 62; Plymouth, n = 48; and Sheffield, n = 43). The large number of centres recruiting small numbers of participants, together with the relatively large block size (6–8), led to a small imbalance in the numbers included randomised to the placebo (n = 388) and rifampicin groups (n = 370).
Baseline characteristics
Baseline characteristics were well-balanced between randomised groups (Tables 1 and 2).
| Factor | Treatment group | Total (N = 758)a | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370)a | ||
| Male, n (%) | 246 (63.4) | 249 (67.3) | 495 (65.3) |
| Age at last birthday (years), median (IQR) | 66 (51–76) | 64 (49–76) | 65 (50–76) |
| Charlson Comorbidity Index score,a median (IQR) | 2 (0–3) | 1 (0–3) | 2 (0–3) |
| Cancer (N = 756), n (%) | 60 (15.5) | 66 (17.8) | 126 (16.6) |
| Chronic lung disease (N = 756), n (%) | 42 (10.8) | 48 (13.0) | 90 (11.9) |
| Congestive heart disease (N = 756), n (%) | 40 (10.3) | 42 (11.4) | 82 (10.8) |
| Moderate or severe liver disease (N = 755), n (%) | 5 (1.3) | 5 (1.4) | 10 (1.3) |
| Moderate or severe renal disease (N = 755), n (%) | 80 (20.6) | 58 (15.7) | 138 (18.2) |
| Diabetes,a n (%) | 119 (30.7) | 109 (29.5) | 228 (30.1) |
| Active injecting drug use (N = 751), n (%) | 41 (10.6) | 42 (11.4) | 83 (10.9) |
| Weight (kg) (N = 755), median (IQR) | 76.0 (65.0–90.0) | 76.0 (64.0–89.0) | 76.0 (64.0–90.0) |
| Admitted to ICU,a n (%) | 36 (9.3) | 34 (9.2) | 70 (9.2) |
| CRP level (mg/l) (N = 755),b mean (SE) | 163 (5.2) | 166 (5.3) | 164 (3.7) |
| White blood cell count (× 109/l) (N = 752), median (IQR) | 9.5 (6.7–13.4) | 9.5 (7.1–13.1) | 9.5 (6.9–13.2) |
| Neutrophil count (× 109/l) (N = 752), median (IQR) | 7.3 (4.7–11.0) | 7.4 (4.9–10.7) | 7.3 (4.8–10.9) |
| Lymphocyte count (× 109/l) (N = 751), median (IQR) | 1.0 (0.7–1.5) | 1.0 (0.7–1.5) | 1.0 (0.7–1.5) |
| SOFA score (points),a median (IQR) | 2 (1–4) | 2 (1–4) | 2 (1–4) |
| Vascular catheter in situ (N = 744), n (%) | 102 (26.8) | 89 (24.5) | 191 (25.7) |
| Surgery in the last 30 days (N = 756), n (%) | 53 (13.7) | 37 (10.1) | 90 (11.9) |
| Days between first new symptom caused by S. aureus and randomisation,a median (IQR) | 4 (3–6) | 4 (3–6) | 4 (3–6) |
| Days between drawing of first positive blood culture and randomisation,a median (IQR) | 3 (2–3) | 3 (2–4) | 3 (2–3) |
| Hours of active antibiotic therapy before randomisation, median (IQR) | 63 (42–75) | 60 (41–76) | 62 (42–75) |
| Blood culture positive at randomisation, n (%) | 69/326 (21.2) | 88/316 (27.8) | 157/642 (24.5) |
| Factor | Treatment group, n (%) | Total (N = 758),a n (%) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370)a | ||
| Mode of acquisition of infectiona | |||
| Community acquired | 240 (61.9) | 245 (66.2) | 485 (64.0) |
| Nosocomial infection (onset ≥ 48 hours after admission) | 76 (19.6) | 56 (15.1) | 132 (17.4) |
| Health-care associated (all other) | 72 (18.6) | 68 (18.4) | 140 (18.5) |
| MRSA | 21 (5.4) | 26 (7.0) | 47 (6.2) |
| Rifampicin-resistant infection at randomisation (N = 750)b | 0 | 0 | 0 |
| Main focus/foci of infectiona,c | |||
| Native heart valve | 16 (4.1) | 17 (4.6) | 33 (4.4) |
| Native joint | 34 (8.8) | 29 (7.8) | 63 (8.3) |
| Prosthetic heart valve/jointd | 5 (1.3) | 9 (2.4) | 14 (1.8) |
| Implanted vascular device (other than i.v. catheter) | 23 (5.9) | 13 (3.5) | 36 (4.7) |
| Deep tissue infection/abscess | 94 (24.2) | 82 (22.2) | 176 (23.2) |
| Central or peripheral i.v. catheter | 67 (17.3) | 63 (17.0) | 130 (17.2) |
| Skin/soft tissue (excluding wounds) | 66 (17.0) | 72 (19.5) | 138 (18.2) |
| Surgical wound | 15 (3.9) | 10 (2.7) | 25 (3.3) |
| Pneumonia or urinary tract infection | 30 (7.7) | 30 (8.1) | 60 (7.9) |
| Not established | 67 (17.3) | 72 (19.5) | 139 (18.3) |
| Any deep-seated focuse | 159 (41.0) | 142 (38.4) | 301 (39.7) |
| Likely portal of entry of S. aureus into the bloodstreamc | |||
| Clinically apparent skin or soft tissue infection unrelated to a surgical intervention | 131 (33.8) | 124 (33.5) | 255 (33.6) |
| Infected surgical wound within last 3 months, with or without associated prosthesis | 19 (4.9) | 19 (5.1) | 38 (5.0) |
| Peripheral vascular catheter (including arterial line) | 23 (5.9) | 26 (7.0) | 49 (6.5) |
| Central vascular catheter (including PICC line) | 50 (12.9) | 42 (11.4) | 92 (12.1) |
| Other implanted vascular device (e.g. pacemaker, stent, graft) | 15 (3.9) | 12 (3.2) | 27 (3.6) |
| Respiratory | 16 (4.1) | 13 (3.5) | 29 (3.8) |
| Per-urethral or suprapubic urinary catheter | 7 (1.8) | 8 (2.2) | 15 (2.0) |
| Recent (within 1 week of bacteraemia) urological surgery | 1 (0.3) | 3 (0.8) | 4 (0.5) |
| Not known (absence of any of the above) | 110 (28.4) | 108 (29.2) | 218 (28.8) |
| Injecting drug user | 8 (2.1) | 9 (2.4) | 17 (2.2) |
| Corticosteroid injection into joint | 4 (1.0) | 2 (0.5) | 6 (0.8) |
| Other | 2 (0.5) | 3 (0.8) | 5 (0.7) |
| Not completed (missing data) | 2 (0.5) | 1 (0.3) | 3 (0.4) |
A total of 495 (65.3%) participants were men (see Table 1). The median age was 65 years (IQR 50–76 years), median weight was 76.0 kg (IQR 64.0–90.0 kg) and the median Charlson Comorbidity Index score was 2 (IQR 0–3). Diabetes (30.1%), renal disease (18.2%), cancer (16.6%) and chronic lung disease (11.9%) were all common comorbidities. A total of 83 (10.9%) participants were active injecting drug users, 70 (9.2%) participants were in an ICU, 90 (11.9%) participants had undergone surgery in the past 30 days, and 127 (16.8%) participants had consent provided by a legal representative because of incapacity. Reflecting disease severity, mean CRP level was 164 mg/l [standard error (SE) 3.7 mg/l] and a median Sequential Organ Failure Assessment (SOFA) score was 2 points (IQR 1–4 points).
At randomisation, participants had already received a median of 62 hours (IQR 42–75 hours) of active antibiotics, with their first blood culture taken a median of 3 days (IQR 2–3 days) previously and their first symptoms occurring a median of 4 days (IQR 3–6 days) previously. A total of 157 out of 642 (24.5%) participants still had a positive blood culture on the day of randomisation.
A total of 485 (64.0%) infections were community acquired, with only 132 (17.4%) infections being nosocomial; 47 (6.2%) were caused by MRSA. No patients were known to have rifampicin-resistant S. aureus bacteraemia at randomisation.
The initial focus was deep in 301 (39.7%) patients, including 33 (4.4%) with endocarditis and 14 (1.8%) with infected prostheses. A total of 130 (17.2%) infections were due to infected central/peripheral lines, and 138 (18.2%) were associated with skin/soft tissue infections. Another type of focus was identified in 49 (6.5%) participants and not established in 139 (18.3%) participants.
In 255 (33.6%) participants, the most likely portal of entry of S. aureus into the bloodstream was a clinically apparent skin or soft tissue infection unrelated to a surgical intervention. Central or peripheral lines were the most likely portal of entry in 141 (18.6%) participants, although 191 (25.7%) had a vascular catheter in situ at randomisation. For 218 (18.6%) participants, the portal of entry was unknown.
Follow-up and treatment received
Overall, completeness of scheduled visits was high up to 14 days. Excluding visits after death or discharge, day 3 visits were missed in 10 out of 372 (2.7%) participants in the placebo group versus 12 out of 350 (3.4%) participants in the rifampicin group, day 7 visits were missed in 15 out of 337 (4.5%) participants in the placebo group versus 16 out of 311 (5.1%) participants in the rifampicin group, day 10 visits were missed in 22 out of 293 (7.5%) participants in the placebo group versus 26 out of 262 (9.9%) participants in the rifampicin group, and day 14 visits were missed in 9 out of 230 (3.9%) participants in the placebo group versus 13 out of 204 (6.4%) participants in the rifampicin group. Completeness dropped after 14 days when patients started to be discharged, for example visits were missed in 21 out of 149 (14.1%) participants in the placebo group versus 19 out of 134 (14.2%) participants in the rifampicin group at day 21, 23 out of 115 (20.0%) participants in the placebo group versus 23 out of 93 (24.7%) participants in the rifampicin group at day 28, and 25 out of 89 (28.1%) participants in the placebo group versus 19 out of 58 (32.8%) participants in the rifampicin group at day 35.
A total of 22 (2.9%) participants withdrew consent. At the 12-week visit, only 39 (5.1%) participants had unknown vital status and 65 (8.6%) were not assessed for signs/symptoms of S. aureus infection (including consent withdrawals).
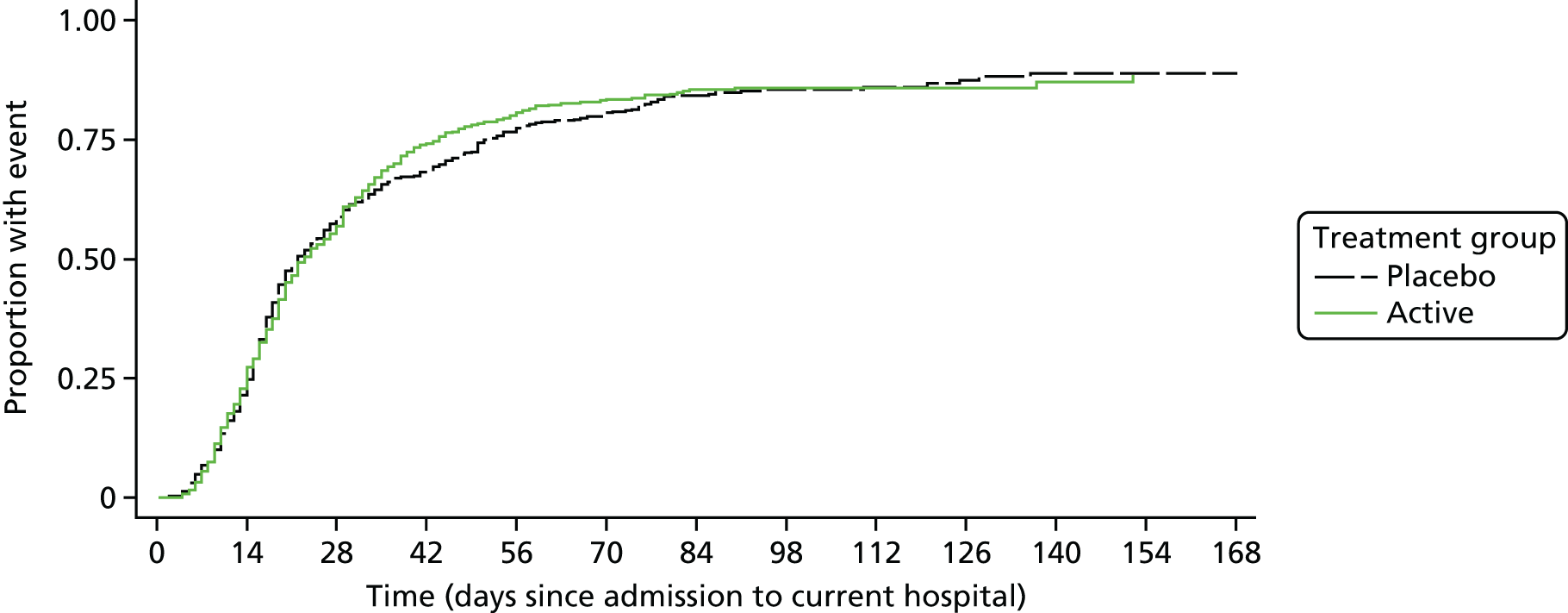
A total of 23 (3.0%) participants were still in hospital at 12 weeks [15 (3.9%) participants in the placebo group vs. 8 (2.2%) participants in the rifampicin group; p = 0.17). The median initial hospitalisation duration was 21 days (IQR 14–50 days) versus 22 days (IQR 13–43 days) in placebo and rifampicin groups, respectively (p = 0.80) (Figure 3). A total of 132 (39.8%) participants in the placebo group versus 138 (44.8%) participants in the rifampicin group were discharged on outpatient parental antibiotic therapy (p = 0.35). A total of 94 (24.2%) participants in the placebo group versus 83 (22.4%) participants in the rifampicin group were readmitted post discharge and before 12 weeks (p = 0.56), spending a median of 9 nights (IQR 4–20 nights) and 10 nights (IQR 3–20 nights) in hospital post original discharge, respectively. Any admission was considered relating to S. aureus bacteraemia in 16 (4.1%) participants in the placebo group and 9 (2.4%) participants in the rifampicin group (p = 0.19).
FIGURE 3.
Days from admission to current hospital to original post-randomisation discharge. p = 0.80 subhazard regression. Note: death treated as competing risk.
A total of 744 (98.2%) participants initiated blinded trial drug a median of 4.3 hours (IQR 2.3–7.8 hours) after randomisation. Reasons for not initiating blinded trial drug were patient decision (n = 7), increasing liver enzyme levels (n = 2), starting on open-label rifampicin (n = 2), withdrawn for palliation (n = 1), incorrectly believing that the bacteraemia was rifampicin resistant (n = 1) and unable to access i.v. trial drug from trials pharmacy at weekend (n = 1).
A total of 96 (12.7%) participants initiated i.v. trial drug rather than oral trial drug (Table 3). A total of 595 (78.5%) participants initiated 900 mg daily rather than 600 mg daily and 362 (52.2%) participants twice daily rather than once daily. The median dose was 11.1 mg/kg (IQR 10.0–12.9 mg/kg). The trial drug was initiated a median of 68 hours (IQR 48–85 hours) after starting active antibiotics for the current infection. The trial drug was continued for a median of 12.6 days (IQR 6.0–13.2 days) for participants in the rifampicin group versus 13.0 days (IQR 11.3–13.5 days) in participants in the placebo group (p < 0.0001; discontinuations primarily because of antibiotic-modifying AEs and drug–drug interactions, see Safety). A total of 60 (15.5%) participants in the placebo group versus 51 (15.6%) participants in the rifampicin group received the i.v. trial drug. Percentages reporting missing any doses of trial drug ranged from 9.5% to 16.2% but did not differ between randomised groups (Figure 4; global p = 0.71).
| Factor | Treatment group | Total (N = 758) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | ||
| Never initiated trial drug, n (%) | 8 (2.1) | 6 (1.6) | 14 (1.8) |
| Initiated i.v. trial drug, n (%) | 51 (13.1) | 45 (12.2) | 96 (12.7) |
| Initiated oral trial drug, n (%) | 329 (84.8) | 319 (86.2) | 648 (85.5) |
| Initiated trial drug once daily, n (%) | 175 (45.1) | 173 (46.8) | 348 (45.9) |
| Initiated trial drug twice daily, n (%) | 205 (52.8) | 191 (51.6) | 396 (52.2) |
| Initiated 600 mg of trial drug daily, n (%) | 74 (19.1) | 75 (20.3) | 149 (19.7) |
| Initiated 900 mg of trial drug daily, n (%) | 306 (78.9) | 289 (78.1) | 595 (78.5) |
| Initial total daily dose (mg/kg) (N = 741), median (IQR) | 11.2 (9.9–12.9) | 11.0 (10.0–12.7) | 11.1 (10.0–12.9) |
| Hours from starting active antibiotics to starting trial drug, median (IQR) | 69 (49–85) | 68 (46–85) | 68 (48–85) |
| Hours from randomisation to initiation of randomised treatment, median (IQR) | 4.2 (2.3–7.6) | 4.3 (2.3–8.0) | 4.3 (2.3–7.8) |
| Days on trial drug, median (IQR) | 13.0 (11.3–13.5) | 12.6 (6.0–13.2) | 12.8 (7.9–13.4) |
| Total duration of the study drug (days), n (%) | |||
| 0 | 8 (2.1) | 6 (1.6) | 14 (1.8) |
| < 3 | 18 (4.6) | 22 (5.9) | 40 (5.3) |
| 3–5 | 28 (7.2) | 57 (15.4) | 85 (11.2) |
| 6–9 | 24 (6.2) | 43 (11.6) | 67 (8.8) |
| 10–13 | 49 (12.6) | 42 (11.4) | 91 (12.0) |
| 14 | 255 (65.7) | 197 (53.2) | 452 (59.6) |
| > 14 | 6 (1.5) | 3 (0.8) | 9 (1.2) |
| Ever received i.v. trial drug, n (%) | 60 (15.5) | 56 (15.1) | 116 (15.3) |
FIGURE 4.
Percentage reporting missing one or more doses of trial drugs since the previous scheduled visit.
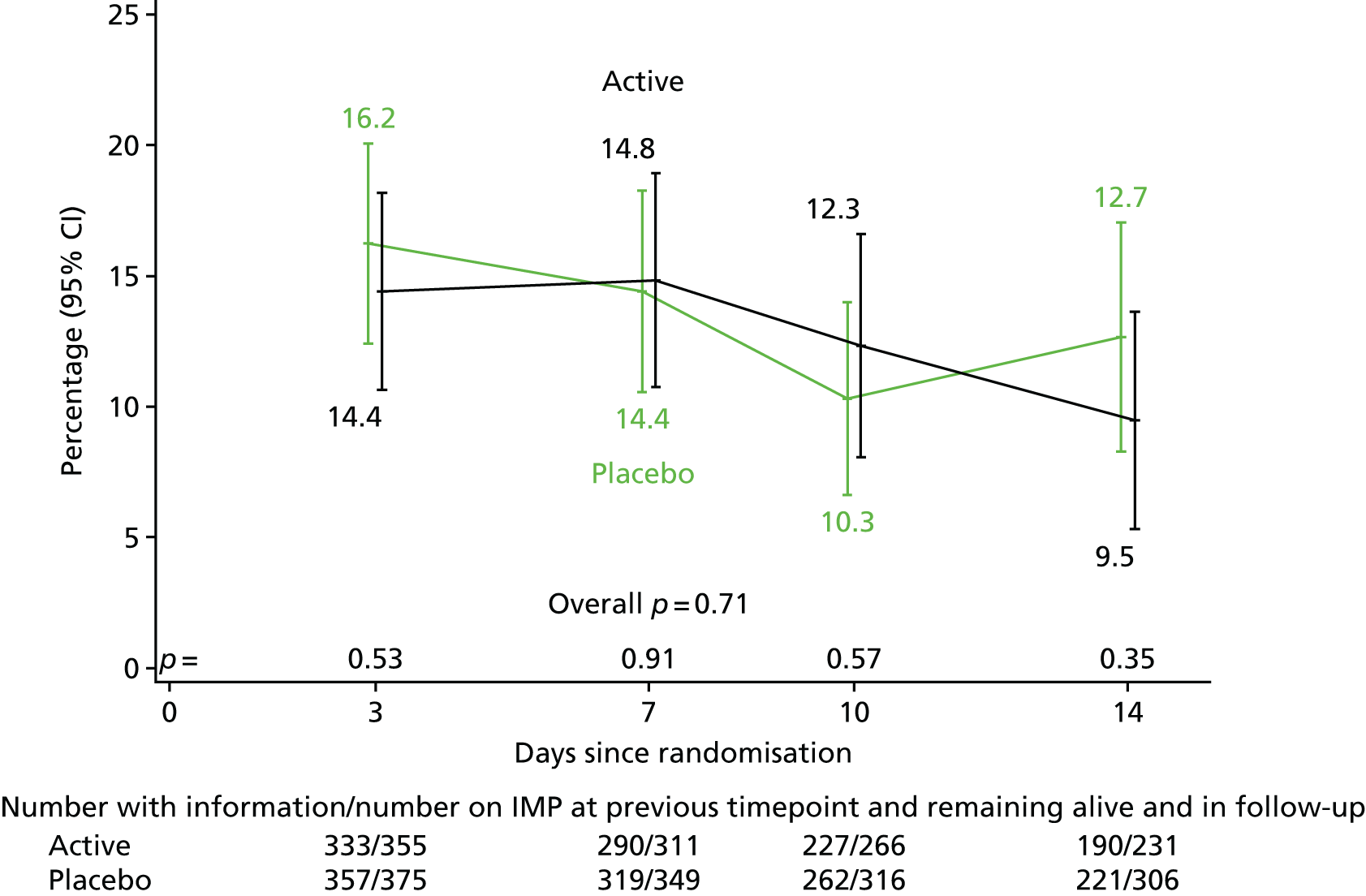
A substantial variety of ‘backbone’ active antibiotics were used (Table 4; details in Table 28, Appendix 2). Flucloxacillin was given in 619 (81.7%) participants, and vancomycin or teicoplanin in 380 (50.1%) participants at some point in the primary treatment course, with no evidence of difference between randomised groups (p = 0.44 and p = 0.34, respectively). Stat (one-off) doses of gentamicin or amikacin were used in 199 (26.3%) participants (p = 0.89). There was no evidence that the number of antibiotics used [median of 3 (IQR 2–4)] or the total duration of active anti-staphylococcal treatment (including therapy received before randomisation) (median 29 days (IQR 18–45 days)] differed between groups (p = 0.98 and 0.64, respectively) (see Table 4). Post-randomisation active anti-staphylococcal treatment was taken for a median of 27 days (IQR 15–41 days) in the placebo group versus 26 days (IQR 15–43 days) in the rifampicin group.
| Factor | Treatment group | Total (N = 758) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | ||
| ‘Backbone’ active antibiotic treatment,a n (%) | |||
| Flucloxacillin | 321 (82.7) | 298 (80.5) | 619 (81.7) |
| Co-amoxiclavulante | 122 (31.4) | 107 (28.9) | 229 (30.2) |
| Piperacillin/tazobactam | 115 (29.6) | 102 (27.6) | 217 (28.6) |
| Vancomycin/teicoplanin | 188 (48.5) | 192 (51.9) | 380 (50.1) |
| Cephalosporin | 110 (28.4) | 104 (28.1) | 214 (28.2) |
| Fluoroquinolone | 47 (12.1) | 46 (12.4) | 93 (12.3) |
| Macrolide | 30 (7.7) | 28 (7.6) | 58 (7.7) |
| Clindamycin | 23 (5.9) | 36 (9.7) | 59 (7.8) |
| Tetracycline | 29 (7.5) | 26 (7.0) | 55 (7.3) |
| Gentamicin/amikacin | 101 (26.0) | 98 (26.5) | 199 (26.3) |
| Stat gentamicin/amikacin | 95 (24.5) | 87 (23.5) | 182 (24.0) |
| Carbapenem | 38 (9.8) | 35 (9.5) | 73 (9.6) |
| Other antibioticb | 52 (13.4) | 52 (14.1) | 104 (13.7) |
| Number of antibiotics received during the S. aureus infection episode (excluding study drug), median (IQR) | 3 (2–4) | 3 (2–4) | 3 (2–4) |
| Days of antibiotic treatment for the S. aureus infection episode (days), median (IQR) | 30 (18–44) | 29 (17–45) | 29 (18–45) |
| Rifampicin used open label, n (%) | 52 (13.4) | 32 (8.6) | 84 (11.1) |
| Initiated < 14 days from randomisationc | 25 (6.4) | 18 (4.9) | 43 (5.7) |
| Initiated ≥ 14 days from randomisation | 27 (7.0) | 14 (3.8) | 41 (5.4) |
A total of 32 (8.6%) participants in the rifampicin group versus 52 (13.4%) participants in the placebo group used open-label rifampicin at some point after randomisation (p = 0.04). Median time from randomisation to initiation of open-label rifampicin was 14 days (IQR 7–18 days) (see Table 4). There was a trend to slightly fewer participants initiating open-label rifampicin from 14 days onwards [i.e. after stopping trial drug; 14 (3.8%) participants in the rifampicin group vs. 27 (7.0%) participants in the placebo group; p = 0.053]. Open-label rifampicin was used in participants with a range of original infection foci (Table 5). The median duration of open-label rifampicin was 25 days (IQR 13–45 days) in participants in the placebo group versus 32 days (IQR 26–48 days) in participants in the rifampicin group. A total of 60 (15.5%) participants in the placebo group received antibiotics after the primary course versus 34 (9.2%) participants in the rifampicin group (p = 0.01).
| Infection focus | Treatment group, n (%) | Total (N = 84), n (%) | |
|---|---|---|---|
| Placebo (N = 52) | Rifampicin (N = 32) | ||
| Central venous line (including PICC line) | 1 (1.9) | 2 (6.3) | 3 (3.6) |
| Implanted vascular device (e.g. pacemaker, stent, graft) | 8 (15.4) | 0 (0.0) | 8 (9.5) |
| Infected intravascular thrombus | 2 (3.8) | 3 (9.4) | 5 (6.0) |
| Native heart valve | 6 (11.5) | 2 (6.3) | 8 (9.5) |
| Prosthetic heart valve | 1 (1.9) | 2 (6.3) | 3 (3.6) |
| Native joint | 1 (1.9) | 5 (15.6) | 6 (7.1) |
| Prosthetic joint | 0 (0.0) | 1 (3.1) | 1 (1.2) |
| Vertebral bone/disc | 13 (25.0) | 8 (25.0) | 21 (25.0) |
| Epidural or intraspinal empyema | 4 (7.7) | 1 (3.1) | 5 (6.0) |
| Deep tissue infection or abscess | 6 (11.5) | 3 (9.4) | 9 (10.7) |
| Surgical wound | 3 (5.8) | 0 (0.0) | 3 (3.6) |
| Skin/soft tissue (excluding wounds) | 6 (11.5) | 3 (9.4) | 9 (10.7) |
| Pneumonia | 2 (3.8) | 1 (3.1) | 3 (3.6) |
| Othera | 6 (11.5) | 0 (0.0) | 6 (7.1) |
| Not established | 6 (11.5) | 9 (28.1) | 15 (17.9) |
A total of 159 participants in the placebo group versus 142 participants in the rifampicin group had a deep focus that was drained/removed in 35 (22.0%) versus 29 (20.4%) participants, and a median (IQR) of 5 (2–12) and 3 (1–6) days from randomisation respectively (Table 6). A total of 88 participants in the placebo group versus 76 participants in the rifampicin group had an intravascular device that was removed in 62 (70.5%) versus 60 (78.9%) participants, and a median (IQR) of 2 (0–3) and 1 (0–2) days prior to randomisation, respectively.
| Factor | Treatment group | Total (N = 758) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | ||
| Any deep-seated focusa | 159 | 142 | 301 |
| Drained/removed, n (%) | 35 (22.0) | 29 (20.4) | 64 (21.3) |
| Median days from randomisation to drainage/removal (IQR) | 5 (2–12) | 3 (1–6) | 4 (2–10) |
| Not removed, n (%) | 118 (74.2) | 109 (76.8) | 227 (75.4) |
| Not known, n (%) | 6 (3.8) | 4 (2.8) | 10 (3.3) |
| Non-device-related focus | 233 | 222 | 455 |
| Drained/removed, n (%) | 39 (16.7) | 36 (16.2) | 75 (16.5) |
| Median days from randomisation to drainage/removal (IQR) | 4 (2–11) | 4 (2–8) | 4 (2–10) |
| Not removed, n (%) | 187 (80.3) | 179 (80.6) | 366 (80.4) |
| Not known, n (%) | 7 (3.0) | 7 (3.2) | 14 (3.1) |
| Intravascular device | 88 | 76 | 164 |
| Removed, n (%) | 62 (70.5) | 60 (78.9) | 122 (74.4) |
| Median days from randomisation to removal (IQR) | –2 (–3 to 0) | –1 (–2 to 0) | –1 (–2 to 0) |
| Not removed, n (%) | 25 (28.4) | 15 (19.7) | 40 (24.4) |
| Not known, n (%) | 1 (1.1) | 1 (1.3) | 2 (1.2) |
| Non-vascular prosthetic implant/device | 5 | 9 | 14 |
| Removed, n (%) | 0 (0.0) | 2 (22.2) | 2 (14.3) |
| Median days from randomisation to removal (IQR) | – | 7 (2–11) | 7 (2–11) |
| Not removed, n (%) | 5 (100.0) | 7 (77.8) | 12 (85.7) |
Unblinding and blinding assessment
At least one individual was unblinded for 14 participants (rifampicin group, n = 9; placebo group, n = 5). In two cases this was only of a non-trial physician and ward pharmacist, respectively, for participant safety. In three further cases this was of the research nurse only, but no other members of the clinical or research teams.
At the final 12-week visit, physicians and participants were asked which treatment they believed that they had received. A total of 203 out of 243 (83.5%) physicians of participants randomised to rifampicin reported that they genuinely had no idea versus 249 out of 279 (89.2%) participants randomised to the placebo group (p = 0.08). A total of 32 (13.2%) and 17 (6.1%) participants, respectively, guessed the correct allocation. In contrast, 113 out of 199 (56.8%) participants randomised to the rifampicin group reported that they genuinely had no idea versus 159 out of 229 (69.4%) participants randomised to the placebo group (p = 0.007). A total of 72 (36.2%) and 35 (15.3%) participants, respectively, guessed the correct allocation.
Primary end point
By 12 weeks, bacteriological failure/recurrence or death occurred in 62 (16.8%) participants in the rifampicin group versus 71 (18.3%) participants in the placebo group [absolute risk difference (RD) –1.4%, 95% CI –7.0% to 4.3%; HR 0.96, 95% CI 0.68 to 1.35; p = 0.81] (Figure 5a). In exploratory post hoc analyses, comparing rifampicin with placebo there were 4 (1.1%) versus 5 (1.3%) failures (competing risks p = 0.82), 3 (0.8%) versus 16 (4.1%) recurrences (competing risks p = 0.01) and 55 (14.9%) versus 50 (12.9%) deaths without bacteriological failure/recurrence respectively (competing risks p = 0.30) (Table 7). The NNT to prevent one bacteriologically confirmed recurrence was 29 participants.
FIGURE 5.
Bacteriological failure/recurrence or death (a) overall (HR 0.96, 95% CI 0.68 to 1.35; p = 0.81) and (b) according to three priority subgroups. Note: see Figures 6 and 7 for other subgroup analyses. © The Author(s). Published by Elsevier Ltd. This is an open access article published under the CC BY 4.0 licence.
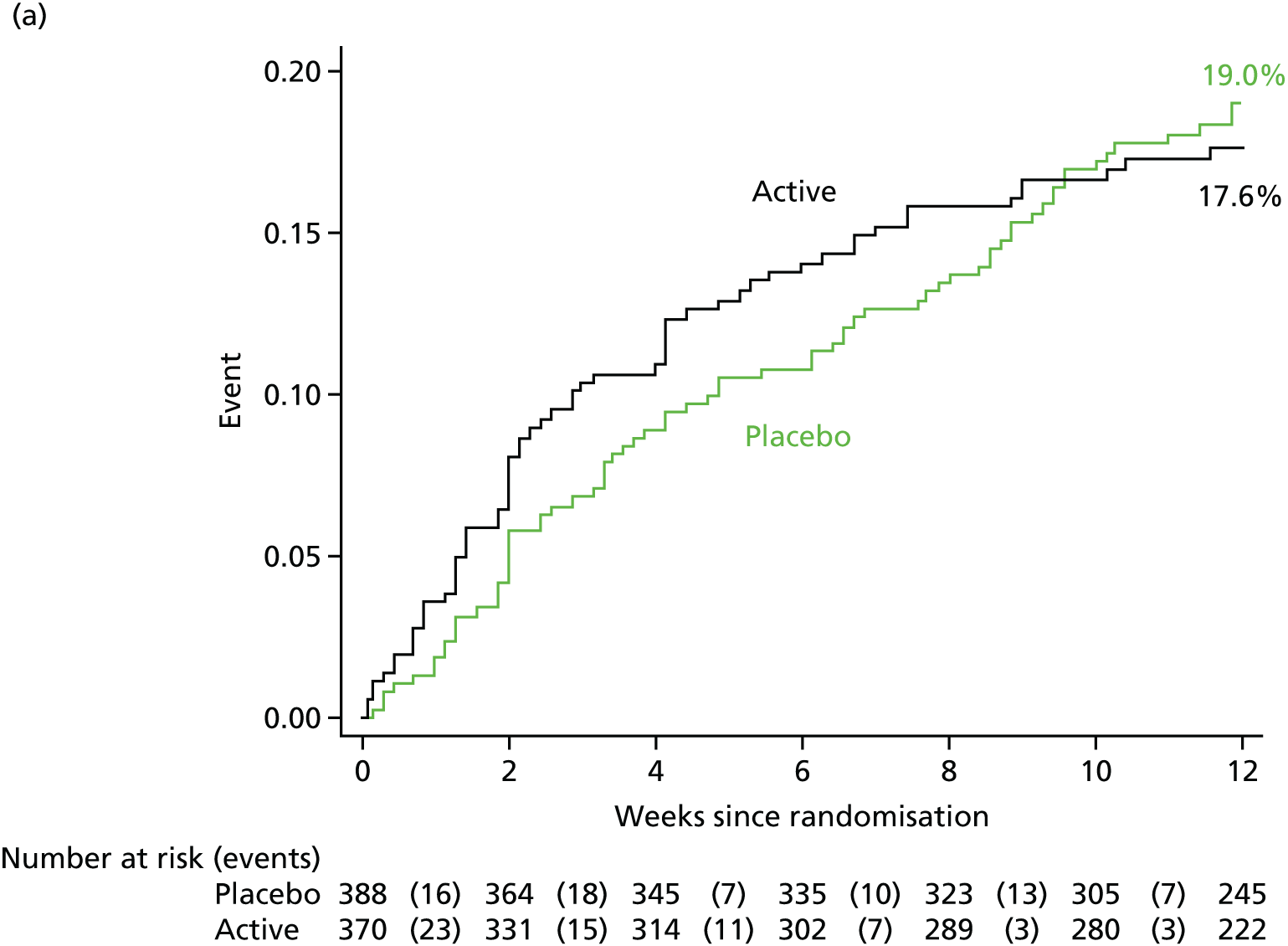
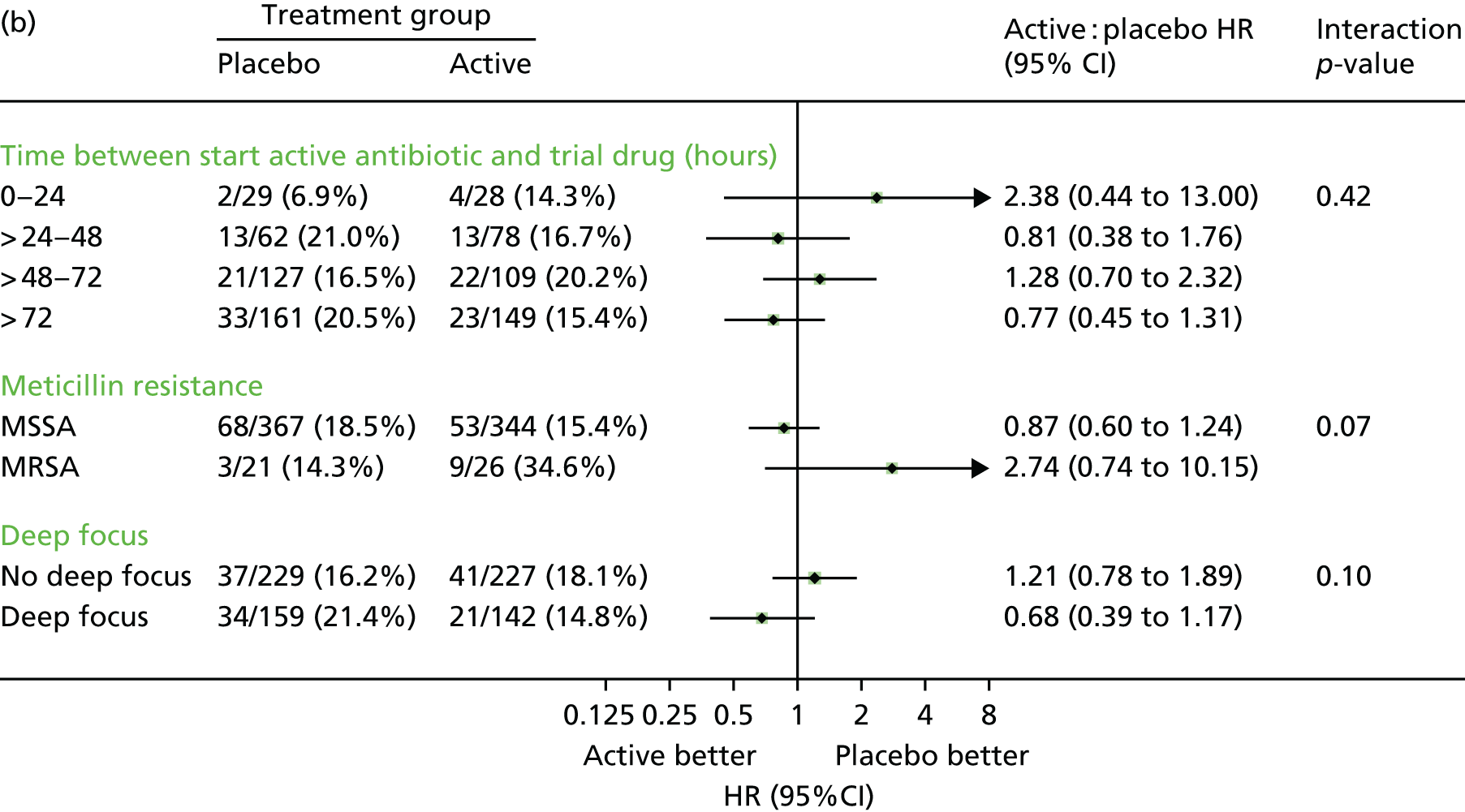
| Events and ERC adjudication | Failure or recurrence | Deaths (all), n (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Bacteriological, n (%) | p-value | Clinical, n (%) | p-value | |||||
| Placebo | Rifampicin | Placebo | Rifampicin | Placebo | Rifampicin | |||
| Total randomised | 388 | 370 | – | 388 | 370 | – | 388 | 370 |
| Total events | 71 (18.3) | 62 (16.8) | 0.81 | 86 (22.2) | 76 (20.5) | 0.84 | 56 (14.4) | 56 (15.1) |
| Failure | 5 (1.3) | 4 (1.1) | 0.82 | 25 (6.4) | 23 (6.2) | 0.97 | ||
| Failure due to slow resolution | 3 (0.8) | 1 (0.3) | 17 (4.4) | 10 (2.7) | ||||
| Recurrence | 16 (4.1) | 3 (0.8) | 0.01 | 23 (5.9) | 8 (2.2) | 0.01 | ||
| Death without either failure or recurrence | 50 (12.9) | 55 (14.9) | 0.30 | 38 (9.8) | 45 (12.2) | 0.22 | ||
| Total failures/recurrences (first two columns) or S. aureus-related deaths (third column): attributed by end-point review committee to | 21 (100) | 7 (100) | 48 (100) | 31 (100) | 32 (100) | 36 (100) | ||
| Failure of antibiotics | 1 (5) | 0 | 3 (6) | 1 (3) | 1 (3) | 3 (8) | ||
| Failure of source management | 17 (81) | 3 (43) | 38 (79) | 24 (77) | 21 (66) | 18 (50) | ||
| Not recognised | 9 (43) | 2 (29) | 12 (25) | 5 (16) | 3 (9) | 4 (11) | ||
| Recognised, not actively managed | 5 (24) | 1 (14) | 16 (33) | 14 (45) | 8 (25) | 8 (22) | ||
| Recognised, actively managed still failed/recurred | 3 (14) | 0 | 10 (21) | 5 (16) | 10 (31) | 6 (17) | ||
| Not possible to distinguish | 3 (14) | 4 (57) | 7 (15) | 6 (19) | 10 (31) | 15 (42) | ||
| Death a consequence of late presentation | – | – | – | – | 3 (9) | 11 (31) | ||
A total of 242 (65.4%) participants in the rifampicin group versus 290 (74.7%) participants in the placebo group were included in the per-protocol population [received active rifampicin/placebo for ≥ 80% of days from start of trial drug to 14 days subsequently, death or discontinuation of active antibiotics (not including trial drug), whichever came earliest]. By 12 weeks, 39 (16.1%) participants in the rifampicin group versus 49 (16.9%) participants in the placebo group experienced bacteriological failure/recurrence or died (absolute RD –0.8%, 95% CI –7.3 to 5.6; HR 1.00, 95% CI 0.65 to 1.52; p = 0.99). An exploratory post hoc analysis was also done, excluding participants in either group who started open-label rifampicin at any time during follow-up. A total of 225 (60.1%) participants in the rifampicin group versus 262 (67.5%) participants in the placebo group were included in this post hoc per-protocol population. By 12 weeks, 37 (16.4%) participants in the rifampicin group versus 37 (14.1%) participants in the placebo group experienced bacteriological failure/recurrence or died (absolute RD 2.3%, 95% CI –4.3 to 8.8; HR 1.23, 95% CI 0.78 to 1.93; p = 0.38).
Of the 28 failures/recurrences in which S. aureus was isolated from a sterile site, paired baseline and failure/recurrence isolates were stored for 11 (39%) participants. All failure/recurrence isolates were whole genome sequenced and within 12 single nucleotide variants of the baseline isolate [median 1 (IQR 1–6) (range 0–12) single nucleotide variants].
Subgroup analyses according to the three most important characteristics [time between starting active antibiotics and trial drug, meticillin resistance, and foci of infection (deep versus not deep)] suggested no heterogeneity in lack of effect of rifampicin (pheterogeneity 0.42, 0.07, 0.10, respectively, see Figure 5b. The rifampicin effect varied significantly according to the initial antibiotic given at randomisation, with some suggestion of benefit in those with meticillin-sensitive infection treated with flucloxacillin alone (pheterogeneity = 0.01, Figure 6), but across none of 16 other subgroup analyses (pheterogeneity > 0.05, Figure 7). At the suggestion of a reviewer, subgroup analyses were also considered by diabetes (pheterogeneity = 0.37), weight (pheterogeneity = 0.13), body mass index (BMI) (pheterogeneity = 0.58) and dose in mg/kg (pheterogeneity = 0.42).
FIGURE 6.
Five other priority subgroup analyses for bacteriological failure/recurrence or death through 12 weeks (primary end point). Note: presenting class-level and antibiotic-level categorisation of initial active antibiotics (as per the statistical analysis plan). See Figure 5b for the three other priority subgroup analyses defined in the statistical analysis plan [time between starting active antibiotics and trial drug, meticillin resistance and foci of infection (deep versus not deep)]. All eight priority subgroup analyses were prespecified in the protocol and the statistical analysis plan.
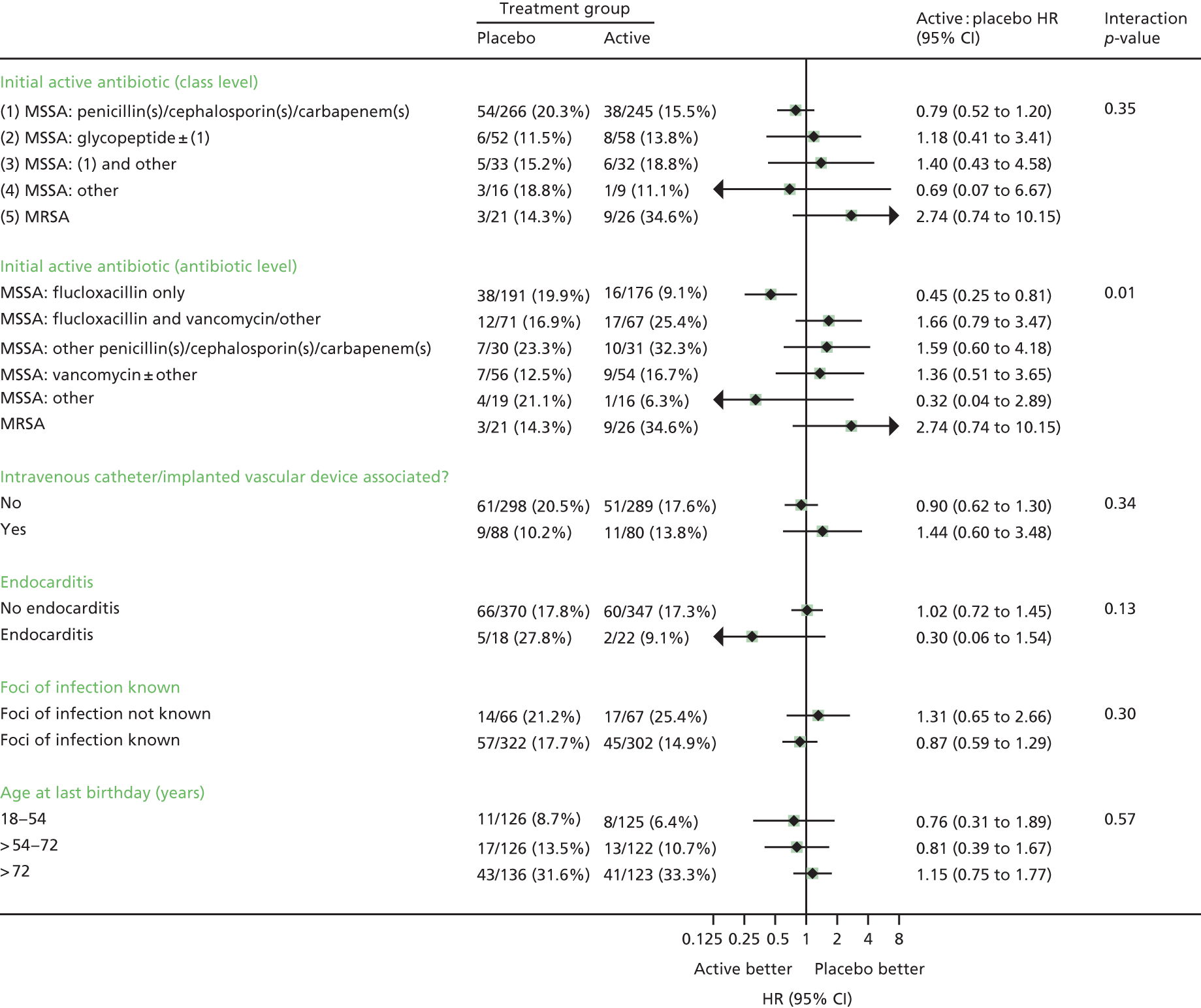
FIGURE 7.
Twelve other subgroup analyses for bacteriological failure/recurrence or death through 12 weeks (primary end point). Bc, positive blood culture; first/gent, first administered/gentamicin; first+, first positive blood culture. a, Subgroup analysis prespecified in the statistical analysis plan but not the protocol. b, p = 0.07 using continuous interactions (splines); p = 0.01 using continuous interaction (linear). c, Additional subgroup analysis not in protocol or statistical analysis plan.
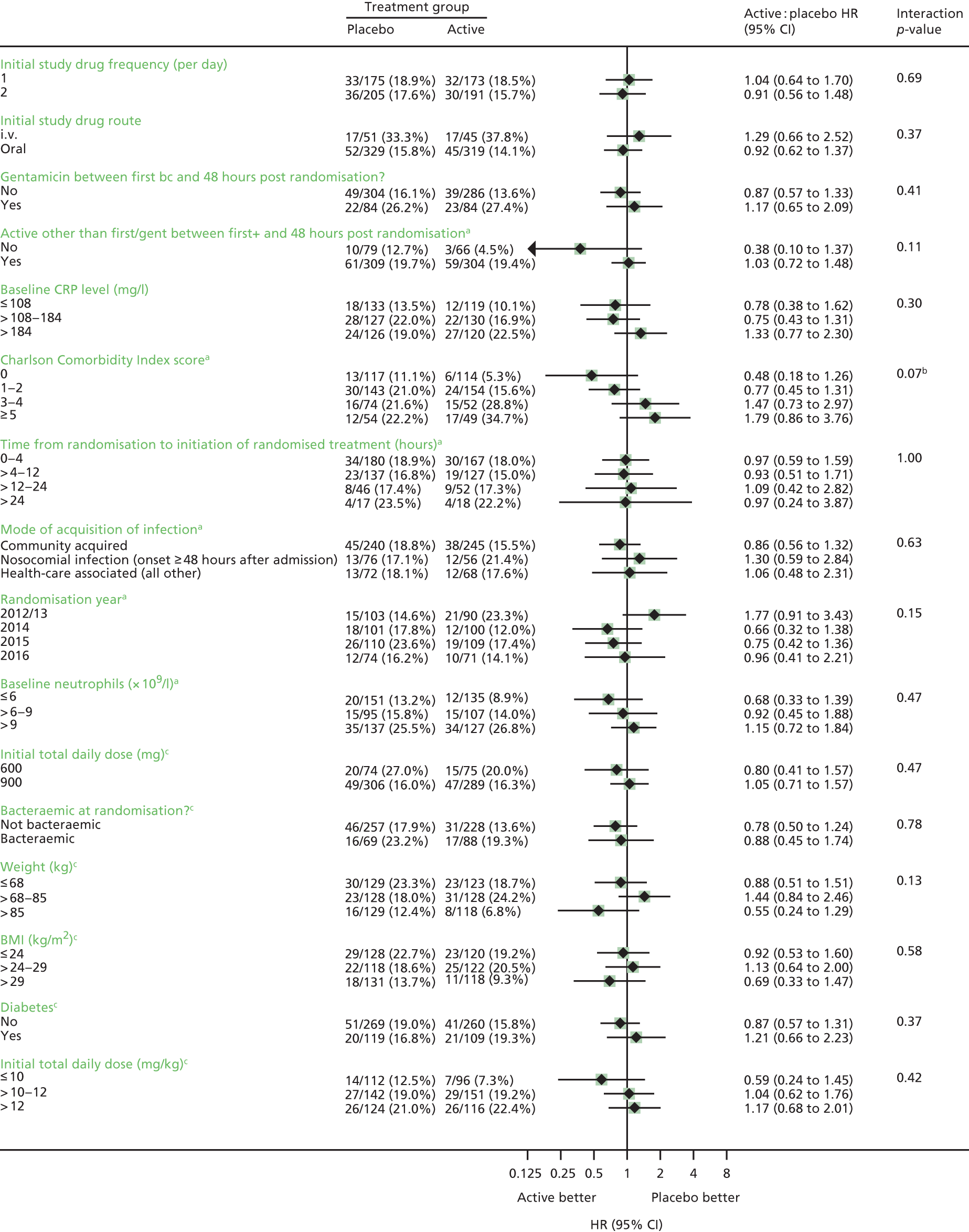
Secondary end points
Clinically defined failure/recurrence or death occurred in 76 (20.5%) participants in the rifampicin group versus 86 (22.2%) participants in the placebo group (RD –1.4%, 95% CI –7.4% to 4.7%; HR 0.97, 95% CI 0.71 to 1.32; p = 0.84 (Figure 8). In exploratory post hoc analyses comparing rifampicin and placebo there were 23 (6.2%) versus 25 (6.4%) failures (competing risks p = 0.97), 8 (2.2%) versus 23 (5.9%) recurrences (competing risks p = 0.01) and 45 (12.2%) versus 38 (9.8%) deaths without clinically defined failure/recurrence, respectively (competing risks p = 0.22) (see Table 7). The NNT to prevent one clinically confirmed recurrence was 26 participants.
FIGURE 8.
Clinically defined failure/recurrence or death. HR 0.97, 95% CI 0.71 to 1.32; p = 0.84. © The Author(s). Published by Elsevier Ltd. This is an open access article published under the CC BY 4.0 licence.
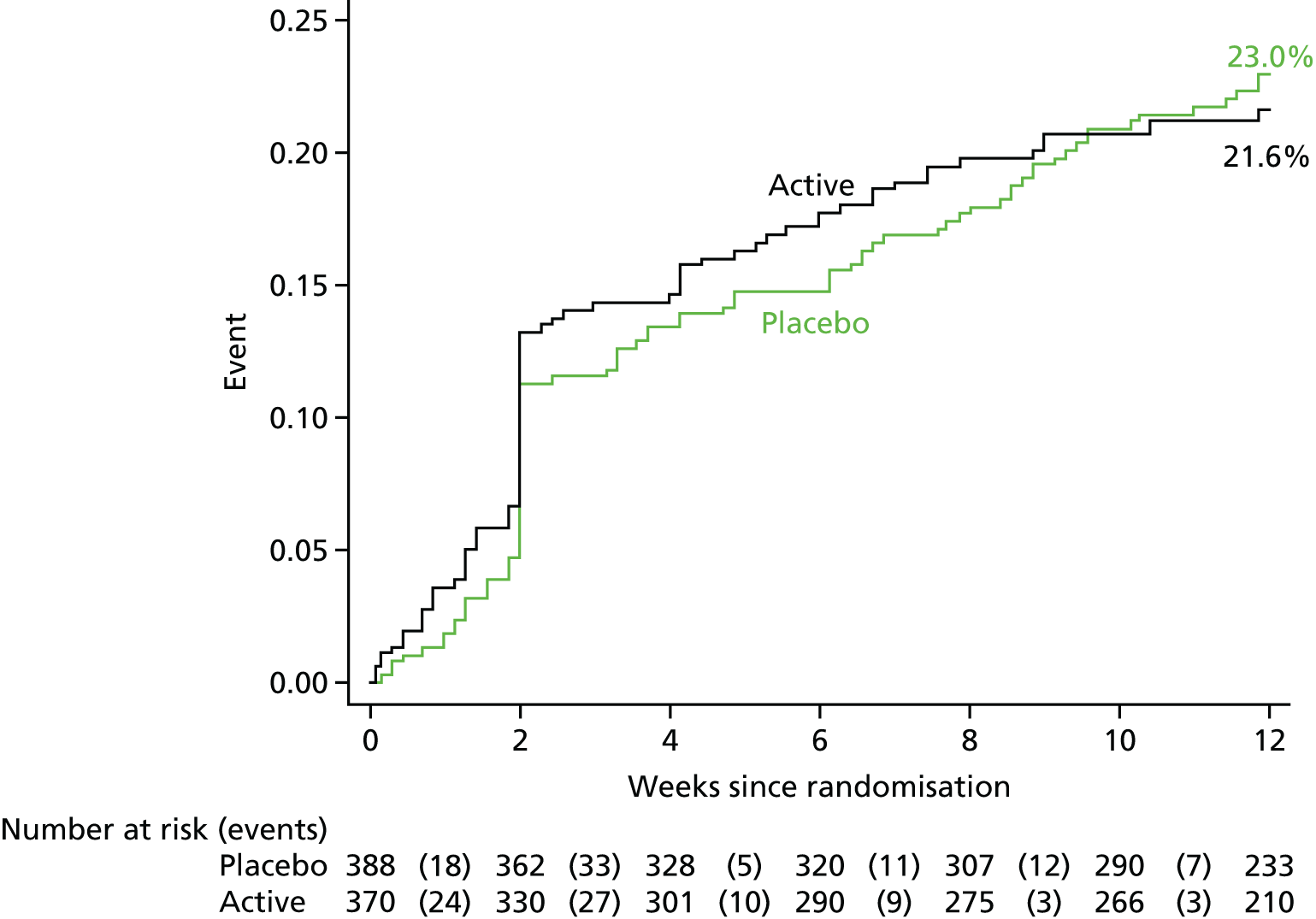
The end-point review committee adjudicated that failure of infection focus management was implicated in 38 out of 48 (79%) on placebo versus 24 out of 31 (77%) failures/recurrences on rifampicin (see Table 7). Of these failures of infection focus management, there were five participants in the placebo group versus 12 participants in the rifampicin group for whom the focus was not recognised, 16 participants in the placebo group versus 14 participants in the rifampicin group in which the focus was recognised but not actively managed (e.g. because it was in an inaccessible site, or other patient characteristics made intervention impossible) and 10 participants in the placebo group versus five participants in the rifampicin group for whom the focus was recognised and actively managed, but despite this failure/recurrence still occurred. Failure of antibiotic therapy was implicated in the failure/recurrence in only 3 (6%) placebo versus 1 (3%) rifampicin failures/recurrences, with the cause being impossible to distinguish in the remaining 7 (15%) versus 6 (19%), respectively.
By 12 weeks, 56 (15.1%) participants in the rifampicin group versus 56 (14.4%) participants in the placebo group had died (RD 1.0%, 95% CI –4.3% to 6.2%; HR 1.10, 95% CI 0.76 to 1.60; p = 0.60) (Figure 9). A total of 25 (6.8%) participants in the rifampicin group versus 17 (4.4%) participants in the placebo group died before 2 weeks (HR 1.60, 95% CI 0.86 to 2.95; p = 0.13). Fourteen deaths in the rifampicin group versus 16 deaths in the placebo group were adjudicated definitely S. aureus related, 14 deaths in the rifampicin group versus 12 deaths in the placebo group were probably S. aureus related, and 8 deaths in the rifampicin group versus 4 deaths in the placebo group were possibly S. aureus related (see Appendix 2, Table 29). A total of 18 deaths in the rifampicin group versus 23 deaths in the placebo group were not attributed to S. aureus (remainder unattributable) (overall p = 0.64).
FIGURE 9.
Mortality through 12 weeks. (Week 2: HR 1.60, 95% CI 0.86 to 2.95; p = 0.13. Week 12: HR 1.10, 95% CI 0.76 to 1.60; p = 0.60.) © The Author(s). Published by Elsevier Ltd. This is an open access article published under the CC BY 4.0 licence.
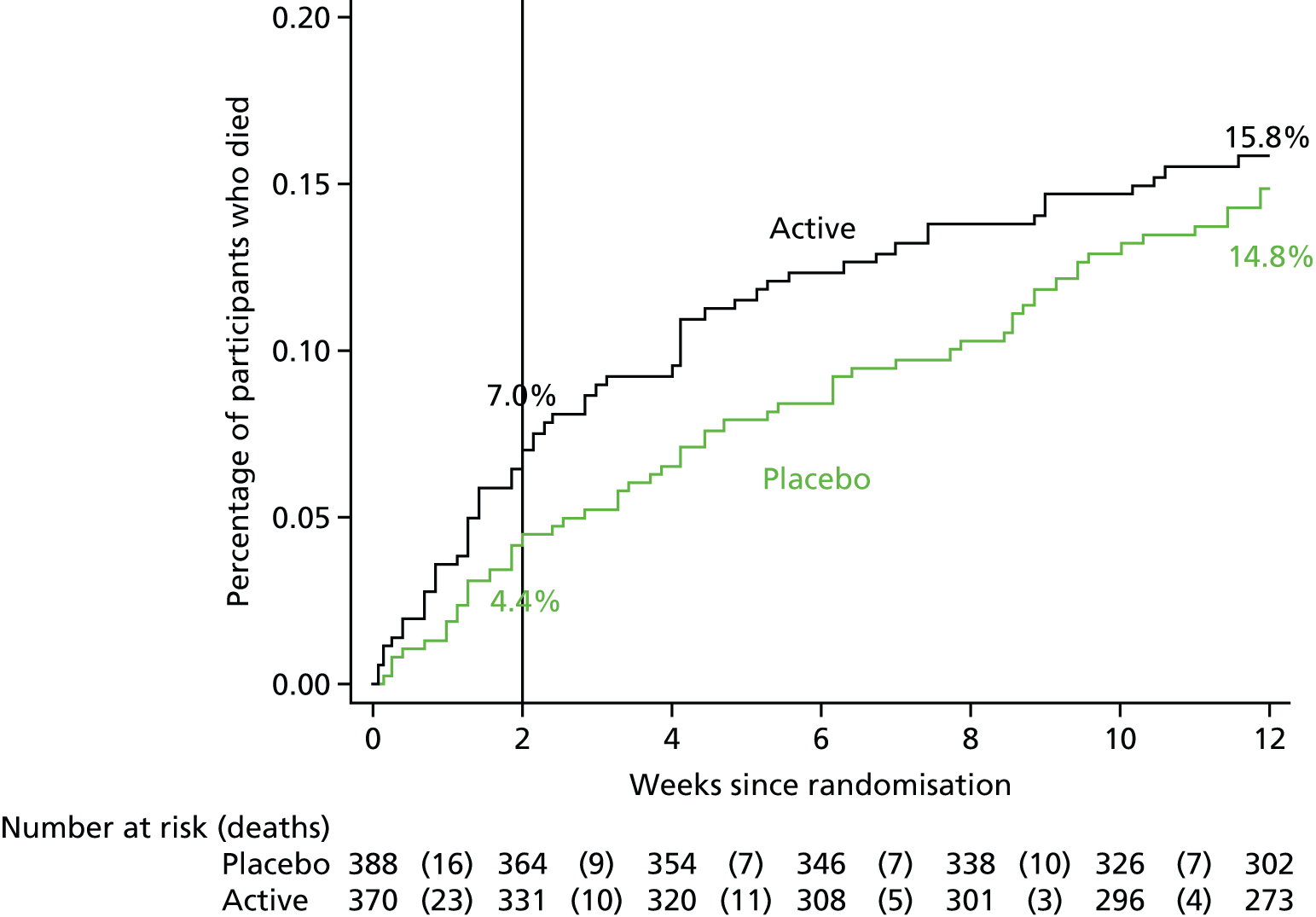
As for clinically defined and bacteriologically confirmed failures/recurrences, the end-point review committee adjudicated that failure of infection focus management was implicated in most S. aureus-related deaths, 21 out of 32 (66%) on placebo versus 18 out of 36 (50%) on rifampicin (see Table 7). Of these failures of infection focus management, there were three participants in the placebo group and four participants in the rifampicin group for whom the focus was not recognised, eight participants in the placebo group and eight participants in the rifampicin group for whom the focus was recognised but not actively managed, and 10 participants in the placebo group and six participants in the rifampicin group for whom the focus was recognised and actively managed, but despite this the participant still died from S. aureus. Failure of antibiotic therapy was implicated in only one (3%) S. aureus-related death in the placebo group versus three (8%) S. aureus-related deaths in the rifampicin group, with the relationship to antibiotics/focus management being impossible to distinguish in the remaining 10 (31%) and 15 (42%) participants, respectively. Three (9%) S. aureus-related deaths in the placebo group versus 11 (31%) S. aureus-related deaths in the rifampicin group were considered to have occurred as a consequence of late presentation to health care (i.e. were not preventable).
There was no difference in longer-term (post week 12) survival between the groups, based on consented updates of vital status from routine electronic health records (p = 0.69) (Figure 10).
FIGURE 10.
Mortality over the longer term. (HR 0.95, 95% CI 0.72 to 1.24; p = 0.69.)
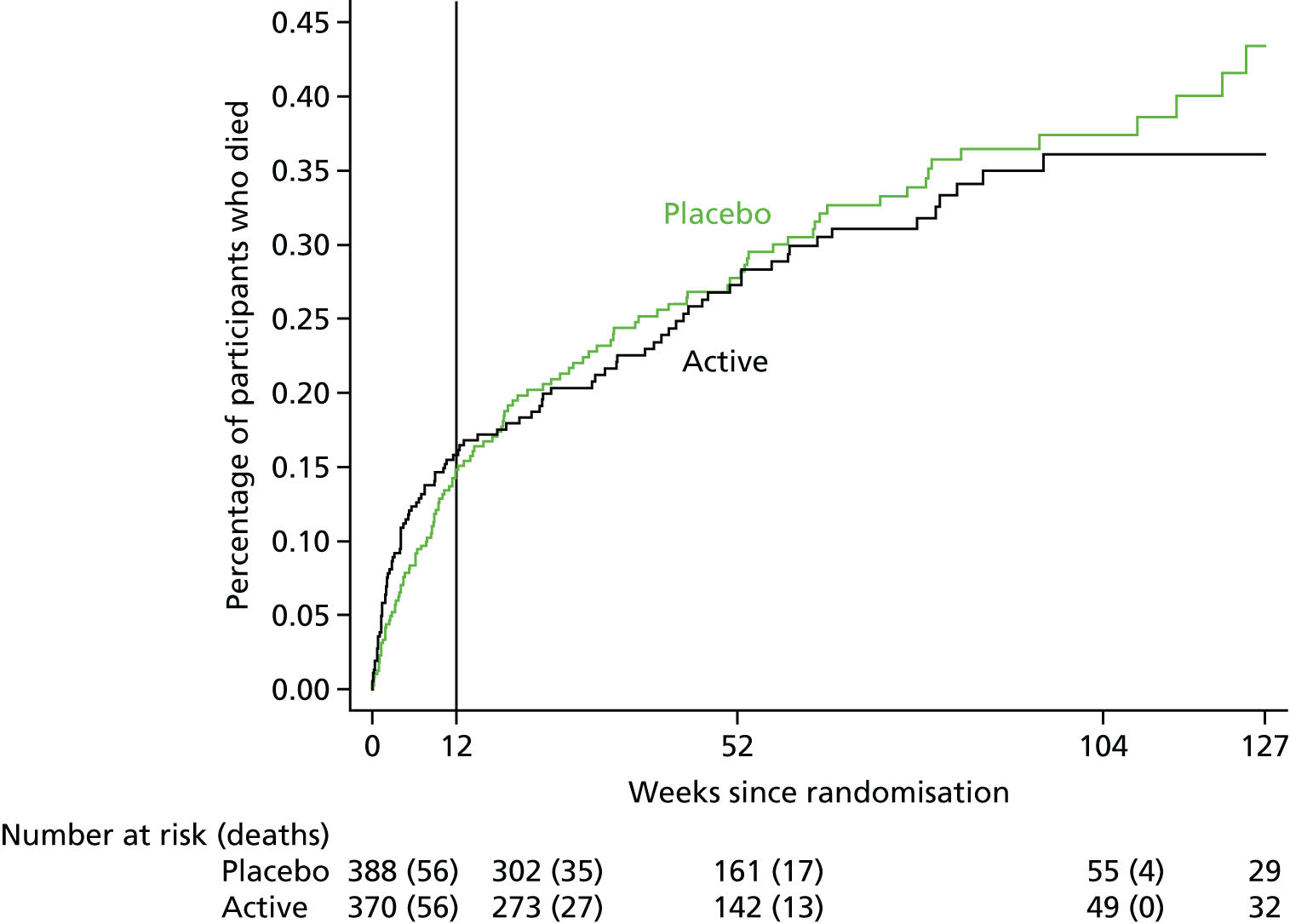
Two (0.5%) participants in the rifampicin group developed new rifampicin-resistant S. aureus bacteraemia at 7 and 42 days after randomisation (p = 0.24). One occurred on day 7 (followed by rifampicin discontinuation on day 11 and bacteriological failure on day 14), and the other on day 42 (prescribed 14 days of rifampicin and bacteriological recurrence on day 42). One additional participant had rifampicin-resistant S. aureus isolated from a permanent pacemaker wire removed on day 1 (within 4 hours of the first dose of trial drug). The screening blood culture had isolated a rifampicin-sensitive S. aureus. Further blood cultures were sterile for the remainder of follow-up. Following whole-genome sequencing, the rifampicin-resistant pacemaker isolate was 11 single nucleotide polymorphisms from the screening isolate and another isolate taken from the pacemaker on day 1, whereas these last two isolates did not differ genetically, suggesting a diversity between isolates of > 3 days in origin, and thus suggesting that the patient had a mixed infection with both rifampicin-resistant and rifampicin-susceptible strains that were not detected at screening.
There was no evidence that duration of bacteraemia was significantly shorter in those randomised to the rifampicin group (Figure 11; global p = 0.66). Eighty-eight patients in the rifampicin group had positive blood cultures at enrolment. Of these 88, only one failed bacteriologically, none had bacteriological recurrence and none developed rifampicin-resistant infection. Eight failed clinically (including the one who failed bacteriologically) and two had clinical recurrence.
FIGURE 11.
Persistence of bacteraemia. Global test of difference between groups across days 3 and 7: p = 0.66. Test of difference between groups at day 3 in those bacteraemic at day 0: p = 0.36.
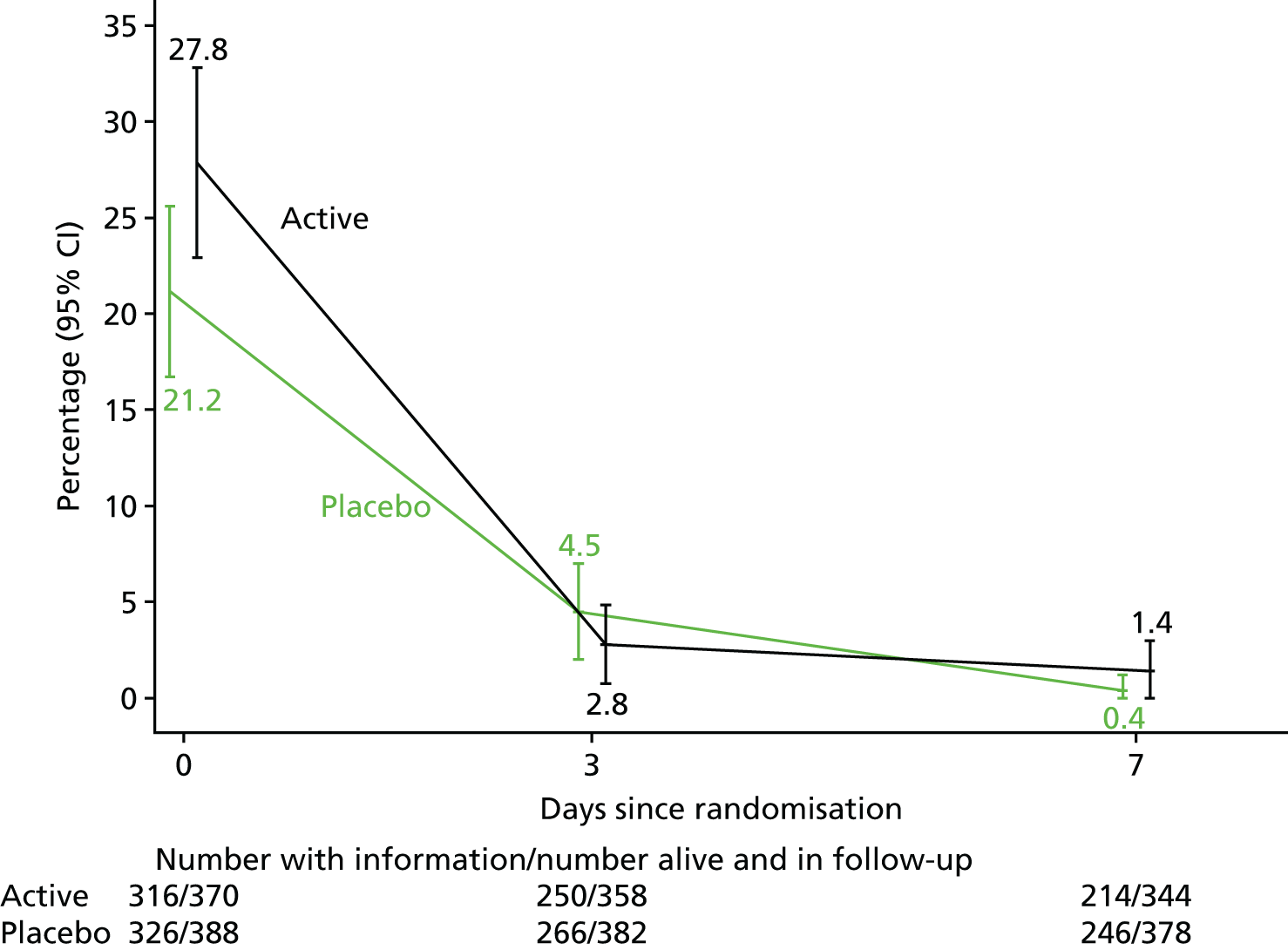
The CRP level declined significantly in both the rifampicin and the placebo groups, but decreases were smaller in participants in the rifampicin group (global p = 0.001, Figure 12).
FIGURE 12.
C-reactive protein levels over 2 weeks from randomisation. Note that p-values compare change from baseline across randomised groups.
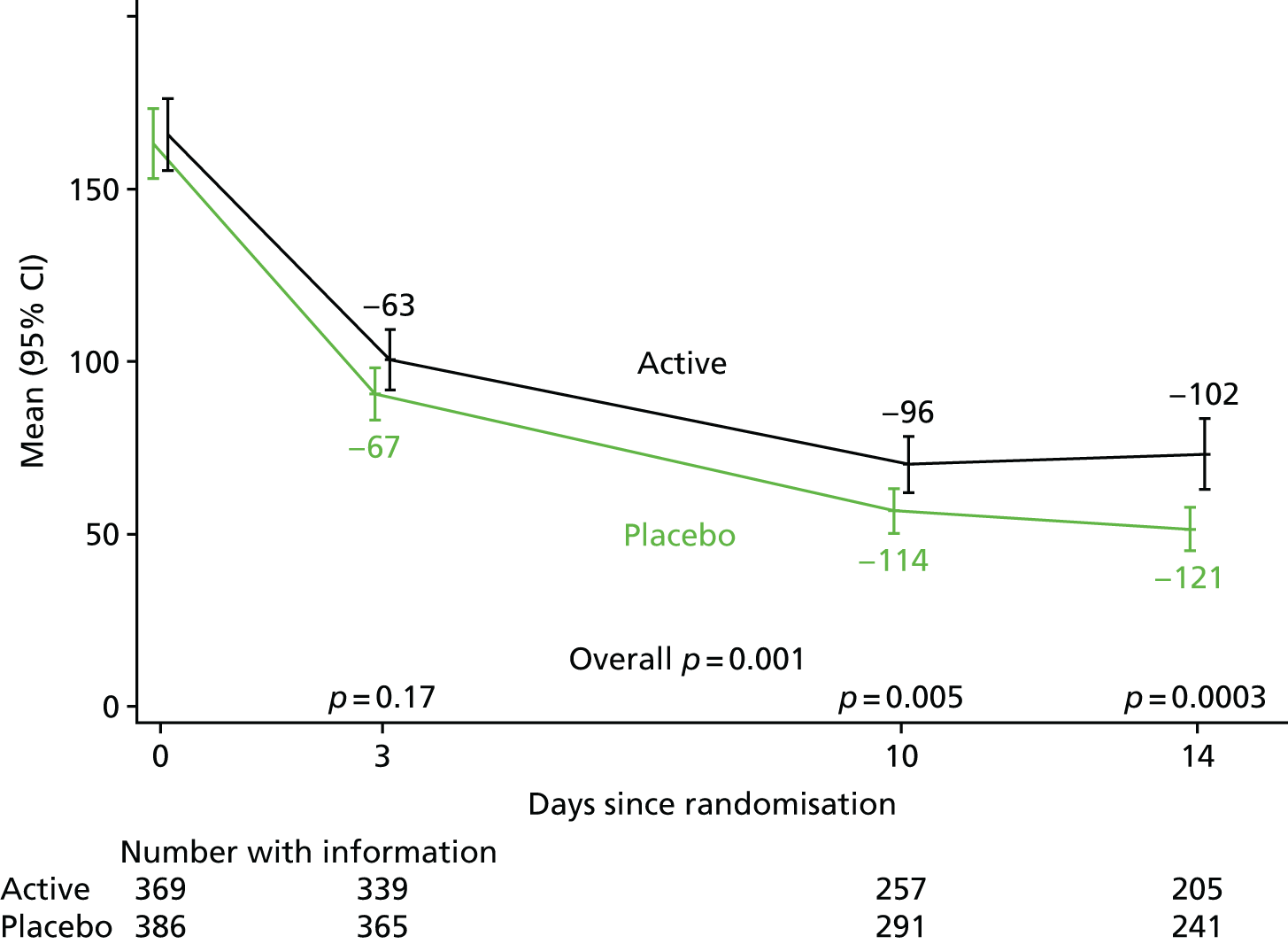
Safety
By 12 weeks, 101 (27.3%) participants in the rifampicin group versus 94 (24.2%) participants in the placebo group experienced 112 versus 116 SAEs, respectively (HR 1.21, 95% CI 0.92 to 1.61; p = 0.17) (Figure 13 and Table 8; see also Appendix 2, Table 30). The most common type of SAE was related to infections and infestations, and the vast majority were because of fatal events caused by S. aureus bacteraemia (non-fatal S. aureus-related AEs were exempted from AE reporting in the protocol to avoid double-counting disease failure/recurrence events).
FIGURE 13.
Time to first SAE. (p = 0.17 log-rank test.)
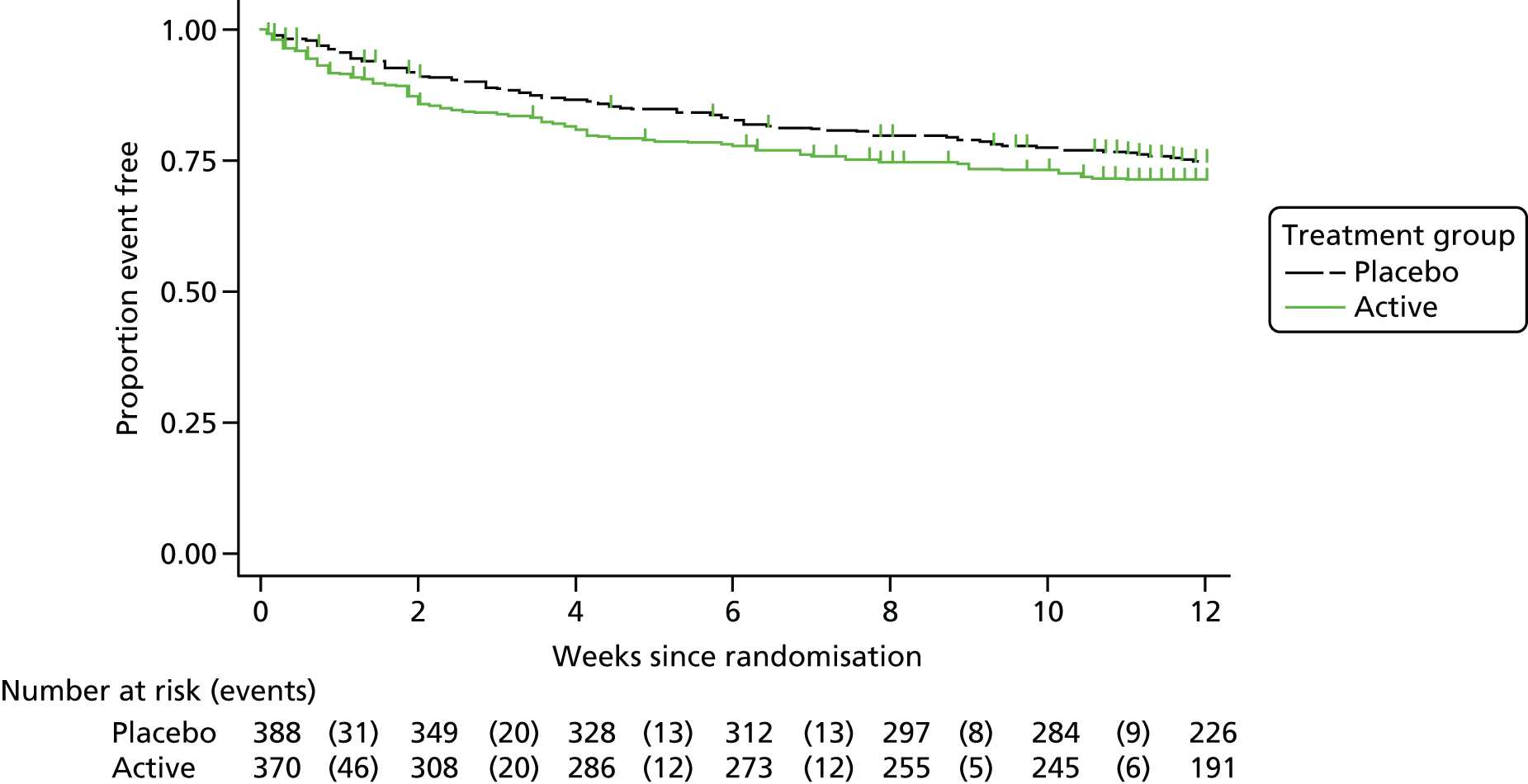
| SAEs | Treatment group | Total (N = 758) | p-value | |
|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | |||
| Any SAE | 94 (24.2); 116 | 101 (27.3); 112 | 195 (25.7); 228 | 0.36 |
| Infections and infestations | 39 (10.1); 40 | 37 (10.0); 38 | 76 (10.0); 78 | 1.00 |
| Cardiac disorders | 13 (3.4); 15 | 5 (1.4); 6 | 18 (2.4); 21 | 0.09 |
| Vascular disorders | 2 (0.5); 2 | 4 (1.1); 4 | 6 (0.8); 6 | 0.44 |
| Respiratory, thoracic and mediastinal disorders | 12 (3.1); 12 | 6 (1.6); 6 | 18 (2.4); 18 | 0.23 |
| Gastrointestinal disorders | 7 (1.8); 7 | 10 (2.7); 12 | 17 (2.2); 19 | 0.47 |
| Hepatobiliary disorders | 0 (0.0); 0 | 2 (0.5); 2 | 2 (0.3); 2 | 0.24 |
| Skin and subcutaneous tissue disorders | 1 (0.3); 1 | 1 (0.3); 1 | 2 (0.3); 2 | 1.00 |
| Renal and urinary disorders | 4 (1.0); 4 | 10 (2.7); 10 | 14 (1.8); 14 | 0.11 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 7 (1.8); 7 | 11 (3.0); 12 | 18 (2.4); 19 | 0.34 |
| Congenital, familial and genetic disorders | 1 (0.3); 1 | 0 (0.0); 0 | 1 (0.1); 1 | 1.00 |
| General disorders and administration site conditions | 12 (3.1); 12 | 11 (3.0); 11 | 23 (3.0); 23 | 1.00 |
| Investigations | 0 (0.0); 0 | 1 (0.3); 1 | 1 (0.1); 1 | 0.49 |
| Injury, poisoning and procedural complications | 5 (1.3); 5 | 3 (0.8); 3 | 8 (1.1); 8 | 0.73 |
| Blood and lymphatic system disorders | 1 (0.3); 1 | 1 (0.3); 1 | 2 (0.3); 2 | 1.00 |
| Metabolism and nutrition disorders | 1 (0.3); 1 | 3 (0.8); 3 | 4 (0.5); 4 | 0.36 |
| Psychiatric disorders | 2 (0.5); 2 | 0 (0.0); 0 | 2 (0.3); 2 | 0.50 |
| Nervous system disorders | 5 (1.3); 6 | 2 (0.5); 2 | 7 (0.9); 8 | 0.45 |
Two participants in the rifampicin group with pre-existing liver disease experienced non-fatal hepatic failure.
One 47-year-old female required prolongation of hospitalisation for acute hepatic failure (grade 3) with, raised international normalised ratio (INR) (grade 2), ascites (grade 3) and acute renal failure (grade 3), which developed on ICU following 5 days of rifampicin (at a dose of 900 mg daily) with flucloxacillin. The participant had pre-existing hepatitis C and chronic liver disease. Acute hepatic and renal failure were considered to have been triggered by sepsis. The participant recovered.
One 51-year-old female required prolongation of hospitalisation for decompensated liver disease (grade 3) with ascites (grade 3) following 14 days’ rifampicin (initially on a dose of 900 mg daily) with flucloxacillin. The participant did not mention liver disease at screening/enrolment and there was nothing in her medical notes regarding any past history of liver problems. When she developed decompensated liver disease with ascites, it was discovered that she had a previous diagnosis of non-alcoholic steatosis at another hospital several years previously, but was no longer being followed up. The participant recovered.
A total of 129 (34.9%) participants in the rifampicin group versus 131 (33.8%) participants in the placebo group experienced 209 versus 193 grade 3/4 AEs, respectively, (HR 1.12, 95% CI 0.88 to 1.43; p = 0.36) (Figure 14 and Table 9; see also Appendix 2, Table 31). Most notable was a trend towards more renal grade 3/4 AEs with rifampicin, which occurred in 19 (5.1%) participants in the rifampicin group and 9 (2.3%) participants in the placebo group (p = 0.053); 17 and 6 of which, respectively, were acute kidney injury.
FIGURE 14.
Time to first grade 3 or 4 AE. (p = 0.36 log-rank test.)
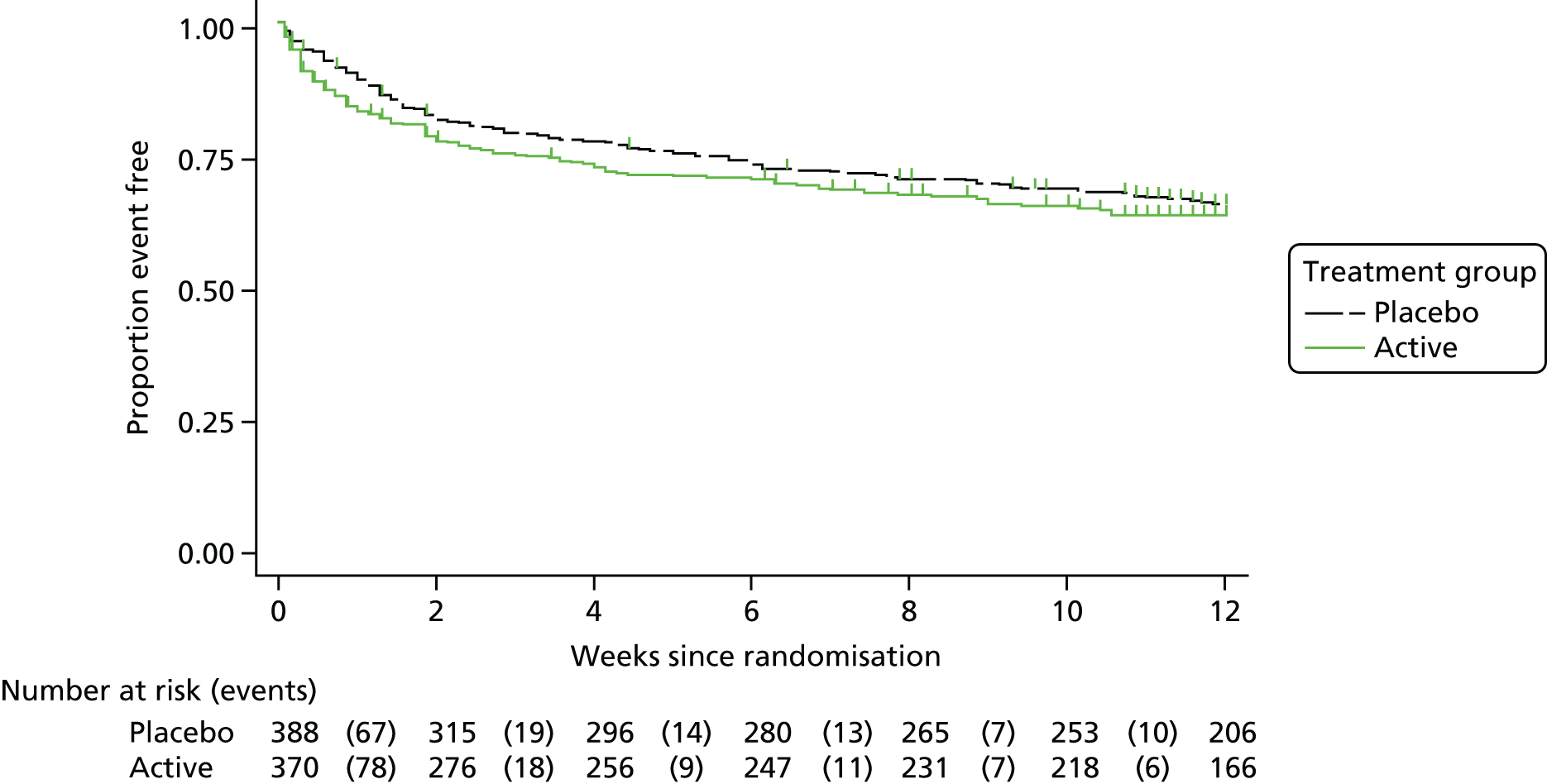
| Grade 3 or 4 AEs | Treatment group | Total (N = 758) | p-value | |
|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | |||
| Any grade 3 or 4 AE | 131 (33.8); 193 | 129 (34.9); 209 | 260 (34.3); 402 | 0.76 |
| Infections and infestations | 45 (11.6); 53 | 40 (10.8); 48 | 85 (11.2); 101 | 0.82 |
| Cardiac disorders | 15 (3.9); 17 | 6 (1.6); 8 | 21 (2.8); 25 | 0.08 |
| Vascular disorders | 7 (1.8); 7 | 5 (1.4); 5 | 12 (1.6); 12 | 0.77 |
| Respiratory, thoracic and mediastinal disorders | 16 (4.1); 17 | 10 (2.7); 11 | 26 (3.4); 28 | 0.32 |
| Gastrointestinal disorders | 21 (5.4); 24 | 29 (7.8); 40 | 50 (6.6); 64 | 0.19 |
| Hepatobiliary disorders | 0 (0.0); 0 | 3 (0.8); 3 | 3 (0.4); 3 | 0.12 |
| Skin and subcutaneous tissue disorders | 7 (1.8); 7 | 5 (1.4); 5 | 12 (1.6); 12 | 0.77 |
| Musculoskeletal and connective tissue disorders | 2 (0.5); 2 | 0 (0.0); 0 | 2 (0.3); 2 | 0.50 |
| Renal and urinary disorders | 9 (2.3); 9 | 19 (5.1); 20 | 28 (3.7); 29 | 0.053 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 7 (1.8); 7 | 11 (3.0); 12 | 18 (2.4); 19 | 0.34 |
| Reproductive system and breast disorders | 0 (0.0); 0 | 1 (0.3); 1 | 1 (0.1); 1 | 0.49 |
| Congenital, familial and genetic disorders | 1 (0.3); 1 | 0 (0.0); 0 | 1 (0.1); 1 | 1.00 |
| General disorders and administration site conditions | 11 (2.8); 11 | 12 (3.2); 12 | 23 (3.0); 23 | 0.83 |
| Investigations | 6 (1.5); 6 | 11 (3.0); 16 | 17 (2.2); 22 | 0.22 |
| Injury, poisoning and procedural complications | 6 (1.5); 6 | 5 (1.4); 5 | 11 (1.5); 11 | 1.00 |
| Surgical and medical procedures | 0 (0.0); 0 | 1 (0.3); 1 | 1 (0.1); 1 | 0.49 |
| Blood and lymphatic system disorders | 3 (0.8); 3 | 5 (1.4); 6 | 8 (1.1); 9 | 0.50 |
| Metabolism and nutrition disorders | 3 (0.8); 3 | 5 (1.4); 6 | 8 (1.1); 9 | 0.50 |
| Psychiatric disorders | 5 (1.3); 5 | 5 (1.4); 6 | 10 (1.3); 11 | 1.00 |
| Nervous system disorders | 11 (2.8); 14 | 4 (1.1); 4 | 15 (2.0); 18 | 0.12 |
| Eye disorders | 1 (0.3); 1 | 0 (0.0); 0 | 1 (0.1); 1 | 1.00 |
A total of 63 (17.0%) participants in the rifampicin group versus 39 (10.1%) participants in the placebo group experienced 89 versus 52 antibiotic-modifying AEs, respectively, (subdistribution HR 1.78, 95% CI 1.20 to 2.65; p = 0.004) (Figure 15 and Table 10; see also Appendix 2, Table 32). Gastrointestinal disorders (24 vs. 8 participants, respectively, p = 0.003) and renal/urinary disorders (8 vs. 1 participants, respectively, p = 0.02) were more common in the rifampicin group, as were events classified as general disorders and administration site conditions (13 vs. 4 participants, respectively), which included some drug interactions (see below).
FIGURE 15.
Time to first antibiotic-modifying AE. (p = 0.004 subhazard regression.)
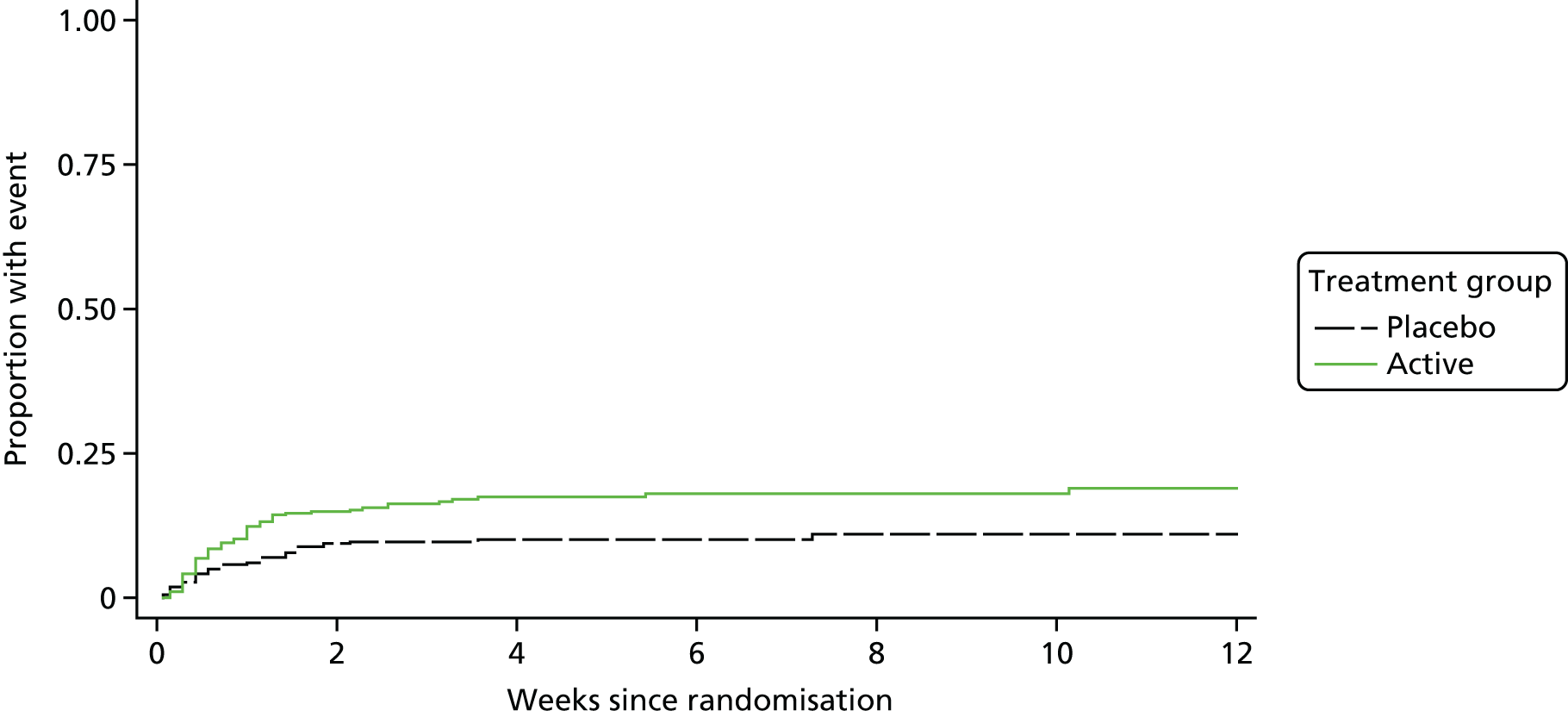
| Antibiotic-modifying AEs | Treatment group | Total (N = 758) | p-value | |
|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | |||
| Any antibiotic-modifying AE | 39 (10.1); 52 | 63 (17.0); 89 | 102 (13.5); 141 | 0.006 |
| Infections and infestations | 3 (0.8); 3 | 5 (1.4); 5 | 8 (1.1); 8 | 0.50 |
| Respiratory, thoracic and mediastinal disorders | 2 (0.5); 4 | 0 (0.0); 0 | 2 (0.3); 4 | 0.50 |
| Gastrointestinal disorders | 8 (2.1); 9 | 24 (6.5); 32 | 32 (4.2); 41 | 0.003 |
| Hepatobiliary disorders | 0 (0.0); 0 | 2 (0.5); 2 | 2 (0.3); 2 | 0.24 |
| Skin and subcutaneous tissue disorders | 7 (1.8); 9 | 8 (2.2); 9 | 15 (2.0); 18 | 0.80 |
| Renal and urinary disorders | 1 (0.3); 2 | 8 (2.2); 10 | 9 (1.2); 12 | 0.02 |
| General disorders and administration site conditions | 4 (1.0); 4 | 13 (3.5); 13 | 17 (2.2); 17 | 0.03 |
| Investigations | 12 (3.1); 13 | 12 (3.2); 14 | 24 (3.2); 27 | 1.00 |
| Injury, poisoning and procedural complications | 1 (0.3); 1 | 0 (0.0); 0 | 1 (0.1); 1 | 1.00 |
| Blood and lymphatic system disorders | 1 (0.3); 1 | 3 (0.8); 3 | 4 (0.5); 4 | 0.36 |
| Metabolism and nutrition disorders | 2 (0.5); 3 | 0 (0.0); 0 | 2 (0.3); 3 | 0.50 |
| Psychiatric disorders | 1 (0.3); 2 | 0 (0.0); 0 | 1 (0.1); 2 | 1.00 |
| Nervous system disorders | 1 (0.3); 1 | 1 (0.3); 1 | 2 (0.3); 2 | 1.00 |
A total of 24 (6.5%) participants in the rifampicin group versus 6 (1.5%) participants in the placebo group experienced drug interactions with antibiotics or other drugs (p = 0.0005). This led to the discontinuation of trial drug in 13 participants in the rifampicin group versus 4 participants in the placebo group (p = 0.03), grade 1/2 AEs in 14 versus 3 participants, respectively (p = 0.006), and grade 3/4 AEs in five versus two participants, respectively (p = 0.27).
There was no evidence of differences between groups in changes in ALT (global p = 0.18, Figure 16) or alkaline phosphatase (global p = 0.11, Figure 17). Bilirubin increased significantly in the rifampicin group at day 3 (p < 0.0001; global p < 0.0001; Figure 18). Very few participants experienced grade 3 or 4 elevations in these laboratory parameters (Table 11).
FIGURE 16.
Alanine transaminase levels over 2 weeks from randomisation. Note that p-values compare change from baseline across randomised groups.
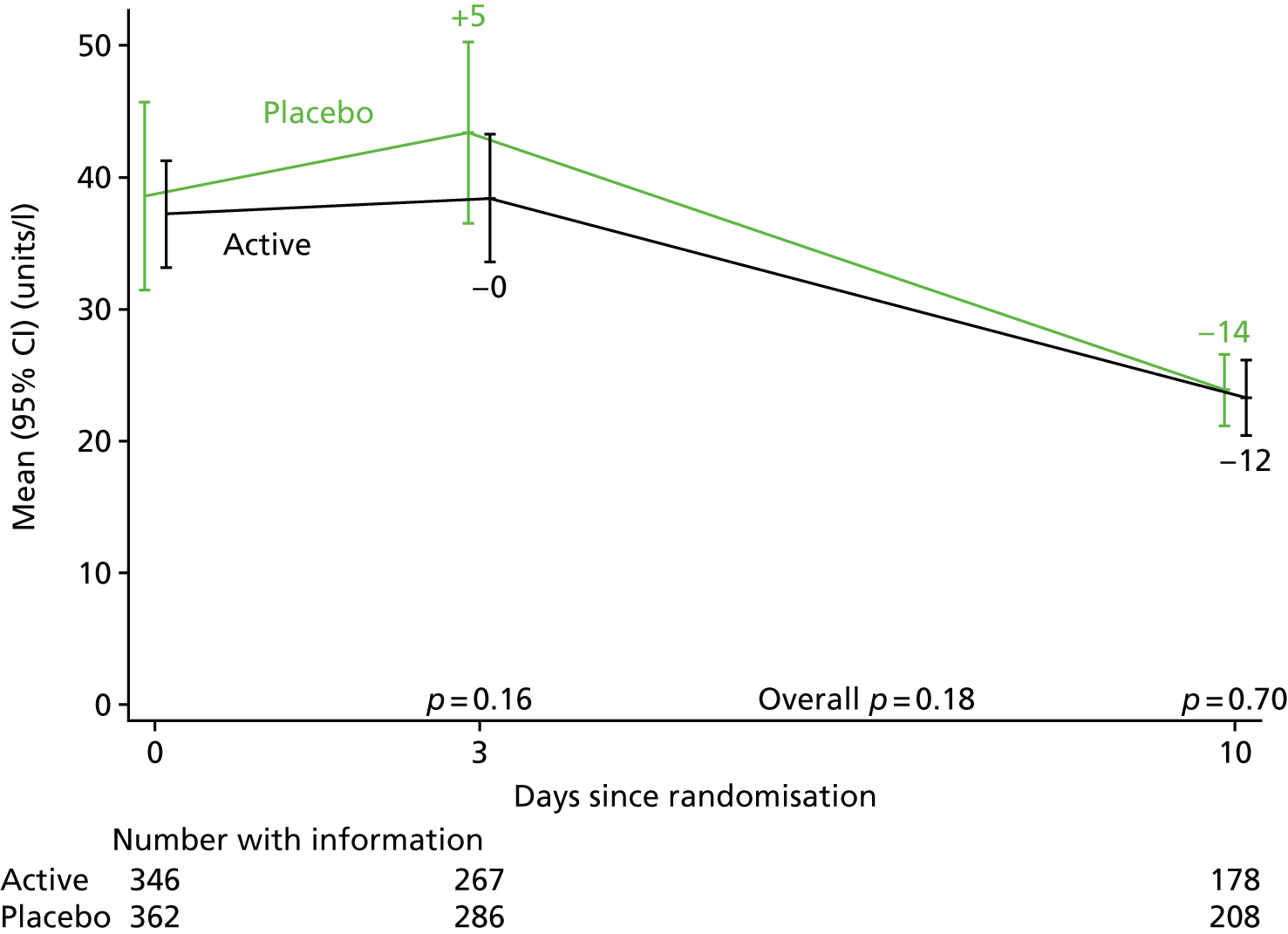
FIGURE 17.
Alkaline phosphatase levels over 2 weeks from randomisation. Note that p-values compare change from baseline across randomised groups.
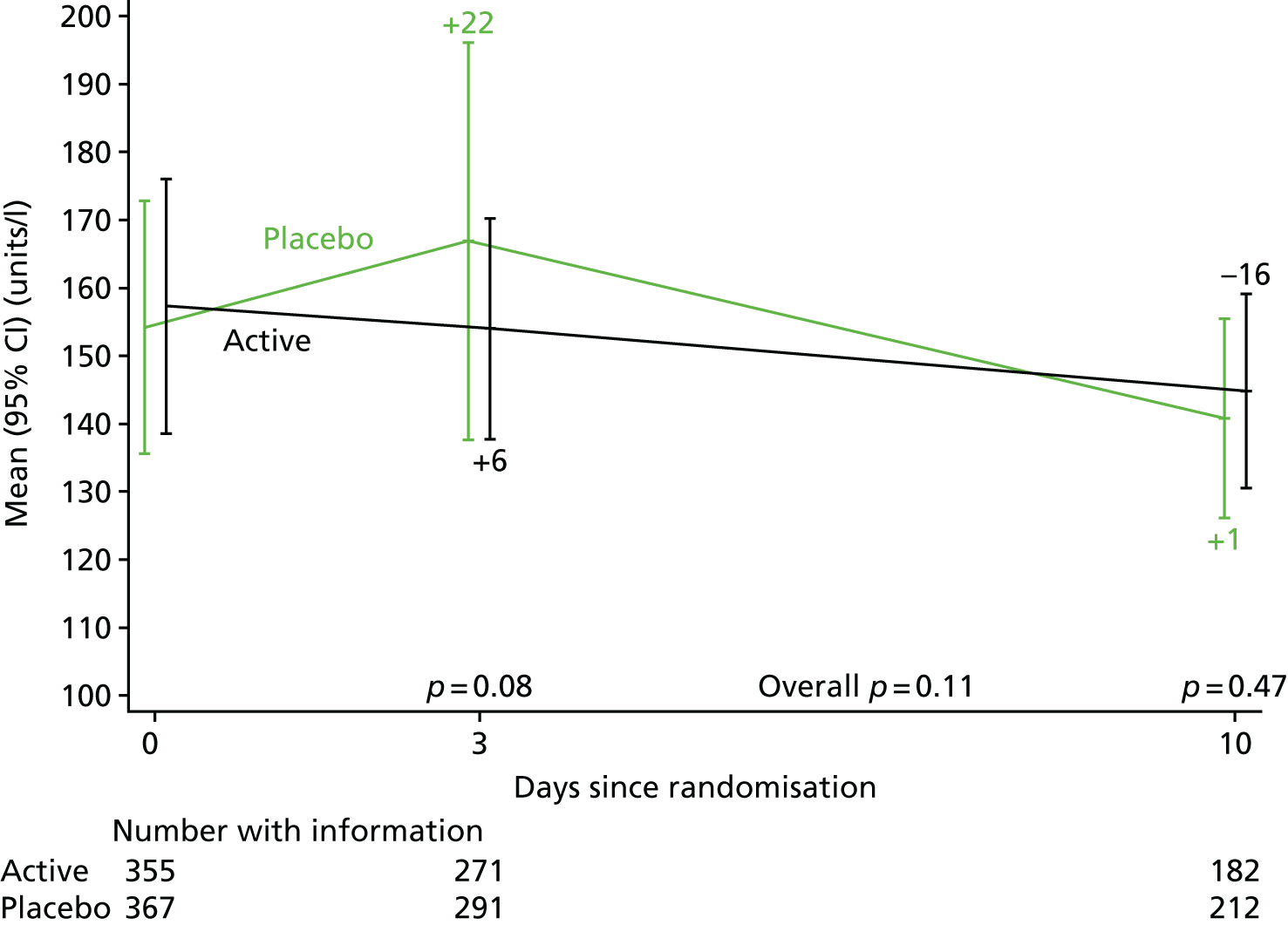
FIGURE 18.
Bilirubin levels over 2 weeks from randomisation. Note that p-values compare change from baseline across randomised groups.
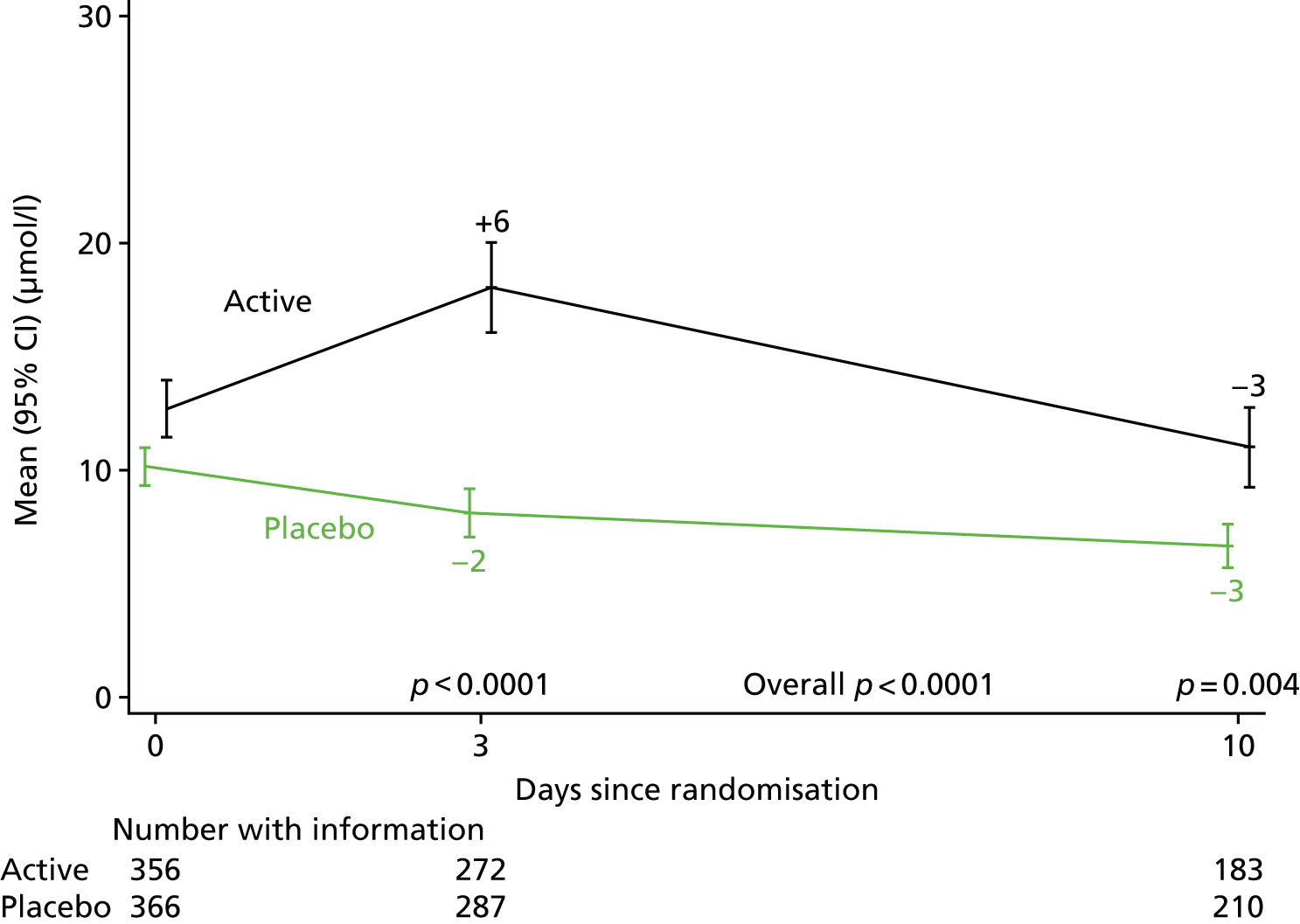
| Toxicity | Treatment group | Total | |
|---|---|---|---|
| Placebo | Rifampicin | ||
| ALT levels: day 0, n (%) | |||
| Normal | 274 (74.5) | 268 (75.1) | 542 (74.8) |
| > ULN – 3.0 × ULN (grade 1) | 85 (23.1) | 81 (22.7) | 166 (22.9) |
| > 3.0 – 5.0 × ULN (grade 2) | 6 (1.6) | 5 (1.4) | 11 (1.5) |
| > 5.0 – 20.0 × ULN (grade 3) | 2 (0.5) | 3 (0.8) | 5 (0.7) |
| > 20.0 × ULN (grade 4) | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| ALT levels: day 3, n (%) | |||
| Normal | 202 (70.6) | 203 (76.0) | 405 (73.2) |
| > ULN – 3.0 × ULN (grade 1) | 70 (24.5) | 50 (18.7) | 120 (21.7) |
| > 3.0 – 5.0 × ULN (grade 2) | 8 (2.8) | 12 (4.5) | 20 (3.6) |
| > 5.0 – 20.0 × ULN (grade 3) | 6 (2.1) | 2 (0.7) | 8 (1.4) |
| ALT levels: day 10, n (%) | |||
| Normal | 182 (87.5) | 160 (89.9) | 342 (88.6) |
| > ULN – 3.0 × ULN (grade 1) | 24 (11.5) | 17 (9.6) | 41 (10.6) |
| > 3.0 – 5.0 × ULN (grade 2) | 2 (1.0) | 1 (0.6) | 3 (0.8) |
| Alkaline phosphatase levels: day 0, n (%) | |||
| Normal | 267 (71.8) | 252 (69.2) | 519 (70.5) |
| > ULN – 2.5 × ULN (grade 1) | 90 (24.2) | 101 (27.7) | 191 (26.0) |
| > 2.5 – 5.0 × ULN (grade 2) | 13 (3.5) | 9 (2.5) | 22 (3.0) |
| > 5.0 – 20.0 × ULN (grade 3) | 2 (0.5) | 2 (0.5) | 4 (0.5) |
| Alkaline phosphatase levels: day 3, n (%) | |||
| Normal | 196 (67.4) | 175 (64.6) | 371 (66.0) |
| > ULN – 2.5 × ULN (grade 1) | 82 (28.2) | 91 (33.6) | 173 (30.8) |
| > 2.5 – 5.0 × ULN (grade 2) | 11 (3.8) | 2 (0.7) | 13 (2.3) |
| > 5.0 – 20.0 × ULN (grade 3) | 1 (0.3) | 3 (1.1) | 4 (0.7) |
| > 20.0 × ULN (grade 4) | 1 (0.3) | 0 (0.0) | 1 (0.2) |
| Alkaline phosphatase levels: day 10, n (%) | |||
| Normal | 149 (70.3) | 119 (65.4) | 268 (68.0) |
| > ULN – 2.5 × ULN (grade 1) | 57 (26.9) | 58 (31.9) | 115 (29.2) |
| > 2.5 – 5.0 × ULN (grade 2) | 6 (2.8) | 5 (2.7) | 11 (2.8) |
| Bilirubin levels: day 0, n (%) | |||
| Normal | 341 (91.7) | 309 (85.1) | 650 (88.4) |
| > ULN – 1.5 × ULN (grade 1) | 14 (3.8) | 31 (8.5) | 45 (6.1) |
| > 1.5 – 3.0 × ULN (grade 2) | 17 (4.6) | 17 (4.7) | 34 (4.6) |
| > 3.0 – 10.0 × ULN (grade 3) | 0 (0.0) | 6 (1.7) | 6 (0.8) |
| Bilirubin levels: day 3, n (%) | |||
| Normal | 270 (94.1) | 190 (69.9) | 460 (82.3) |
| > ULN – 1.5 × ULN (grade 1) | 11 (3.8) | 35 (12.9) | 46 (8.2) |
| > 1.5 – 3.0 × ULN (grade 2) | 3 (1.0) | 35 (12.9) | 38 (6.8) |
| > 3.0 – 10.0 × ULN (grade 3) | 3 (1.0) | 12 (4.4) | 15 (2.7) |
| Bilirubin levels: day 10, n (%) | |||
| Normal | 200 (95.2) | 162 (88.5) | 362 (92.1) |
| > ULN – 1.5 × ULN (grade 1) | 6 (2.9) | 7 (3.8) | 13 (3.3) |
| > 1.5 – 3.0 × ULN (grade 2) | 4 (1.9) | 12 (6.6) | 16 (4.1) |
| > 3.0 – 10.0 × ULN (grade 3) | 0 (0.0) | 2 (1.1) | 2 (0.5) |
Chapter 4 Trial participation qualitative substudy
Experiences of being approached for trial participation, the consenting process and trial participation
The overall objective of this substudy was to identify patient and personal legal representative barriers to recruitment. The substudy was led by Jennifer Bostock, the trial PPI representative. The substudy had two components: the first involved patients/legal representatives who did not consent to trial recruitment, and the second involved patients/legal representatives who did consent to trial recruitment.
Patient/legal representatives who did not consent to trial recruitment
The overall objective of this substudy was to identify patient and legal representative barriers to recruitment, to:
-
aid learning about why patients/legal representatives did not consent to being in this trial and whether or not there are any improvements that can be made to the information-giving and/or consent process that may encourage greater participation in a future similar study
-
give patients/legal representatives choosing not to join the study a voice so that researchers can learn of any unintended barriers in the way that information is given and/or consent taken when recruiting patients with serious illness.
At the time that they did not consent to the study, patients/legal representatives from all participating NHS trusts were given a short, completely anonymous questionnaire with a Freepost envelope, which could be completed at any time in the future and posted directly to the MRC Clinical Trials Unit (CTU) at UCL. Health-care professionals involved in consenting patients to the ARREST trial and who were asked to act as legal representatives but did not consent for the patient to join the study were provided a parallel questionnaire.
At the Guy’s and St Thomas’ centre, at the end of the questionnaire, participants/legal representatives were offered the option of being interviewed by the ARREST trial PPI advisor. If they agreed to be interviewed, they were asked to provide their name and contact details, and this would indicate consent for an interview. The aim was to get experiences from approximately three participants and approximately three personal legal representatives (who were not health-care professionals) not providing consent to join the trial. However, this would continue to get experiences from up to 10 participants and legal representatives if new views and experiences continued to be expressed (i.e. had not reached saturation). The interview guide followed the questions in the questionnaire, seeking to obtain a more complete narrative of experiences around each aspect.
Patient/legal representatives who did consent to trial recruitment
The overall objective was to sample views on experiences of trial participation, that is, to assess what participants or their (personal) legal representatives liked, and what they did not like and think could have been done better. This was to:
-
gain valuable insight into the experience of participating in such a trial, the reasoning behind participation and the pros and cons of being involved
-
gain an understanding of the ‘patient perspective’ and how this might inform future trials to improve them, and potentially how (at the time) the ongoing conduct of the ARREST trial could be improved
-
examine the process of consent and information-giving at the time of consenting the patient and whether or not there were any barriers that might be improved to aid recruitment in future
-
run as a parallel narrative alongside the feedback from clinicians and researchers involved in the study to explore differences and commonalities, and pool suggestions for improvements for future studies.
This was an interview study conducted at one centre, Guy’s and St Thomas’ NHS Trust. As participants were typically very unwell when they joined the study, the approach to each patient to discuss the interview study and seek additional consent was made at a variable time after randomisation depending on clinical status. For most patients, this was between 2 and 3 weeks from randomisation, when their clinical status had improved and discharge was being planned. However, it could have been at any time up to their final 12-week ARREST trial follow-up visit. The research nurse provided an additional information sheet to ask if they would be willing to have a short (20–30 minute) semistructured interview about their experiences of trial participation with the ARREST trial PPI advisor (not a member of the trial team). If they agreed and provided consent for this additional interview, then the ARREST trial PPI advisor conducted the interview on the telephone at a time that was convenient for the participant. Participants who gave consent originally or subsequently and legal representatives who gave consent for relative/friend participation were approached.
The aim was to get experiences from approximately three participants and approximately three personal legal representatives (who were not health-care professionals). However, this would continue to get experiences from up to 10 participants and legal representatives if new views and experiences continued to be expressed (i.e. had not reached saturation).
The interview was semistructured. The first set of questions explored how participants/legal representatives viewed the process of recruitment:
-
Did you feel able to ask questions about the study?
-
Did you feel that your questions were answered satisfactorily?
-
Did you feel you had enough time to make up your mind?
-
What made it hard to agree to join the study? Were there things that the study team could have done differently to make the decision-making process easier?
The second set of questions explored how participants/legal representatives viewed trial participation:
-
Did you feel that you understood what was happening to you/your relative while you were in the study?
-
After you had joined, did you wish you had not?
-
What made it hard to continue to be in the study? Were there things that the study team could have done differently to make being in the study easier?
-
If a friend told you they had been asked to join a study, what kind of things would you tell them to find out about? Would you recommend they join (and why/why not)?
Any additional questions would directly relate to the objectives described above (i.e. why they joined, what they liked/disliked, what could have done better/differently, their experience of consent, what would make them consider/not consider joining another trial in future).
Findings
The study revealed two findings: first, there was a disappointing uptake of both questionnaire completion and interview. A total of 20 questionnaires were sent and only seven people responded and three patients/legal representatives were interviewed. Although it was expected that the study would be challenging and unorthodox in such a trial (especially seeking to explore views of those who did not consent), it was not anticipated that uptake would be as low as it was.
For patients who did not consent, the reasons given were ‘everything else going on was too much’ (patients A, B, C, D and E), ‘could not make a decision either way’ (patients A, B and E), ‘best to play safe’ (patients A, C, D and E), ‘felt too ill/tired’ (patients A, B, C, D and E), and ‘did not have enough time to decide’ (patients A, D and E). Another added, that they ‘would have liked to take part but the side effects were too risky and I didn’t want to take any risks. Everything was explained really well, sorry I couldn’t help’ (patient B). The same patient said that ‘more time to decide’ was very important and would have improved the likelihood of him participating. Another patient who gave similar reasons said that they would have been more likely to have participated if the ‘information sheet was shorter’ (patient C).
One questionnaire was sent back without any questions being answered but with a narrative arguing that it was not appropriate for patients who were ‘very unwell in A&E [accident and emergency] to be hassled by a research nurse about studies that are going on’. The patient went on to state:
I fully understand and appreciate trials take place and have taken part in clinical trials. Timing is the key and explaining when people feel a bit more human and can think straight about partaking when they have had time to read and digest it.
Patient F
Although this was only one patient, it is important that all studies seeking to recruit those with serious illness do so in a manner that is sensitive to the needs of patients. The fact that only one patient used the questionnaire or interview as an opportunity to complain in this way is evidence of the careful and considerate method of recruitment displayed by the recruiting staff.
Reasons given by personal legal representatives as to why they did not want their relative to participate were: ‘felt too worried’, ‘too much responsibility’, ‘worried that my relative might GET the study drug’ and ‘worried about side effects and liver problems’ (legal representative Z).
For patients who did consent, the reasons given were: ‘I didn’t really need time to think about it, I was ill and so it’s all rather difficult’, and ‘I don’t remember what the information sheet was like but XXXX explained it all to me and I don’t think I had any questions, but I’m sure she would have answered them if I had some’ (patient G).
Another said:
It’s not about the information, I just thought well I’ve got nothing to lose, but I did ask them to come back the next day and I thought about it, asked some people and still came to the same conclusion that I had nothing to lose so the next day I just signed up.
Patient H
The reasons given by personal legal representatives as to why they consented to their relative participating were ‘to help my mum and perhaps other people, it’s a 50/50 chance of her getting the medicine or the placebo and I just thought she might be helped’ (legal representative Y). On the information given they said:
But to be honest I don’t think I even read that sheet. Well I suppose the stuff about safety they told me about and I read it, it wasn’t difficult to understand. I just signed it and they were helpful the people who told me about it.
Legal representative Y
What has been learned?
Despite the limited responses, it is possible to draw some lessons for the future from this small study. These are:
-
Although there are some things that researchers cannot change, there are some things that they can change. The ‘felt too ill/tired’ and ‘had too much going on’ might be decreased if researchers delayed recruitment until the patients feel a little better (although this was not possible in the present trial given the requirement for < 96 hours of treatment).
-
Similarly, ‘not enough time to decide’ is something that can changed, and ‘information sheet too long’ can also be altered.
-
More subtle and challenging adaptations might come when consideration is given to comments, such as ‘worried that my relative might get the study drug’. Although honesty is paramount, promoting the reason for the trial and the reason why this medicine is being studied might help sway the balance in favour of the risk being worth taking.
Conclusion
It is unusual for a trial such as this to explore these issues and it is ethically challenging to gain approval to conduct a study approaching patients/representatives who did not consent to participate in the primary study. However, it was deemed important both by the trial team for this and future research, and by the PPI advisor for the benefit of patients and their representatives. Having gained ethics approval for this study and having learned lessons on how to improve in future, the authors are confident that other research will benefit from the lessons, methods and findings of this small study. A number of practical suggestions were made based on the findings and were presented at an investigator meeting by the PPI advisor. It is hoped that these suggestions and the model for this substudy will be used by those at the meeting and their wider research networks.
Chapter 5 Economic and health-related quality-of-life consequences of S. aureus bacteraemia, and effect of treatment with adjunctive rifampicin
Introduction
The ARREST trial was designed to evaluate the efficacy of adjunctive rifampicin in reducing bacteriologically confirmed failure/recurrence of S. aureus bacteraemia or death in 12 weeks. The trial did not provide evidence that rifampicin improves the composite primary end point. However, analyses of the components of this composite primary end point suggested that adjunctive rifampicin reduced the risk of disease recurrence. Nevertheless, the trial did not find any impact of rifampicin on short- or long-term mortality (secondary outcomes). Rifampicin also significantly complicated other drug treatment. Hence, clinically, adjunctive rifampicin was not considered to provide overall benefit over standard antibiotic therapy in adults with S. aureus bacteraemia.
The pragmatic design of this trial means that the population included is clinically relevant, and non-comparative findings can be considered generalisable. The clinical results highlight the severity of S. aureus bacteraemia and also show the high degree of heterogeneity in the patient population. In this component of the analyses, the trial evidence on the HRQoL and economic consequences of a S. aureus bacteraemia episode in this patient population are described, which can inform the burden to patients (in terms of HRQoL) and to health systems (in terms of health system costs). Heterogeneity was also explored by evaluating determinants of costs and HRQoL. Quantifying the burden of S. aureus bacteraemia allows better informed future evaluations of alternative treatment and prevention strategies, a research area that has been highlighted. 38 Although the literature on the economic impact of S. aureus bacteraemia is substantial, particularly for meticillin-resistant strains (such as MRSA), the evidence it is based on is poor and often does not rely on any empirical data. 39
Although evaluation of the costs and HRQoL impact of S. aureus bacteraemia, and potential determinants of these, are the main focus of the analyses, the effect of adjunctive rifampicin on HRQoL and cost outcomes was also investigated, given the primary aim of the trial. From the results of the clinical analyses it can be hypothesised that rifampicin adjunctive treatment may be associated with cost savings and improvements in HRQoL via the small but significant reduction in bacteriologically and clinically defined disease recurrences (hypothesised to arise from the sterilisation of deep infection foci). The trial data will be used to determine the potential cost-effectiveness of adjunctive rifampicin treatment, and given the likely high degree of uncertainty, the value of further research will be determined.
Methods
Cost and health outcomes for patients with S. aureus bacteraemia were evaluated using data from the ARREST trial. Health outcomes were measured as QALYs. The QALY combines survival and HRQoL into a single metric, in which time spent with poorer HRQoL is downweighted. Costs considered in analysis were those incurred by the NHS and Personal Social Services, as recommended by the National Institute for Health and Care Excellence (NICE). 40 Costs and QALYs were only measured for 84 days (i.e. 12 weeks) from the date of randomisation, which was also the maximum duration of active follow-up (longer follow-up through electronic health records was done only for mortality). When considering determinants of costs and QALYs, the effect of adjunctive rifampicin was evaluated, which allowed for a cost-effectiveness analysis to be conducted. Given the short time horizon, neither costs nor health benefits were discounted. The analysis was conducted using the statistical software R version 3.4.1 (The R Foundation for Statistical Computing, Vienna, Austria).
Details of each component of analyses (analysis of health benefits, analysis of costs and analysis of cost-effectiveness and value of information) are presented in the following sections. These are followed by a description of the statistical methods used.
Costs
Data on the use of S. aureus bacteraemia-related health-care resources were collected during the trial and served as the basis for the calculation of total costs included in this analysis. Data related to three different resource use categories:
-
All antibiotic therapy received from randomisation in the active follow-up period (84 days), including trial drug and any other antibiotic therapy used.
-
First admissions and readmissions to secondary care and length of stay, including investigations and procedures undertaken while hospitalised.
-
Consultations with health-care providers (in primary or secondary care) after hospital discharge from first hospital admission.
See Appendix 3 for the resource use questions on the eCRFs. Costs for each trial participant were calculated as the product of health resources used during the trial follow-up period and the relevant NHS unit costs. Unit costs were based on the NHS Reference Costs 2013 to 2014,41 NHS Reference Costs 2015 to 2016,42 the Unit Costs of Health and Social Care 2016,43 the British National Formulary44 and relevant literature. All values are in Great British pounds and were, when required, updated to 2016 prices [hospital and community health services index provided by the Personal Social Services Research Unit (PSSRU) 2016]. 43
Antibiotic therapy
Antibiotic regimens used during the trial were costed using information on the agent, dose, frequency and route of administration. For rifampicin, this information was recorded in trial drug logs by health-care professionals until the earlier of 14 days or cessation of ‘backbone’ antibiotics. Time (in days) from initiation to end of randomised treatment was estimated and used to estimate the overall trial drug cost per patient during follow-up period. The use of other antibiotics was recorded by health-care professionals in treatment logs completed at each change in therapy until end of follow-up, or death. Time (in days) on other antibiotics was also estimated and was mainly informed by administration ‘start’ and ‘stop’ information. Only antibiotics taken after randomisation were considered. As for the trial drug, estimated time (in days) on other antibiotics only considered time from randomisation.
Appendix 2, Table 33, lists, for all antibiotic therapies costed in the trial, including the trial drug, the unit costs by dose and route of administration. Appendix 2 ,Table 34 lists antibiotic therapies by dose and route for which a unit cost was not obtained.
Admissions to secondary care
With regard to hospital inpatient stay, health resource utilisation was recorded by study personnel at weekly clinical assessments until discharge, and then at the final 84-day follow-up visit. These include days spent in wards, including ICUs, or high-dependency units (HDUs), or investigations and procedures [e.g. computed tomography (CT) scan, magnetic resonance imaging (MRI) scan, positron emission tomography (PET) scan]. Haematology and biochemistry test results were only collected at specific time points, thus were not included. The use of other drugs and the consequences of drug–drug interactions or AEs were not collected. Hospital readmission information provided at the final day-84 visit was also considered; this included readmission as hospital day cases, readmissions with hospital stay to hospital ward, ICU or HDU, together with all procedures undertaken after rehospitalisation. As trial patients were expected to have a long stay in hospital in their initial hospitalisation (i.e. number of days from admission to hospital to first post-enrolment discharge), unit costs from non-elective long-stay tariffs were used. This analysis used only days in hospital after randomisation (in contrast with Chapter 3, which looked at duration of the entire admission). The unit costs used to calculate the cost of secondary care-related health consumption in the trial are summarised in Appendix 2, Table 35.
Consultations with health-care providers
Data on the number of consultations with health-care providers were available for discharged patients from participant-reported questionnaires at the final 84-day follow-up. For the period since discharge, each trial participant recorded the number of GP consultations (either at a doctor’s surgery or at home) and number of hospital outpatient visits with a doctor or nurse, separating the number of those that were S. aureus bacteraemia related from those that were not. All health consultations reported were included in the economic analysis. The unit costs used to cost these are again summarised in Appendix 2, Table 35.
Health-related quality of life
The health outcome used was total QALYs over 84 days (i.e. period of active follow-up). Data on the EuroQol-5 Dimensions, three-level version (EQ-5D-3L) instrument, a widely recognised and validated HRQoL descriptive system,45,46 were collected at baseline and at 7, 14 and 84 days. The recent five-response version of the EQ-5D, the EuroQol-5 Dimensions, five-level version, and associated UK-specific valuation set were not fully available at the start of this study.
The EQ-5D-3L questionnaire has five questions, each relating to a different health dimension: mobility, self-care, ability to undertake usual activity, pain and anxiety/depression. Each question allows three possible responses: no problems, moderate problems and severe problems. Based on their answers, participants can be classified as 1 of 243 possible health states, plus death and unconscious health states. A separate algorithm was then applied to identify the impact of the particular health state on HRQoL, that is, a weight, when full health assumes a value of 1, death a value of 0, and where values < 0 represent health states worse than death. The algorithm used to generate the weights was based on a population study that elicited societal preferences using a time trade-off technique (a technique that, for instance, asks participants how many years in the current health state they would be willing to ‘trade off’ for a shorter period in full health). 47,48
In this study, QALYs were estimated using the area under the curve method with interpolation of EQ-5D-3L index scores measured at the beginning and end of each time interval. Hence, for each study participant, and when sufficient data were available, a QALY estimate was obtained considering the product of the mean EQ-5D-3L index score during the interval and the duration of the interval. 49
Statistical methods of analyses
Missingness
Given that the population recruited into the trial contained a proportion of critically ill patients, non-negligible missingness on the EQ-5D data was expected. It is typical of trials in very sick participants, such as the ARREST trial, to recruit or have during the follow-up period, a non-negligible proportion of individuals in a coma. To these patients [n = 80 (10.6%) at baseline; n = 48 at 7 days; n = 38 at 14 days; and n = 4 at 84 days], a HRQoL weight of –0.402 was assigned. 49,50 Some patients were also reported to be unable/unwilling to provide EQ-5D answers [n = 20 (2.6%) at baseline; n = 18 at 7 days; n = 12 at 14 days; and n = 10 at 84 days]. These patients were assumed to have a HRQoL weight value of –0.261, corresponding to the bottom decile of the EQ-5D index score distribution of all trial patients for which a EQ-5D index score was available. As a sensitivity analysis EQ-5D answers for unable/unwilling patients were kept missing.
In the estimation of QALYs over the 84-day period, interpolation between adjacent assessments was used. When EQ-5D information was missing at 7 and/or 14 days, interpolation used the other (non-missing) assessments. Non-optimal imputation techniques, such as last observation carried forward/backward, were not implemented owing to the clear observed differences between mean EQ-5D data at 7 days and baseline and between mean EQ-5D data at 14 days and 84 days.
Missing values of the outcome variable QALYs over the active follow-up period (i.e. 84 days) that could not be interpolated as earlier were dealt with formally using multiple imputation,51 a statistical technique that imputes with uncertainty based on the observed characteristics of patients or of the disease (i.e. an assumption of missing at random). This technique imputes with uncertainty by creating, at a first stage, several plausible imputed data sets and, in a second stage, by combining results obtained from each. Thus, in the first stage, missing values on a covariate of interest are replaced by imputed values using predictions from a model that uses a set of covariates deemed relevant to predict the variable of interest based on those observations that were not missing. In the second stage, statistical regression methods are fitted to each of the imputed data sets and analysis results are integrated into a single, pooled result. The Multivariate Imputation by Chained Equations R package52 using predictive mean matching was used. 53
The data collection tool on health-care resource use did not allow distinguishing between no consumption and missing reporting of consumption of health resources. However, given that resource use was collected by investigators in the study, true missingness was assumed negligible and, hence, no consumption of health resources was assumed when data were missing.
Estimating adjusted mean costs and quality-adjusted life-years
Total costs and imputed QALYs were independently regressed on a set of baseline covariates, including treatment group and other potential predictor or treatment-effect modifiers that could be relevant for subgroup analyses. The variables defined for the trial’s subgroup analyses (both the prespecified set and the additional set) were also considered for inclusion here by the clinical advisors to the trial. The final set of covariates was:
-
age (categorical, 1: 18–54 years; 2: > 54–72 years; and 3: > 72 years)
-
sex (binary, 1: male; 0: female)
-
body mass index (categorical, 1: 18.5–24.9 kg/m2; 2: 25.0–29.9 kg/m2; 3: 30.0–39.9 kg/m2; and 4: ≥ 40 kg/m2)
-
mode of acquisition of infection (categorical, 1: community acquired; 2: nosocomial infection; and 3: health-care associated)
-
Charlson Comorbidity Index score (categorical, 1: 0; 2: 1–2; 3: 3–4; and 4: ≥ 5)
-
neutrophil count (categorical, 1: < 6 × 109/l; 2: 6–9 × 109/l; and 3: > 9 × 109/l)
-
deep infection foci (binary, 1: yes; 0: no)
-
endocarditis (binary, 1: yes; 0: no)
-
MRSA (binary, 1: yes; 0: no)
-
comatose (binary, 1: yes; 0: no)
-
randomised group.
Continuous variables were categorised using the same thresholds as used in subgroup analyses. In addition, the baseline EQ-5D index score was used in the QALY regression as patients’ baseline utilities are likely to be highly correlated with their QALY estimates over the follow-up period and, thus, baseline utility imbalances need to be accounted for. 54
Five scenarios were analysed: the first, a tentative scenario [models total costs (TCs) and total QALYs (TQ) for costs and QALYs, respectively], assessed the impact of randomised treatment alone in explaining the outcome variables. The second, the base case, retained all covariates irrespective of their importance to explain the outcome (model 1C for TCs and model 1Q for total QALYs). The third scenario follows from the second, but retains/excludes covariates from the full covariate set to select the model of lowest Akaike information criterion (AIC). 55 The result is the most parsimonious model based on the AIC statistic, a measure of model quality and goodness of fit (model 1Cp for TCs and model 1Qp for total QALYs). A fourth scenario extends the base case to include interactions with randomised treatment and explore treatment effect modifiers (models 2C and 2Q for TCs and QALYs, respectively). Finally, and similarly to scenario three, the most parsimonious interaction model based on the AIC statistic is obtained (model 2Cp for TCs and model 2Qp for total QALYs). The scenarios with and without randomised treatment interactions may have different implications for policy, which will be examined.
The TQs and TCs captured during 84 days were regressed using a generalised linear modelling (GLM)56 framework, which accounts for the characteristics of the data (i.e. continuously distributed data are potentially skewed). Alternative distributions and link functions were tested, and the best fitting based on AIC was chosen. 55 To determine cost-effectiveness, predicted TCs and TQs were evaluated for the mean characteristics of all patients in the trial.
Note that the effect of randomised treatment was modelled independently for costs and health effects, although it is likely that some correlation exists. This should be considered in the interpretation of findings.
Cost-effectiveness and decision uncertainty
To ascertain the cost-effectiveness of a health-care intervention relative to another, expected health benefits need to be considered against any additional costs expected to be incurred. The fact that a particular technology imposes additional costs means that other activities (that could be financed by these costs) are not undertaken, and this has health consequences for other patients: the health opportunity costs. If the health gains associated with the technology compensate the health opportunity costs imposed by its additional costs, then using the technology brings net benefits to the NHS and could be recommended for use.
Health opportunity costs are often evaluated from the additional costs imposed by particular technologies using a cost-effectiveness threshold (lambda, λ). Currently, NICE sets the threshold at £20,000–30,000 per QALY gained (although recent work undertaken by the University of York has estimated this to be somehow lower – approximately £13,000 per QALY gained57). A new technology is considered cost-effective in relation to existing technologies if the net health benefit (NHB) is:
where λ, ΔB and ΔC represent, respectively, the cost-effectiveness threshold, the incremental benefits and incremental costs.
Decision-makers may decide on the provision of services using expected cost-effectiveness findings. However, given the nature of the underlying evidence used, such expectation is not known with certainty. It is hence important that the consequences of uncertainty, and the extent to which it has an impact on the adoption decision, are investigated to inform whether or not further research is needed. 58,59 Uncertainty here stems from the fact that all analyses are based on data collected within this trial, based on a sample of patients and hence generating uncertain estimates of population parameters. The cost-effectiveness analysis can, however, consider such uncertainty over expected costs and benefits (i.e. parameter uncertainty), and evaluate whether the decision to adopt (or reject) the technology is also uncertain (i.e. if the incremental net benefit crosses zero).
To propagate uncertainty in cost-effectiveness analyses (i.e. conduct a probabilistic analysis), Monte Carlo simulation methods are commonly used. 60 With a large number of simulations (sampled 10,000 times in this work) it is possible to examine the effect on costs and effects hence, on cost-effectiveness results when the underlying variables are allowed to vary simultaneously across a plausible range according to predefined distributions. Given total costs and benefits were modelled independently, their predicted distributions were also assumed to be independent. However, costs and benefits were individually modelled using a multivariate regression approach and, therefore, simulating the regression coefficients’ variance–covariance matrix was considered in a multivariate normal framework. 59
Decisions that are uncertain have expected consequences for the NHS (as well as any attempt to delay or reverse them). 59,61 Acquiring more evidence to support the decision is expected to mitigate these risks, and hence quantifying the risks of uncertainty can inform the value of further evidence collection. The risks and consequences of uncertainty can be quantified using a simple extension of probabilistic analyses called expected value of perfect information (EVPI). 59 The EVPI determines the maximum amount that the health-care system should be willing to pay for more information. In the event that the new evidence demonstrates the current decision to be wrong, the decision can be reversed, benefiting prospective patients. Individual- and population-level EVPI estimates were estimated at the commonly used cost-effectiveness thresholds referred to earlier.
Subgroup analysis
Together with base-case and scenario analyses, which explored cost-effectiveness in the whole patient population with S. aureus bacteraemia, subgroup analyses were also implemented. Subgroup analyses are important as an intervention can prove to be cost-effective for one subgroup of the population and not for another. This might be because the baseline risk of events may differ, or because treatment effects or cost implications are different across subgroups (i.e. treatment effect-modifying factors). Thus, there may be population health gains from stratifying treatment decisions based on subgroup membership. These analyses explored subgroups based on the regression covariates, namely: age, mode of acquisition of infection, Charlson Comorbidity Index score, BMI, deep infection foci, neutrophils and coma status.
Results
A total of 758 participants were recruited: 388 were randomly allocated to receive standard antibiotic therapy (placebo) and 370 to receive adjunctive rifampicin. Baseline characteristics of participants by treatment group can be found in Table 12. Note that one rifampicin participant withdrew shortly after randomisation without an enrolment form having been completed. This patient has been excluded from all tables after baseline as they had no post-baseline data, leaving the number in the rifampicin group as 369 rather than 370 in Chapter 3.
| Baseline characteristica | Treatment group, n (%) | Total (N = 758)b, n (%) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370)b | ||
| Sex: male | 246 (63.4) | 249 (67.3) | 495 (65.3) |
| Age | |||
| At last birthday (years), mean (median, min.–max.) | 63.0 (66.0, 20.0–100.0) | 61.4 (64.0, 18.0–94.0) | 62.2 (65.0, 18.0–100.0) |
| 18–53 years | 126 (32.5) | 125 (33.9) | 251 (33.2) |
| 54–71 years | 126 (32.5) | 122 (33.1) | 248 (32.8) |
| ≥ 72 years | 136 (35.1) | 123 (33.3) | 259 (34.2) |
| BMI (kg/m2), mean (median, min.–max.) | 27.6 (26.4, 15.2–58.5) | 27.2 (26.3, 12.1–73.6) | 27.4 (26.3, 12.1–73.6) |
| < 18.4 | 24 (6.2) | 21 (5.7) | 45 (5.9) |
| 18.5–24.9 | 129 (33.2) | 128 (34.7) | 257 (33.9) |
| 25.0–29.9 | 111 (28.6) | 113 (30.6) | 224 (29.6) |
| 30.0–39.9 | 90 (23.2) | 77 (20.9) | 167 (22.1) |
| ≥ 40 | 23 (5.9) | 21 (5.7) | 44 (5.8) |
| Mode of acquisition of infection | |||
| Community acquired | 240 (61.9) | 245 (66.4) | 485 (64.1) |
| Nosocomial infection (onset ≥ 48 hours after admission) | 76 (19.6) | 56 (15.2) | 132 (17.4) |
| Health-care associated (all other) | 72 (18.6) | 68 (18.4) | 140 (18.5) |
| Mean Charlson Comorbidity Index score, median (min.–max.) | 2.10 (2.00, 0.00–9.0) | 1.97 (1.00, 0.00–11.0) | 2.04 (2.00, 0.00–11.0) |
| 0 | 117 (30.2) | 114 (30.9) | 231 (30.5) |
| 1–2 | 143 (36.9) | 154 (41.7) | 297 (39.2) |
| 3–4 | 74 (19.1) | 52 (14.1) | 126 (16.6) |
| ≥ 5 | 54 (13.9) | 49 (13.3) | 103 (13.6) |
| Neutrophils (× 109/l), mean (median, min.–max.) | 8.9 (7.30, 0.00–64.40) | 9.25 (7.40, 0.00–83.70) | 9.06 (7.30, 0.00–83.70) |
| < 6 | 151 (38.9) | 135 (36.6) | 286 (37.8) |
| 6–9 | 95 (24.5) | 107 (29.0) | 202 (26.7) |
| > 9 | 137 (35.3) | 127 (34.4) | 264 (34.9) |
| Meticillin resistance | 21 (5.4) | 26 (7.0) | 47 (6.2) |
| Deep infection foci | 159 (41.0) | 142 (38.5) | 301 (39.8) |
| Comatose status | 43 (11.1) | 37 (10.0) | 80 (10.6) |
| Endocarditis | 18 (4.6) | 22 (6.0) | 40 (5.3) |
Resource use and costs
Table 13 provides summary statistics on the trial drug and all other antibiotic therapies received after randomisation during the trial active follow-up period. Fourteen patients (1.8%) never initiated the trial drug. Active antibiotic therapies administered included flucloxacillin (n = 597, 80.9%), ceftriaxone (n = 164, 22.2%) and vancomycin (n = 144, 19.5%). Open-label rifampicin was used in 52 (13.4%) and 32 (8.7%) patients in the placebo and rifampicin groups, respectively.
| Patients | Treatment group, n (%) | Total (N = 757) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 369) | ||
| Trial drug administration during active follow-up period | 380 (97.9) | 364 (98.4) | 744 (98.3) |
| Antibiotic therapy administration during active follow-up period | |||
| Any antibiotic | 382 (98.5) | 356 (96.5) | 738 (97.5) |
| Flucloxacillin | 315 (82.5) | 282 (79.2) | 597 (80.9) |
| Ceftriaxone | 81 (21.2) | 83 (23.3) | 164 (22.2) |
| Vancomycin | 79 (20.7) | 65 (18.3) | 144 (19.5) |
| Piperacillin/tazobactam | 62 (16.2) | 57 (16.0) | 119 (16.1) |
| Gentamicin | 45 (11.8) | 40 (11.2) | 85 (11.5) |
| Rifampicin | 52 (13.6) | 32 (9.0) | 84 (11.4) |
| Teicoplanin | 36 (9.4) | 41 (11.5) | 77 (10.4) |
| Co-amoxiclavulante | 46 (12) | 25 (7.0) | 71 (9.6) |
| Meropenem | 30 (7.9) | 24 (6.7) | 54 (7.3) |
| Clindamycin | 24 (6.3) | 29 (8.1) | 53 (7.2) |
| Ciprofloxacin | 29 (7.6) | 22 (6.2) | 51 (6.9) |
| Metronidazole | 24 (6.3) | 14 (3.9) | 38 (5.1) |
| Daptomycin | 13 (3.4) | 22 (6.2) | 35 (4.7) |
| Doxycycline | 16 (4.2) | 16 (4.5) | 32 (4.3) |
| Linezolid | 13 (3.4) | 12 (3.4) | 25 (3.4) |
| Levofloxacin | 12 (3.1) | 11 (3.1) | 23 (3.1) |
| Trimethoprim | 19 (5.0) | 1 (0.3) | 20 (2.7) |
| Amoxicillin | 10 (2.6) | 5 (1.4) | 15 (2.0) |
| Other antibioticsa | 67 (17.5) | 47 (13.2) | 114 (15.4) |
A summary of the secondary care health resources utilised during trial active follow-up period (i.e. from randomisation to 84 days of follow-up) is provided in Table 14, and of consultations with health-care providers in Table 15.
| Secondary care health resourcea | Treatment group | Total (N = 757) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 369) | ||
| Hospital visits | |||
| Total hospital stay from randomisation to first dischargeb | |||
| Mean, number of procedures (SD) days | 23.9 (21.2) | 20.5 (17.9) | 22.3 (19.7) |
| Ward | |||
| Mean, number of procedures (SD) days | 23.4 (20.4) | 20.2 (17.3) | 21.8 (19.0) |
| Number of patients (%) | 388 (100.0) | 367 (99.5) | 755 (99.7) |
| ITU | |||
| Mean, number of procedures (SD) days | 16.7 (20.9) | 14.4 (10.6) | 15.6 (16.4) |
| Number of patients (%) | 12 (3.1) | 11 (3.0) | 23 (3.0) |
| HDU | |||
| Mean, number of procedures (SD) days | 1.8 (0.9) | 1.3 (0.5) | 1.5 (0.7) |
| Number of patients (%) | 4 (1.0) | 7 (1.9) | 11 (1.5) |
| Total hospital readmissions, n (%) | 94 (24.2) | 83 (22.5) | 177 (23.4) |
| Hospital readmission (day case),c n (%) | 4 (1.0) | 5 (1.4) | 9 (1.2) |
| Hospital readmission (critical care),c n (%) | 6 (1.5) | 4 (1.1) | 10 (1.3) |
| Hospital readmission (ward),c n (%) | 90 (23.2) | 80 (21.7) | 170 (22.5) |
| Hospital readmission with overnight stay (in ward, ICU or HDU) | |||
| Mean, number of procedures (SD) days | 15.9 (19.0) | 13.9 (13.5) | 14.9 (16.7) |
| Number of patients (%) | 92 (23.7) | 81 (22.0) | 173 (22.9) |
| Hospital procedures, including other | |||
| Radiologically guided biopsy/aspirate/abscess drainage | |||
| Mean, number of procedures (SD) | 1.4 (1.0) | 1.7 (1.9) | 1.6 (1.6) |
| Number of patients (%) | 25 (6.4) | 32 (8.7) | 57 (7.5) |
| Surgical drainage/removal of non-device-related focus | |||
| Mean, number of procedures (SD) | 1.2 (0.4) | 1.4 (1.1) | 1.3 (0.8) |
| Number of patients (%) | 42 (10.8) | 32 (8.7) | 74 (9.8) |
| Surgical removal of infected prosthetic device | |||
| Mean, number of procedures (SD) | 1.1 (0.4) | 1.0 (0.0) | 1.1 (0.2) |
| Number of patients (%) | 7 (1.8) | 8 (2.2) | 15 (2.0) |
| Cardiac surgery for S. aureus endocarditis | |||
| Mean, number of procedures (SD) | 1.4 (0.9) | 1.0 (0.0) | 1.2 (0.6) |
| Number of patients (%) | 5 (1.3) | 6 (1.6) | 11 (1.5) |
| Insertion of Hickman line | |||
| Mean, number of procedures (SD) | 1.0 (0.0) | 1.0 (0.0) | 1.0 (0.0) |
| Number of patients (%) | 5 (1.3) | 6 (1.6) | 11 (1.5) |
| Other procedures | |||
| Mean, number of procedures (SD) | 1.53 (1.0) | 1.45 (0.9) | 1.49 (1.0) |
| Number of patients (%) | 78 (20.1) | 62 (16.8) | 140 (18.5) |
| Hospital investigations, including other | |||
| Ultrasound scan (other than echocardiogram) | |||
| Mean, number of procedures (SD) | 1.9 (1.1) | 1.6 (1.1) | 1.7 (1.1) |
| Number of patients (%) | 112 (28.9) | 125 (22.9) | 237 (31.3) |
| CT scan | |||
| Mean, number of procedures (SD) | 1.9 (2.1) | 1.73 (1.5) | 1.83 (1.8) |
| Number of patients (%) | 145 (37.4) | 128 (34.7) | 273 (36.1) |
| MRI scan | |||
| Mean, number of procedures (SD) | 1.7 (1.1) | 1.6 (0.9) | 1.7 (1.0) |
| Number of patients (%) | 127 (32.7) | 107 (29.0) | 234 (30.9) |
| PET scan | |||
| Mean, number of procedures (SD) | 1.0 (0.0) | 1.0 (0.0) | 1.0 (0.0) |
| Number of patients (%) | 3 (0.8) | 4 (1.1) | 7 (0.9) |
| PET CT scan | |||
| Mean, number of procedures (SD) | 1.1 (0.4) | 1.0 (0.0) | 1.1 (0.2) |
| Number of patients (%) | 7 (1.8) | 10 (2.7) | 17 (2.2) |
| Bone scan | |||
| Mean, number of procedures (SD) | 1.0 (0.0) | 1.3 (0.5) | 1.13 (0.4) |
| Number of patients (%) | 9 (2.3) | 6 (1.6) | 15 (2.0) |
| White blood cell scan | |||
| Mean, number of procedures (SD) | 2.0 (N/A) | N/A (N/A) | 2.0 (N/A) |
| Number of patients (%) | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Other investigations | |||
| Mean, number of procedures (SD) | 2.5 (3.1) | 2.1 (1.7) | 2.3 (2.6) |
| Number of patients (%) | 31 (8.0) | 26 (7.0) | 57 (7.5) |
| Consultations with health-care providers | Treatment group | Total (n = 757) | |
|---|---|---|---|
| Placebo (n = 388) | Rifampicin (n = 369) | ||
| All hospital outpatient visits within follow-up period | |||
| Mean (SD) | 4.6 (6.1) | 4.6 (5.4) | 4.6 (5.8) |
| Number of patients (%) | 162 (41.8) | 154 (41.7) | 316 (41.7) |
| All general practitioner visits within follow-up period | |||
| Mean (SD) | 2.9 (3.1) | 3.1 (3.4) | 3.0 (3.3) |
| Number of patients (%) | 137 (35.3) | 138 (37.4) | 275 (36.3) |
All trial patients spent time in hospital, either in the ward or in a critical care unit, with a mean length of stay of 22.3 days post randomisation [standard deviation (SD) 19.7 days]. Patients in the placebo group spent a mean 3.2 days more in the hospital ward than patients in the rifampicin group. Approximately 4% (n = 33) of trial patients spent time in a critical care unit. Patients using these units had a mean stay of 11.0 days (SD 14.5 days). A total of 177 (23%) patients were readmitted to hospital (as a day case, to a general ward or to a critical care unit) for any reason. Once readmitted to hospital to a general ward or critical care unit, patients in both group spent a mean of 15 days hospitalised (placebo group: mean 15.9 days, SD 19.0 days, n = 92; rifampicin group: mean 13.9 days, SD 13.5 days, n = 81). The number of hospital procedures and investigations undertaken were fairly balanced between treatment groups and across the different items. The most common hospital procedures were surgical drainage/removal of non-device-related foci [n = 74, 9.8%, with a mean of 1.3 (SD 0.8) procedures per patient] and radiologically guided biopsy/aspirate/abscess drainages [n = 57, 7.5%, with a mean of 1.6 (SD 1.6) procedures per patient]. The most common hospital investigations included CT scans [n = 273, 36.0%, with a mean of 1.8 (SD 1.8) procedures per patient), ultrasound scans (other than echocardiogram) (n = 237, 31.3%, with a mean of 1.7 (SD 1.1) procedures per patient] and MRI scans [n = 234, 30.9%, with a mean of 1.6 (SD 1.0) procedures per patient]. A total of 316 (41.7%) trial patients had at least one hospital outpatient visit (see Table 15), and 275 (36.3%) trial patients had a GP visit.
Total costs
Descriptive, unadjusted results
The unadjusted costs per category are shown in Table 16. The item with largest mean unadjusted cost was hospital stay in critical care on first admission (£14,625, SD £20,272, n = 34), followed by hospital stay in critical care on readmission (£9034, SD £8036, n = 10) and then by hospital procedures (£7001, SD £6936, n = 279).
| Unadjusted cost (£)a | Treatment group | Total (n = 757) | |
|---|---|---|---|
| Placebo (n = 388) | Rifampicin (n = 369) | ||
| Treatment costs | |||
| Trial drugb | |||
| Mean (SD) | 0.0 | 30.7 (59.4) | N/A (N/A) |
| Number of patients (%) | 380 (97.9) | 364 (98.4) | 744 (98.3) |
| All antibiotic therapy | |||
| Mean (SD) | 862.1 (1841.8) | 836.0 (1114.5) | 849.2 (1525.8) |
| Number of patients (%) | 351 (90.5) | 342 (92.7) | 693 (91.5) |
| Secondary care health resources utilised | |||
| Hospital first admission | |||
| Hospital ward stay | |||
| Mean (SD) | 6973.2 (6073.4) | 6025.4 (5164.6) | 6512.5 (5666.1) |
| Number of patients (%) | 388 (100.0) | 367 (99.5) | 755 (99.7) |
| Hospital stay in critical care (ICU or HDU) | |||
| Mean (SD) | 17,241.3 (25,719.7) | 12,299.0 (14,209.8) | 14,624.8 (20,272.4) |
| Number of patients (%) | 16 (4.1) | 18 (4.9) | 34 (4.5) |
| Hospital readmission | |||
| Hospital ward stay | |||
| Mean (SD) | 4680.8 (5659.4) | 4092.0 (4038.6) | 4403.7 (4957.6) |
| Number of patients (%) | 90 (23.2) | 80 (21.7) | 170 (22.5) |
| Hospital stay in critical care (ICU or HDU) | |||
| Mean (SD) | 9472.5 (9556.2) | 8375.3 (6367.6) | 9033.6 (8035.6) |
| Number of patients (%) | 6 (1.5) | 4 (1.1) | 10 (1.3) |
| Day case | |||
| Mean (SD) | 481.3 (192.5) | 385.1 (0) | 427.9 (128.4) |
| Number of patients (%) | 4 (1.0) | 5 (1.4) | 9 (1.2) |
| Hospital procedures | |||
| Mean (SD) | 7079.4 (6810.4) | 6920.0 (7088.4) | 7001.1 (6936.2) |
| Number of patients (%) | 142 (36.6) | 137 (37.1) | 279 (36.9) |
| Hospital investigations | |||
| Mean (SD) | 423.0 (449.2) | 367.9 (398.5) | 395.6 (425.2) |
| Number of patients (%) | 249 (64.2) | 246 (66.7) | 495 (65.4) |
| Consultations with health-care providers | |||
| Hospital outpatient visits | |||
| Mean (SD) | 624.6 (833.4) | 626.1 (734.8) | 625.3 (785.6) |
| Number of patients (%) | 162 (41.8) | 154 (41.7) | 316 (41.7) |
| General practitioner visits | |||
| Mean (SD) | 104.3 (111.6) | 110.1 (123.1) | 107.2 (117.3) |
| Number of patients (%) | 137 (35.3) | 138 (37.4) | 275 (36.3) |
| Total costs over the follow-up period | |||
| Mean (SD) | 12,861.3 (12,753.1) | 11,497.8 (10,116.0) | 12,196.6 (11,555.7) |
| Number of patients (%) | 388 (100.0) | 369 (100.0) | 757 (100.0) |
For most categories, the mean unadjusted cost was fairly similar between treatment groups. However, and generally, mean unadjusted cost for hospital stay in the rifampicin group was lower than in the placebo group. The mean unadjusted cost of hospital ward stay on first admission was greater (by approximately 16%) in the placebo group (£6973, SD £6074, n = 388) than in the rifampicin group (£6025, SD £5165, n = 367, as two participants allocated to rifampicin were only ever on ICU/HDU). Similarly, mean unadjusted costs relating to hospital stay in critical care was also higher in the placebo group than in the rifampicin group, although < 5% (n = 34) of trial patients were admitted to hospital in these circumstances.
Mainly driven by greater hospital stay, the unadjusted total cost over the active follow-up period for the placebo group was estimated to be mean £1364 higher than in the rifampicin group (placebo group: £12,861, SD £12,753 vs. rifampicin: £11,498, SD £10,116).
Unadjusted costs per treatment group are, alternatively, presented by time intervals in Table 17. As some health-care resource consumption within the active follow-up period had no associated date, because either assessment date or form date were not available, the mean unadjusted costs of unspecified date are also presented.
| Unadjusted cost (£) | Treatment group | Total (n = 757) | |
|---|---|---|---|
| Placebo (n = 388) | Rifampicin (n = 369) | ||
| From baseline to day 14 | |||
| Mean (SD) (£) | 6293.1 (8259.30) | 5879.7 (7606.40) | 6088.6 (7945.20) |
| Number of patients (%) | 380 (97.9) | 369 (100.0) | 749 (98.9) |
| Days 15 to 84 | |||
| Mean (SD) (£) | 5310.5 (8574.30) | 4523.8 (6855.70) | 4927.0 (7789.00) |
| Number of patients (%) | 285 (73.5) | 247 (66.9) | 532 (70.3) |
| Day unspecified, within follow-up periodb | |||
| Mean (SD) (£) | 2130.9 (4643.70) | 1952.1 (3661.90) | 2045.8 (4201.30) |
| Number of patients (%) | 192 (49.5) | 169 (45.8) | 361 (47.7) |
During the first 2 weeks after randomisation, similar estimated costs were observed between treatment groups with mean unadjusted costs of approximately £5880 and £6293, respectively, for the rifampicin and placebo groups. During the following 10 weeks and until the end of active follow-up, the health care allocated to the placebo group was estimated to cost mean £787 more than the care required by patients in the rifampicin group (rifampicin: £4524 vs. placebo: £5311). Similarly, unadjusted mean costs of health care during active follow-up period but of no specified date were higher in the placebo group relative to the rifampicin group.
Adjusted results
Base-case model (model 1C)
A series of distributional and functional assumptions were modelled. Models assuming observed data followed a gamma distribution with a log-link function produced the lowest AIC statistics (highlighted in bold) for the different scenarios (see Appendix 2, Table 36). Note that smaller AIC values indicate better model quality of fit. Thus, for the base case (model 1C) and the treatment interactions model (model 2C), a gamma distribution with a log-link function was chosen.
The results of the regression models total costs and 1C are presented in Table 18. The results of model 1Cp, the most parsimonious model based on AIC using the covariate set of model 1C, are also presented.
| Model specification | Model | ||
|---|---|---|---|
| TC | 1C | 1Cp | |
| Type of regression model | Gamma, log-link | Gamma, log-link | Gamma, log-link |
| Equation | Log-total costs | Log-total costs | Log-total costs |
| Covariates (baseline) | Coefficient (SE) | Coefficient (SE) | Coefficient (SE) |
| Randomised treatment (1: rifampicin; 0: placebo) | –0.11 (0.07) | –0.10 (0.07) | – |
| Age (years) | |||
| 54–71 | – | 0.08 (0.08) | – |
| ≥ 72 | – | 0.04 (0.08) | – |
| Sex (1: male; 0: female) | – | –0.06 (0.07) | – |
| Acquisition | |||
| Nosocomial infection | – | 0.35 (0.09)*** | 0.37 (0.09)*** |
| Health-care associated | – | 0.06 (0.09) | 0.07 (0.09) |
| Charlson Comorbidity Index score | |||
| 1 or 2 | – | 0.11 (0.08) | – |
| 3 or 4 | – | 0.01 (0.11) | – |
| ≥ 5 | – | 0.06 (0.11) | – |
| BMI (kg/m2) | |||
| 18.5–24.9 | – | –0.23 (0.14) | – |
| 25.0–29.9 | – | –0.09 (0.15) | – |
| 30.0–39.9 | – | –0.14 (0.15) | – |
| ≥ 40 | – | –0.11 (0.19) | – |
| Deep focus (1 – yes; 0 – no) | – | 0.36 (0.07)*** | 0.35 (0.07)*** |
| Endocarditis (1 – yes; 0 – no) | – | 0.50 (0.15)** | 0.43 (0.16)** |
| Meticillin resistance | – | 0.18 (0.14) | – |
| Neutrophils (×109/l) | |||
| 6–9 | – | 0.12 (0.08) | 0.09 (0.08) |
| > 9 | – | 0.30 (0.08)*** | 0.29 (0.08)*** |
| Comatose (1 – yes; 0 – no) | – | 0.28 (0.11)* | 0.27 (0.11)* |
| Intercept | 9.46 (0.05)*** | 9.08 (0.16)*** | 8.97 (0.07)*** |
| Observations | 757 | 730 | 730 |
Results showed that no evidence exists that indicate that health-care costs differed between the rifampicin and placebo groups (p-value = 0.14 in model 1C). Note that, given the non-linear specification of the model, coefficients are interpreted multiplicatively rather than additively. Thus, and for instance, to obtain predicted total costs with model 1C, a patient at the reference category for all factors is associated with expected costs of £8752 [calculated as exp(9.08)]. For the rifampicin group, total expected costs are £7956, calculated as:
Patients with nosocomial infections, with deep foci infection, endocarditis, a neutrophil count of > 6 × 109/l and in a coma had significantly higher health-care costs than those in the respective reference categories (community-acquired infections, without deep foci, without endocarditis, a neutrophil count of < 6 × 109/l, with consciousness). Model 1Cp retained only the above-mentioned variables, reinforcing that this reduced covariate set is sufficient to explain variation in health-care resource consumption.
Model 1C predictions can be found in Table 19, first set of results. For the mean patient in the trial (see Table 12) across all covariates used in the regression, the weighted mean predicted total cost for the placebo group was £1092 higher than in the rifampicin group (rifampicin group, £11,050, SE £510; vs. placebo group, £12,142, SE £546). Model 1Cp total cost predictions were similar, in magnitude, to model 1C.
| Cost predictions (£) | Model | Treatment group | |
|---|---|---|---|
| Placebo | Rifampicin | ||
| Mean predicted total costs (95% CI) | 1C | 12,142 (11,194 to 13,249) | 11,050 (10,089 to 12,068) |
| Median predicted total costs (IQR) | 12,129 (11,778 to 12,500) | 11,040 (10,708 to 11,389) | |
| Mean predicted total cost difference (95% CI) | –1092 (–2564 to –371.7) | ||
| Mean predicted total costs (95% CI) | 2C | 11,969 (10,962 to 13,040) | 10,900 (9947 to 11,925) |
| Median predicted total costs (IQR) | 11,952 (11,604 to 12,321) | 10,889 (10,556 to 11,233) | |
| Mean predicted total cost difference (95% CI) | 1068 (–2510 to 392) | ||
Scenario analysis: consideration of treatment effect modifiers (model 2C)
Results of model 2C can be found in Table 20. The scenario analysis using a model with treatment interactions (model 2C), irrespective of their statistical significance, showed that, in general, the associations observed in model 1C persisted. Note that the BMI category of 18.5–24.9 kg/m2 was now associated with lower health-care costs relative to the reference BMI category (< 18.5 kg/m2). The predicted total costs for a patient at the reference category for all other factors in the rifampicin group in model 2C are exp(–0.43 + 9.23) = £6635, whereas for the placebo group these are exp(9.23) = £10,240.
| Model specification | Model | |
|---|---|---|
| 2C | 2Cp | |
| Type of regression model | Gamma, log-link | |
| Equation | Log-total costs | |
| Covariates (baseline) | Coefficient (SE) | Coefficient (SE) |
| Randomised treatment (1: rifampicin; 0: placebo) | –0.43 (0.31) | –0.16 (0.11) |
| Age (years) | ||
| 54–71 | –0.05 (0.11) | –0.05 (0.12) |
| ≥ 72 | 0.05 (0.12) | 0.08 (0.11) |
| Sex (1: male; 0: female) | –0.12 (0.09) | – |
| Acquisition | ||
| Nosocomial infection | 0.38 (0.12)* | 0.36 (0.09)*** |
| Health-care associated | 0.11 (0.12) | 0.09 (0.09) |
| Charlson Comorbidity Index score | ||
| 1 or 2 | 0.13 (0.11) | – |
| 3 or 4 | 0.02 (0.14) | – |
| ≥ 5 | 0.25 (0.15)† | – |
| BMI (kg/m2) | ||
| 18.5–24.9 | –0.41 (0.20) | – |
| 25.0–29.9 | –0.35 (0.20)† | – |
| 30.0–39.9 | –0.28 (0.20) | – |
| ≥ 40 | –0.41 (0.26) | – |
| Deep focus (1 – yes; 0 – no) | 0.49 (0.10)*** | 0.33 (0.07)*** |
| Endocarditis (1 – yes; 0 – no) | 0.43 (0.22)† | 0.48 (0.16)** |
| Meticillin resistance | 0.22 (0.21) | – |
| Neutrophils (× 109/l) | ||
| 6–9 | 0.15 (0.12) | 0.09 (0.08) |
| > 9 | 0.30 (0.11)** | 0.29 (0.08)*** |
| Comatose (1 – yes; 0 – no) | 0.16 (0.15) | 0.25 (0.11)* |
| Treatment × age, 54–71 years | 0.25 (0.16)† | 0.27 (0.16)† |
| Treatment × age, ≥ 72 years | –0.05 (0.17) | –0.06 (0.16) |
| Treatment × sex (1 – male; 0 – female) | 0.11 (0.14) | – |
| Treatment × acquisition, nosocomial infection | –0.06 (0.18) | – |
| Treatment × acquisition, health care associated | –0.06 (0.18) | – |
| Treatment × Charlson Comorbidity Index score of 1 or 2 | –0.06 (0.16) | – |
| Treatment × Charlson Comorbidity Index score of 3 or 4 | 0.005 (0.21) | – |
| Treatment × Charlson Comorbidity Index score of ≥ 5 | –0.36 (0.21)† | – |
| Treatment × BMI of 18.5–24.9 kg/m2 | 0.37 (0.28) | – |
| Treatment × BMI of 25.0–29.9 kg/m2 | 0.51 (0.29) | – |
| Treatment × BMI of 30.0–39.9 kg/m2 | 0.27 (0.30)† | – |
| Treatment × BMI of ≥ 40 kg/m2 | 0.61 (0.37) | – |
| Treatment × deep focus (1 – yes; 0 – no) | –0.25 (0.14)† | – |
| Treatment × endocarditis (1 – yes; 0 – no) | 0.19 (0.31) | – |
| Treatment × meticillin resistance | –0.10 (0.28) | – |
| Treatment × neutrophils, 6–9 109/l | –0.04 (0.16) | – |
| Treatment × neutrophils, > 9 109/l | 0.02 (0.15) | – |
| Treatment × comatose (1 – yes; 0 – no) | 0.17 (0.22) | – |
| Intercept | 9.23 (0.22)*** | 9.00 (0.10)*** |
| AIC | 15,105 | 15,080 |
| Observations | 730 | 730 |
Model 2Cp restricted model 2C to the covariates and potential effect modifiers that represent the most parsimonious model. Results for this model are also shown in Table 20. This model produced similar findings to the model 1Cp, with the exception that age and randomised treatment interaction with age were now retained. Patients in the rifampicin group and in the age category between 54 and 71 years were associated with higher health-care costs [£8602, calculated as exp(9.00–0.16–0.05 + 0.27)] than those in the placebo group [£7726, calculated as exp(9.00–0.05)]. The predicted total costs for a patient at the reference category for all other factors in the rifampicin group in model 2Cp was exp(9.00–0.16) = £6908, whereas for the placebo group this was exp(9.00) = £8128.
Model 2C weighted total cost predictions considering the mean patient in the trial across all covariates can be found in Table 19. Overall, total cost predictions are similar to the ones obtained in model 1C, the base-case model. Model 2Cp total cost predictions (not shown) for the placebo group were £1239 higher than in the rifampicin group. Total cost predictions for each patient subgroup and randomised treatment are presented in Cost-effectiveness and decision uncertainty.
Health benefits
Utility and quality-adjusted life-years (unadjusted and not using multiple imputation)
At baseline, there were approximately 10% (n = 80) comatose patients and 3% (n = 20) of patients unable or unwilling to provide answers to the EQ-5D questionnaire owing to their poor health. Descriptive statistics on HRQoL at different assessment times can be found in Table 21. Observed EQ-5D scores by domain/level and by time period can be found in Appendix 2, Table 37.
| Unadjusted EQ-5D index scorea | Treatment group | Total (N = 757) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 369) | ||
| Baseline | |||
| Number of patients, n (%) | 381 (98.2) | 365 (98.9) | 746 (98.5) |
| Number responded, n (%) | 329 (84.8) | 317 (85.7) | 646 (85.2) |
| Number in coma, n (%) | 43 (11.1) | 37 (10.0) | 80 (10.5) |
| Number unwilling/unable, n (%) | 9 (2.3) | 11 (3.0) | 20 (2.6) |
| Mean of unadjusted EQ-5D index score (SD)a | 0.09 (0.35) | 0.12 (0.34) | 0.10 (0.34) |
| Day 7 | |||
| Number of patients, n (%) | 314 (80.9) | 293 (79.4) | 608 (80.3) |
| Number responded, n (%) | 283 (72.9) | 258 (69.9) | 542 (71.6) |
| Number in coma, n (%) | 24 (6.2) | 24 (6.5) | 48 (6.3) |
| Number unwilling/unable, n (%) | 7 (1.8) | 11 (3.0) | 18 (2.4) |
| Number died, n (%) | 7 (1.8) | 13 (3.5) | 20 (2.6) |
| Mean of unadjusted EQ-5D index score (SD)a | 0.19 (0.34) | 0.19 (0.35) | 0.19 (0.34) |
| Day 14 | |||
| Number of patients, n (%) | 240 (61.9) | 213 (57.7) | 453 (59.8) |
| Number responded, n (%) | 215 (55.4) | 188 (50.9) | 403 (53.2) |
| Number in coma, n (%) | 20 (5.2) | 18 (4.9) | 38 (5.0) |
| Number unwilling/unable, n (%) | 5 (1.3) | 7 (1.9) | 12 (1.6) |
| Number died, n (%) | 17 (4.4) | 25 (6.8) | 42 (5.5) |
| Mean of unadjusted EQ-5D index score (SD)a | 0.20 (0.34) | 0.17 (0.32) | 0.19 (0.33) |
| Day 84 | |||
| Number of patients, n (%) | 280 (72.2) | 251 (68.0) | 531 (70.1) |
| Number responded, n (%) | 273 (70.4) | 244 (66.1) | 516 (68.2) |
| Number in coma, n (%) | 2 (0.5) | 2 (0.5) | 4 (0.5) |
| Number unwilling/unable, n (%) | 5 (1.5) | 5 (1.4) | 10 (1.5) |
| Number died, n (%) | 56 (14.4) | 56 (15.2) | 112 (14.8) |
| Mean of unadjusted EQ-5D index score (SD)a | 0.29 (0.31) | 0.32 (0.28) | 0.30 (0.29) |
Descriptive statistics of the EQ-5D index scores (unadjusted) show that the mean score is fairly balanced across treatment groups, irrespective of time point of assessment. The baseline unadjusted mean EQ-5D index score was 0.10 (SD 0.34, n = 746), reflecting the very poor quality of life of patients affected with S. aureus bacteraemia. At 7 days, the unadjusted mean EQ-5D index score was 0.19 (SD 0.34, n = 608) and at 14 days this was also 0.19 (SD 0.33, n = 453). At this assessment point, 42 (5.5%) patients had died and hence were allocated an EQ-5D index score of 0. In interpreting these figures, care is needed, as 40% fewer patients completed the EQ-5D questionnaire at 14 days. At the end of the active follow-up (84 days), the mean unadjusted EQ-5D index score was 0.30 (SD 0.29, n = 531). Again, at this point 112 (14.8%) patients were deceased and received an EQ-5D index score of 0. Although only about 70% (n = 531, including values allocated for deceased/comatose/unable to answer patients as per Statistical methods of analyses, Missingness, denoted ‘hard’ imputations subsequently) of the total number of patients who were recruited into the trial completed an EQ-5D at 84 days, it shows that the selective group of patients for whom a EQ-5D index score at 84 days was obtained had a better (higher) mean EQ-5D score than the remaining individuals, at baseline. Distributions of EQ-5D index score at baseline, 7, 14 and 84 days (not using multiple imputation, but including hard imputations for coma/unwilling/unable to complete and death) are shown in Appendix 4, Figure 22.
The unadjusted total QALYs (over the whole of the follow-up period, including hard imputed values as per Statistical methods of analyses, Missingness) are presented in Table 22. It is highlighted again that results correspond to only about 70% of the sample as information for remaining patients was missing. Given that the period of assessment is 84 days (i.e. 3 months = one-quarter of 1 year), the maximum total QALYs that were observed is 0.25. Thus, the distribution of total QALYs will be inherently be both left and right truncated. Mean unadjusted total QALYs were similar between treatment groups, with a total mean QALY of 0.06 (SE 0.06). The distribution of total unadjusted total QALYs, including hard imputed values as per Statistical methods of analyses, Missingness, can be seen in Figure 19.
| Unadjusted total QALYs | Treatment group | Total (n = 757) | |
|---|---|---|---|
| Placebo (n = 388) | Rifampicin (n = 369) | ||
| Mean (SD) | 0.054 (0.063) | 0.059 (0.059) | 0.057 (0.061) |
| Number of patients (%) | 275 (70.9) | 249 (67.3) | 524 (69.1) |
FIGURE 19.
Distribution of (a) total QALYs; and (b) imputed total QALYs from one randomly selected imputed data set using multiple imputation techniques.
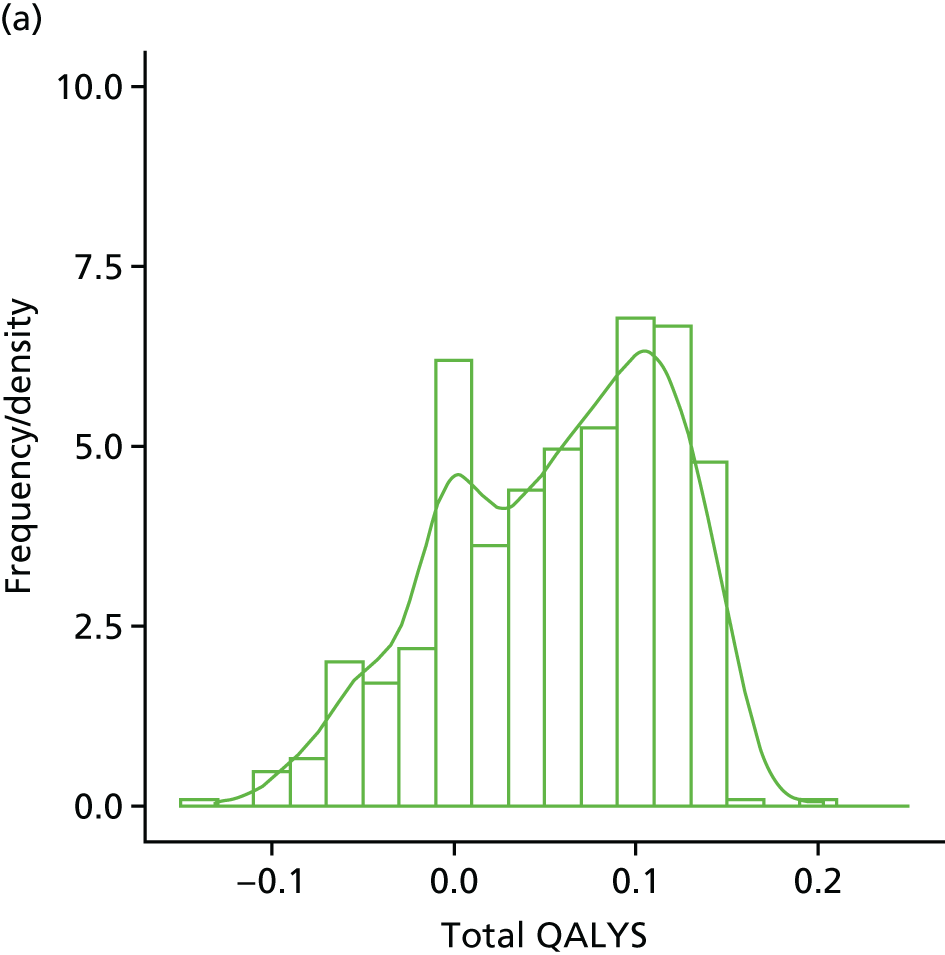
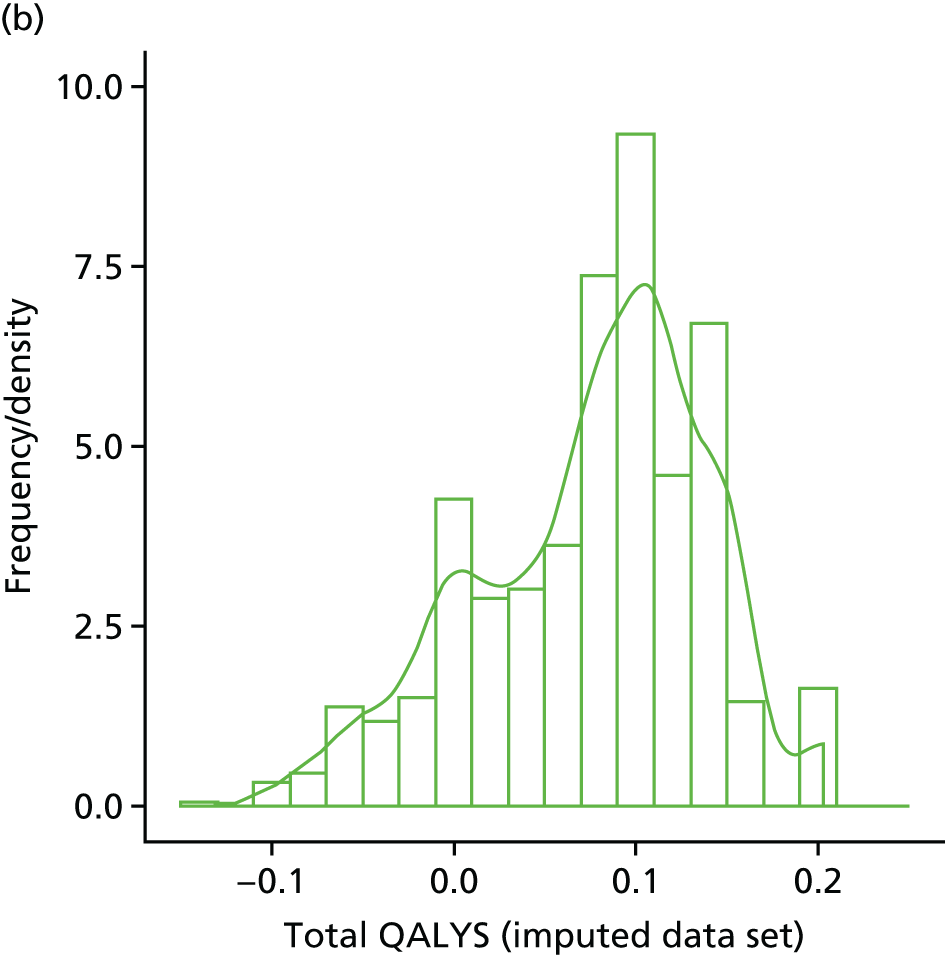
Quality-adjusted life-years (using multiple imputation)
A multiple imputation procedure was used to impute missing total QALYs at 84 days, which occurred in approximately 31% of the sample. Following a suggestion from the literature, in which the number of imputations should be similar to the percentage of cases that are incomplete,62,63 30 imputations of 20 iterations each were performed. Mode of acquisition of infection, Charlson Comorbidity Index score, BMI, deep infection foci, endocarditis, neutrophil count, coma status and EQ-5D index score were the baseline patient characteristics used as predictors by the imputation model. This process generated 30 different data sets with a calculated imputed outcome variable (i.e. total QALYs at 84 days). The distribution of total QALYs at 84 days for one of the imputed data sets, randomly chosen, can be seen in Figure 19. On these multiple imputed data sets, alternative GLM models for the total QALYs at 84 days were considered. As for the cost data, the null model, a model with only randomised treatment (model TQ) and a model with all covariates were implemented (model 1Q). A regression model assuming a normally distributed outcome with identity link (i.e. ordinary least squares model) was chosen (AIC statistic in model 1Q: –2109). Other modelling distributional assumptions tested either did not run or did not converge.
Base-case model (model 1Q)
The results of the base-case model (model 1Q) are shown in Table 23. These results are complemented with results of the model considering randomised treatment only (model TQ, first column). Regression models presented are linear and, therefore, additive, so coefficients can be interpreted directly to assess their impact on the outcome variable. In the model 1Q, being randomised to rifampicin was associated with slightly higher total QALYs (mean 0.004, SE 0.004) than being randomised to placebo, although this association was not statistically significant (p = 0.40, similar for model TQ). As expected, the EQ-5D index score at baseline was one of the main predictors of total QALYs accrued over 84 days, with one unit higher baseline EQ-5D estimated to be associated with higher total QALYs (model 1Q: mean difference of 0.06, 95% CI 0.05 to 0.08). Conversely, those aged ≥ 72 years (model 1Q: –0.043, 95% CI –0.072 to –0.014), those with any comorbidities as indicated in the Charlson Comorbidity Index score (gradient from –0.015, 95% CI –0.027 to –0.003 for index scores of 1–2, and up to –0.024, 95% CI –0.041 to –0.006, for higher index scores) and those in a coma (–0.020, 95% CI –0.037 to –0.004) were associated with significantly lower total QALYs. These covariates, together with meticillin resistance and neutrophil count, were retained in model 1Qp, showing that the latter are also relevant to explain variation in total QALYs.
| Model specification | Model | ||
|---|---|---|---|
| TQ | 1Q | 1Qpa | |
| Type of regression model | OLS | OLS | OLS |
| Equation | Total QALYs (imputed) | Total QALYs (imputed) | Total QALYs (imputed) |
| Covariates (baseline) | Coefficient (SE) | Coefficient (SE) | Coefficient (SE) |
| EQ-5D index baseline score | – | 0.064 (0.008)*** | 0.064 (0.008)*** |
| Randomised treatment (1 – rifampicin; 0 – placebo) | 0.007 (0.005) | 0.004 (0.004) | – |
| Age (years) | |||
| 54–71 | – | –0.026 (0.020) | –0.027 (0.020)*** |
| ≥ 72 | – | –0.043 (0.014)** | –0.044 (0.014)** |
| Sex (1 – male; 0 – female) | – | 0.004 (0.005) | – |
| Acquisition | |||
| Nosocomial infection | – | –0.005 (0.006) | – |
| Health care associated | – | –0.001 (0.006) | – |
| Charlson Comorbidity Index score | |||
| 1 or 2 | – | –0.015 (0.006)** | –0.015 (0.006)* |
| 3 or 4 | – | –0.019 (0.008)** | –0.020 (0.007)** |
| ≥ 5 | – | –0.024 (0.009)** | –0.024 (0.009)** |
| BMI (kg/m2) | |||
| 18.5–24.9 | – | 0.005 (0.010) | – |
| 25.0–29.9 | – | 0.005 (0.010) | – |
| 30.0–39.9 | – | 0.010 (0.011) | – |
| ≥ 40 | – | 0.004 (0.013) | – |
| Deep focus (1 – yes; 0 – no) | – | 0.0004 (0.005) | – |
| Endocarditis (1 – yes; 0 – no) | – | 0.003 (0.011) | – |
| Meticillin resistance | – | –0.027 (0.020) | –0.024 (0.021) |
| Neutrophils | |||
| 6–9 109/l | – | 0.004 (0.006) | 0.005 (0.006) |
| > 9 109/l | – | –0.010 (0.006)† | –0.011 (0.006)† |
| Comatose (1 – yes; 0 – no) | – | –0.020 (0.008)* | –0.020 (0.008)* |
| Intercept | 0.076 (0.009)*** | 0.104 (0.012)*** | 0.115 (0.006)*** |
| Observations | 757 | 724 | 724 |
For the mean patient in the trial across all covariates used in the regression, the weighted mean predicted total QALYs for the placebo group were similar in the rifampicin and the placebo groups (Table 24, first set of results). As expected, and because of model linearity, the difference in predicted total QALYs between treatment groups was estimated to be mean 0.004 (SE 0.004). Results of the sensitivity analysis on patients unable/unwilling to provide EQ-5D answers can be found in Appendix 2, Tables 38–40.
| HRQoL predictions (QALYs) | Treatment group | |
|---|---|---|
| Placebo | Rifampicin | |
| Model 1Q | ||
| Mean predicted total QALYs (SE) | 0.077 (0.008) | 0.080 (0.009) |
| Median predicted total QALYs (IQR) | 0.077 (0.071–0.082) | 0.080 (0.074–0.086) |
| Mean predicted total QALYs difference (SE) | 0.004 (0.004) | |
| Model 2Q | ||
| Mean predicted total QALYs (SE) | 0.076 (0.010) | 0.080 (0.013) |
| Median predicted total QALYs (IQR) | 0.076 (0.070–0.083) | 0.080 (0.071–0.088) |
| Mean predicted total QALYs difference (SE) | 0.004 (0.003) | |
Scenario analysis: consideration of treatment effect modifiers (model 2Q)
As for total costs, a scenario analysis was implemented for total QALYs (model 2Q) considering treatment interactions. The results of this scenario analysis can be found in Table 25. In general, similar results to model 1Q were obtained. Following model 1Q, model 2Q did not find treatment to be significantly associated with total QALYs. Model 2Qp results (also in Table 25) show that the most parsimonious model retained the following covariates as important to explain the outcome variable: EQ-5D baseline score, age, Charlson Comorbidity Index score, meticillin resistance, neutrophil count and coma status. Thus, randomised treatment and randomised treatment interactions were not selected for the most parsimonious model based on AIC statistics. As for model 1Q, and considering the mean trial patient, as no statistically significant difference was found between treatment groups, both groups had similar mean total QALYs (see Table 24, second set of results). Total QALY predictions for each patient subgroup and randomised treatment are presented in Cost-effectiveness and decision uncertainty.
| Model specification | Model | |
|---|---|---|
| 2Q | 2Qp | |
| Type of regression model | OLS | OLS |
| Equation | Total QALYs (imputed) | Total QALYs (imputed) |
| Covariates (baseline) | Coefficient (SE) | Coefficient (SE) |
| EQ-5D index score | 0.064 (0.011)*** | 0.064 (0.008)*** |
| Randomised treatment (1 – rifampicin; 0 – placebo) | 0.016 (0.022) | – |
| Age (years) | ||
| 54–71 | –0.028 (0.020) | –0.027 (0.020) |
| ≥ 72 | –0.041 (0.014)** | –0.044 (0.014)** |
| Sex (1 – male; 0 – female) | 0.005 (0.007) | – |
| Acquisition | ||
| Nosocomial infection | –0.002 (0.008) | – |
| Health care associated | –0.005 (0.009) | – |
| Charlson Comorbidity Index score | ||
| 1 or 2 | –0.011 (0.008) | –0.015 (0.006)* |
| 3 or 4 | –0.017 (0.010)† | –0.020 (0.007)** |
| ≥ 5 | –0.012 (0.010) | –0.024 (0.009)** |
| BMI (kg/m2) | ||
| 18.5–24.9 | –0.0003 (0.014) | – |
| 25.0–29.9 | 0.004 (0.014) | – |
| 30.0–39.9 | 0.015 (0.014) | – |
| ≥ 40 | 0.015 (0.018) | – |
| Deep focus (1 – yes; 0 – no) | 0.008 (0.007) | – |
| Endocarditis (1 – yes; 0 – no) | –0.005 (0.016) | – |
| Meticillin resistance | –0.020 (0.028) | –0.024 (0.021) |
| Neutrophils | ||
| 6–9 × 109/l | 0.010 (0.009) | 0.005 (0.006) |
| > 9 × 109/l | –0.018 (0.008)* | –0.011 (0.006)† |
| Comatose (1 – yes; 0 – no) | –0.020 (0.012)† | –0.020 (0.008)* |
| Treatment × EQ-5D index score | 0.003 (0.016) | – |
| Treatment × age, 54–71 years | 0.005 (0.011) | – |
| Treatment × age, ≥ 72 years | –0.007 (0.011) | – |
| Treatment × sex (1 – male; 0 – female) | –0.003 (0.009) | – |
| Treatment × acquisition, nosocomial infection | –0.010 (0.012) | – |
| Treatment × acquisition, health care associated | 0.008 (0.013) | – |
| Treatment × Charlson Comorbidity Index score of 1–2 | –0.007 (0.011) | – |
| Treatment × Charlson Comorbidity Index score of 3–4 | –0.003 (0.016) | – |
| Treatment × Charlson Comorbidity Index score of ≥ 5 | –0.024 (0.016) | – |
| Treatment × BMI of 18.5–24.9 kg/m2 | 0.009 (0.020) | – |
| Treatment × BMI of 25.0–29.9 kg/m2 | 0.003 (0.020) | – |
| Treatment × BMI of 30.0–39.9 kg/m2 | –0.009 (0.021) | – |
| Treatment × BMI of ≥ 40 kg/m2 | –0.020 (0.026) | – |
| Treatment × deep focus (1 – yes; 0 – no) | –0.018 (0.011)† | – |
| Treatment × endocarditis (1 – yes; 0 – no) | 0.019 (0.021) | – |
| Treatment × meticillin resistance | –0.013 (0.022) | – |
| Treatment × neutrophils, 6–9 × 109/l | –0.007 (0.011) | – |
| Treatment × neutrophils, > 9 × 109/l | 0.015 (0.011) | – |
| Treatment × comatose (1 – yes; 0 – no) | –0.001 (0.017) | – |
| Intercept | 0.098 (0.015)*** | 0.115 (0.006)*** |
| Observations | 724 | 724 |
Cost-effectiveness and decision uncertainty
The results presented on the analysis of costs and health outcomes (QALYs) showed that participants randomised to receive rifampicin for the treatment of S. aureus bacteraemia were expected to attain higher QALYs over the duration of the trial, and were expected to incur lower costs than those participants allocated to receive the placebo. These results were not, however, statistically significant.
Considering the mean total costs and mean total QALYs at face value, adjunctive rifampicin could promote relevant cost savings over an 84-day time horizon without compromising health outcomes, and that actually there may be even positive, although small, implications for total QALYs (Table 26). This means that rifampicin dominates placebo, that is, it costs less but provides additional health benefits compared with placebo. If releasing £20,000 to the NHS is assumed to result in 1 additional QALY (the cost-effectiveness threshold), the mean incremental net health benefit (INHB) of adjunctive rifampicin is approximately 0.06 QALYs (SE 0.04 QALYs).
| Cost-effectiveness outcomes | Treatment group, mean (SE) | |
|---|---|---|
| Placebo | Rifampicin | |
| Base-case results (using results from regression models 1C and 1Q) | ||
| Predicted total costs (£) | 12,142 (546.0) | 11,050 (509.7) |
| Predicted total QALYs | 0.077 (0.008) | 0.080 (0.009) |
| Incremental predicted total costs (£) | –1092 (749.8) | |
| Incremental predicted total QALYs | 0.004 (0.004) | |
| ICER (£/QALY gained) | Rifampicin dominates (i.e. costs less and has positive health benefits in relation to placebo) | |
| Incremental net health benefita | ||
| £13,000/QALY | 0.088 (0.058) | |
| £20,000/QALY | 0.058 (0.038) | |
| £30,000/QALY | 0.040 (0.025) | |
| Probability of being cost-effectivea | ||
| £13,000/QALY | 0.07 | 0.93 |
| £20,000/QALY | 0.06 | 0.94 |
| £30,000/QALY | 0.06 | 0.94 |
| Scenario analysis results (using results from regression models 2C and 2Q) | ||
| Predicted total costs (£) | 11,969 (535.8) | 10,900 (500.2) |
| Predicted total QALYs | 0.076 (0.010) | 0.080 (0.013) |
| Incremental predicted total costs (£) | –1068 (726.6) | |
| Incremental predicted total QALYs | 0.004 (0.003) | |
| ICER (£/QALY gained) | Rifampicin dominates (i.e. costs less and has positive health benefits in relation to placebo) | |
| Incremental net health benefita | ||
| £13,000/QALY | 0.086 (0.056) | |
| £20,000/QALY | 0.057 (0.037) | |
| £30,000/QALY | 0.039 (0.024) | |
| Probability of being cost-effectivea | ||
| £13,000/QALY | 0.06 | 0.94 |
| £20,000/QALY | 0.06 | 0.94 |
| £30,000/QALY | 0.06 | 0.94 |
Figure 20 shows the cost-effectiveness plane using results from the Monte Carlo simulation to represent uncertainty over incremental mean costs and QALYs (joint density plot). Each green dot represents a simulated incremental cost and QALY pair; the cloud of green points is representative of the uncertainty around the cost-effectiveness outcomes. The blue dot displays the mean incremental costs and effects. The majority of green points lie in the fourth quadrant of negative incremental costs and positive incremental benefits. The ARREST trial data show that rifampicin is likely to be less costly and is associated with small QALY gains (although uncertain) in the 84 days of follow-up (post randomisation). This suggests that rifampicin is cost-effective with very little associated uncertainty, [i.e. a very high probability of being cost-effective (93–94%)]. Similar results to the base case were found for the scenario analysis (see Table 26).
FIGURE 20.
Cost-effectiveness plane for the base-case results.
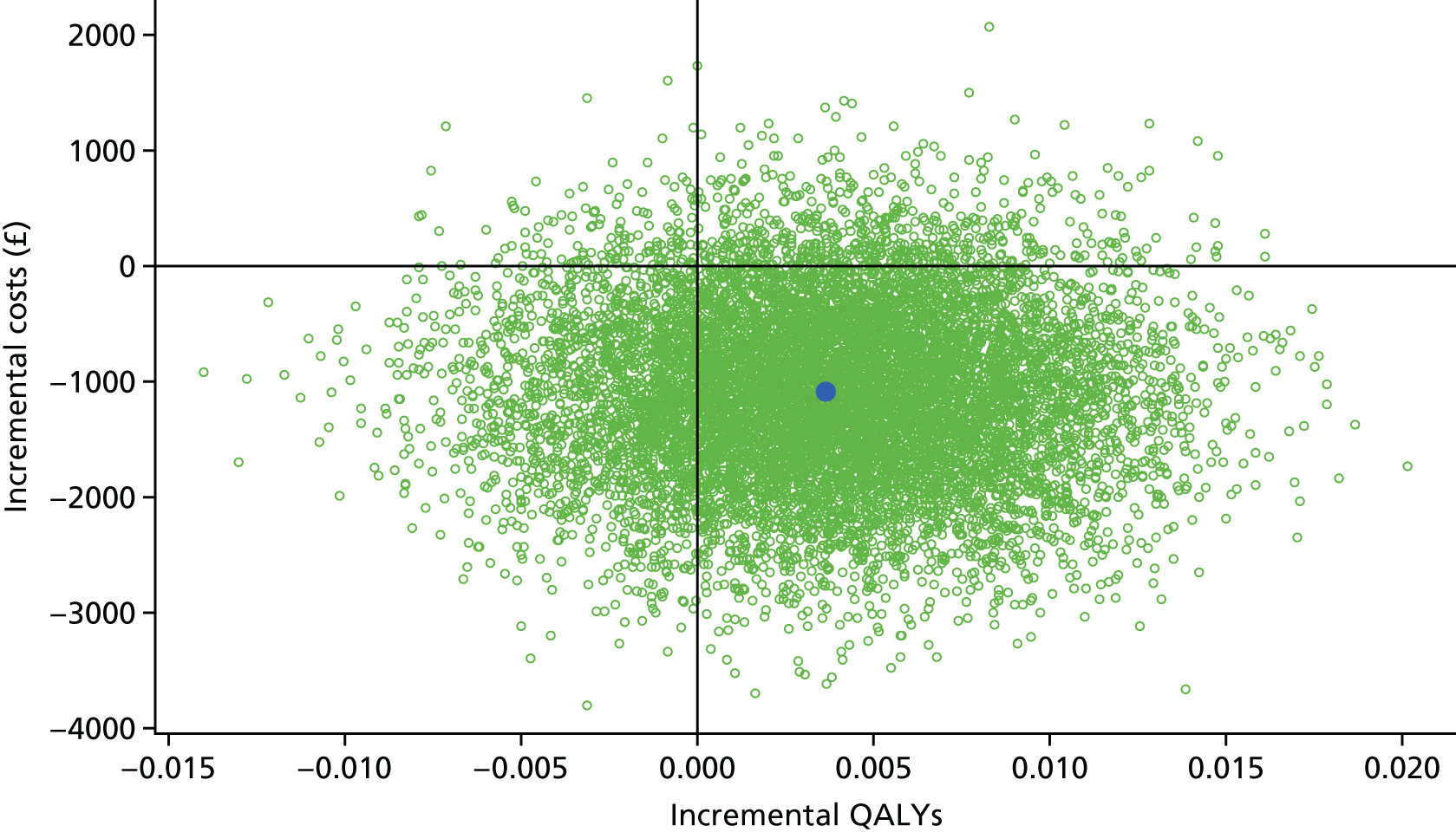
Considering a technology time horizon of 10 years and an annual effective population of 12,500 patients per year in the UK,2 the EVPI for the MRSA/MSSA bacteraemia population is estimated to be approximately £2M at the commonly used cost-effectiveness thresholds. This estimate represents the maximum amount that the health-care system should be willing to pay for further information and resolve identified uncertainties. At the individual level and for the same cost-effectiveness threshold values, the EVPI is estimated to be approximately £20 per MRSA/MSSA patient.
Subgroup analysis
This subgroup analysis uses the exploratory models with interactions (models 2C and 2Q) presented earlier to evaluate the evidence of the ARREST trial on whether or not costs and QALY impacts of a S. aureus episode differ with patient characteristics, and to evaluate whether or not there are subgroups for which the cost-effectiveness profile of rifampicin differs.
In terms of the costs of a S. aureus episode, the results of this analysis (Table 27) suggest that patients presenting with a nosocomial infection, low BMI, deep infection foci, endocarditis and MRSA have higher episode costs (> £15,000 per episode) than their counterparts. With regard to QALYs, a high neutrophil count (> 9 × 109/l), having MRSA or being aged > 72 years at presentation were factors associated with lower overall QALYs. With respect to the impact of baseline patient characteristics on the cost-effectiveness of rifampicin, the subgroup analysis suggests that, for most subgroups, rifampicin offers net health gains in relation to placebo. Those for whom rifampicin may not offer net health gains in relation to placebo are those aged between 54 and 72 years, those with a BMI between 25.0 and 29.9 kg/m2 and > 40 kg/m2, those with endocarditis and those in a coma. Note that this analysis is exploratory and findings should be interpreted with care.
| Cost-effectiveness outcomes, by subgroupsa | Treatment group, mean (SE) | INHBb | PCE rifampicinc | |||
|---|---|---|---|---|---|---|
| Placebo | Rifampicin | |||||
| Costs (£) | QALYs | Incr. costs (£) | Incr. QALYs | |||
| Age (years) | ||||||
| 18–54 | 11,991 (975) | 0.099 (0.006) | –1773 (1287) | 0.004 (0.006) | 0.092 | 0.93 |
| 54–72 | 11,364 (918) | 0.071 (0.021) | 1135 (1360) | 0.009 (0.009) | –0.047 | 0.25 |
| > 72 | 12,637 (1013) | 0.059 (0.014) | –2388 (1316) | –0.002 (0.009) | 0.117 | 0.96 |
| Sex | ||||||
| Female | 12,958 (984) | 0.073 (0.011) | –1944 (1315) | 0.006 (0.005) | 0.102 | 0.94 |
| Male | 11,492 (651) | 0.078 (0.010) | –634 (885) | 0.002 (0.005) | 0.034 | 0.78 |
| Mode of acquisition of infection | ||||||
| Community acquired | 10,998 (640) | 0.078 (0.011) | –767 (869) | 0.004 (0.004) | 0.041 | 0.83 |
| Nosocomial infection | 16,066 (1665) | 0.075 (0.012) | –1970 (2373) | –0.006 (0.011) | 0.094 | 0.78 |
| Health-care associated | 12,291 (1317) | 0.072 (0.011) | –1499 (1764) | 0.012 (0.012) | 0.087 | 0.83 |
| Charlson Comorbidity Index score | ||||||
| 0 | 10,989 (924) | 0.085 (0.011) | –233 (1312) | 0.010 (0.009) | 0.020 | 0.62 |
| 1 or 2 | 12,517 (909) | 0.074 (0.010) | –1014 (1213) | 0.003 (0.007) | 0.054 | 0.81 |
| 3 or 4 | 11,225 (1215) | 0.068 (0.013) | –147 (1880) | 0.007 (0.013) | 0.013 | 0.56 |
| > 5 | 14,089 (1700) | 0.073 (0.015) | –4414 (2084) | –0.013 (0.011) | 0.209 | 0.98 |
| BMI (kg/m2) | ||||||
| < 18.5 | 17,016 (3153) | 0.071 (0.018) | –6471 (3739) | 0.003 (0.021) | 0.322 | 0.97 |
| 18.5–24.9 | 11,179 (860) | 0.070 (0.009) | –1072 (1175) | 0.012 (0.008) | 0.066 | 0.87 |
| 25.0–29.9 | 11,826 (982) | 0.075 (0.011) | 393 (1414) | 0.006 (0.007) | –0.015 | 0.42 |
| 30.0–39.9 | 12,723 (1169) | 0.086 (0.011) | –2378 (1560) | –0.006 (0.008) | 0.113 | 0.93 |
| ≥ 40 | 11,222 (2098) | 0.085 (0.021) | 1645 (3255) | –0.017 (0.019) | –0.099 | 0.27 |
| Deep infection foci | ||||||
| No | 9855 (605) | 0.073 (0.010) | 56 (850) | 0.011 (0.005) | 0.007 | 0.57 |
| Yes | 16,111 (1148) | 0.081 (0.011) | –3479 (1513) | –0.007 (0.005) | 0.167 | 0.99 |
| Endocarditis | ||||||
| No | 11,703 (537) | 0.076 (0.010) | –1153 (730) | 0.003 (0.003) | 0.060 | 0.95 |
| Yes | 18,325 (4080) | 0.071 (0.016) | 1654 (5768) | 0.022 (0.023) | –0.056 | 0.42 |
| Meticillin resistance | ||||||
| No | 11,805 (540) | 0.077 (0.011) | –984 (741) | 0.004 (0.004) | 0.053 | 0.92 |
| Yes | 15,029 (3083) | 0.058 (0.028) | –2669 (3780) | –0.009 (0.021) | 0.127 | 0.75 |
| Neutrophil count | ||||||
| < 6 × 109/l | 10,998 (640) | 0.078 (0.011) | –767 (869) | 0.004 (0.004) | 0.041 | 0.83 |
| 6–9 × 109/l | 24 (1707) | 0.087 (0.017) | –1404 (2208) | –0.004 (0.011) | 0.065 | 0.72 |
| > 9 × 109/l | 14,944 (1793) | 0.060 (0.010) | –769 (2513) | 0.019 (0.010) | 0.055 | 0.67 |
| Coma | ||||||
| No | 11,769 (558) | 0.078 (0.010) | –1245 (751) | 0.004 (0.004) | 0.066 | 0.96 |
| Yes | 13,945 (2002) | 0.058 (0.014) | 899 (2975) | 0.003 (0.015) | –0.043 | 0.39 |
Discussion
The ARREST trial aimed to determine whether or not adjunctive rifampicin improved outcomes following S. aureus bacteraemia, but found no evidence of an effect either on resolution of bacteraemia or on mortality (design and effectiveness results of the trial are reported in detail in Chapters 3 and 4, respectively). In this chapter, evaluating the cost and HRQoL implications of S. aureus bacteraemia using the trial data were focused on first.
This first set of analyses found that an episode of S. aureus bacteraemia costs, on average, £12,197 over 12 weeks (unadjusted results). The cost categories that contribute the most to costs (descriptive analyses) are length of stay (primary hospital admission and readmissions) and procedures undertaken in hospital. Determinants of higher episode costs (variables evaluated at baseline), evident from the trial population, were whether or not the primary infection was nosocomial (episode costs 41% higher), whether or not there was a deep focus primary infection (episode costs 43% higher), whether or not the patient had endocarditis (episode costs 65% higher), whether or not the patient had a high neutrophil count (> 9 × 109/l, episode costs 34% higher), and whether or not the patient was comatose (episode costs 33% higher). For example, for an infection classified as having a deep focus, the mean costs of the episode were estimated at £12,514, whereas for infections without a deep focus the mean costs were £8752. In the ARREST trial population, neither age, sex, BMI, Charlson Comorbidity Index score nor meticillin resistance were found to determine costs at standard levels of standard statistical significance.
Analyses indicate that adjunctive rifampicin may save 10% of episode costs, although this result was not statistically significant at the standard 95% level (p = 0.14). Descriptive, unadjusted, analyses suggest that these savings start in the first 14 days of treatment (unadjusted difference in the first 14 days was £413), but that are perhaps most relevant after 14 days (unadjusted difference of £787). Because the trial was not powered on this outcome, the relevance of this finding (had a larger sample size been recruited) is unclear. However, this result is consistent with the reduction in recurrences, which occurred in a small proportion of participants, but significantly fewer in the rifampicin group (1%) than in the placebo group (4%).
As expected in this population of acutely ill patients, very low values of the EQ-5D score were observed at baseline (mean EQ-5D score of 0.10). A high proportion of patients were comatose, and a high proportion of individuals had health states that the valuation algorithm ascribed as worse than death (i.e. returning negative EQ-5D score values). Unadjusted figures show, however, that mean HRQoL score was significantly higher at 84 days (mean 0.30). The measure of benefit in the adjusted analysis considered QALYs over 84 days. QALYs are often the recommended measure of benefit for societal decision-makers, as they are generic and thus allow comparisons to be made across different treatments, conditions and patient populations. Given the high mortality and low HRQoL that this population is subjected to, total QALYs over the 84 days were on average 0.077 per patient, only 33% of the maximum innings for this period (0.23 QALYs or 84 out of 365 days). Determinants of QALYs in the sample were baseline EQ-5D score (0.0064 QALYs lost for every 0.1-point decrease in baseline EQ-5D), older age (up to 0.044 QALY loss), higher Charlson Comorbidity Index score (up to 0.024 QALY loss) and being comatose (mean QALY loss of 0.020). As opposed to total costs, deep foci infection did not affect total QALYs. After adjustment, the effect of rifampicin on total QALYs was positive (0.004 QALYs) but not statistically significant (SE 0.004 QALYs). Given the lack of statistical significance, the relevance of the finding that rifampicin has a positive (but small) effect on total QALYs is unclear; however, it is in accordance with the reduction in recurrences in the rifampicin group.
Public Health England has conducted mandatory enhanced surveillance of MRSA bacteraemia since October 2005 and of MSSA bacteraemia since January 2011. From April 2017 to March 2017, 823 cases of MRSA and 11,486 cases of MSSA were reported in England. 2 At the episode cost determined in the ARREST trial, these incidence figures imply a £150M burden to the NHS.
Based on the analyses from ARREST, adjunctive rifampicin could result in cost savings and negligibly small gains in mean QALYs. The cost savings possibly arise from reductions in hospital stay and readmissions in the short term. In cost-effectiveness terms, adjunctive rifampicin could be said to dominate placebo. The within-trial economic analysis, however, excluded potentially important outcomes, such as resistance arising from increased use of rifampicin and the clinical consequences of its drug–drug interactions (which may not have been captured fully in the analyses as costs of non-antibiotic drugs were not included, nor were costs of monitoring tests, e.g. for toxicity). This was a pragmatic decision because patients enrolled in this trial had wide range of underlying conditions and will have required a very large number of other drugs. A decision was therefore made not to try to record all of these other drugs on case report forms (CRFs), which would make them impossible to cost. Similarly, it was difficult to know what quantitative data to record to assess drug interactions. Rather than collect a large amount of free text to try to code and risk missing different items for different episodes, a pragmatic decision was made to not include these on CRFs either. Moreover, the ARREST trial was conducted under experimental conditions, and, despite providing unbiased estimates of treatment effects, practice may not be as homogeneous, and, hence, further research could confirm whether or not any predicted cost savings would be effectively realised in practice.
Chapter 6 Discussion
A large, multicentre, pragmatic, placebo-controlled trial was conducted that randomised 758 adults with S. aureus bacteraemia. The trial was designed to determine whether or not rifampicin, added to standard ‘backbone’ antibiotics for up to 14 days, reduced bacteriologically confirmed failure/recurrence or death by 12 weeks. No evidence was found that rifampicin affected any of the composite primary or secondary efficacy measures including mortality, the duration of bacteraemia or the development of rifampicin-resistant S. aureus. Rifampicin was, however, associated with a small but significant reduction in bacteriologically and clinically defined disease recurrences.
The population included in the trial represented the severity and heterogeneity of S. aureus bacteraemia. Participants were mostly older adults (median age 65 years), many of whom had a number of comorbidities (median Charlson Comorbidity Index score of 2). A substantial minority (9.2%) were enrolled in an ICU, reflecting the severity of the infection. Substantial improvements in hospital infection prevention and control over the last decade in the UK meant that most (64.0%) infections were acquired in the community, with only 17.4% being nosocomial in origin (acquired > 48 hours after hospital admission). Similarly, the UK has witnessed a major decline in MRSA infections over the same period and only 6.2% of patients had bacteraemia caused by MRSA. 64 A deep infection focus, denoting a complicated infection, was present at baseline in 301 (39.7%) patients, around half with endocarditis, orthopaedic or intravascular devices, or osteoarticular infections, and 139 (18.3%) patients had no established infection focus. Therefore, a substantial proportion of patients had what are generally considered as uncomplicated infections, in which there is a single, superficial and easily removable infection focus (an infected intravascular catheter, for example) without evidence of deep infection foci.
One of the key findings from the trial is the enormous variation in the choice and duration of ‘backbone’ antibiotics (see Appendix 2, Table 28). The majority of participants (81.7%), however, received flucloxacillin (an anti-staphylococcal penicillin) at some point in their primary treatment. In the UK and Australia, flucloxacillin is the recommended first-line anti-staphylococcal penicillin for MSSA infections, whereas other agents, such as nafcillin and cloxacillin, are recommended in the USA. There is no evidence supporting clinically relevant differential anti-staphylococcal activity between these antibiotics,65,66 and, therefore, the authors believe that the results are generalisable across countries regardless of their chosen anti-staphylococcal penicillin. A total of 50.1% of patients received a glycopeptide at some point in their primary treatment, probably reflecting ongoing concerns about MRSA infections despite the overall low rates, particularly given the severity of disease in many of the trial participants. The use of other antibiotics (including open-label rifampicin) and the total duration of active antibiotic therapy (median 29 days) were similar between randomised groups. Fewer rifampicin- than placebo-treated participants were restarted on antibiotics after the primary treatment course, which may reflect the lower recurrence rate in the placebo group. The variety of antibiotics received demonstrates the utter infeasibility of conducting a trial restricting therapy to one single backbone antibiotic. Furthermore, had only one standard antibiotic regimen been used, clinicians could legitimately argue that the effect of rifampicin might be different on another backbone antibiotic. All antibiotic regimens were chosen by infection specialists, taking into account individual patient allergy and concomitant medication. No evidence was found of variation in the lack of effect of rifampicin by initial treatment class. Therefore, the authors consider that the results are more generalisable than would have been obtained from one single regimen.
Planned subgroup analysis did not identify a subpopulation of participants who clearly benefited from the addition of rifampicin. There was a suggestion that the effect of rifampicin may have varied according to the antibiotics used at randomisation, with any benefit restricted to those with MSSA infection treated with flucloxacillin alone. However, this result has uncertain clinical significance. There was no evidence of benefit if flucloxacillin was used with vancomycin or another antibiotic, or if antibiotic class was used to define subgroups, findings that are inconsistent with an isolated effect of flucloxacillin. With 20 subgroups analysed, one statistically significant association may have occurred by chance. Many infection specialists might have predicted that rifampicin might benefit those with deep, complicated infection the most, and possibly those with disease caused by MRSA. No such associations were found, although only a small minority of participants had MRSA bacteraemia. Indeed, if anything, those with MRSA bacteraemia did worse with rifampicin than the placebo (see Figure 5).
It was hypothesised that the early addition of rifampicin to standard antibiotic treatment would enhance the early killing of S. aureus and thereby improve outcomes. The trial inclusion criteria therefore required rifampicin to be initiated anywhere from 0 to 96 hours after initiating active antibiotics for the infection. Given that most patients with S. aureus bacteraemia are very unwell and require immediate empirical antibiotic therapy, and it takes at least 36 hours to culture and identify S. aureus from blood cultures, it is unsurprising that participants received a median of 62 hours of other active antibiotics before treatment with rifampicin. This may have represented a clinically meaningful delay in initiating rifampicin treatment, which could have affected efficacy. However, there was no evidence of such an effect considering time from randomisation to initiation of rifampicin/placebo as either categorical subgroups or as a continuous interaction factor (see Figure 5). Additional subgroup analysis needs to be interpreted carefully, given the number of tests performed,67 and we do not believe they should be highlighted as clinically significant findings within the conclusions of the study.
The authors believe that the study results refute the hypothesis that adjunctive rifampicin enhances S. aureus killing in blood and thereby reduces the risk of dissemination and death. 16 Both randomised groups had similar rates of bacterial clearance in blood, and there was no evidence of difference in all-cause mortality over the short (2 weeks), medium (12 weeks) or even in the longer term (> 52 weeks). Even the 50% of deaths that were adjudicated as definitely/probably because of S. aureus (50%) occurred similarly in rifampicin and placebo groups. However, the observed mortality in the trial was lower than that observed in many recent observational studies. For example, a recent large multicentre case series reported substantially higher 12-week mortality (29.2%)68 than observed here (14.8%). The few RCTs that have been reported in this disease (e.g. the trial of daptomycin in S. aureus bacteraemia69) tend to report lower mortality. This probably reflects differences in the populations between observational and interventional studies. It is possible that the most severely unwell patients, who are expected to die quickly, are less likely to enter interventional studies. Indeed, in the ARREST trial there were 129 patients who either died or were considered too unwell for active treatment and, therefore, did not join the trial (see Figure 2). Mortality would have nearly doubled had they joined the trial and died. But there may be other reasons for the lower mortality observed in the ARREST trial. Regular infection specialist consults were mandatory for the trial, which may have reduced mortality. Infection consults have been associated with improved S. aureus bacteraemia outcomes in observational studies. 70
Another hypothesis, that rifampicin enhances the sterilisation of deep infection foci and thus reduces disease recurrences, is, at least partially, supported by the findings. 71 A small but statistically significant reduction in recurrences was found in the rifampicin group, suggesting some biological activity of the drug. However, the clinical significance of such a small reduction is unclear. The NNTs to prevent bacteriologically and clinically defined recurrences were 29 and 26 patients, respectively. More importantly, prevention of recurrences did not affect either short- or long-term mortality (see Figures 9 and 10). Of note, the independent, blinded end-point review committee adjudicated that recurrences were much more likely to have been caused by failure to recognise or remove the primary infection focus than by failure of antibiotic treatment (see Table 7). This observation demonstrates the importance of source management in future research to improve outcomes from S. aureus bacteraemia. Recent strategies that enhance the identification of infection foci by PET scanning have been associated with reduced mortality from S. aureus bacteraemia. 72 Taken together, these findings suggest the need for a multifaceted approach to improving outcomes from S. aureus bacteraemia. Rifampicin may assist in sterilising deep S. aureus infection foci and prevent a few recurrences, but it does not replace the need to define and, when possible, drain or remove the infection focus.
The modest benefit of rifampicin on recurrences (and any resulting cost savings) needs to be balanced against the toxicity of rifampicin and complications surrounding its use, especially in an older population with comorbidities that often require other drug treatments. Predicted drug interactions or pre-existing liver disease prevented 306 out of 2896 (10.6%) screened subjects from being randomised. Although there was no evidence of differences between groups in the proportions with SAEs, significantly more antibiotic-modifying AEs and drug interactions occurred in participants in the rifampicin group than in the placebo group. The AEs were predominantly gastrointestinal disorders and, interestingly, renal impairment. Rifampicin was associated with acute kidney injury in 17 participants compared with six participants in the placebo group. Although these numbers are small, and renal impairment is a recognised toxicity of rifampicin in the summary of product characteristics, this is an important aspect of its use that is rarely considered by clinicians. In contrast, drug-induced liver injury was predicted to be common but turned out to be extremely rare, possibly because patients vulnerable to liver injury were not enrolled.
The strengths of the ARREST trial include its placebo-controlled, multicentre pragmatic design. This ensures that it provides generalisable, clinically relevant findings to clinicians and patients within the NHS. It is also the largest trial ever conducted examining S. aureus bacteraemia treatment. It does, however, have important limitations that reflect the many challenges of performing trials in acutely unwell patients with severe bacterial infections, and the current UK trial funding arena. 73 The heterogeneous nature of this severe disease, and the requirement to randomise patients within 96 hours of the start of antibiotic therapy because of the underlying hypothesis, led to a large number of ineligible patients and meant recruitment was slower than anticipated. Only 26.6% (770/2896) of those screened were enrolled; the most common reason was having already received > 96 hours of antibiotics in around one-third of those not enrolled (664 patients, 31.2%). Furthermore, 232 (10.9%) screened patients were not randomised because rifampicin was considered mandatory. This information was available only as a reason for ineligibility with no additional details, but anecdotally prosthetic device-related infections were common in these patients. The clinical effect of rifampicin may potentially have been reduced as a consequence of excluding these patients, reducing the relevance of the findings to those with bacteraemia associated with infected prostheses, for whom rifampicin may have more benefit. 34
A proportion of patients initiated open-label rifampicin or stopped blinded trial drug early, predominantly for drug–drug interactions or AEs. Such deviation from intended treatment would be expected in normal clinical practice and, therefore, the intention-to-treat comparison of the groups probably reflects the effectiveness of rifampicin more widely. However, there was also no evidence of benefit from rifampicin in the per-protocol population who received ≥ 80% of expected doses. Outcome ascertainment was very high, with only a small number (≈9%) of patients in whom vital status and/or signs and symptoms could not be ascertained at the 12-week follow-up visit. The total number randomised in error and lost to follow-up or withdrawing consent was very close to the 10% incorporated in the sample size calculation.
A far more critical limitation to the timely completion of this trial was the heterogeneity in the trials support network in the UK, which is far more suited to recruiting large numbers of chronically unwell individuals from a small number of fixed clinics than recruiting acutely unwell individuals who present sporadically at varying times of day and night and require a great deal of care in explaining research at a time of acute illness. Some centres received excellent support and were able to recruit larger numbers. Others received, for example, research nurse support on 2 fixed days of the week, regardless of when patients presented acutely unwell, or were unable to access promised support when patients did present, because research staff were committed to fixed clinics at the time. Thus, in many centres the burden of recruiting patients and conducting research visits fell to the PI, typically a consultant microbiologist or infection specialist who took this on outside their day-to-day work. There are clearly enormous challenges for research networks in supporting trials in acute, relatively uncommon and sporadic diseases, but their severity, with one in six patients dying in this trial, highlights the importance of finding a way to do this. Even more frustrating was the system of ‘targets’, which are extremely difficult to assess in acute illnesses such as S. aureus bacteraemia. One of the top recruiting sites was forced to close to recruitment early, despite the trial struggling as a whole to meet its recruitment targets, because their individual site target had been met and the local research office was required to move its resources to other studies to avoid being penalised. The unintended consequences of rigid adherence to targets, which are really impossible to specify with any degree of confidence in acutely presenting complaints such as S. aureus bacteraemia, was an increase in the total time the trial took to recruit. Particularly when randomised controlled trials are competing with observational studies for research support, there needs to be a better way to encourage sites that are able to recruit to trials to do so beyond arbitrary targets.
Originally, the trial was powered to detect an absolute difference of 10% in bacteriological failure/recurrence or death from 35% to 25% and a 7% absolute reduction in mortality from 16% to 9%, based on results from a small systematic review. 74 Slow recruitment meant that the mortality coprimary end point was moved to a secondary end point, consequently reducing the sample size needed to detect the 10% absolute reduction in bacteriological failure/recurrence or death because the two-sided alpha (false positive) increased from 0.025 (two coprimary outcomes) to 0.05 (one primary outcome). The 758 eligible participants included are more than double the number in the largest previous trial in S. aureus bacteraemia,25 and increase the total numbers with S. aureus bacteraemia recruited in randomised trials over the last 50 years by 50%. The 95% CI around the estimates of the difference between rifampicin and placebo lie within 7.5%, smaller than the 10% non-inferiority margins recommended by licensing authorities for antibiotic trials and commonly used in other infections such as HIV. This would have been considered to conclusively demonstrate non-inferiority of rifampicin had an active comparator been used. Although the trial was designed to test the superiority of rifampicin, it thus provides convincing evidence of non-inferiority of rifampicin to placebo; that is, convincing evidence of lack of benefit. A small minority (13%) used open-label rifampicin in the placebo group, but per-protocol analyses confirmed this well-estimated lack of benefit of rifampicin over placebo.
It was found that an episode of S. aureus bacteraemia costs, on average, £12,197 over 12 weeks. These costs were driven primarily by length of stay and procedures undertaken in hospital. Last year (April 2016–March 2017) there were 12,309 episodes of S. aureus bacteraemia reported within the NHS in England. 75 Therefore, it is estimated that S. aureus costs the NHS around £150M each year.
Interventions that reduced these costs would be welcome. On the basis of the clinical data provided by the trial, it was concluded that rifampicin was of no overall clinical benefit to individuals with S. aureus bacteraemia. However, the cost-effectiveness analysis suggested that adjunctive rifampicin may have a possible health economic benefit to the NHS. Rifampicin was estimated to save 10% of episode costs (p = 0.14). Most of these savings related to small reductions in length of hospital stay, especially after the first 14 days of treatment. These reductions probably relate to the small but significant reductions on recurrences associated with the use of rifampicin over placebo (1% vs. 4%; p = 0.01).
Important limitations to the cost-effectiveness analysis include the missing costs of rifampicin toxicity (including monitoring for toxicity) and drug–drug interactions in the analysis. These important clinical complications of rifampicin treatment were highlighted in the clinical data but were not captured in the cost-effectiveness analysis. In addition, the widespread use of rifampicin would undoubtedly lead to the increased prevalence of rifampicin resistance among S. aureus and other medically important bacteria. These costs could be substantial, especially if it caused a rise in rifampicin-resistant M. tuberculosis infections, and have not been considered. In short, on balance, the authors do not believe that the possible cost savings of rifampicin to the NHS should outweigh the lack of overall clinical benefit to an individual with S. aureus bacteraemia. In support of this position is the lack of a significant effect of rifampicin on QALYs.
The ARREST trial was developed with the assistance of the Healthcare-associated Infection Service Users Research Forum, and Jennifer Bostock, the PPI representative. Ms Bostock advised on the inclusion of incapacitated adults and the application of the Mental Capacity Act 2005,36 and the information provided to patients. The information sheets, consent forms and recruitment processes were developed in collaboration with the SURF, and Ms Bostock to help ensure that they communicated the risks and benefits clearly and appropriately. There were sensitive ethics issues that arose at ethics review and the PPI representative was instrumental in helping the team gain ethics approval. Furthermore, when it was necessary for the trial team to request an extension to the study from the funders, Ms Bostock accompanied them to the meeting and helped put the case as to why the trial was important to patients/relatives and the public. The panel remarked that it was the first time they had seen a public member attend such a meeting. It reflected the trial team’s commitment to PPI and the creative use to which they engaged the ‘expertise’ of Ms Bostock. Ms Bostock was also a member of the ARREST Trial Steering Committee.
It was Ms Bostock’s idea to run a qualitative substudy within the main trial (see Chapter 4). The study was designed, developed and delivered by her with assistance from the trial team. It was deemed important that the PPI representative was responsible for this aspect of the trial as it was felt that there would be a better response rate and more honest answers if the person conducting the study was independent and had a ‘public voice’. The substudy was small in scope and had limited findings, but it was an unusual inclusion in a trial of this nature.
The PPI played, and will continue to play, an active role in disseminating the trial’s results. Ms Bostock has both reviewed and co-authored some of the main academic outputs from the study and the main conference presentations of the results, as well as a leaflet presenting results to patients and their GPs [www.journalslibrary.nihr.ac.uk/programmes/hta/1010425/#/ (accessed 4 June 2018)]. It is important to the entire trial team that dissemination goes beyond the traditional academic and health-care professional communities to others, including patient groups and the wider public. With this in mind, the team agreed that having a creative approach to dissemination to engage with patients, the public and policy-makers may benefit this process. An example of this creative approach is provided by an infographic, designed by Will Everett (Science Communications Officer, MRC CTU, UCL, 2017), which summarises the findings of the trial for dissemination (Figure 21). This was reviewed and revised by the PPI advisor and will be used to showcase the trial and results to patients and the public after publication. In addition, Ms Bostock and other members of the trial team were interviewed for a podcast aimed at clinicians [see https://soundcloud.com/user-110325996–105034477/arrest-podcast-v03/s-J4lta (accessed 4 June 2018)]. The interviewees discussed the results of the study and the implications for health-care workers, patients and the public. Ms Bostock and her wider network will continue to disseminate the results of the study to relevant audiences via her links with MRSA Action UK, The Healthcare Infection Society, The Infection Prevention Society, The Patients Association, The Research Design Service PPI Advisory Group and The Biomedical Research Centre PPI Advisory Group.
FIGURE 21.
The ARREST trial infographic.

From Ms Bostock’s perspective, the trial has succeeded in involving patients and the public in a way that is rare in clinical trials of medicines. The team’s support, patience and willingness to adapt and change the trial in response to public input have benefited both the public representative and the trial. Being involved in the trial has enabled Ms Bostock to develop skills and understanding of clinical trials and PPI, which she will use to benefit other research, and it is hoped that researchers conducting similar trials will adopt some of the methods used in the ARREST trial as a model of good PPI practice.
Summary and future research
In summary, the ARREST trial provides high-quality data when there are almost none. The clinical management of infectious diseases and, in particular, the treatment of many common severe bacterial infections, lacks high-quality clinical trial evidence. The situation is especially stark for S. aureus bacteraemia, probably the commonest life-threatening community- and hospital-acquired infection worldwide. But although the ARREST trial addresses some of these inadequacies, it also leaves many questions unanswered as to how to improve outcomes from S. aureus bacteraemia.
The ARREST trial has exposed two possible windows in which to intervene. The first is in the acute phase, when S. aureus can be cultured from the bloodstream and a severe inflammatory response (or ‘sepsis’) can have rapidly fatal consequences. The interventions in this early phase are those targeted at more rapid recognition or diagnosis of S. aureus bacteraemia, and those that might enhance bacterial killing and control the detrimental effects of the inflammatory response. Future research therefore might investigate novel molecular techniques, perhaps based on rapid next-generation sequencing, to identify S. aureus from the blood and predict drug susceptibility such that effective antibiotic treatment can be given quickly. The question of whether or not intensified antibiotic therapy – be it different drugs, doses, or drug combinations – might speed bloodstream sterilisation very early in the infection and thereby improve outcomes, has not been resolved by the ARREST trial, although it has delineated the considerable challenges of conducting a trial to address this question. Likewise, early control of the inflammatory response, using corticosteroids or newer drugs targeted at specific molecules in the inflammatory cascade, might reduce early mortality and would be amenable to testing by clinical trials.
However, given the challenges experienced conducting this trial, this is probably not the most important priority for future studies. Rather, the second interventional window in S. aureus bacteraemia is more easily accessible to triallists than the first window and, as the health economic analysis suggests, may also bring substantial cost savings. It opens after around 72–96 hours of active antibiotic treatment, once the acute phase is over, and concerns interventions to prevent, detect and manage the longer-term complications of the S. aureus bacteraemia, including disease recurrence. These complications include endocarditis, vertebral osteomyelitis, and other deep and potentially occult infection foci. As recent PET scan studies have shown,72 perhaps the most promising strategies that should be prioritised for future research are those that aim to speed the detection of these complications and improve their antibiotic and surgical management; these could have major impacts on outcome and costs. A clinical trial investigating these strategies against current standards of care would be both feasible and likely to have a major impact on clinical practice.
Chapter 7 Conclusions
Adjunctive rifampicin did not improve outcomes from S. aureus bacteraemia, with the exception of a modest reduction in disease recurrence, which may be associated with reduced costs. Given that (1) the clinical effect had no impact on short- or longer-term mortality, (2) rifampicin significantly complicated other drug treatment, and (3) widespread rifampicin use risks increasing resistance among S. aureus and other bacteria (e.g. M. tuberculosis), it is considered that, despite potential cost savings, adjunctive rifampicin provides no overall benefit over standard antibiotic therapy in adults with S. aureus bacteraemia.
Acknowledgements
The authors would like to thank all of the patients, their families and the staff from the participating centres in the ARREST trial. They would particularly like to thank Annabelle South and Will Everitt for assistance with dissemination activities, including the ARREST trial infographic (see Figure 21).
The ARREST co-applicants
Professor Tim Peto, Dr Gerraint Davies, Dr Martin Llewelyn, Professor Peter Wilson, Dr Duncan Wyncoll and Marta Soares.
The ARREST Trial Steering Committee
Dr Adrian Martineau (chairperson), Dr Geoff Scott, Dr Achim Kaasch, Ms Jennifer Bostock, Professor Guy Thwaites, Professor A Sarah Walker, Dr Gavin Barlow and Dr Susan Hopkins.
The ARREST Trial Management Group
Professor Guy Thwaites, Professor A Sarah Walker, Fleur Hudson, Janet Cairns, Denise Ward, Alex Szubert, Helen Webb, Charlotte Russell, Chiara Borg, Brooke Jackson, Damilola Otiko, Lindsey Masters, Zaheer Islam, Carlos Diaz and Debbie Johnson.
Data Monitoring Committee
Professor David Lalloo (chairperson), Professor Mark Wilcox and Professor Doug Altman.
End-point review committee
Professor Tim Peto and Dr Graham Cooke.
Recruiting sites
Oxford University Hospitals NHS Foundation Trust: M Scarborough, M Kamfose, A de Veciana, NC Gordon, L Peto, G Pill, T Clarke, L Watson, B Young, D Griffiths, A Vaughn, L Anson, E Liu, S Perera, L Rylance-Knight, C Cantell and R Moroney.
Guy’s and St Thomas’ NHS Foundation Trust: JD Edgeworth, G Thwaites, K Bisnauthsing, A Querol-Rubiera, C Gibbs, A Patel, C Hemsley, AL Goodman, D Wyncoll, J Biswas, J Fitzpatrick, L Roberts, J Millard, N Stone, A Cape, L Hurley and C Kai Tam.
The Royal Liverpool and Broadgreen University Hospitals NHS Trust: E Nsutebu, M Hoyle, K Maitland, L Trainor, H Reynolds, J Harrison, J Anson, J Lewis, J Folb, L Goodwin, N Beeching, S Dyas, H Winslow, E Foote, P Roberts, P Natarajan, A Chrdle, M Fenech and H Allsop.
Plymouth Hospitals NHS Trust: R Tilley, R Austin-Hutchison, L Barrett, K Brookes, L Carwithen, A Conbeer, R Cunningham, C Eglinton, R Fok, H Gott, S Hughes, L Jones, M Kalita, A King, L March, M Marner, T Mynes, A Plant, S Price, J Sercombe, A Stolton, M Wallis, M West, J Westcott, C Williams, R Wosley and L Yabsley.
Sheffield Teaching Hospitals NHS Foundation Trust: J Greig, L Butland, J Sorrell, T Mitchell, A Alli, J Meiring, B Masake, C Rowson, L Smart, L Makey, S Moll, J Cunningham, K Ryalls, K Burchall, J Middle, Y Jackson, D Swift, J Cole, B Subramanian, F Okhuoya, M Edwards, C Bailey, R Warren, G Islam, M Ankcorn, S Birchall, P Jones, J Humphries, S Booth and C Evans.
Portsmouth Hospitals NHS Trust: S Wyllie, A Flatt, L Strakova, M Hayes, S Valentine, C James, M Wands, N Cortes, N Khan, R Porter, Z Martin, K Yip, H Preedy, H Chesterfield, T Dobson and C Walker.
Brighton and Sussex University Hospitals NHS Trust: M Llewelyn, A Dunne, L Latter, A Porges, J Price, J Paul, L Behar, L Robinson, A Murray, J Fitzpatrick, T Sargent, C Ridley, L Ortiz-Ruiz de Gordoa, D Gilliam, C McPherson, S Matthews, E Foreman, R Jarghese, A Beddoe, S Martin, S Shaw, D Wlazly, M Cole, A Gihawi and K Cole.
Cambridge University Hospitals NHS Foundation Trust: ME Török, T Gouliouris, L Bedford, RB Saunderson, I Mariolis, R Bousfield, I Ramsay, D Greaves, S Aliyu, K Cox, L Mlemba, L Whitehead, N Vyse and M Bolton.
South Tees Hospitals NHS Foundation Trust: J Williams, P Lambert, D Chadwick, K Baillie, M Cain, R Bellamy, J Wong, J Thompson, H Vassallo, A Skotnicka, A Boyce and A Donnelly.
University College London Hospitals NHS Foundation Trust: P Wilson, G FitzGerald, V Dean, K Warnes, A Reyes, S Rahman, L Tsang, J Williams and S Morris-Jones.
Royal Free London NHS Foundation Trust: S Hopkins, E Witness, O Brady, E Woodford, T Pettifer, A McCadden, B Marks, S Collier, D Mack, S Warren, C Brown, A Lyons, S Taiyari, S Mepham, A Sweeney and L Brown.
Royal Devon and Exeter NHS Foundation Trust: C Auckland, A Potter, J Mandiza, M Hough, S Williams, C Renton, F Walters, M Nadolski, M Hough, A Evans, P Tarrant, S Williams, K Curley, S Whiteley, J Halpin, M Hutchings, S Todd, C Lohan, T Chapter, E Folland, A Colville, K Marden, M Morgan, R Fok, R Porter and M Baxter.
The Leeds Teaching Hospital NHS Trust: J Minton, S Rippon, M Cevik, J Chapman, T Kemp, R Vincent, D Osborne, T Platt, J Calderwood, B Cook, C Bedford, L Galloway-Browne, N Abberley, K Attack and J Allen.
Aintree University Hospital NHS Foundation Trust: P Lal, M Harrison, S Stevenson, C Brooks, P Harlow, J Ewing, S Cooper, R Balancio-Tolentino, L O’Neil, R Tagney and D Shackcloth.
St George’s Healthcare NHS Trust: T Planche, J Fellows, R Millett, J Studham, C de Souza, G Howell, H Greaves, E Foncel, R Kurup, J Briggs, M Smith, C Suarez, G Sorrentino, A Scobie, A Houston, F Ahmad, A Breathnach, R Chahuan and K Wilkins.
Blackpool Teaching Hospitals NHS Foundation Trust: A Guleri, N Waddington, R Sharma, P Flegg, V Kollipara, M Alam, A Potter, S Donaldson, C Armer and J Frudd.
King’s College Hospital NHS Foundation Trust: D Jeyaratnam, M Joy, A Mathews, SK Glass, A Ajayi, A Fife, S Qaiser, S Sheehan, S Muñoz Villaverde, NO Yogo, I De Abreu, G Notcheva, J Flanagan, C Watson, E Sais, A Adedayo, V Chu, G Shaw, MA Graver, R Palmer, D Palmer, S Haile, J Gordon, C Kai Tam, Mandar K and W Szypura.
Heart of England NHS Foundation Trust: N Jenkins, J Marange, V Shabangu, K Moore, J Lyons, M Munang, M Sangombe, E Moran and A Hussain.
University Hospitals of Leicester NHS Trust: M Wiselka, A Lewszuk, S Batham, K Ellis, L Bahadur, H White, M Pareek, A Sahota, S Coleman, H Pateman, A Kotecha, C Sim and A Rosser.
County Durham and Darlington NHS Foundation Trust: D Nayar, J Deane, R Nendick, C Aldridge, A Clarke, M Wood, A Marshall, L Stephenson, T Matheson-Smith, J Sloss, K Potts, J Malkin, L Ftika and V Raviprakash.
University Hospital Southampton NHS Foundation Trust: J Sutton, A Malachira, M Kean, K Criste, K Gladas, C Andrews, C Hutchison, E Adams, J Andrews, B Romans, N Ridley, M Ekani, J Mitchell, N Smith, T Clark, S Glover, R Reed, T Yam, H Burton and R Said.
Wirral University Teaching Hospital NHS Foundation Trust: D Harvey, A Janvier, R Jacob, C Smalley and A Fair.
Dartford and Gravesham NHS Trust: A Gonzalez-Ruiz, S Lord, K Ripalda, H Wooldridge, L Cotter, G Cardoso, E Strachan, G Kaler, A Mohamoodally, E Lawrence, Z Prime and R Abrahams.
Newcastle Upon Tyne Hospitals NHS Foundation Trust: DA Price, L Rigden, L Shewan, K Cullen, I Emmerson, K Martin, H Wilson, C Higham, K L Taylor, E Ong, B Patel, H Bond, J Gradwell and J Widdrington.
North Cumbria University Hospitals: C Graham, S Prentice, S Thornthwaite, U Poultney, H Crowther, H Fairlamb, E Hetherington, C Brewer, S Banerjee, C Hamson and A McSkeane.
Bradford Teaching Hospitals NHS Foundation Trust: P McWhinney, P Sharratt, J Thorpe, S Kimachia, H Wilson, B Jeffs, L Masters, J Wilson, J Platt and L Burgess.
Salford Royal NHS Foundation Trust: P Chadwick, A Jeans, C Keatley, A Moran, Z Swann, K Pagett, A Peel and J Howard.
Royal United Hospital Bath NHS Trust: S Meisner, K Maloney, A Masdin and L Wright.
Hull and East Yorkshire Hospitals NHS Trust: G Barlow, S Crossman, V Lowthorpe, E Moore, P Moss, A Parkin, A Wolstencroft, B Warner, C Tarbotton, A Eyre, A Anderson, T Burdett and A Driffill.
Contributions of authors
Guy E Thwaites (Professor of Infectious Diseases) drafted the report.
Matthew Scarborough (Consultant in Infectious Diseases and Microbiology) helped draft the report.
Alexander Szubert (Medical Statistician) performed the analysis.
Pedro Saramago Goncalves (Health Economist) designed and implemented the cost-effectiveness analysis and drafted Chapter 5.
Marta Soares (Health Economist) designed the cost-effectiveness analysis and drafted Chapter 5.
Jennifer Bostock (Public and Patient Representative) drafted the PPI sections.
Emmanuel Nsutebu (Consultant in Infectious Diseases) helped draft the report.
Robert Tilley (Consultant Microbiologist) helped draft the report.
Richard Cunningham (Consultant Microbiologist) helped draft the report.
Julia Greig (Consultant in Infectious Diseases) helped draft the report.
Sarah A Wyllie (Consultant Microbiologist) helped draft the report.
Peter Wilson (Consultant Microbiologist) helped draft the report.
Cressida Auckland (Consultant Microbiologist) helped draft the report.
Janet Cairns (Clinical Trials Manager) helped compile data and draft the report.
Denise Ward (Clinical Trials Manager) helped compile data and draft the report.
Pankaj Lal (Consultant Microbiologist) helped draft the report.
Achyut Guleri (Consultant Microbiologist) helped draft the report.
Neil Jenkins (Consultant in Infectious Diseases and Microbiology) helped draft the report.
Julian Sutton (Consultant in Infectious Diseases and Microbiology) helped draft the report.
Martin Wiselka (Consultant in Infectious Diseases and General Medicine) helped draft the report.
Gonzalez-Ruiz Armando (Consultant Microbiologist) helped draft the report.
Clive Graham (Consultant Microbiologist) helped draft the report.
Paul R Chadwick (Consultant Microbiologist) helped draft the report.
Gavin Barlow (Consultant Infectious Diseases) helped draft the report.
N Claire Gordon (Infectious Diseases/Microbiology Trainee) helped draft the report.
Bernadette Young (Infectious Diseases/Microbiology Trainee) sequenced and analysed bacterial isolates.
Sarah Meisner (Consultant Microbiologist) helped draft the report.
Paul McWhinney (Consultant Microbiologist) helped draft the report.
David A Price (Consultant Microbiologist) helped draft the report.
David Harvey (Consultant Microbiologist) helped draft the report.
Deepa Nayar (Consultant Microbiologist) helped draft the report.
Dakshika Jeyaratnam (Consultant Microbiologist) helped draft the report.
Timothy Planche (Consultant in Infectious Diseases) helped draft the report.
Jane Minton (Consultant in Infectious Diseases) helped draft the report.
Fleur Hudson (Clinical Trials Manager) helped compile data and draft the report.
Susan Hopkins (Consultant in Infectious Diseases and Microbiology) helped draft the report.
John Williams (Consultant infectious diseases) helped draft the report.
M Estee Török (Consultant in Infectious Diseases and Microbiology) helped draft the report.
Martin J Llewelyn (Professor of Infectious Diseases) helped draft the report.
Jonathan D Edgeworth (Consultant Microbiologist) helped draft the report.
A Sarah Walker (Professor of Medical Statistics) analysed data and drafted the report.
Publications
Thwaites GE, Scarborough M, Szubert A, Nsutebu E, Tilley R, Greig J, et al. Adjunctive rifampicin for Staphylococcus aureus bacteraemia (ARREST): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2018;391:668–78.
Thwaites G, Auckland C, Barlow G, Cunningham R, Davies G, Edgeworth J, et al. Adjunctive rifampicin to reduce early mortality from Staphylococcus aureus bacteraemia (ARREST): study protocol for a randomised controlled trial. Trials 2012;13:241.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Thwaites G, Auckland C, Barlow G, Cunningham R, Davies G, Edgeworth J, et al. Adjunctive rifampicin to reduce early mortality from Staphylococcus aureus bacteraemia (ARREST): study protocol for a randomised controlled trial. Trials 2012;13. https://doi.org/10.1186/1745-6215-13-241.
- Public Health England . Staphylococcus Aureus: Guidance, Data and Analysis 2017. www.gov.uk/government/collections/staphylococcus-aureus-guidance-data-and-analysis (accessed 27 September 2017).
- Wyllie DH, Crook DW, Peto TE. Mortality after Staphylococcus aureus bacteraemia in two hospitals in Oxfordshire, 1997–2003: cohort study. BMJ 2006;333. https://doi.org/10.1136/bmj.38834.421713.2F.
- Elliott TS, Foweraker J, Gould FK, Perry JD, Sandoe JA. Working Party of the British Society for Antimicrobial Chemotherapy . Guidelines for the antibiotic treatment of endocarditis in adults: report of the Working Party of the British Society for Antimicrobial Chemotherapy. J Antimicrob Chemother 2004;54:971-81. https://doi.org/10.1093/jac/dkh474.
- Gemmell CG, Edwards DI, Fraise AP, Gould FK, Ridgway GL, Warren RE. Hospital Infection Society and Infection Control Nurses Association . Guidelines for the prophylaxis and treatment of methicillin-resistant Staphylococcus aureus (MRSA) infections in the UK. J Antimicrob Chemother 2006;57:589-608. https://doi.org/10.1093/jac/dkl017.
- Mermel LA, Allon M, Bouza E, Craven DE, Flynn P, O’Grady NP, et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009;49:1-45. https://doi.org/10.1086/599376.
- Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis 2011;52:285-92. https://doi.org/10.1093/cid/cir034.
- Naber CK, Baddour LM, Giamarellos-Bourboulis EJ, Gould IM, Herrmann M, Hoen B, et al. Clinical consensus conference: survey on Gram-positive bloodstream infections with a focus on Staphylococcus aureus. Clin Infect Dis 2009;48:260-70. https://doi.org/10.1086/598185.
- Cadena J, Restrepo MI. Methicillin-resistant Staphylococcus aureus guidelines: a myriad of open questions. Clin Infect Dis 2011;53:97-8. https://doi.org/10.1093/cid/cir278.
- Buniva G, Pagani V, Carozzi A. Bioavailability of rifampicin capsules. Int J Clin Pharmacol Ther Toxicol 1983;21:404-9.
- Perlroth J, Kuo M, Tan J, Bayer AS, Miller LG. Adjunctive use of rifampin for the treatment of Staphylococcus aureus infections: a systematic review of the literature. Arch Intern Med 2008;168:805-19. https://doi.org/10.1001/archinte.168.8.805.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2011.
- Fowler VG, Olsen MK, Corey GR, Woods CW, Cabell CH, Reller LB, et al. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med 2003;163:2066-72. https://doi.org/10.1001/archinte.163.17.2066.
- Khatib R, Johnson LB, Fakih MG, Riederer K, Khosrovaneh A, Shamse Tabriz M, et al. Persistence in Staphylococcus aureus bacteremia: incidence, characteristics of patients and outcome. Scand J Infect Dis 2006;38:7-14. https://doi.org/10.1080/00365540500372846.
- Khatib R, Johnson LB, Sharma M, Fakih MG, Ganga R, Riederer K. Persistent Staphylococcus aureus bacteremia: incidence and outcome trends over time. Scand J Infect Dis 2009;41:4-9. https://doi.org/10.1080/00365540802441711.
- Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus?. Nat Rev Microbiol 2011;9:215-22. https://doi.org/10.1038/nrmicro2508.
- Yancey RJ, Sanchez MS, Ford CW. Activity of antibiotics against Staphylococcus aureus within polymorphonuclear neutrophils. Eur J Clin Microbiol Infect Dis 1991;10:107-13. https://doi.org/10.1007/BF01964421.
- Carryn S, Chanteux H, Seral C, Mingeot-Leclercq MP, Van Bambeke F, Tulkens PM. Intracellular pharmacodynamics of antibiotics. Infect Dis Clin North Am 2003;17:615-34. https://doi.org/10.1016/S0891-5520(03)00066-7.
- Mandell GL. Interaction of intraleukocytic bacteria and antibiotics. J Clin Invest 1973;52:1673-9. https://doi.org/10.1172/JCI107348.
- Saginur R, Stdenis M, Ferris W, Aaron SD, Chan F, Lee C, et al. Multiple combination bactericidal testing of staphylococcal biofilms from implant-associated infections. Antimicrob Agents Chemother 2006;50:55-61. https://doi.org/10.1128/AAC.50.1.55-61.2006.
- Mandell GL. Uptake, transport, delivery, and intracellular activity of antimicrobial agents. Pharmacotherapy 2005;25:130S-3S. https://doi.org/10.1592/phco.2005.25.12part2.130S.
- Barcia-Macay M, Seral C, Mingeot-Leclercq MP, Tulkens PM, Van Bambeke F. Pharmacodynamic evaluation of the intracellular activities of antibiotics against Staphylococcus aureus in a model of THP-1 macrophages. Antimicrob Agents Chemother 2006;50:841-51. https://doi.org/10.1128/AAC.50.3.841-851.2006.
- Menzies D, Dion MJ, Rabinovitch B, Mannix S, Brassard P, Schwartzman K. Treatment completion and costs of a randomized trial of rifampin for 4 months versus isoniazid for 9 months. Am J Respir Crit Care Med 2004;170:445-9. https://doi.org/10.1164/rccm.200404-478OC.
- Schrenzel J, Harbarth S, Schockmel G, Genné D, Bregenzer T, Flueckiger U, et al. A randomized clinical trial to compare fleroxacin-rifampicin with flucloxacillin or vancomycin for the treatment of staphylococcal infection. Clin Infect Dis 2004;39:1285-92. https://doi.org/10.1086/424506.
- Ruotsalainen E, Jarvinen A, Koivula I, Kauma H, Rintala E, Lumino J, et al. Levofloxacin does not decrease mortality in Staphylococcus aureus bacteraemia when added to the standard treatment: a prospective and randomized clinical trial of 381 patients. J Intern Med 2006;259:179-90. https://doi.org/10.1111/j.1365-2796.2005.01598.x.
- Khanlari B, Elzi L, Estermann L, Weisser M, Brett W, Grapow M, et al. A rifampicin-containing antibiotic treatment improves outcome of staphylococcal deep sternal wound infections. J Antimicrob Chemother 2010;65:1799-806. https://doi.org/10.1093/jac/dkq182.
- Riedel DJ, Weekes E, Forrest GN. Addition of rifampin to standard therapy for treatment of native valve infective endocarditis caused by Staphylococcus aureus. Antimicrob Agents Chemother 2008;52:2463-7. https://doi.org/10.1128/AAC.00300-08.
- Lai CC, Tan CK, Lin SH, Liao CH, Huang YT, Hsueh PR. Emergence of rifampicin resistance during rifampicin-containing treatment in elderly patients with persistent methicillin-resistant Staphylococcus aureus bacteremia. J Am Geriatr Soc 2010;58:1001-3. https://doi.org/10.1111/j.1532-5415.2010.02842.x.
- Ju O, Woolley M, Gordon D. Emergence and spread of rifampicin-resistant, methicillin-resistant Staphylococcus aureus during vancomycin-rifampicin combination therapy in an intensive care unit. Eur J Clin Microbiol Infect Dis 2006;25:61-2. https://doi.org/10.1007/s10096-005-0063-1.
- Van der Auwera P, Klastersky J, Thys JP, Meunier-Carpentier F, Legrand JC. Double-blind, placebo-controlled study of oxacillin combined with rifampin in the treatment of staphylococcal infections. Antimicrob Agents Chemother 1985;28:467-72. https://doi.org/10.1128/AAC.28.4.467.
- Van der Auwera P, Meunier-Carpentier F, Klastersky J. Clinical study of combination therapy with oxacillin with rifampicin for staphylococcal infections. Rev Infect Dis 1983;5:S515-22. https://doi.org/10.1093/clinids/5.Supplement_3.S515.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991;115:674-80. https://doi.org/10.7326/0003-4819-115-9-674.
- Jung YJ, Koh Y, Hong SB, Chung JW, Ho Choi S, Kim NJ, et al. Effect of vancomycin plus rifampicin in the treatment of nosocomial methicillin-resistant Staphylococcus aureus pneumonia. Crit Care Med 2010;38:175-80. https://doi.org/10.1097/CCM.0b013e3181b9ecea.
- Rieg S, Joost I, Weiß V, Peyerl-Hoffmann G, Schneider C, Hellmich M, et al. Combination antimicrobial therapy in patients with Staphylococcus aureus bacteraemia – a post hoc analysis in 964 prospectively evaluated patients. Clin Microbiol Infect 2017;23. https://doi.org/10.1016/j.cmi.2016.08.026.
- Thwaites GE. United Kingdom Clinical Infection Research Group (UKCIRG) . The management of Staphylococcus aureus bacteremia in the United Kingdom and Vietnam: a multi-centre evaluation. PLOS ONE 2010;5. https://doi.org/10.1371/journal.pone.0014170.
- Mental Capacity Act 2005. London: The Stationery Offfice; 2005.
- ICH Harmonised Tripartite Guideline: Clinical Safety Data Management: Definitions and Standards for Expedited Reporting (E2A). Geneva: International Conference on Harmonisation of Technical Requirements for Regulation of Pharmaceuticals for Human Use; 1994.
- Lawes T, Edwards B, López-Lozano JM, Gould I. Trends in Staphylococcus aureus bacteraemia and impacts of infection control practices including universal MRSA admission screening in a hospital in Scotland, 2006-2010: retrospective cohort study and time-series intervention analysis. BMJ Open 2012;2. https://doi.org/10.1136/bmjopen-2011-000797.
- Gould IM, Reilly J, Bunyan D, Walker A. Costs of healthcare-associated methicillin-resistant Staphylococcus aureus and its control. Clin Microbiol Infect 2010;16:1721-8. https://doi.org/10.1111/j.1469-0691.2010.03365.x.
- Guide to the Methods of Technology Appraisal 2013. London: NICE; 2013.
- NHS Reference Costs 2013 to 2014. London: DHSC; 2014.
- NHS Reference Costs 2015 to 2016. London: DHSC; 2016.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2016. Canterbury: Personal Social Services Research Unit, University of Kent; 2016.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2017.
- Kind P. Quality of Life and Pharmacoeconomics in Clinical Trials. Lippincott-Raven: Riverwoods, IL; 1996.
- Brazier J. Measuring and Valuing Health Benefits for Economic Evaluation. Oxford: Oxford University Press; 2007.
- Kind P, Hardman G, Macran S. Population Norms for EQ-5D. York: Centre for Health Economics, University of York; 1999.
- Dolan P, Gudex C, Kind P, Williams A. A Social Tariff for EuroQol: Results From a UK General Population Survey. York: Centre for Health Economics, University of York; 1995.
- Glick H. Economic Evaluation in Clinical Trials. Oxford: Oxford University Press; 2007.
- Shah HA, Dritsaki M, Pink J, Petrou S. Psychometric properties of Patient Reported Outcome Measures (PROMs) in patients diagnosed with Acute Respiratory Distress Syndrome (ARDS). Health Qual Life Outcomes 2016;14. https://doi.org/10.1186/s12955-016-0417-7.
- Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ 2009;338. https://doi.org/10.1136/bmj.b2393.
- van Buuren S, Groothuis-Oudshoorn K. MICE: Multivariate Imputation by Chained Equations in R. J Stat Softw 2011;45:1-67. https://doi.org/10.18637/jss.v045.i03.
- Little RJA. Missing-data adjustments in large surveys. Journal of Business &Amp; Economic Statistics 1988;6:287-96.
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96. https://doi.org/10.1002/hec.944.
- Sakamoto Y, Ishiguro M, Kitagawa G. Akaike Information Criterion Statistics. Tokyo: D Reidel Publishing Company; 1986.
- Dobson AJ, Barnett AG. An Introduction to Generalized Linear Models. Boca Raton, FL: CRC Press; 2008.
- Claxton K, Martin S, Soares M, Rice N, Spackman E, Hinde S, et al. Methods for the estimation of the National Institute for Health and Care Excellence cost-effectiveness threshold. Health Technol Assess 2015;19. https://doi.org/10.3310/hta19140.
- Drummond M. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2005.
- Briggs AH, Claxton K, Sculpher MJ. Decision Modelling for Health Economic Evaluation. Oxford: Oxford University Press; 2006.
- Briggs AH, Gray AM. Handling uncertainty in economic evaluations of healthcare interventions. BMJ 1999;319:635-8. https://doi.org/10.1136/bmj.319.7210.635.
- Claxton K. The irrelevance of inference: a decision-making approach to the stochastic evaluation of health care technologies. J Health Econ 1999;18:341-64. https://doi.org/10.1016/S0167-6296(98)00039-3.
- Bodner TE. What improves with increased missing data imputations?. Struct Equ Modeling 2008;15:651-75. https://doi.org/10.1080/10705510802339072.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. https://doi.org/10.1002/sim.4067.
- Duerden B, Fry C, Johnson AP, Wilcox MH. The control of meticillin-resistant Staphylococcus aureus blood stream infections in England. Open Forum Infect Dis 2015;2. https://doi.org/10.1093/ofid/ofv035.
- Loubet P, Burdet C, Vindrios W, Grall N, Wolff N, Yasdanpanah Y, et al. Cefazolin versus anti-staphylococcal penicillins for treatment of meticillin-susceptible Staphylococcus aureus bacteraemia: a narrative review. Clin Microbiol Infect 2017;24:124-32.
- Watanakunakorn C. A general survey of antibiotic treatment of staphylococcal septicaemia and endocarditis. Scand J Infect Dis Suppl 1983;41:151-7.
- Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine – reporting of subgroup analyses in clinical trials. N Engl J Med 2007;357:2189-94. https://doi.org/10.1056/NEJMsr077003.
- Kaasch AJ, Barlow G, Edgeworth JD, Fowler VG, Hellmich M, Hopkins S, et al. Staphylococcus aureus bloodstream infection: a pooled analysis of five prospective, observational studies. J Infect 2014;68:242-51. https://doi.org/10.1016/j.jinf.2013.10.015.
- Fowler VG, Boucher HW, Corey GR, Abrutyn E, Karchmer AW, Rupp ME, et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006;355:653-65. https://doi.org/10.1056/NEJMoa053783.
- Vogel M, Schmitz RP, Hagel S, Pletz MW, Gagelmann N, Scherag A, et al. Infectious disease consultation for Staphylococcus aureus bacteremia – a systematic review and meta-analysis. J Infect 2016;72:19-28. https://doi.org/10.1016/j.jinf.2015.09.037.
- Sendi P, Zimmerli W. Antimicrobial treatment concepts for orthopaedic device-related infection. Clin Microbiol Infect 2012;18:1176-84. https://doi.org/10.1111/1469-0691.12003.
- Berrevoets MAH, Kouijzer IJE, Aarntzen EHJG, Janssen MJR, De Geus-Oei LF, Wertheim HFL, et al. 18F-FDG PET/CT optimizes treatment in Staphylococcus aureus bacteremia and is associated with reduced mortality. J Nucl Med 2017;58:1504-10. https://doi.org/10.2967/jnumed.117.191981.
- Harris PN, McNamara JF, Lye DC, Davis JS, Bernard L, Cheng AC, et al. Proposed primary endpoints for use in clinical trials that compare treatment options for bloodstream infection in adults: a consensus definition. Clin Microbiol Infect 2017;23:533-41. https://doi.org/10.1016/j.cmi.2016.10.023.
- Russell CD, Lawson McLean A, Saunders C, Laurenson IF. Adjunctive rifampicin may improve outcomes in Staphylococcus aureus bacteraemia: a systematic review. J Med Microbiol 2014;63:841-8. https://doi.org/10.1099/jmm.0.072280-0.
- Public Health England . Staphylococcus Aureus: Guidance, Data and Analysis 2017. www.gov.uk/government/collections/staphylococcus-aureus-guidance-data-and-analysis (accessed 4 June 2018).
- Chetter IC, Oswald AV, Fletcher M, Dumville JC, Culluma NA. Survey of patients with surgical wounds healing by secondary intention; an assessment of prevalence, aetiology, duration and management. J Tissue Viabil 2017;26.
Appendix 1 The ARREST trial protocol changes
The final protocol is available at www.journalslibrary.nihr.ac.uk/programmes/hta/1010425#/.
Version 1.01
Original version approved by the London-Westminster Research Ethics Committee (REC).
Version 1.02
Amended safety reporting text, as requested by the Medicines and Healthcare Products Regulatory Agency:
-
page 3, SAE reporting: ‘within 1 working day’ changed to ‘within 24 hours’
-
page 44, 7.3 investigator responsibilities: ‘within 1 working day’ changed to ‘within 24 hours’
-
page 46, 7.3.1.E notification: ‘within 1 working day’ changed to ‘within 24 hours’.
Version 2.0
Amended TSC chairperson, as requested by the Health Technology Assessment:
-
page i, authorisation: Professor Jeremy Farrar changed to Dr Adrian Martineau
-
page 72, appendix I: Dr Adrian Martineau named as chairperson.
Major edits:
-
page ix, 55, 56: addition of UCL Hospitals to sites enrolling patients to intensive PK/PD study
-
page ix, 38, 57: DNA and RNA stored for later genetic testing by storing 2.5 ml of blood in PAXgene Blood RNA tube at day 0 (no change in total volume of blood taken at that time point)
-
page ix, 38, 56, 57: clarification of sample storage and subsequent archiving
-
page 25, 32: clarification that rifampicin susceptibility testing by disc test or Vitek is acceptable
-
page 76, 77, Patient information sheets: text changed to explain that DNA and RNA will be stored for later genetic testing.
Minor edits:
-
pages i and v: added Clinical Trials Authorisation and Medical Research Ethics Committee (MREC) reference numbers
-
page iii, MRC CTU staff: added clinical project manager
-
page iv, site PIs: amended typographical errors in site names
-
page vi, duration: changed to patient ‘recruited’ rather than ‘randomised’ over 3 years
-
page vii, trial schema: ‘consenting’ changed to ‘consented’
-
page 14, abbreviations: King’s College London (KCL) and King’s Health Partners (KHP) Clinical Trials Office (CTO) added
-
page 23, 2.3 trial centres: amended typographical errors in site names
-
page 27, 4.1 randomisation practicalities: amended CRF names. Amended text to clarify sample storage.
-
page 30, treatment schedule: clarified that those ≥ 60 kg will receive 900 mg of rifampicin.
-
page 31, 5.3.1 dispensing: amended text, ‘will be prescribed by the local pharmacist’ changed to ‘will be dispensed by the local pharmacist’
-
page 44, 7.2.1 pregnancy: amended typographical error, ‘rifampicin in safe’ changed to ‘rifampicin is safe’
-
page 48, 8.1 risk assessment: Quality Management Committee changed to Research Governance Committee
-
page 58 and 63, site compliance and Finance: agreement between site, KCL and MRC
-
page 81, 83, appendix III: version and date of patient information sheets and consent forms changed to version 2.0, 23 August 2012. Reformatted optional consent boxes
-
page 85, appendix IV: added MREC reference number.
Version 3.0
Reasons for substantial amendment: (1) remove KCL as co-sponsor and (2) add four new trial sites:
-
page i: remove KCL logo
-
pages ii and vii, sponsor: remove KCL
-
pages iv and v, site PIs: Drs Sutton, Guleri, Minton and Munthali
-
page 24, 2.3 trial centres: Southampton, Blackpool, Leeds and Coventry
-
page 63, indemnity: remove King’s College London
-
page 66, 15.6: remove paragraph ‘Role of Study Sponsor’.
Major edits to intensive and sparse PK/PD substudy information:
-
Pages ix–xi, trial assessment schedule (h), (i), (k): changed time point of PK/PD lithium heparin blood collections from day 0 to day 1. Clarified total volume of blood to be collected. Added reference to PK/PD laboratory manual.
-
Page x, (j): clarified procedure for intensive PK/PD component studies.
-
Pages 56–58, 11.1–11.1.2: updated sampling time points and procedures.
-
Page 81, appendix II, Intensive substudy: updated sampling time points.
-
Page 82, appendix II, Sparse substudy: updated sampling time points.
-
Major edits to bacterial genetic substudy information:
-
Pages ix and x, trial assessment schedule (g): single nasal bacterial swab added at baseline.
-
Page 58, 11.2: nasal swab added to study details. Clarification that bacteria will be archived in Oxford and Brighton.
-
Page 77, appendix II, What will happen if I take part?, point 1: updated to include baseline nasal swab.
Minor edits:
-
Page iii: updated randomisation summary and SAE reporting summary.
-
Page iii, MRC CTU Staff: added Trial Statistician Alex Szubert.
-
Page iv, chief investigator: updated CI address and e-mail address.
-
Page v, site PIs: updated Sheffield PI to Dr Julia Greig.
-
Page x, (h): amended typographical errors ‘University of Liverpool’ and ‘Davies’.
-
Page 23, 2.2: amended pack name and document name.
-
Pages 26, 3.4: ‘Trial register’ changed to ‘Screening & Randomisation Register’.
-
Page 28, 4 randomisation: added text to clarify randomisation via the ARREST trial database.
-
Pages 30, 32, 35 and 51, 5.1, 5.2.2, 5.2.3, 5.3.1, 5.7, 9.1: updated name of Clinical Trials Supplier to Sharp Clinical Services (formerly Bilcare Global Clinical Supplies).
-
Page 31, 5.2.3 blinding issues, second paragraph, last sentence: added text to clarify that infusion volumes used in the ICU may be altered in accordance with local standard practices and the product’s SPC.
-
Page 31, 5.3 treatment schedule, third paragraph: changed ‘intended’ to ‘initial’.
-
Page 32, 5.3.1 dispensing, second paragraph, last sentence: added text to clarify that temperature monitoring of treatment packs is not required after dispensing.
-
Page 32, 5.3.1 dispensing, fourth paragraph: changed ‘Dispensing Log’ to ‘Accountability Log’, and added ‘temperature monitoring’.
-
Page 34, 5.5.2 unblinding by the MRC CTU: updated Trial Manager contact number.
-
Page 38, 6 assessments and follow-up, second paragraph: added text to clarify that the final follow-up visit may take place over the telephone.
-
Page 41, 6.6 early stopping of follow-up, first paragraph: amended CRF name.
-
Page 47, 7.3.2 notification procedure, points 1, 2 and 3: updated text to clarify notification practicalities using the ARREST trial database.
-
Page 49, 8.2 central monitoring: amended typographical error ‘Case Report Forms’.
-
Page 51, 9.1 method of randomisation: changed ‘website randomisation service’ to ‘the ARREST trial database’. Updated name of Delegation Log.
-
Page 52, 9.2 outcome measures, last paragraph: added ‘clinical’.
-
Page 53, 9.4 interim monitoring and analyses, fifth sentence: update text to clarify Haybittle-Peto rules in the context of this trial.
-
Page 53, 9.5 analysis plan (brief), second paragraph: added more detailed text regarding analysis of end points.
-
Page 54, 9.5 analysis plan (brief), third paragraph: changed ‘intended’ to ‘initial’.
-
Page 65, 15.1–15.2: added ‘as required’ to clarify Trial Management Team and Trial Management Group membership.
-
Page 77, appendix II, What will happen if I take part?, point 3, second paragraph, second sentence: amended error in text by removing day 7 time point.
-
Page 79, appendix II, storing samples: added more detailed name of storage facility.
-
Pages 84 and 86, appendix III: updated protocol version number and date. Corrected typographical errors in legal representative consent form.
Version 4.0
Reasons for substantial amendment: addition of substudy – experiences of being approached for trial participation, the consenting process and trial participation.
Major edits: addition of new substudy –
-
page 56: section 11 text amended to include participation and consenting substudy and number of ancillary studies changed from two to three
-
page 59–61: section 11.3 new section and text added to give details of the participation and consenting substudy
-
page 95: appendix VII questionnare for non-consenting patients and legal representatives who are not health-care professionals added
-
page 98: appendix VIII questionnaire for non-consenting health-care professionals added
-
page 100: appendix IX information sheet for consenting patients or legal representatives added.
Minor edits:
Throughout document: change of name of co-ordinating site from MRC CTU to MRC CTU at UCL.
Throughout document: change of e-mail address for co-ordinating centre from arrest@ctu.mrc.ac.uk to mrcctu.arrest@ucl.ac.uk.
-
Page ii: infections theme corrected to ARREST trial team.
-
Page iii: co-ordinating site generic telephone number removed.
-
Page iii and iv: MRC CTU at UCL staff titles, names, e-mail addresses and telephone numbers updated.
-
Page iv: site PIs table removed.
-
Page v: protocol version and date updated.
-
Page vi: trial manager name updated; Project Lead title amended.
-
Page vii: amended trials schema error – day 10 added to follow up visits.
-
Page viii: added text to Trial assessment schedule footnote to clarify that if a patient is discharged before day 14 but is attending outpatient appointments then blood samples should be collected if possible.
-
Page 16: MRC CTU amended to MRC CTU at UCL; percutaneous endoscopic gastrostomy (PEG) and UCL added to abbreviations table.
-
Page 24: section 2.3 trial centres removed.
-
Page 25: section 3.1 added text to clarify that stat doses should be excluded from the 96 hour active antibiotic therapy in inclusion criteria.
-
Page 31: section 5.3 added text to clarify that it is not permissible for IMP capsules to be opened and administered via PEG.
-
Page 36: section 5.8 added text to clarify that if a patient receives additional doses of IMP for > 24 hours (i.e. 15 days of trial treatment) then this must be reported as a protocol deviation.
-
Page 39: section 6.1 added text to clarify that if a patient is discharged before day 14 but is attending outpatient appointments then blood samples should be collected, if possible.
-
Page 44: section 7.1.2 moved bullet points describing AEs that should be reported to page 44, section 7.1 in order to prevent confusion. Added text to clarify that disease-related events that are not fatal are exempt from being reported as SAEs.
-
Page 49: amended text to clarify that site initiation visits may occur either as a site visit or by WebEx (Cisco, San Jose, CA, USA) teleconference.
-
Page 66: section 13 MRC indemnity text removed, UCL insurance text added.
-
Page 79: appendix I Professor Jeremy Farrar removed from TSC membership, Dr Achim Kaasch added; trial statistician title amended.
-
Page 80–86: appendix II friend/relative amended to friend/relative/patient to clarify that a doctor primarily responsible for a patient’s medical treatment may also act as a legal representative.
-
Page 87: appendix II what is a legal representative information sheet. Text added to clarify that a doctor primarily responsible for a patient’s medical treatment may also act as a Legal Representative.
-
Page 88: appendix III trial participant consent form. Updated protocol and version number. ‘Doctor’s signature’ changed to ‘Signature of person delegated to take consent’ since some trusts allow nurses to take consent.
-
Page 90: appendix III Legal Representative consent form. Updated protocol and version number. Friend/relative amended to friend/relative/patient to clarify that a doctor primarily responsible for a patient’s medical treament may also act as a legal representative. ‘Doctor’s signature’ changed to ‘Signature of person delegated to take consent’ since some Trusts allow nurses to take consent. Box for participant’s name added.
Version 4.1
Minor edits requested by REC.
-
Page 92: appendix IV, GP letter amended to clarify that consent may have been granted by a legal representative. Version number and date of GP letter updated.
Protocol version number and date updated throughout protocol.
Version 5.0
Reason for substantial amendment: sample size reduced and co-primary end point (all-cause mortality up to 14 days) reassigned as a secondary end point at the request of the funder.
Major edits: sample size reduced and co-primary end point (all-cause mortality up to 14 days) reassigned as a secondary end point.
-
Page v, summary of trial: ‘all-cause mortality up to 14 days from randomisation’ removed as a primary end point, added as a secondary outcome measure.
-
Page vi, summary of trial: number of patients to be studied changed from ‘940’ to ‘770’.
-
Page viii, trial schema updated to reflect change in sample size and end points.
-
Section 1.5, hypothesis and objectives: ‘all-cause mortality up to 14 days from randomisation removed as a primary objective, ‘evaluating the impact of rifampicin on all-cause mortality up to 14 days from randomisation’ added as a secondary objective.
-
Section 3.3, number of patients: number of patients to be studied changed from ‘940 patients (470 in each treatment arm)’ to ‘770 patients (385 in each treatment arm)’; ‘enrolled over a target of 3 years’ changed to ‘enrolled over a target of 3.5 years’.
-
Section 6.2, procedures for assessing efficacy: ‘all-cause mortality up to 14 days from randomisation’ removed as a primary end point, added as a secondary outcome measure.
-
Section 9.2, outcome measures: ‘all-cause mortality up to 14 days from randomisation’ removed as a primary end point, added as a secondary outcome measure. Text modified to clarify.
-
Section 9.3, sample size: updated to reflect sample size change from ‘940’ to ‘770’.
-
Section 9.5, analysis plan (brief): ‘all-cause mortality up to 14 days from randomisation’ removed as a primary end point.
Minor edits:
-
Page vi, summary of trial: ‘experiences of being approached for trial participation, the consenting process and trial participation’ added to Ancillary/substudies section (omitted in error in version 4.1 of protocol).
-
Section 5.3.1, ‘the ward nurse’ changed to ‘the ward or the ARREST trial research nurse’ to clarify that the ARREST trial nurse may take the prescription form to the pharmacy.
-
Section 5.3.2, unblinding by MRC CTU at UCL: trial manager telephone number corrected.
-
Section 9.4, interim monitoring analyses: changed from ‘the DMC will in general meet’ to ‘the DMC will meet at least once ber year’ for clarification.
-
Appendix VII, substudy questionnare: ‘relative/spouse’, ‘relative’ ‘friend/relative’ have been corrected to ‘relative/friend’ for consistency.
Appendix 2 Supplementary tables
| Active antibiotic therapy for the current infection | Treatment group, n (%) | Total (N = 758), n (%) | |
|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | ||
| Classa | |||
| Beta-lactam | 350 (90.2) | 321 (86.8) | 671 (88.5) |
| Penicillin | 335 (86.3) | 308 (83.2) | 643 (84.8) |
| Cephalosporin | 110 (28.4) | 104 (28.1) | 214 (28.2) |
| Carbapenem | 38 (9.8) | 35 (9.5) | 73 (9.6) |
| Aminoglycoside | 108 (27.8) | 103 (27.8) | 211 (27.8) |
| Glycopeptide | 188 (48.5) | 192 (51.9) | 380 (50.1) |
| Fluoroquinolone | 47 (12.1) | 46 (12.4) | 93 (12.3) |
| Macrolide | 30 (7.7) | 28 (7.6) | 58 (7.7) |
| Tetracycline | 29 (7.5) | 26 (7.0) | 55 (7.3) |
| Fusidic acid | 9 (2.3) | 6 (1.6) | 15 (2.0) |
| Co-trimoxazole | 6 (1.5) | 7 (1.9) | 13 (1.7) |
| Linezolid | 14 (3.6) | 13 (3.5) | 27 (3.6) |
| Chloramphenicol | 3 (0.8) | 1 (0.3) | 4 (0.5) |
| Lincosamide | 26 (6.7) | 38 (10.3) | 64 (8.4) |
| Lipopeptide | 13 (3.4) | 23 (6.2) | 36 (4.7) |
| Rifamycin | 53 (13.7) | 33 (8.9) | 86 (11.3) |
| Glycylcycline | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Sulfonamide | 23 (5.9) | 10 (2.7) | 33 (4.4) |
| Combinations of classesa | |||
| Aminoglycoside, glycopeptide | 3 (0.8) | 2 (0.5) | 5 (0.7) |
| Aminoglycoside, glycopeptide, co-trimoxazole, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, fluoroquinolone | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Aminoglycoside, glycopeptide, fluoroquinolone, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Aminoglycoside, glycopeptide, fluoroquinolone, lipopeptide | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Aminoglycoside, glycopeptide, fluoroquinolone, macrolide, fusidic acid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Aminoglycoside, glycopeptide, fluoroquinolone, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, lincosamide, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, linezolid, lincosamide, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, macrolide, fusidic acid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Aminoglycoside, glycopeptide, macrolide, linezolid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Aminoglycoside, glycopeptide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Aminoglycoside, glycopeptide, tetracycline | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Carbapenem, aminoglycoside, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Carbapenem, glycopeptide, fluoroquinolone, macrolide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Carbapenem, glycopeptide, fluoroquinolone, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Carbapenem, glycopeptide, linezolid, lipopeptide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cephalosporin | 4 (1.0) | 1 (0.3) | 5 (0.7) |
| Cephalosporin, aminoglycoside | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Cephalosporin, aminoglycoside, glycopeptide | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Cephalosporin, aminoglycoside, glycopeptide, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, aminoglycoside, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cephalosporin, carbapenem, glycopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, carbapenem, glycopeptide, fluoroquinolone, chloramphenicol, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, carbapenem, glycopeptide, macrolide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cephalosporin, carbapenem, glycopeptide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, glycopeptide | 1 (0.3) | 4 (1.1) | 5 (0.7) |
| Cephalosporin, glycopeptide, fluoroquinolone | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, glycopeptide, fluoroquinolone, co-trimoxazole, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cephalosporin, glycopeptide, fluoroquinolone, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cephalosporin, glycopeptide, fluoroquinolone, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, glycopeptide, lipopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, glycopeptide, macrolide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cephalosporin, glycopeptide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cephalosporin, macrolide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Co-trimoxazole, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Fluoroquinolone | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide | 11 (2.8) | 13 (3.5) | 24 (3.2) |
| Glycopeptide, co-trimoxazole | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, co-trimoxazole, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, fluoroquinolone | 2 (0.5) | 1 (0.3) | 3 (0.4) |
| Glycopeptide, fluoroquinolone, lincosamide | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Glycopeptide, fluoroquinolone, macrolide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, fluoroquinolone, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, fluoroquinolone, tetracycline, linezolid, lipopeptide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, fusidic acid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, lincosamide | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Glycopeptide, lincosamide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, linezolid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, linezolid, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, linezolid, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, lipopeptide | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Glycopeptide, macrolide | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Glycopeptide, macrolide, fusidic acid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, rifamycin | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Glycopeptide, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, tetracycline, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, tetracycline, lincosamide, rifamycin, sulfonamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, tetracycline, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, tetracycline, lipopeptide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Glycopeptide, tetracycline, lipopeptide, rifamycin, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Glycopeptide, tetracycline, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin | 62 (16.0) | 55 (14.9) | 117 (15.4) |
| Penicillin, aminoglycoside | 12 (3.1) | 12 (3.2) | 24 (3.2) |
| Penicillin, aminoglycoside, fluoroquinolone | 2 (0.5) | 0 (0.0) | 2 (0.3) |
| Penicillin, aminoglycoside, fluoroquinolone, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, fluoroquinolone, macrolide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, fluoroquinolone, macrolide, linezolid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, fluoroquinolone, tetracycline, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide | 21 (5.4) | 18 (4.9) | 39 (5.1) |
| Penicillin, aminoglycoside, glycopeptide, chloramphenicol | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, fluoroquinolone | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, aminoglycoside, glycopeptide, fluoroquinolone, linezolid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, fluoroquinolone, lipopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, fluoroquinolone, macrolide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, fluoroquinolone, rifamycin | 3 (0.8) | 0 (0.0) | 3 (0.4) |
| Penicillin, aminoglycoside, glycopeptide, fluoroquinolone, tetracycline, fusidic acid, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, fusidic acid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, lincosamide | 2 (0.5) | 1 (0.3) | 3 (0.4) |
| Penicillin, aminoglycoside, glycopeptide, linezolid, rifamycin, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, macrolide | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, aminoglycoside, glycopeptide, rifamycin | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, aminoglycoside, glycopeptide, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, tetracycline, chloramphenicol | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, tetracycline, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, glycopeptide, tetracycline, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, lincosamide | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Penicillin, aminoglycoside, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, lipopeptide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, macrolide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, aminoglycoside, macrolide, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, macrolide, rifamycin, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, macrolide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, aminoglycoside, rifamycin | 3 (0.8) | 1 (0.3) | 4 (0.5) |
| Penicillin, aminoglycoside, sulfonamide | 5 (1.3) | 2 (0.5) | 7 (0.9) |
| Penicillin, aminoglycoside, tetracycline | 3 (0.8) | 0 (0.0) | 3 (0.4) |
| Penicillin, aminoglycoside, tetracycline, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem | 5 (1.3) | 4 (1.1) | 9 (1.2) |
| Penicillin, carbapenem, aminoglycoside | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, carbapenem, aminoglycoside, glycopeptide | 3 (0.8) | 4 (1.1) | 7 (0.9) |
| Penicillin, carbapenem, aminoglycoside, glycopeptide, co-trimoxazole | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, carbapenem, aminoglycoside, glycopeptide, fluoroquinolone | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Penicillin, carbapenem, aminoglycoside, glycopeptide, fluoroquinolone, co-trimoxazole | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, aminoglycoside, glycopeptide, macrolide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, aminoglycoside, glycopeptide, rifamycin | 2 (0.5) | 0 (0.0) | 2 (0.3) |
| Penicillin, carbapenem, aminoglycoside, tetracycline, lipopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, fluoroquinolone, linezolid, lipopeptide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, fluoroquinolone, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, carbapenem, glycopeptide | 3 (0.8) | 5 (1.4) | 8 (1.1) |
| Penicillin, carbapenem, glycopeptide, fluoroquinolone, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, glycopeptide, fluoroquinolone, macrolide, co-trimoxazole | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, glycopeptide, glycylcycline | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, carbapenem, glycopeptide, lincosamide | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Penicillin, carbapenem, glycopeptide, linezolid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, glycopeptide, tetracycline, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, carbapenem, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, carbapenem, lipopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, macrolide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, carbapenem, sulfonamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin | 24 (6.2) | 24 (6.5) | 48 (6.3) |
| Penicillin, cephalosporin, aminoglycoside | 7 (1.8) | 6 (1.6) | 13 (1.7) |
| Penicillin, cephalosporin, aminoglycoside, co-trimoxazole | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, fluoroquinolone | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, fluoroquinolone, linezolid, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, fluoroquinolone, macrolide, tetracycline | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, glycopeptide | 8 (2.1) | 8 (2.2) | 16 (2.1) |
| Penicillin, cephalosporin, aminoglycoside, glycopeptide, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, glycopeptide, rifamycin | 2 (0.5) | 1 (0.3) | 3 (0.4) |
| Penicillin, cephalosporin, aminoglycoside, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, macrolide | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Penicillin, cephalosporin, aminoglycoside, macrolide, tetracycline, fusidic acid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, aminoglycoside, tetracycline | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, cephalosporin, carbapenem, aminoglycoside | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Penicillin, cephalosporin, carbapenem, aminoglycoside, glycopeptide, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem, aminoglycoside, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem, fluoroquinolone | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem, glycopeptide | 3 (0.8) | 2 (0.5) | 5 (0.7) |
| Penicillin, cephalosporin, carbapenem, glycopeptide, fluoroquinolone, lincosamide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem, glycopeptide, fluoroquinolone, macrolide, tetracycline, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem, glycopeptide, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, carbapenem, glycopeptide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, co-trimoxazole, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, fluoroquinolone | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, cephalosporin, fluoroquinolone, lipopeptide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, fluoroquinolone, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, fluoroquinolone, rifamycin, sulfonamide | 2 (0.5) | 0 (0.0) | 2 (0.3) |
| Penicillin, cephalosporin, fusidic acid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, fusidic acid, co-trimoxazole | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, glycopeptide | 22 (5.7) | 17 (4.6) | 39 (5.1) |
| Penicillin, cephalosporin, glycopeptide, fluoroquinolone | 2 (0.5) | 3 (0.8) | 5 (0.7) |
| Penicillin, cephalosporin, glycopeptide, fluoroquinolone, linezolid, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, glycopeptide, fluoroquinolone, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, glycopeptide, lincosamide | 2 (0.5) | 0 (0.0) | 2 (0.3) |
| Penicillin, cephalosporin, glycopeptide, linezolid, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, glycopeptide, macrolide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, glycopeptide, rifamycin | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Penicillin, cephalosporin, glycopeptide, tetracycline | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, lincosamide | 3 (0.8) | 2 (0.5) | 5 (0.7) |
| Penicillin, cephalosporin, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, cephalosporin, macrolide | 2 (0.5) | 3 (0.8) | 5 (0.7) |
| Penicillin, cephalosporin, macrolide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, cephalosporin, rifamycin | 2 (0.5) | 1 (0.3) | 3 (0.4) |
| Penicillin, cephalosporin, tetracycline, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, co-trimoxazole, lincosamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, fluoroquinolone | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Penicillin, fluoroquinolone, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, fluoroquinolone, lincosamide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, fluoroquinolone, linezolid, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, fluoroquinolone, rifamycin | 3 (0.8) | 0 (0.0) | 3 (0.4) |
| Penicillin, fluoroquinolone, sulfonamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, fluoroquinolone, tetracycline, co-trimoxazole, linezolid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, fluoroquinolone, tetracycline, fusidic acid | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, fluoroquinolone, tetracycline, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, fluoroquinolone, tetracycline, rifamycin | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Penicillin, fusidic acid | 3 (0.8) | 0 (0.0) | 3 (0.4) |
| Penicillin, fusidic acid, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, glycopeptide | 31 (8.0) | 29 (7.8) | 60 (7.9) |
| Penicillin, glycopeptide, chloramphenicol | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, glycopeptide, fluoroquinolone | 1 (0.3) | 3 (0.8) | 4 (0.5) |
| Penicillin, glycopeptide, fluoroquinolone, lincosamide | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Penicillin, glycopeptide, fluoroquinolone, lipopeptide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, glycopeptide, fluoroquinolone, macrolide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, fluoroquinolone, macrolide, lipopeptide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, fluoroquinolone, rifamycin | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Penicillin, glycopeptide, fusidic acid, rifamycin, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, glycopeptide, lincosamide | 0 (0.0) | 4 (1.1) | 4 (0.5) |
| Penicillin, glycopeptide, lincosamide, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, lincosamide, rifamycin | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, linezolid, rifamycin, glycylcycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, glycopeptide, lipopeptide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, macrolide | 7 (1.8) | 1 (0.3) | 8 (1.1) |
| Penicillin, glycopeptide, rifamycin | 3 (0.8) | 1 (0.3) | 4 (0.5) |
| Penicillin, glycopeptide, sulfonamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, glycopeptide, tetracycline | 2 (0.5) | 3 (0.8) | 5 (0.7) |
| Penicillin, glycopeptide, tetracycline, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, lincosamide | 3 (0.8) | 5 (1.4) | 8 (1.1) |
| Penicillin, lincosamide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, linezolid | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, linezolid, lipopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, lipopeptide | 0 (0.0) | 3 (0.8) | 3 (0.4) |
| Penicillin, lipopeptide, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, macrolide | 3 (0.8) | 8 (2.2) | 11 (1.5) |
| Penicillin, macrolide, tetracycline | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, rifamycin | 2 (0.5) | 1 (0.3) | 3 (0.4) |
| Penicillin, sulfonamide | 6 (1.5) | 4 (1.1) | 10 (1.3) |
| Penicillin, tetracycline | 3 (0.8) | 2 (0.5) | 5 (0.7) |
| Penicillin, tetracycline, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, tetracycline, linezolid, lincosamide | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Penicillin, tetracycline, lipopeptide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, tetracycline, rifamycin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, tetracycline, rifamycin, sulfonamide | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Penicillin, tetracycline, sulfonamide | 2 (0.5) | 0 (0.0) | 2 (0.3) |
| Antibiotic | |||
| Cefazolin | 1 (0.3) | 2 (0.5) | 3 (0.4) |
| Temocillin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Flucloxacillin | 321 (82.7) | 298 (80.5) | 619 (81.7) |
| Vancomycin | 137 (35.3) | 139 (37.6) | 276 (36.4) |
| Teicoplanin | 57 (14.7) | 64 (17.3) | 121 (16.0) |
| Amikacin | 5 (1.3) | 4 (1.1) | 9 (1.2) |
| Amoxicillin | 11 (2.8) | 13 (3.5) | 24 (3.2) |
| Azithromycin | 0 (0.0) | 2 (0.5) | 2 (0.3) |
| Benzylpenicillin | 5 (1.3) | 5 (1.4) | 10 (1.3) |
| Cefaclor | 0 (0.0) | 1 (0.3) | 1 (0.1) |
| Cefadrine | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Cefalexin | 3 (0.8) | 0 (0.0) | 3 (0.4) |
| Cefotaxime | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Ceftazidime | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Ceftriaxone | 92 (23.7) | 92 (24.9) | 184 (24.3) |
| Cefuroxime | 10 (2.6) | 7 (1.9) | 17 (2.2) |
| Chloramphenicol | 3 (0.8) | 1 (0.3) | 4 (0.5) |
| Ciprofloxacin | 24 (6.2) | 29 (7.8) | 53 (7.0) |
| Clarithromycin | 25 (6.4) | 25 (6.8) | 50 (6.6) |
| Clindamycin | 23 (5.9) | 36 (9.7) | 59 (7.8) |
| Co-amoxiclavulante | 122 (31.4) | 107 (28.9) | 229 (30.2) |
| Co-trimoxazole | 5 (1.3) | 7 (1.9) | 12 (1.6) |
| Daptomycin | 13 (3.4) | 21 (5.7) | 34 (4.5) |
| Doxycycline | 26 (6.7) | 23 (6.2) | 49 (6.5) |
| Ertapenem | 2 (0.5) | 3 (0.8) | 5 (0.7) |
| Erythromycin | 3 (0.8) | 3 (0.8) | 6 (0.8) |
| Fusidic acid | 9 (2.3) | 6 (1.6) | 15 (2.0) |
| Gentamicin | 98 (25.3) | 96 (25.9) | 194 (25.6) |
| Levofloxacin | 11 (2.8) | 12 (3.2) | 23 (3.0) |
| Linezolid | 11 (2.8) | 12 (3.2) | 23 (3.0) |
| Meropenem | 32 (8.2) | 31 (8.4) | 63 (8.3) |
| Moxifloxacin | 2 (0.5) | 2 (0.5) | 4 (0.5) |
| Ofloxacin | 1 (0.3) | 0 (0.0) | 1 (0.1) |
| Piperacillin/tazobactam | 115 (29.6) | 102 (27.6) | 217 (28.6) |
| Rifampicin | 45 (11.6) | 32 (8.6) | 77 (10.2) |
| Ticarcillin/clavulanate | 3 (0.8) | 0 (0.0) | 3 (0.4) |
| Tigecycline | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Trimethoprim | 17 (4.4) | 10 (2.7) | 27 (3.6) |
| Cause | Treatment group, n | Total (N = 112), n | |
|---|---|---|---|
| Placebo (N = 56) | Rifampicin (N = 56) | ||
| Definitely attributed to S. aureus | 16 | 14 | 30 |
| Probably attributed to S. aureus | 12 | 14 | 26 |
| Possibly attributed to S. aureus | 4 | 8 | 12 |
| Unlikely to be attributed to S. aureus | 23 | 18 | 41 |
| Attribution to S. aureus undeterminable | 1 | 2 | 3 |
| Not attributed to S. aureus | 23 | 18 | 41 |
| Infections and infestations | 5 | 4 | 9 |
| Pneumonia | 2 | 1 | 3 |
| Pneumonia, acute myeloid leukaemia, nasopharyngeal cancer | 0 | 1 | 1 |
| Pneumonia, chronic obstructive pulmonary disease | 1 | 0 | 1 |
| Pneumonia, renal failure | 1 | 0 | 1 |
| Urosepsis, lung disorder | 1 | 0 | 1 |
| Biliary sepsis | 0 | 1 | 1 |
| Serratia infection, pleural infection, nosocomial infection, chronic obstructive pulmonary disease | 0 | 1 | 1 |
| Cardiac disorders | 2 | 4 | 6 |
| Cardiac failure | 1 | 2 | 3 |
| Cardiac failure, myocardial infarction | 0 | 1 | 1 |
| Myocardial ischaemia | 1 | 0 | 1 |
| Myocardial ischaemia, renal failure | 0 | 1 | 1 |
| Vascular disorders | 1 | 0 | 1 |
| Peripheral ischaemia, colorectal cancer metastatic | 1 | 0 | 1 |
| Respiratory, thoracic and mediastinal disorders | 2 | 2 | 4 |
| Pneumonia aspiration, peripheral vascular disorder | 0 | 1 | 1 |
| Pulmonary embolism, colon cancer metastatic | 0 | 1 | 1 |
| Pulmonary embolism, death | 1 | 0 | 1 |
| Pulmonary oedema, peripheral vascular disorder | 1 | 0 | 1 |
| Gastrointestinal disorders | 2 | 3 | 5 |
| Diarrhoea | 1 | 0 | 1 |
| Gastrointestinal haemorrhage, angiodysplasia | 0 | 1 | 1 |
| Pancreatitis, intestinal perforation | 1 | 0 | 1 |
| Pancreatitis acute, end-stage renal disease | 0 | 1 | 1 |
| Upper gastrointestinal haemorrhage | 0 | 1 | 1 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 5 | 5 | 10 |
| Chronic myeloid leukaemia | 1 | 1 | 2 |
| Lymphoma, cardiac failure, diabetes mellitus | 0 | 1 | 1 |
| Plasma cell myeloma, endocarditis | 1 | 0 | 1 |
| Prostate cancer metastatic | 0 | 1 | 1 |
| Lung cancer metastatic | 0 | 1 | 1 |
| Renal cancer metastatic | 1 | 0 | 1 |
| Breast cancer metastatic | 1 | 1 | 2 |
| Prostate cancer | 1 | 0 | 1 |
| General disorders and administration site conditions | 3 | 0 | 3 |
| Death, cerebrovascular disorder | 1 | 0 | 1 |
| Death, dementia | 1 | 0 | 1 |
| Sudden death | 1 | 0 | 1 |
| Nervous system disorders | 3 | 0 | 3 |
| Cerebral haemorrhage | 1 | 0 | 1 |
| Cerebrovascular accident | 1 | 0 | 1 |
| Dementia Alzheimer’s type | 1 | 0 | 1 |
| Attributed to S. aureus | 32 | 36 | 68 |
| Infections and infestations | 29 | 28 | 57 |
| Endocarditis, staphylococcal bacteraemia | 2 | 1 | 3 |
| Osteomyelitis, staphylococcal bacteraemia | 0 | 1 | 1 |
| Pneumonia | 0 | 1 | 1 |
| Pneumonia, decubitus ulcer, acute myocardial infarction | 0 | 1 | 1 |
| Pneumonia, staphylococcal bacteraemia | 4 | 3 | 7 |
| Pneumonia, staphylococcal bacteraemia, chronic obstructive pulmonary disease | 0 | 1 | 1 |
| Sepsis | 0 | 4 | 4 |
| Sepsis, renal failure, chronic obstructive pulmonary disease | 1 | 0 | 1 |
| Sepsis, staphylococcal bacteraemia | 0 | 1 | 1 |
| Staphylococcal bacteraemia | 3 | 2 | 5 |
| Staphylococcal bacteraemia, alcoholism, diabetes mellitus | 1 | 0 | 1 |
| Staphylococcal bacteraemia, bone cancer metastatic, colorectal cancer metastatic | 1 | 0 | 1 |
| Staphylococcal bacteraemia, brain injury | 1 | 0 | 1 |
| Staphylococcal bacteraemia, carcinoid tumour | 0 | 1 | 1 |
| Staphylococcal bacteraemia, death | 1 | 0 | 1 |
| Staphylococcal bacteraemia, dementia | 0 | 1 | 1 |
| Staphylococcal bacteraemia, device-related infection, endocarditis | 0 | 1 | 1 |
| Staphylococcal bacteraemia, endocarditis | 4 | 2 | 6 |
| Staphylococcal bacteraemia, endocarditis, atrioventricular block complete | 0 | 1 | 1 |
| Staphylococcal bacteraemia, endocarditis, gastrointestinal carcinoma | 1 | 0 | 1 |
| Staphylococcal bacteraemia, graft infection, endocarditis | 0 | 1 | 1 |
| Staphylococcal bacteraemia, intervertebral discitis | 0 | 1 | 1 |
| Staphylococcal bacteraemia, lung cancer metastatic | 1 | 0 | 1 |
| Staphylococcal bacteraemia, lung neoplasm malignant | 0 | 1 | 1 |
| Staphylococcal bacteraemia, mediastinal abscess, coronary artery bypass | 1 | 0 | 1 |
| Staphylococcal bacteraemia, parotitis, prostate cancer metastatic | 0 | 1 | 1 |
| Staphylococcal bacteraemia, peripheral artery aneurysm | 1 | 0 | 1 |
| Staphylococcal bacteraemia, pneumonia | 1 | 1 | 2 |
| Staphylococcal bacteraemia, pneumonia aspiration | 0 | 1 | 1 |
| Staphylococcal bacteraemia, psoas abscess | 1 | 0 | 1 |
| Staphylococcal bacteraemia, pyelonephritis | 1 | 0 | 1 |
| Staphylococcal bacteraemia, pyomyositis, extradural abscess | 1 | 0 | 1 |
| Staphylococcal bacteraemia, renal failure, extradural abscess | 1 | 0 | 1 |
| Staphylococcal bacteraemia, soft tissue infection | 1 | 0 | 1 |
| Staphylococcal bacteraemia, tongue neoplasm malignant stage unspecified, alcohol abuse | 0 | 1 | 1 |
| Arthritis bacterial, osteomyelitis, staphylococcal bacteraemia | 1 | 0 | 1 |
| Cardiac disorders | 0 | 2 | 2 |
| Cardiac failure, endocarditis, staphylococcal bacteraemia | 0 | 1 | 1 |
| Cardiopulmonary failure | 0 | 1 | 1 |
| Respiratory, thoracic and mediastinal disorders | 0 | 1 | 1 |
| Pneumothorax, pneumonia, staphylococcal bacteraemia | 0 | 1 | 1 |
| Gastrointestinal disorders | 0 | 2 | 2 |
| Intestinal ischaemia, staphylococcal bacteraemia | 0 | 1 | 1 |
| Intestinal ischaemia, staphylococcal bacteraemia, Endocarditis | 0 | 1 | 1 |
| Renal and urinary disorders | 3 | 0 | 3 |
| Renal failure, spinal cord compression, staphylococcal bacteraemia | 1 | 0 | 1 |
| Renal tubular acidosis, staphylococcal bacteraemia | 1 | 0 | 1 |
| Acute kidney injury, pseudomonal bacteraemia | 1 | 0 | 1 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 0 | 3 | 3 |
| Mycosis fungoides, staphylococcal bacteraemia | 0 | 1 | 1 |
| Lung cancer metastatic | 0 | 1 | 1 |
| Metastatic carcinoma of the bladder | 0 | 1 | 1 |
| Attribution to S. aureus undeterminable | 1 | 2 | 3 |
| Infections and infestations | 0 | 1 | 1 |
| Pneumonia, chronic obstructive pulmonary disease | 0 | 1 | 1 |
| Cardiac disorders | 1 | 0 | 1 |
| Left ventricular failure, lung disorder, sepsis | 1 | 0 | 1 |
| General disorders and administration site conditions | 0 | 1 | 1 |
| Death | 0 | 1 | 1 |
| SAEs | Treatment group | Total (N = 758) | p-value | |
|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | |||
| MedDRA code | ||||
| Any | 94 (24.2) 116 | 101 (27.3) 112 | 195 (25.7) 228 | 0.36 |
| Infections and infestations | 39 (10.1) 40 | 37 (10.0) 38 | 76 (10.0) 78 | 1.00 |
| Cellulitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Clostridium difficile colitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Diarrhoea infectious | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Endocarditis | 3 (0.8) 3 | 2 (0.5) 2 | 5 (0.7) 5 | |
| Endocarditis, graft infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Gastroenteritis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Lower respiratory tract infection | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pneumonia | 12 (3.1) 12 | 10 (2.7) 10 | 22 (2.9) 22 | |
| Pneumonia, osteomyelitis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pneumonia, sepsis | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Pneumonia, staphylococcal bacteraemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pneumonia, staphylococcal bacteraemia, confusional state, cellulitis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Postoperative wound infection | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pyelonephritis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Sepsis | 3 (0.8) 3 | 4 (1.1) 4 | 7 (0.9) 7 | |
| Septic shock, necrotising fasciitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Subcutaneous abscess | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Urinary tract infection | 2 (0.5) 2 | 2 (0.5) 2 | 4 (0.5) 4 | |
| Urosepsis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Groin abscess | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Abscess limb | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Staphylococcal bacteraemia | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Staphylococcal bacteraemia, cardiac tamponade | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Arthritis bacterial | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Arthritis bacterial, pneumonia, intervertebral discitis, staphylococcal bacteraemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Escherichia bacteraemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Staphylococcal sepsis | 3 (0.8) 3 | 2 (0.5) 2 | 5 (0.7) 5 | |
| Staphylococcal sepsis, tongue neoplasm | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Biliary sepsis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Intervertebral discitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pseudomonas infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Extradural abscess | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Device-related infection | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Staphylococcal parotitis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Candida infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Disseminated varicella zoster vaccine virus infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Cardiac disorders | 13 (3.4) 15 | 5 (1.4) 6 | 18 (2.4) 21 | 0.09 |
| Atrial fibrillation, acute kidney injury, thrombocytopenia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Atrioventricular block complete | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Cardiac arrest | 5 (1.3) 7 | 1 (0.3) 1 | 6 (0.8) 8 | |
| Cardiac arrest, pulmonary embolism | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cardiac failure | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cardiac failure congestive | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cardiac tamponade | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cardiorespiratory arrest | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Myocardial infarction | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Myocardial ischaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Tachycardia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Ventricular tachycardia | 0 (0.0) 0 | 1 (0.3) 2 | 1 (0.1) 2 | |
| Ischaemic cardiomyopathy | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Vascular disorders | 2 (0.5) 2 | 4 (1.1) 4 | 6 (0.8) 6 | 0.44 |
| Hypotension | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Peripheral ischaemia | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Deep-vein thrombosis | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Respiratory, thoracic and mediastinal disorders | 12 (3.1) 12 | 6 (1.6) 6 | 18 (2.4) 18 | 0.23 |
| Asthma | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Chronic obstructive pulmonary disease | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Dyspnoea | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Dyspnoea, pulmonary oedema | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Epistaxis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hypoxia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pleural effusion | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pneumonia aspiration | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Pulmonary embolism | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pulmonary oedema | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Pulmonary oedema, acute kidney injury, staphylococcal bacteraemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Respiratory arrest | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Respiratory failure | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Gastrointestinal disorders | 7 (1.8) 7 | 10 (2.7) 12 | 17 (2.2) 19 | 0.47 |
| Colitis ulcerative | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Diarrhoea | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Diarrhoea, vomiting | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Dyspepsia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Gastrointestinal angiodysplasia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Gastrointestinal haemorrhage | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Intestinal ischaemia | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Intestinal obstruction | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Nausea | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pancreatitis | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Pancreatitis acute | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Upper gastrointestinal haemorrhage | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Vomiting | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Lower gastrointestinal haemorrhage | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Hepatobiliary disorders | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | 0.24 |
| Hepatic failure | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Skin and subcutaneous tissue disorders | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | 1.00 |
| Erythema | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Rash vesicular | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Renal and urinary disorders | 4 (1.0) 4 | 10 (2.7) 10 | 14 (1.8) 14 | 0.11 |
| Renal tubular acidosis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Renal impairment | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Acute kidney injury | 0 (0.0) 0 | 7 (1.9) 7 | 7 (0.9) 7 | |
| Acute kidney injury, hypernatraemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Acute kidney injury, respiratory failure | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Acute kidney injury, sepsis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Acute kidney injury, urinary retention | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Acute kidney injury, vomiting | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 7 (1.8) 7 | 11 (3.0) 12 | 18 (2.4) 19 | 0.34 |
| Bladder cancer | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Breast cancer | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Chronic lymphocytic leukaemia | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Chronic myeloid leukaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Colon cancer | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Lung adenocarcinoma | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Mycosis fungoides | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Prostate cancer metastatic | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Lung cancer metastatic | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Colorectal cancer metastatic | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Breast cancer metastatic | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Lung neoplasm malignant | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Renal cell carcinoma | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hepatocellular carcinoma | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Congenital, familial and genetic disorders | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | 1.00 |
| Sickle cell anaemia, osteomyelitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| General disorders and administration site conditions | 12 (3.1) 12 | 11 (3.0) 11 | 23 (3.0) 23 | 1.00 |
| Asthenia | 2 (0.5) 2 | 2 (0.5) 2 | 4 (0.5) 4 | |
| Chest pain | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Death | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Drug withdrawal syndrome | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pain | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pyrexia | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Pyrexia, urinary tract infection | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| General physical health deterioration | 3 (0.8) 3 | 0 (0.0) 0 | 3 (0.4) 3 | |
| General physical health deterioration, urinary tract infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Multiple organ dysfunction syndrome | 3 (0.8) 3 | 1 (0.3) 1 | 4 (0.5) 4 | |
| Multiple organ dysfunction syndrome, peritonitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Multiple organ dysfunction syndrome, sepsis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Investigations | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | 0.49 |
| Alanine aminotransferase increased | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Injury, poisoning and procedural complications | 5 (1.3) 5 | 3 (0.8) 3 | 8 (1.1) 8 | 0.73 |
| Fall | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Femoral neck fracture | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Subdural haematoma | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Vascular pseudoaneurysm | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Crush injury | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Toxicity to various agents | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Stoma obstruction | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Blood and lymphatic system disorders | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | 1.00 |
| Disseminated intravascular coagulation | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Sickle cell anaemia with crisis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Metabolism and nutrition disorders | 1 (0.3) 1 | 3 (0.8) 3 | 4 (0.5) 4 | 0.36 |
| Diabetes mellitus | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Diabetic ketoacidosis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Hyperglycaemia | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Psychiatric disorders | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | 0.50 |
| Agitation | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Confusional state | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Nervous system disorders | 5 (1.3) 6 | 2 (0.5) 2 | 7 (0.9) 8 | 0.45 |
| Cerebral haemorrhage | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cerebrovascular accident | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Generalised tonic–clonic seizure | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Seizure | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Syncope | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Alcoholic seizure | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Neurological symptom | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| AE | Treatment group | Total (N = 758) | p-value | |
|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | |||
| MedDRA code | ||||
| Any | 131 (33.8) 193 | 129 (34.9) 209 | 260 (34.3) 402 | 0.76 |
| Infections and infestations | 45 (11.6) 53 | 40 (10.8) 48 | 85 (11.2) 101 | 0.82 |
| Cellulitis | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Diarrhoea infectious | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Endocarditis | 3 (0.8) 3 | 3 (0.8) 3 | 6 (0.8) 6 | |
| Infection | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Lower respiratory tract infection | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Necrotising fasciitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Osteomyelitis | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Peritonitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pneumonia | 17 (4.4) 18 | 13 (3.5) 13 | 30 (4.0) 31 | |
| Postoperative wound infection | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pyelonephritis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Sepsis | 5 (1.3) 5 | 6 (1.6) 6 | 11 (1.5) 11 | |
| Septic shock | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Systemic candida | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Urinary tract infection | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Urosepsis | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Groin abscess | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Abscess limb | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Staphylococcal bacteraemia | 2 (0.5) 2 | 5 (1.4) 5 | 7 (0.9) 7 | |
| Pulmonary sepsis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Arthritis bacterial | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Escherichia bacteraemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Staphylococcal sepsis | 3 (0.8) 3 | 3 (0.8) 3 | 6 (0.8) 6 | |
| Graft infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Biliary sepsis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Intervertebral discitis | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Pseudomonas infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Extradural abscess | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Device-related infection | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Staphylococcal parotitis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Candida infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Disseminated varicella zoster vaccine virus infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Cardiac disorders | 15 (3.9) 17 | 6 (1.6) 8 | 21 (2.8) 25 | 0.08 |
| Atrial fibrillation | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Atrioventricular block complete | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Cardiac arrest | 6 (1.5) 8 | 1 (0.3) 1 | 7 (0.9) 9 | |
| Cardiac failure | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cardiac failure congestive | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cardiac tamponade | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Cardiorespiratory arrest | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Cardiogenic shock | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Myocardial infarction | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Myocardial ischaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Tachycardia | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Ventricular tachycardia | 0 (0.0) 0 | 1 (0.3) 2 | 1 (0.1) 2 | |
| Ischaemic cardiomyopathy | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Acute coronary syndrome | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Vascular disorders | 7 (1.8) 7 | 5 (1.4) 5 | 12 (1.6) 12 | 0.77 |
| Hypotension | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Peripheral ischaemia | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Thrombophlebitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Thrombophlebitis superficial | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Deep-vein thrombosis | 2 (0.5) 2 | 2 (0.5) 2 | 4 (0.5) 4 | |
| Extremity necrosis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Respiratory, thoracic and mediastinal disorders | 16 (4.1) 17 | 10 (2.7) 11 | 26 (3.4) 28 | 0.32 |
| Aspiration | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Asthma | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Chronic obstructive pulmonary disease | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Dyspnoea | 2 (0.5) 2 | 3 (0.8) 3 | 5 (0.7) 5 | |
| Epistaxis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hypoxia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pleural effusion | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pneumonia aspiration | 3 (0.8) 3 | 1 (0.3) 1 | 4 (0.5) 4 | |
| Pulmonary embolism | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Pulmonary oedema | 2 (0.5) 3 | 5 (1.4) 5 | 7 (0.9) 8 | |
| Respiratory arrest | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Respiratory failure | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Gastrointestinal disorders | 21 (5.4) 24 | 29 (7.8) 40 | 50 (6.6) 64 | 0.19 |
| Abdominal pain | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Colitis ulcerative | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Diarrhoea | 7 (1.8) 7 | 11 (3.0) 13 | 18 (2.4) 20 | |
| Gastrointestinal angiodysplasia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Gastrointestinal haemorrhage | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Intestinal ischaemia | 0 (0.0) 0 | 3 (0.8) 3 | 3 (0.4) 3 | |
| Intestinal obstruction | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Nausea | 1 (0.3) 1 | 3 (0.8) 3 | 4 (0.5) 4 | |
| Pancreatitis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Pancreatitis acute | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Swollen tongue | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Upper gastrointestinal haemorrhage | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Vomiting | 10 (2.6) 10 | 10 (2.7) 12 | 20 (2.6) 22 | |
| Lower gastrointestinal haemorrhage | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Hepatobiliary disorders | 0 (0.0) 0 | 3 (0.8) 3 | 3 (0.4) 3 | 0.12 |
| Hepatic failure | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Jaundice | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Skin and subcutaneous tissue disorders | 7 (1.8) 7 | 5 (1.4) 5 | 12 (1.6) 12 | 0.77 |
| Erythema | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Rash | 6 (1.5) 6 | 4 (1.1) 4 | 10 (1.3) 10 | |
| Rash vesicular | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Musculoskeletal and connective tissue disorders | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | 0.50 |
| Arthralgia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Groin pain | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Renal and urinary disorders | 9 (2.3) 9 | 19 (5.1) 20 | 28 (3.7) 29 | 0.053 |
| Haematuria | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Renal failure | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Renal tubular acidosis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Urinary retention | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Renal impairment | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Acute kidney injury | 6 (1.5) 6 | 17 (4.6) 17 | 23 (3.0) 23 | |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 7 (1.8) 7 | 11 (3.0) 12 | 18 (2.4) 19 | 0.34 |
| Bladder cancer | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Breast cancer | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Chronic lymphocytic leukaemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Chronic myeloid leukaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Colon cancer | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Lung adenocarcinoma | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Mycosis fungoides | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Prostate cancer, metastatic | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Lung cancer, metastatic | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Colorectal cancer, metastatic | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Breast cancer, metastatic | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Lung neoplasm, malignant | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Tongue neoplasm | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Renal cell carcinoma | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hepatocellular carcinoma | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Reproductive system and breast disorders | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | 0.49 |
| Scrotal swelling | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Congenital, familial and genetic disorders | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | 1.00 |
| Sickle cell anaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| General disorders and administration site conditions | 11 (2.8) 11 | 12 (3.2) 12 | 23 (3.0) 23 | 0.83 |
| Asthenia | 2 (0.5) 2 | 2 (0.5) 2 | 4 (0.5) 4 | |
| Chest pain | 2 (0.5) 2 | 1 (0.3) 1 | 3 (0.4) 3 | |
| Crepitations | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Death | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Drug interaction | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Drug withdrawal syndrome | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Oedema peripheral | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pain | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pyrexia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| General physical health deterioration | 3 (0.8) 3 | 0 (0.0) 0 | 3 (0.4) 3 | |
| Multiple organ dysfunction syndrome | 4 (1.0) 4 | 2 (0.5) 2 | 6 (0.8) 6 | |
| Investigations | 6 (1.5) 6 | 11 (3.0) 16 | 17 (2.2) 22 | 0.22 |
| Alanine aminotransferase increased | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Blood creatinine increased | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Blood glucose increased | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Haemoglobin decreased | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Liver function test abnormal | 4 (1.0) 4 | 4 (1.1) 4 | 8 (1.1) 8 | |
| Neutrophil count decreased | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Platelet count decreased | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Blood alkaline phosphatase increased | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Anticoagulation drug level decreased | 0 (0.0) 0 | 1 (0.3) 2 | 1 (0.1) 2 | |
| Injury, poisoning and procedural complications | 6 (1.5) 6 | 5 (1.4) 5 | 11 (1.5) 11 | 1.00 |
| Fall | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Femoral neck fracture | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Subdural haematoma | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Vascular pseudoaneurysm | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Post-procedural haemorrhage | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Crush injury | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Toxicity to various agents | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Stoma obstruction | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Surgical and medical procedures | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | 0.49 |
| External fixation of fracture | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Blood and lymphatic system disorders | 3 (0.8) 3 | 5 (1.4) 6 | 8 (1.1) 9 | 0.50 |
| Anaemia | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Disseminated intravascular coagulation | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Iron deficiency anaemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Leukopenia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Sickle cell anaemia with crisis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Thrombocytopenia | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Metabolism and nutrition disorders | 3 (0.8) 3 | 5 (1.4) 6 | 8 (1.1) 9 | 0.50 |
| Dehydration | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Diabetes mellitus | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Diabetic ketoacidosis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Electrolyte imbalance | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Hyperglycaemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Hyperkalaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hypernatraemia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Hypokalaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Metabolic acidosis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Psychiatric disorders | 5 (1.3) 5 | 5 (1.4) 6 | 10 (1.3) 11 | 1.00 |
| Agitation | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Confusional state | 2 (0.5) 2 | 4 (1.1) 4 | 6 (0.8) 6 | |
| Delirium | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Disorientation | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hallucination, visual | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Nightmare | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Nervous system disorders | 11 (2.8) 14 | 4 (1.1) 4 | 15 (2.0) 18 | 0.12 |
| Cerebral artery embolism | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cerebral haemorrhage | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Cerebrovascular accident | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Dizziness | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Generalised tonic–clonic seizure | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Presyncope | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Seizure | 3 (0.8) 3 | 0 (0.0) 0 | 3 (0.4) 3 | |
| Somnolence | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Spinal cord compression | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Syncope | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Unresponsive to stimuli | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Alcoholic seizure | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Neurological symptom | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Eye disorders | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | 1.00 |
| Eyelid ptosis | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| AE | Treatment group | Total (N = 758) | p-value | |
|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 370) | |||
| Any | 39 (10.1) 52 | 63 (17.0) 89 | 102 (13.5) 141 | 0.006 |
| Infections and infestations | 3 (0.8) 3 | 5 (1.4) 5 | 8 (1.1) 8 | 0.50 |
| Bacteraemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Lower respiratory tract infection | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Pulmonary tuberculosis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Urinary tract infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Respiratory tract infection | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Spinal cord abscess | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Respiratory, thoracic and mediastinal disorders | 2 (0.5) 4 | 0 (0.0) 0 | 2 (0.3) 4 | 0.50 |
| Pneumonia aspiration | 1 (0.3) 2 | 0 (0.0) 0 | 1 (0.1) 2 | |
| Respiratory failure | 1 (0.3) 2 | 0 (0.0) 0 | 1 (0.1) 2 | |
| Gastrointestinal disorders | 8 (2.1) 9 | 24 (6.5) 32 | 32 (4.2) 41 | 0.003 |
| Abdominal pain | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Diarrhoea | 1 (0.3) 1 | 8 (2.2) 8 | 9 (1.2) 9 | |
| Gastrointestinal disorder | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Gastrointestinal haemorrhage | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Nausea | 2 (0.5) 2 | 9 (2.4) 9 | 11 (1.5) 11 | |
| Upper gastrointestinal haemorrhage | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Vomiting | 6 (1.5) 6 | 10 (2.7) 11 | 16 (2.1) 17 | |
| Hepatobiliary disorders | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | 0.24 |
| Cholestasis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Jaundice | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Skin and subcutaneous tissue disorders | 7 (1.8) 9 | 8 (2.2) 9 | 15 (2.0) 18 | 0.80 |
| Blister | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Petechiae | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Rash | 5 (1.3) 7 | 4 (1.1) 5 | 9 (1.2) 12 | |
| Rash macular | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Rash pruritic | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Renal and urinary disorders | 1 (0.3) 2 | 8 (2.2) 10 | 9 (1.2) 12 | 0.02 |
| Nephritis | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Renal impairment | 1 (0.3) 2 | 4 (1.1) 5 | 5 (0.7) 7 | |
| Acute kidney injury | 0 (0.0) 0 | 3 (0.8) 4 | 3 (0.4) 4 | |
| General disorders and administration site conditions | 4 (1.0) 4 | 13 (3.5) 13 | 17 (2.2) 17 | 0.03 |
| Discomfort | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Drug interaction | 2 (0.5) 2 | 8 (2.2) 8 | 10 (1.3) 10 | |
| Malaise | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Oedema | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pain | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Pyrexia | 2 (0.5) 2 | 0 (0.0) 0 | 2 (0.3) 2 | |
| Investigations | 12 (3.1) 13 | 12 (3.2) 14 | 24 (3.2) 27 | 1.00 |
| Alanine aminotransferase increased | 1 (0.3) 1 | 2 (0.5) 2 | 3 (0.4) 3 | |
| Blood bilirubin levels increased | 0 (0.0) 0 | 5 (1.4) 5 | 5 (0.7) 5 | |
| Blood creatinine levels increased | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | |
| Drug level increased | 3 (0.8) 3 | 1 (0.3) 1 | 4 (0.5) 4 | |
| Liver function test abnormal | 4 (1.0) 4 | 1 (0.3) 1 | 5 (0.7) 5 | |
| Blood alkaline phosphatase levels increased | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Inflammatory marker levels increased | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Liver function test increased | 2 (0.5) 2 | 2 (0.5) 4 | 4 (0.5) 6 | |
| Injury, poisoning and procedural complications | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | 1.00 |
| Foreign body aspiration | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Blood and lymphatic system disorders | 1 (0.3) 1 | 3 (0.8) 3 | 4 (0.5) 4 | 0.36 |
| Eosinophilia | 0 (0.0) 0 | 2 (0.5) 2 | 2 (0.3) 2 | |
| Leukopenia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Neutropenia | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Metabolism and nutrition disorders | 2 (0.5) 3 | 0 (0.0) 0 | 2 (0.3) 3 | 0.50 |
| Fluid overload | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hyperkalaemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Hypernatraemia | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Psychiatric disorders | 1 (0.3) 2 | 0 (0.0) 0 | 1 (0.1) 2 | 1.00 |
| Confusional state | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Disorientation | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Nervous system disorders | 1 (0.3) 1 | 1 (0.3) 1 | 2 (0.3) 2 | 1.00 |
| Dizziness | 1 (0.3) 1 | 0 (0.0) 0 | 1 (0.1) 1 | |
| Headache | 0 (0.0) 0 | 1 (0.3) 1 | 1 (0.1) 1 | |
| Antibiotic | Dose | Route | Pack sizea | Cost (£) | |
|---|---|---|---|---|---|
| Pack/vialb | Unitb | ||||
| Rifampicin | 600 mg | i.v. | – | 9.20 | 9.20 |
| 150 mg | p.o. | 100 | 16.41 | 0.16 | |
| 300 mg | p.o. | 100 | 36.63 | 0.37 | |
| Amikacin | 50 mg/ml – 2-ml vial | i.v. | – | 2.07 | 2.07 |
| Amoxicillin | 250 mg | i.v. | – | 0.32 | 0.32 |
| 500 mg | i.v. | – | 0.55 | 0.55 | |
| 1000 mg | i.v. | – | 1.10 | 1.10 | |
| 250 mg | p.o. | 21 | 1.30 | 0.06 | |
| 500 mg | p.o. | 21 | 1.57 | 0.08 | |
| Azithromycin | 250 mg | p.o. | 4 | 1.80 | 0.45 |
| 500 mg | p.o. | 3 | 1.74 | 0.58 | |
| 500 mg | i.v. | – | 9.50 | 9.50 | |
| Aztreonam | 1000 mg | i.v. | – | 9.40 | 9.40 |
| 2000 mg | i.v. | – | 18.82 | 18.82 | |
| Benzylpenicillin | 600 mg | i.v. | – | 0.95 | 0.95 |
| 1200 mg | i.v. | – | 1.89 | 1.89 | |
| 2000 mg | i.v. | – | 2.64 | 2.64 | |
| Cefaclor | 250 mg | p.o. | 21 | 6.80 | 0.32 |
| 500 mg | p.o. | 21 | 7.50 | 0.36 | |
| Cefalexin | 250 mg | p.o. | 28 | 1.46 | 0.05 |
| 500 mg | p.o. | 21 | 1.76 | 0.08 | |
| Cefotaxime | 500 mg | i.v. | – | 2.25 | 2.25 |
| 1000 mg | i.v. | – | 4.20 | 4.20 | |
| 2000 mg | i.v. | – | 8.57 | 8.57 | |
| Cefradine | 250 mg | p.o. | 20 | 2.29 | 0.12 |
| 500 mg | p.o. | 20 | 3.49 | 0.18 | |
| Ceftazidime | 500 mg | i.v. | – | 4.40 | 4.40 |
| 1000 mg | i.v. | – | 7.92 | 7.92 | |
| 2000 mg | i.v. | – | 15.84 | 15.84 | |
| Ceftriaxone | 250 mg | i.v. | – | 2.40 | 2.40 |
| 1000 mg | i.v. | – | 9.58 | 9.58 | |
| 2000 mg | i.v. | – | 19.18 | 19.18 | |
| Cefuroxime | 250 mg | i.v. | – | 0.94 | 0.94 |
| 750 mg | i.v. | – | 2.34 | 2.34 | |
| 1500 mg | i.v. | – | 4.70 | 4.70 | |
| 125mg | p.o. | 14 | 4.56 | 0.33 | |
| 250 mg | p.o. | 14 | 9.11 | 0.65 | |
| Chloramphenicol | 1% – 4 g | Other | – | 1.08 | 1.08 |
| Ciprofloxacin | 2 mg/ml – 50 ml | i.v. | – | 7.57 | 7.57 |
| 2 mg/ml – 100 ml | i.v. | – | 14.49 | 14.49 | |
| 2 mg/ml – 200 ml | i.v. | – | 19.79 | 19.79 | |
| 100 mg | p.o. | 6 | 1.79 | 0.30 | |
| 250 mg | p.o. | 20 | 1.48 | 0.07 | |
| 500 mg | p.o. | 20 | 1.47 | 0.07 | |
| 750 mg | p.o. | 10 | 8.00 | 0.80 | |
| Clarithromycin | 500 mg | i.v. | – | 9.45 | 9.45 |
| 250 mg | p.o. | 14 | 1.65 | 0.12 | |
| 500 mg | p.o. | 14 | 3.32 | 0.24 | |
| Clindamycin | 150 mg/ml – 2ml | i.v. | – | 5.90 | 5.90 |
| 150 mg/ml – 4 ml | i.v. | – | 11.80 | 11.80 | |
| 75mg | p.o. | 24 | 7.45 | 0.31 | |
| 150 mg | p.o. | 24 | 3.71 | 0.16 | |
| Co-amoxiclavulante, amoxicillan/clavulanate | 600 mg 500/100 | i.v. | – | 1.06 | 1.06 |
| 1200 mg 1000/200 | i.v. | – | 1.06 | 1.06 | |
| 625 mg 500/125 | p.o. | 21 | 3.85 | 0.18 | |
| 375 mg 250/125 | p.o. | 21 | 5.03 | 0.24 | |
| Co-trimoxazole | 96 mg/ml – 5 ml | i.v. | – | 1.78 | 1.78 |
| 480 mg | p.o. | 28 | 3.14 | 0.11 | |
| 960 mg | p.o. | 100 | 23.46 | 0.24 | |
| Daptomycin | 350 mg | i.v. | – | 62.00 | 62.00 |
| 500 mg | i.v. | – | 88.57 | 88.57 | |
| Doxycycline | 50 mg | p.o. | 28 | 1.61 | 0.06 |
| 100 mg | p.o. | 8 | 1.07 | 0.13 | |
| Ertapenem | 1000 mg | i.v. | – | 31.65 | 31.65 |
| Erythromycin | 1000 mg | i.v. | – | 10.98 | 10.98 |
| 250 mg | p.o. | 100 | 18.20 | 0.18 | |
| 500 mg | p.o. | 100 | 36.40 | 0.36 | |
| Fidaxomicin | 200 mg | p.o. | 20 | 1350.00 | 67.50 |
| Flucloxacillin | 250 mg | i.m./i.v. | – | 1.23 | 1.23 |
| 500 mg | i.m./i.v. | – | 2.45 | 2.45 | |
| 1000 mg | i.m./i.v. | – | 4.90 | 4.90 | |
| 250 mg | p.o. | 28 | 1.64 | 0.06 | |
| 500 mg | p.o. | 28 | 2.46 | 0.09 | |
| Fusidic acid | 1% – 5 g | Other | – | 2.69 | 2.69 |
| 250 mg | p.o. | 10 | 6.02 | 0.60 | |
| Gentamicin | 40 mg/ml – 1 ml | i.m. | – | 1.40 | 1.40 |
| 40 mg/ml – 2 ml | i.m. | – | 1.00 | 1.00 | |
| 0.8 mg/ml – 100 ml | i.v. | – | 1.61 | 1.61 | |
| 1 mg/ml – 80 ml | i.v. | – | 1.95 | 1.95 | |
| 3 mg/ml – 80 ml | i.v. | – | 5.95 | 5.95 | |
| 3 mg/ml – 120 ml | i.v. | – | 8.45 | 8.45 | |
| Levofloxacin | 5 mg/ml – 100 ml | i.v. | – | 23.75 | 23.75 |
| 250 mg | p.o. | 5 | 4.99 | 1.00 | |
| 500 mg | p.o. | 5 | 7.80 | 1.56 | |
| Linezolid | 2 mg/ml – 300 ml | i.v. | – | 44.50 | 44.50 |
| 600 mg | p.o. | 10 | 445.00 | 44.50 | |
| Meropenem | 500 mg | i.v. | – | 8.00 | 8.00 |
| 1000 mg | i.v. | – | 16.00 | 16.00 | |
| Metronidazole | 5 mg/ml – 20 ml | i.v. | – | 1.56 | 1.56 |
| 5 mg/ml – 100 ml | i.v. | – | 1.22 | 1.22 | |
| 200 mg | p.o. | 21 | 4.49 | 0.21 | |
| 400 mg | p.o. | 14 | 6.34 | 0.45 | |
| Moxifloxacin | 1.6 mg/ml – 250 ml | i.v. | – | 39.95 | 39.95 |
| 400 mg | p.o. | 5 | 10.95 | 2.19 | |
| Nitrofurantoin | 50 mg | p.o. | 28 | 13.02 | 0.47 |
| 100 mg | p.o. | 28 | 3.53 | 0.13 | |
| Norfloxacin | 400 mg | p.o. | 14 | 12.00 | 0.86 |
| Ofloxacin | 2 mg/ml – 100 ml | i.v. | – | 16.16 | 16.16 |
| Oxytetracycline | 250 mg | p.o. | 28 | 1.14 | 0.04 |
| Phenoxymethylpenicillin | 250 mg | p.o. | 28 | 1.18 | 0.04 |
| Piperacillin/tazobactam | 250 mg – 2.25 g | i.m. | – | 7.65 | 7.65 |
| 500 mg – 4.5 g | i.m. | – | 12.90 | 12.90 | |
| Pivmecillinam | 200 mg | p.o. | 10 | 4.50 | 0.45 |
| Teicoplanin | 200 mg | i.v. | – | 3.93 | 3.93 |
| 400 mg | i.v. | – | 7.32 | 7.32 | |
| Temocillin | 1000 mg | i.v. | – | 25.45 | 25.45 |
| Ticarcillin/clavulanate | 200 mg – 3.2 g | i.v. | – | 5.33 | 5.33 |
| Tigecycline | 50 mg | i.v. | – | 32.31 | 32.31 |
| Trimethoprim | 100 mg | p.o. | 28 | 8.44 | 0.30 |
| 200 mg | p.o. | 14 | 4.37 | 0.31 | |
| Vancomycin | 500 mg | i.v. | – | 6.25 | 6.25 |
| 1000 mg | i.v. | – | 12.50 | 12.50 | |
| Antibiotic | Dose | Route |
|---|---|---|
| Amoxicillin | 1500 mg | Other |
| 42 mg | p.o. | |
| Benzylpenicillin | 10 mg | i.v. |
| Cefazolin | 2000 mg | i.v. |
| Ceftriaxone | 14 mg | i.v. |
| 21 mg | i.v. | |
| 28 mg | i.v. | |
| 100 mg | i.v. | |
| 25 mg | p.o. | |
| Chloramphenicol | 900 mg | i.v. |
| 3100 mg | i.v. | |
| 3600 mg | i.v. | |
| Ciprofloxacin | 2 mg/ml – 50 ml | i.v. |
| 2 mg/ml – 100 ml | i.v. | |
| 2 mg/ml – 200 ml | i.v. | |
| 100 mg | p.o. | |
| 250 mg | p.o. | |
| 500 mg | p.o. | |
| 750 mg | p.o. | |
| Clindamycin | 5 mg | i.v. |
| 2 mg | p.o. | |
| Co-amoxiclavulante | 1 mg | i.v. |
| 2 mg | i.v. | |
| 4 mg | i.v. | |
| 14 mg | i.v. | |
| 30 mg | Other | |
| Doxycycline | 4000 mg | i.v. |
| Erythromycin | 2 mg | i.v. |
| Flucloxacillin | 5 mg | i.v. |
| 50 mg | Other | |
| Gentamicin | 80 mg | Other |
| Vancomycin | 10 mg | Other |
| Health-care resource | Unit cost (£) | Source |
|---|---|---|
| Consultations with health-care providers | ||
| GP visit | 36.00 | PSSRU 15/16;43 per patient contact lasting 9.22 minutes |
| Outpatient visit | 136.00 | PSSRU 15/1643 |
| Hospital ward stay (per day cost) | ||
| ICU | 1344.50 | NHS Reference Costs 2015 to 2016;42 code XC05Z |
| HDU | 994.45 | NHS Reference Costs 2015 to 2016;42 code XC06Z |
| Inpatient stay | 298.14 | NHS Reference Costs 2015 to 2016;42 non-elective excess bed-days – weighted average of the national average unit cost for all excess bed-days HRG codes by the number of excess bed-days |
| Day case | 385.07 | NHS Reference Costs 2015 to 2016;42 code WH07G |
| Investigations and procedures | ||
| Amputation of single limb | 9721.25 | NHS Reference Costs 2015 to 2016;42 code YQ22B |
| Angioplasty | 3265.92 | NHS Reference Costs 2015 to 2016;42 code YR11D |
| Arthroscopy/endoscopy | 5567.65 | NHS Reference Costs 2015 to 2016;42 code FZ42A |
| Aspiration of joint | 2736.71 | NHS Reference Costs 2015 to 2016;42 code YD05Z |
| Aspiration of pleural cavity | 3076.97 | NHS Reference Costs 2015 to 2016;42 code YH30Z |
| Blood transfusion | 161.49 | NHS Reference Costs 2015 to 2016;42 code XD06Z |
| Bone biopsy | 5200.97 | NHS Reference Costs 2015 to 2016;42 code YH31Z |
| Bone marrow extraction | 7205.87 | NHS Reference Costs 2015 to 2016;42 code SA33Z |
| Bone marrow transplant | 9224.90 | NHS Reference Costs 2015 to 2016;42 code SA19A |
| Bone scan | 200.00 | NHS Reference Costs 2015 to 2016;42 code RN16A |
| Bronchoscopy | 2882.15 | NHS Reference Costs 2015 to 2016;42 code DZ69A |
| Cardioverter defibrillator | 15,195.79 | NHS Reference Costs 2015 to 2016;42 code EY02B |
| Central venous catheter – Hickman line | 3737.55 | NHS Reference Costs 2015 to 2016;42 code YR43A |
| Chemotherapy | 282.47 | NHS Reference Costs 2015 to 2016;42 code SB01Z |
| Chronic obstructive pulmonary disease | 4148.15 | NHS Reference Costs 2015 to 2016;42 code DZ64C |
| Colonoscopy | 2692.10 | NHS Reference Costs 2015 to 2016;42 code FZ51Z |
| Coronary artery bypass graft | 13,225.51 | NHS Reference Costs 2015 to 2016;42 code ED23C |
| CT scan | 114.72 | NHS Reference Costs 2015 to 2016;42 code RD24Z |
| Deep-vein thrombosis | 1452.75 | NHS Reference Costs 2015 to 2016;42 code YQ51E |
| Dialysis line change | 181.05 | NHS Reference Costs 2015 to 2016;42 code LD03A |
| Dialysis line change, graft | 187.50 | NHS Reference Costs 2015 to 2016;42 code LD04A |
| Echocardiogram | 71.44 | NHS Reference Costs 2015 to 2016;42 code RD51A |
| Electrocardiogram | 40.35 | NHS Reference Costs 2015 to 2016;42 code EY51Z |
| Endoscopic retrograde cholangiopancreatography | 1774.46 | NHS Reference Costs 2015 to 2016;42 code GB11Z |
| Evacuation of retained products of conception | 2179.18 | NHS Reference Costs 2015 to 2016;42 code MA17C |
| Fistula, graft or shunt procedure | 3660.98 | NHS Reference Costs 2015 to 2016;42 code YQ42Z |
| Fluoroscopy | 129.36 | NHS Reference Costs 2015 to 2016;42 code RD30Z |
| Gastroscopy | 2173.29 | NHS Reference Costs 2015 to 2016;42 code FZ60Z |
| Gastrostomy | 5142.67 | NHS Reference Costs 2015 to 2016;42 code FZ93A |
| Guided biopsy/aspirate/abscess drainage | 4238.06 | NHS Reference Costs 2015 to 2016;42 code YH32Z |
| Heart valve replacement and cardiac surgery for endocarditis | 13,501.17 | NHS Reference Costs 2015 to 2016;42 code ED24C |
| Hip procedure | 3078.07 | NHS Reference Costs 2015 to 2016;42 code HN14E |
| Hypoglycaemic disorder | 1336.45 | NHS Reference Costs 2015 to 2016;42 code KB01E |
| Implantation of pacemaker | 3520.20 | NHS Reference Costs 2015 to 2016;42 code EY08E |
| Infections of bones or joints and debridement | 3499.10 | NHS Reference Costs 2015 to 2016;42 code HD25H |
| Intestine procedure | 5213.46 | NHS Reference Costs 2015 to 2016;42 code FZ67F |
| Laparoscopic procedure bowel | 3618.26 | NHS Reference Costs 2015 to 2016;42 code GA10K |
| Laparoscopic procedure cancer | 5609.66 | NHS Reference Costs 2015 to 2016;42 code MA26C |
| Larynx or pharynx procedure | 2185.67 | NHS Reference Costs 2015 to 2016;42 code CA84B |
| Limb fracture | 2611.15 | NHS Reference Costs 2015 to 2016;42 code HE31C |
| Liver disorder | 3282.22 | NHS Reference Costs 2015 to 2016;42 code GC01D |
| Lower limb procedure | 6246.72 | NHS Reference Costs 2015 to 2016;42 code YQ12D |
| Lumbar puncture | 2533.22 | NHS Reference Costs 2015 to 2016;42 code HC72A |
| Magnetic resonance | 293.25 | NHS Reference Costs 2015 to 2016;42 code RD07Z |
| MRI scan | 208.32 | NHS Reference Costs 2015 to 2016;42 code RD05Z |
| Negative pressure wound therapy | 1228.14 | Chetter et al.,76 based on an average use of NPWT of 37 days at a cost of £31.78/day |
| Partial foot amputation procedure | 4858.81 | NHS Reference Costs 2015 to 2016;42 code YQ26C |
| PET CT scan | 920.24 | NHS Reference Costs 2015 to 2016;42 code RN03A |
| PET scan | 797.26 | NHS Reference Costs 2015 to 2016;42 code RN07A |
| Pleural effusion | 2235.90 | NHS Reference Costs 2015 to 2016;42 code DZ16N |
| Removal of pacemaker | 5676.76 | NHS Reference Costs 2015 to 2016;42 code EY09B |
| Sepsis intervention | 5620.20 | NHS Reference Costs 2015 to 2016;42 code WJ06C |
| Sigmoidoscopy | 3478.73 | NHS Reference Costs 2015 to 2016;42 code FZ54Z |
| Skin graft | 13,031.21 | NHS Reference Costs 2015 to 2016;42 code JB32A |
| Skin procedure | 2439.67 | NHS Reference Costs 2015 to 2016;42 code JC42A |
| Spine procedure | 7595.42 | NHS Reference Costs 2015 to 2016;42 code HC31J |
| Surgical drainage/removal of non-device-related focus | 3740.54 | NHS Reference Costs 2015 to 2016;42 code YF04C |
| Surgical removal of infected prosthetic device | 5557.01 | NHS Reference Costs 2015 to 2016;42 code HE81B |
| Tooth extraction | 3553.38 | NHS Reference Costs 2015 to 2016;42 code CD07B |
| Tracheostomy | 4822.79 | NHS Reference Costs 2015 to 2016;42 code CA63Z |
| Transcatheter aortic valve implantation | 19,990.80 | NHS Reference Costs 2015 to 2016;42 code EY21B |
| Ultrasound scan | 58.59 | NHS Reference Costs 2015 to 2016;42 code RD43Z |
| Ureteric stent removal | 7385.03 | NHS Reference Costs 2015 to 2016;42 code YL12Z |
| Vena cava procedure | 4250.01 | NHS Reference Costs 2015 to 2016;42 code YR22C |
| White blood cell scan | 204.4 | NHS Reference Costs 2015 to 2016;42 code RN13Z |
| Radiography | 30.92 | NHS Reference Costs 2013 to 201441 (updated to 2016 prices) |
| Distributional assumption | Link function | AIC statistic | |||
|---|---|---|---|---|---|
| Null modela | Model TCb | Model 1Cc | Model 2Cd | ||
| Gaussian | Identity | 16,315 | 16,314 | 15,676 | 15,698 |
| Gaussian | Log | 16,315 | 16,314 | 15,664 | 15,678 |
| Inverse Gaussian | 1/mu2 | 15,873 | 15,873 | Model did not fit | Model did not fit |
| Gamma | Identity | 15,725 | 15,724 | Model did not fit | Model did not fit |
| Gamma | Log | 15,725 | 15,724 | 15,088 | 15,105 |
| Observed EQ-5D-3L score | Treatment group | |||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (N = 388) | Rifampicin (N = 369) | |||||||
| L1 (no problems) | L2 (some problems) | L3 (unable) | Missing | L1 (no problems) | L2 (some problems) | L3 (unable) | Missing | |
| Baseline, n (%) | ||||||||
| Mobility | 58 (14.9) | 178 (45.9) | 94 (24.2) | 58 (14.9) | 66 (17.9) | 166 (45.0) | 85 (23.0) | 52 (14.4) |
| Self-care | 120 (30.9) | 140 (36.1) | 70 (18.0) | 58 (14.9) | 124 (33.6) | 133 (36.0) | 60 (16.3) | 52 (14.4) |
| Usual activities | 60 (15.5) | 135 (34.8) | 135 (34.8) | 58 (14.9) | 60 (16.3) | 128 (34.7) | 129 (35.0) | 52 (14.4) |
| Pain/discomfort | 66 (17.0) | 198 (51.0) | 66 (17.0) | 58 (14.9) | 68 (18.4) | 191 (51.8) | 58 (15.7) | 52 (14.4) |
| Anxiety/depression | 131 (33.8) | 150 (38.7) | 48 (12.4) | 59 (15.2) | 148 (40.1) | 136 (36.9) | 33 (8.9) | 52 (14.4) |
| Day 7, n (%) | ||||||||
| Mobility | 84 (21.6) | 136 (35.1) | 58 (14.9) | 80 (20.6) | 82 (22.2) | 117 (31.7) | 46 (12.5) | 93 (25.5) |
| Self-care | 123 (31.7) | 108 (27.8) | 47 (12.1) | 80 (20.6) | 119 (32.2) | 87 (23.6) | 39 (10.6) | 93 (25.5) |
| Usual activities | 76 (19.6) | 110 (28.4) | 92 (23.7) | 80 (20.6) | 69 (18.7) | 95 (25.7) | 81 (22.0) | 93 (25.5) |
| Pain/discomfort | 96 (24.7) | 144 (37.1) | 37 (9.5) | 81 (20.9) | 90 (24.4) | 125 (33.9) | 30 (8.1) | 93 (25.5) |
| Anxiety/depression | 140 (36.1) | 102 (26.3) | 35 (9.0) | 81 (20.9) | 117 (31.7) | 112 (30.4) | 16 (4.3) | 93 (25.5) |
| Day 14, n (%) | ||||||||
| Mobility | 62 (16.0) | 101 (26.0) | 36 (92.8) | 61 (15.7) | 48 (13.0) | 85 (23.0) | 29 (7.9) | 77 (21.1) |
| Self-care | 91 (23.5) | 77 (19.8) | 31 (8.0) | 61 (15.7) | 67 (18.2) | 71 (19.2) | 24 (6.5) | 77 (21.1) |
| Usual activities | 58 (14.9) | 78 (20.1) | 63 (16.2) | 61 (15.7) | 26 (7.0) | 80 (21.7) | 56 (15.2) | 77 (21.1) |
| Pain/discomfort | 73 (18.8) | 105 (27.1) | 20 (5.2) | 62 (16.0) | 58 (15.7) | 87 (23.6) | 17 (4.6) | 77 (21.1) |
| Anxiety/depression | 92 (23.7) | 91 (23.5) | 15 (3.9) | 62 (16.0) | 90 (24.4) | 62 (16.8) | 10 (2.7) | 77 (21.1) |
| Day 84, n (%) | ||||||||
| Mobility | 96 (24.7) | 102 (26.3) | 20 (5.2) | 174 (44.8) | 97 (26.3) | 86 (23.3) | 6 (1.6) | 194 (52.8) |
| Self-care | 132 (34.0) | 73 (18.8) | 13 (3.4) | 174 (44.8) | 124 (33.6) | 56 (15.2) | 9 (2.4) | 194 (52.8) |
| Usual activities | 89 (22.9) | 96 (24.7) | 33 (8.5) | 174 (44.8) | 79 (21.4) | 86 (23.3) | 24 (6.5) | 194 (52.8) |
| Pain/discomfort | 110 (28.4) | 81 (20.9) | 27 (7.0) | 174 (44.8) | 106 (28.7) | 71 (19.2) | 11 (3.0) | 195 (53.1) |
| Anxiety/depression | 134 (34.5) | 65 (16.8) | 19 (4.9) | 174 (44.8) | 116 (31.4) | 62 (16.8) | 10 (2.7) | 195 (53.1) |
| Model specification | Model | |
|---|---|---|
| 1Q SA | 2Q SA | |
| Type of regression model | OLS | |
| Equation | Total QALYs (imputed) | |
| Covariates (baseline) | Coefficient (SE) | Coefficient (SE) |
| EQ-5D index baseline score | 0.073 (0.007)*** | 0.074 (0.010)*** |
| Randomised treatment (1 – rifampicin; 0 – placebo) | –0.006 (0.010) | –0.018 (0.028) |
| Age (years) | ||
| 54–71 | –0.006 (0.005) | –0.010 (0.007) |
| ≥ 72 | –0.017 (0.005)* | –0.017 (0.008)* |
| Sex (1 – male; 0 – female) | –0.007 (0.005) | –0.011 (0.006)† |
| Acquisition | ||
| Nosocomial infection | 0.001 (0.006) | 0.002 (0.008) |
| Health care associated | 0.008 (0.006) | 0.004 (0.008) |
| Charlson Comorbidity Index score | ||
| 1 or 2 | 0.035 (0.008)*** | 0.031 (0.011)** |
| 3 or 4 | 0.006 (0.010) | –0.002 (0.011) |
| ≥ 5 | –0.001 (0.014) | –0.002 (0.015) |
| BMI (kg/m2) | ||
| 18.5–24.9 | 0.006 (0.009) | –0.003 (0.012) |
| 25.0–29.9 | 0.013 (0.010) | 0.010 (0.013) |
| 30.0–39.9 | 0.016 (0.011) | 0.015 (0.013) |
| ≥ 40 | 0.008 (0.012) | 0.010 (0.017) |
| Deep focus (1 – yes; 0 – no) | –0.002 (0.008) | 0.004 (0.010) |
| Endocarditis (1 – yes; 0 – no) | 0.012 (0.017) | 0.010 (0.017) |
| Meticillin resistance | –0.012 (0.008) | –0.003 (0.013) |
| Neutrophils | ||
| 6–9 × 109/l | 0.002 (0.005) | 0.011 (0.007) |
| > 9 × 109/l | –0.014 (0.008)† | –0.019 (0.008)* |
| Comatose (1 – yes; 0 – no) | –0.018 (0.014) | –0.010 (0.012) |
| Treatment × EQ – 5D index score | – | –0.005 (0.015) |
| Treatment × age, 54–71 years | – | 0.009 (0.010) |
| Treatment × age, ≥ 72 years | – | –0.003 (0.011) |
| Treatment × sex (1 – male; 0 – female) | – | 0.007 (0.009) |
| Treatment × acquisition, nosocomial infection | – | –0.004 (0.011) |
| Treatment × acquisition, health-care associated | – | 0.007 (0.011) |
| Treatment × Charlson Comorbidity Index score of 1 or 2 | – | 0.008 (0.016) |
| Treatment × Charlson Comorbidity Index score of 3 or 4 | – | 0.020 (0.016) |
| Treatment × Charlson Comorbidity Index score of ≥ 5 | – | 0.004 (0.015) |
| Treatment × BMI of 18.5–24.9 kg/m2 | – | 0.018 (0.018) |
| Treatment × BMI of 25.0–29.9 kg/m2 | – | 0.007 (0.019) |
| Treatment × BMI of 30.0–39.9 kg/m2 | – | 0.003 (0.019) |
| Treatment × BMI of ≥ 40 kg/m2 | – | –0.004 (0.024) |
| Treatment × deep focus (1 – yes; 0 – no) | – | –0.013 (0.010) |
| Treatment × endocarditis (1 – yes; 0 – no) | – | 0.004 (0.021) |
| Treatment × meticillin resistance | – | –0.018 (0.017) |
| Treatment × neutrophils, 6–9 × 109/l | – | –0.017 (0.011) |
| Treatment × neutrophils, > 9 × 109/l | – | 0.007 (0.011) |
| Treatment × comatose (1 – yes; 0 – no) | – | –0.018 (0.020) |
| Intercept | 0.053 (0.017)*** | 0.060 (0.018)*** |
| Observations | 757 | 724 |
| HRQoL predictions (QALYs) | Model | Treatment group | |
|---|---|---|---|
| Placebo | Rifampicin | ||
| Mean predicted total QALYs (95% CI) | 1Q SA | 0.068 (0.057 to 0.078) | 0.062 (0.040 to 0.083) |
| Median predicted total QALYs (IQR) | 0.068 (0.064–0.071) | 0.061 (0.054–0.069) | |
| Mean predicted total QALYs difference (95% CI) | –0.006 (–0.026 to 0.14) | ||
| Mean predicted total QALYs (95% CI) | 2Q SA | 0.068 (0.006 to 0.079) | 0.062 (0.040 to 0.083) |
| Median predicted total QALYs (IQR) | 0.068 (0.064–0.072) | 0.062 (0.055–0.069) | |
| Mean predicted total QALYs difference (95% CI) | –0.006 (–0.022 to 0.010) | ||
| Cost-effectiveness outcomes, mean (95% CI) | Treatment group | ||
|---|---|---|---|
| Placebo | Rifampicin | ||
| Base-case results (using results from regression models 1C and 1Q SA) | |||
| Predicted total costs (£) | £12,142 (£11,194 to £13,249) | £11,050 (£10,089 to £12,068) | |
| Predicted total QALYs | 0.068 (0.057–0.078) | 0.062 (0.040–0.083) | |
| Incremental predicted total costs (£) | –£1092 (–£2564 to –£371.7) | ||
| Incremental predicted total QALYs | –0.006 (–0.026 to 0.14) | ||
| ICER (£/QALY gained) | 179/631 [i.e. costs less but has negative health benefits in relation to placebo (the higher the ICER, the more cost-effective rifampicin is)] | ||
| INHB | £13,000/QALY | 0.088 (–0.036 to 0.192) | |
| £20,000/QALY | 0.049 (–0.027 to 0.124) | ||
| £30,000/QALY | 0.030 (–0.022 to 0.083) | ||
| Probability of being cost-effectivea | £13,000/QALY | 0.09 | 0.91 |
| £20,000/QALY | 0.11 | 0.89 | |
| £30,000/QALY | 0.13 | 0.87 | |
| Scenario analysis results (using results from regression models 2C and 2Q SA) | |||
| Predicted total costs (£) | £11,969 (£10,962 to £13,040) | £10,900 (£9947 to £11,925) | |
| Predicted total QALYs | 0.068 (0.006 to 0.079) | 0.062 (0.040 to 0.083) | |
| Incremental predicted total costs (£) | –£1068 (–£2510 to £392) | ||
| Incremental predicted total QALYs | –0.006 (–0.022 to 0.010) | ||
| ICER (£/QALY gained) | 183/398 [i.e. costs less but has negative health benefits in relation to placebo (the higher the ICER, the more cost-effective rifampicin is)] | ||
| INHBa | £13,000/QALY | 0.076 (–0.038 to 0.190) | |
| £20,000/QALY | 0.047 (–0.027 to 0.122) | ||
| £30,000/QALY | 0.030 (–0.022 to 0.081) | ||
| Probability of being cost-effectivea | £13,000/QALY | 0.09 | 0.91 |
| £20,000/QALY | 0.11 | 0.89 | |
| £30,000/QALY | 0.12 | 0.88 | |
Appendix 3 Resource use items from the electronic case report forms
Data were entered by staff at each NHS Trust Hospital on to eCRFs on the online ARREST trial database. The design of the eCRFs mirrors that of the emergency paper CRFs.
Appendix 4 Distribution of EuroQol-5 Dimensions index score
FIGURE 22.
(a) Distribution of EQ-5D index score at baseline; (b) distribution of EQ-5D index score at 7 days; (c) distribution of EQ-5D index score at 14 days; and (d) distribution of EQ-5D index score at 84 days (not using multiple imputation, but including hard imputations for coma/unwilling/unable to complete and death).
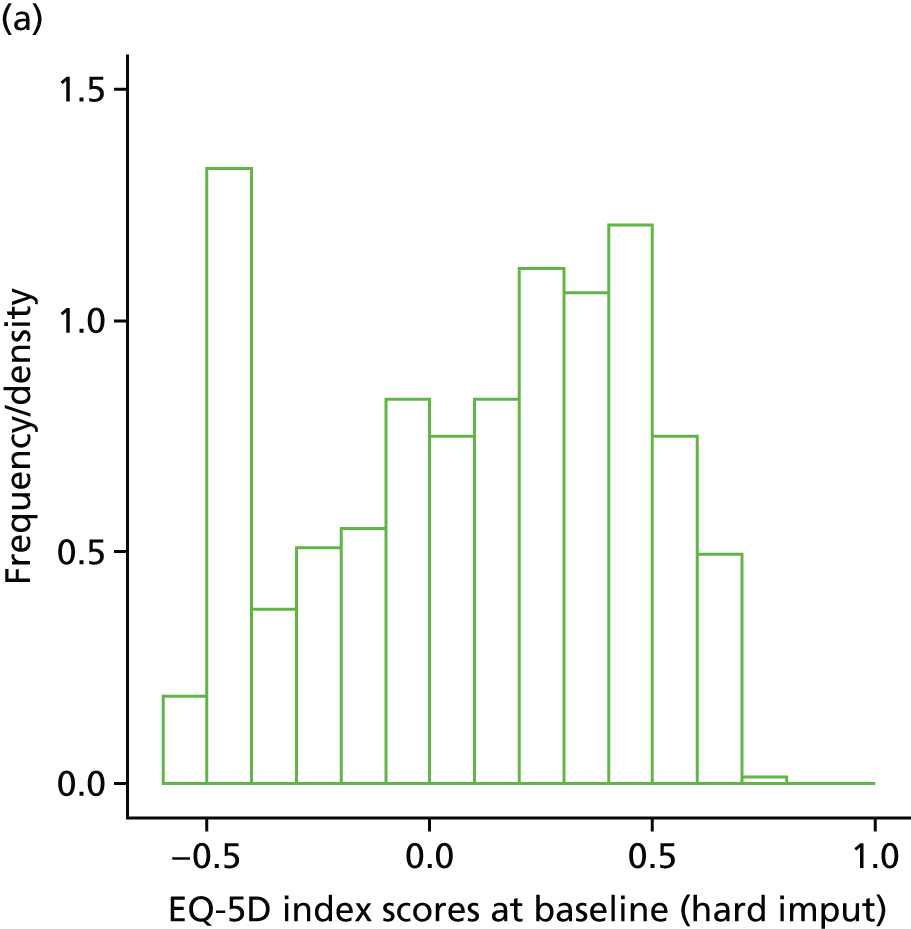
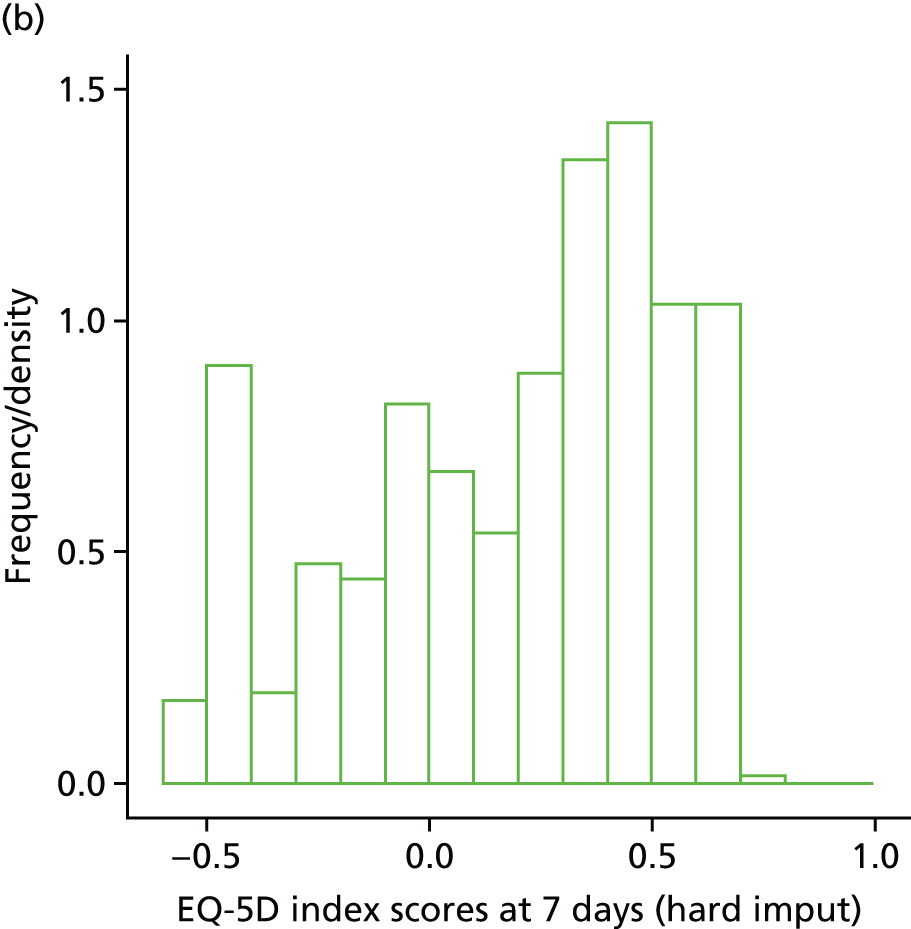
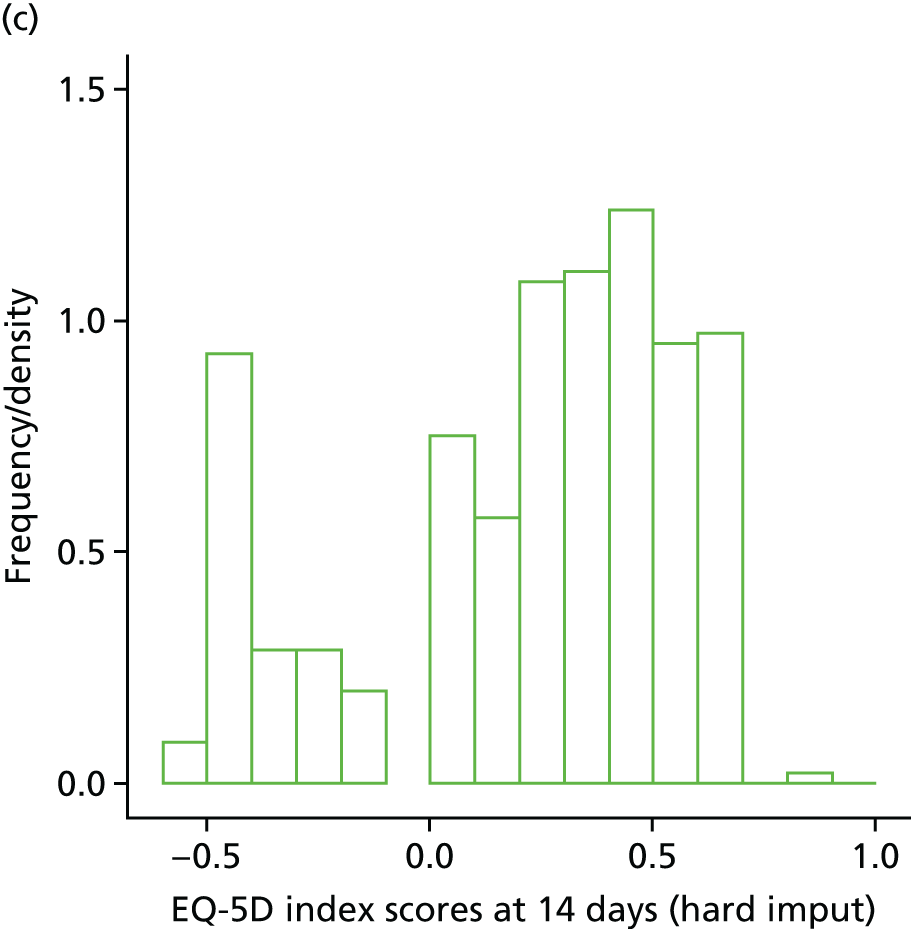
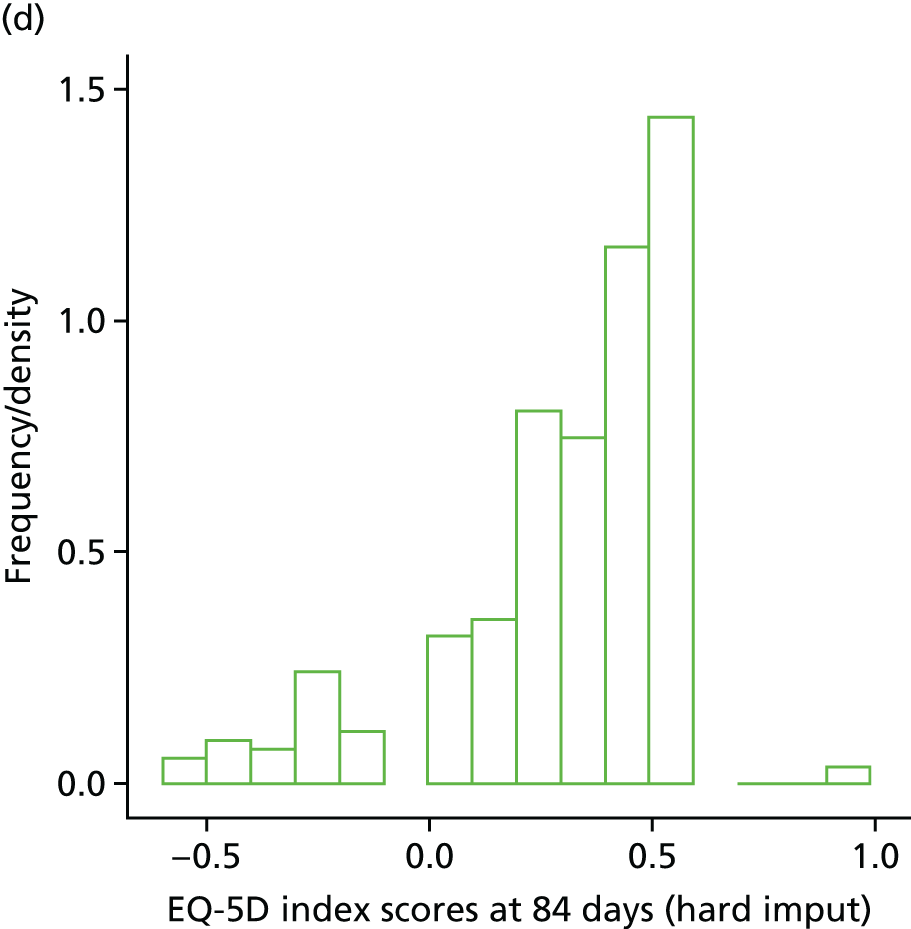
List of abbreviations
- AE
- adverse event
- AIC
- Akaike information criterion
- ALT
- alanine transaminase
- ARREST
- Adjunctive Rifampicin to Reduce Early mortality from STaphylococcus aureus bacteraemia
- AST
- aspartate aminotransferase
- BMI
- body mass index
- CI
- confidence interval
- CRF
- case report form
- CRP
- C-reactive protein
- CT
- computed tomography
- CTU
- Clinical Trials Unit
- DMC
- Data Monitoring Committee
- DNA
- deoxyribonucleic acid
- eCRF
- electronic case report form
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-3L
- EuroQol-5 Dimensions, three-level version
- EVPI
- expected value of perfect information
- GLM
- generalised linear model
- GP
- general practitioner
- HDU
- high-dependency unit
- HIV
- human immunodeficiency virus
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- i.v.
- intravenous
- ICU
- intensive care unit
- IMP
- investigational medicinal product
- INHB
- incremental net health benefit
- IQR
- interquartile range
- MedDRA
- Medical Dictionary for Regulatory Activities
- MRC
- Medical Research Council
- MREC
- Medical Research Ethics Committee
- MRI
- magnetic resonance imaging
- MRSA
- meticillin-resistant Staphylococcus aureus
- MSSA
- meticillin-sensitive Staphylococcus aureus
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NNT
- number needed to treat
- PD
- pharmacodynamic
- PET
- positron emission tomography
- PI
- principal investigator
- PK
- pharmacokinetic
- PPI
- patient and public involvement
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RD
- risk difference
- REC
- Research Ethics Committee
- RNA
- ribonucleic acid
- SAE
- serious adverse event
- SD
- standard deviation
- SE
- standard error
- SOFA
- Sequential Organ Failure Assessment
- SPC
- summary of product characteristics
- SURF
- Service Users Research Forum
- TSC
- Trial Steering Committee
- UCL
- University College London
- ULN
- upper limit of normal