

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 11/129/76. The contractual start date was in April 2013. The draft report began editorial review in May 2017 and was accepted for publication in October 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Simon Gilbody is deputy chairperson of the Health Technology Assessment programme Commissioning Board. Tim J Peters chaired the Medical Research Council–National Institute for Health Research Methodology Research Programme panel from 2007 to 2014. Ian Anderson has received personal fees from the following: Alkermes plc, Lundbeck Ltd, Otsuka Pharmaceutical Ltd, Janssen Ltd and Takeda Pharmaceutical Company Ltd.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Kessler et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Depression is ranked among the top five contributors to the global burden of disease and by 2030 is predicted to be the leading cause of disability in high-income countries. 1 In the UK, people with depression are usually managed in primary care and antidepressants are often the first-line treatment. The number of prescriptions for antidepressants has risen dramatically in recent years, increasing by 6.8% (3.9 million items) between 2014 and 2015. Indeed, antidepressants have shown a greater increase in the volume of prescribing in recent years than drugs for any other therapeutic area, with over 61 million prescriptions being issued in England in 2015, at a cost of £2685M. 2
However, the largest study of sequenced treatment for depression, the STAR*D (Sequenced Treatment Alternatives to Relieve Depression) study,3 found that half of those treated did not experience a reduction of ≥ 50% in depressive symptoms following 12–14 weeks of treatment with a single antidepressant. The reasons for non-response include non-adherence to medication, which may be a result of intolerance. However, a substantial proportion of those who take their antidepressants in an adequate dose and for an adequate period do not experience a clinically meaningful improvement in their depressive symptoms. This can be termed treatment-resistant depression (TRD).
When first-line antidepressant treatments do not work, general practitioners (GPs) can be unsure of what to offer next. The National Institute for Health and Care Excellence (NICE) now advises GPs to reconsider the treatment options if there has been no response after 4–6 weeks of treatment with antidepressant medication. 4 However, there is currently limited evidence to guide management.
Existing evidence on the pharmacological management of treatment-resistant depression
The current NICE guideline4 describes the following pharmacological strategies for sequencing treatments after inadequate response to the initial treatment: switching antidepressants, augmenting medication by adding a drug that is not an antidepressant and combining antidepressants. The guideline4 comments that the evidence for the benefit of switching, either within or between classes, is weak.
Connolly and Thase5 comment that switching antidepressants after an inadequate response is not ‘unequivocally supported by the data, although switching from a SSRI [selective serotonin reuptake inhibitor] to venlafaxine or mirtazapine may offer greater benefits’. Similarly, there is very little evidence on combining two antidepressants.
The evidence for the effectiveness of augmentation with a non-antidepressant is likewise of variable quality. There is some evidence for augmentation with lithium or a thyroid hormone, but mainly in combination with tricyclic antidepressants, which are not prescribed as often today. The use of the atypical antipsychotics to augment the newer antidepressants is better supported by research,6,7 with quetiapine (Seroquel; AstraZeneca plc, Cambridge, UK) and aripiprazole (Abilify®; Otsuka Pharmaceutical Co. Ltd, Tokyo, Japan) the most promising. 8 However, this combination has not, to date, been adopted with any enthusiasm in UK primary care. This may be because of a lack of experience in prescribing them for this indication as they are usually initiated in secondary care. There are also concerns about their adverse events (AEs), including sedation, metabolic syndrome and central obesity and extrapyramidal effects. 9 Indeed, the current NICE guidance recommends that antidepressants should not be combined or augmented without the advice of a consultant psychiatrist. 4
It is possible that GPs would consider adding a second antidepressant, rather than an atypical antipsychotic or lithium, as part of the management of TRD. They are more familiar with these drugs and their starting routines; in addition, there is less concern about their AEs and less need for monitoring. In general, stepwise combination of drug treatments is a standard part of the management of chronic diseases such as asthma and hypertension in primary care and has led to improved clinical outcomes. GPs are comfortable with this model of care and would probably readily adopt this strategy if it were found to be effective. We think that there may be an opportunity to substantially improve the treatment of people with depression in primary care by using antidepressants in combination. However, one of the reasons that this strategy has not been adopted is the lack of convincing evidence for its effectiveness, especially in the primary care setting.
There is a pharmacological rationale for adding a second antidepressant to selective serotonin reuptake inhibitors (SSRIs) or serotonin–noradrenaline reuptake inhibitors (SNRIs) with a different and complementary mode of action. Mirtazapine, an alpha2-adrenoreceptor antagonist, increases central noradrenergic and serotonergic neurotransmission by inhibiting negative feedback from synaptic noradrenaline (NA) acting on presynaptic alpha2 autoreceptors on noradrenergic neurones and alpha2 heteroreceptors on 5-hydroxytryptaminergic (5-HT) neurones. Its mechanism of action is, therefore, different from that of both SSRIs and SNRIs, which inhibit synaptic neurotransmitter reuptake after release. Thus, treatment with mirtazapine in combination with either a SSRI or a SNRI may produce a sustained increase in both 5-HT and NA synaptic availability in terminal fields. A further property of mirtazapine not shared by SSRIs and SNRIs is its affinity for the 5-HT2C receptor, where it acts as an inverse agonist. This mechanism has been linked to specific therapeutic effects. Overall, there is the potential for a synergistic action that could enhance the clinical response compared with the response of those patients receiving only monotherapy. Mirtazapine is now off patent and relatively inexpensive.
Because of its different mechanism of action, there is an argument that switching to mirtazapine alone after SSRI treatment failure might be an effective strategy, rather than subjecting patients to the potential AE burden of a second medication. The STAR*D study compared mirtazapine with nortriptyline in a group of patients who had not responded to two consecutive antidepressant monotherapy regimes. 10 The rates of remission were low for both drugs, suggesting that switching to mirtazapine monotherapy is not the most useful strategy.
In spite of the potential benefit of combining mirtazapine with a SSRI, there is relatively little trial evidence to support this strategy. Carpenter et al. 11 compared the addition of mirtazapine to a SSRI with placebo in a group of 26 patients who had not responded to at least 4 weeks of monotherapy. Although the sample size was very small, the results in terms of effectiveness and tolerability are encouraging,11 but more definitive evidence is required before widespread adoption of this strategy. In patients who have not failed previous treatment, Blier et al. 12,13 reported that mirtazapine in combination with a SSRI gave a greater improvement than monotherapy12 and that it was well tolerated with either a SSRI or a SNRI (venlafaxine),13 with both combinations providing significantly higher remission rates than a SSRI alone. In contrast, a larger study found no benefit from combining antidepressants, including mirtazapine and venlafaxine, over SSRI monotherapy with escitalopram (Cipralex®; Lundbeck, Copenhagen, Denmark), although combined treatment had a higher side-effect burden. 14
Mirtazapine treatment is, however, associated with more weight gain than SSRIs15 and, therefore, as well as assessing the efficacy of its combination with SSRIs, it is important to determine its AE burden, especially when used long term.
Defining treatment-resistant depression
Many definitions of TRD have been proposed. These definitions cover a broad spectrum, ranging from failure to respond to at least 4 weeks of antidepressant medication given at an adequate dose16 to more stringent criteria based on non-response to multiple courses of treatment. 5 A number of staging systems have been proposed, including, most recently, the three-stage model suggested by Conway et al. 17 However, as the authors acknowledge, although the various models may provide guidance in defining TRD, they lack empirical support.
In this study we used a more inclusive definition of TRD, that is, patients who still met the International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10),18 criteria for depression after taking a SSRI or a SNRI antidepressant at an adequate dose [based on the British National Formulary (BNF)19 and advice from psychopharmacology experts] for a minimum of 6 weeks. This definition corresponds to stage I TRD as described by Thase and Rush. 20 It is directly relevant to UK primary care, given the large numbers of non-responders in primary care and the current uncertainty about what course of action to recommend for this group of patients.
Although this 6-week criterion seems a relatively short period to define treatment resistance, many of the patients who satisfy this criterion of ‘non-response’ are suffering from moderate to severe chronic depression. The baseline measures for a recent study of the effectiveness of cognitive–behavioural therapy (CBT) for TRD in primary care, CoBalT (Cognitive behavioural therapy as an adjunct to pharmacotherapy for primary care patients with treatment resistant depression: a randomised controlled trial),21 found that 59% of those recruited had been depressed for > 2 years, 70% had been prescribed their current antidepressant for > 12 months and 28% satisfied the ICD-10 criteria18 for severe depression. These data on chronicity and severity illustrate the extent of the unmet need in this population. 22
It is, therefore, important to undertake a study to investigate the effectiveness of the addition of mirtazapine to SSRIs or to SNRIs in primary care. In the UK, most depression is diagnosed and treated in primary care; this is where most antidepressants are prescribed and where most treatment resistance is encountered. The rise in antidepressant prescribing has continued at a steady rate in the UK despite the introduction of the government initiative Improving Access to Psychological Services (IAPT). Failure to adequately respond to treatment is a substantial problem and there is a need to develop the evidence base for the rational prescribing of antidepressants in primary care. An effective intervention has the potential to have a substantial impact on the health and economic burden associated with this patient group.
Aims and objectives
This trial investigated whether or not combining mirtazapine with SNRI or SSRI antidepressants results in better patient outcomes and more efficient NHS care than SNRI or SSRI therapy alone in TRD. All patients who entered the trial were recruited from primary care and had TRD, defined as meeting ICD-1018 criteria for depression after at least 6 weeks of treatment with either a SSRI or a SNRI antidepressant at an adequate dose.
Our specific aims were to:
-
determine the effectiveness of the addition of the antidepressant mirtazapine to a SSRI or a SNRI in reducing depressive symptoms and improving quality of life (QoL) at 12 and 24 weeks and 12 months (compared with the addition of a placebo)
-
determine the cost-effectiveness of this intervention over 12 months
-
qualitatively explore patients’ views and experiences of taking either two antidepressant medications or an antidepressant and a placebo and identify patients’ reasons for completing or not completing the study, including reasons for withdrawal from the study medication
-
qualitatively explore GPs’ views on prescribing combined antidepressant therapy in this patient group.
Chapter 2 Methods
Study design
The MIR trial was a two-parallel-group, multicentre, pragmatic, placebo-controlled randomised trial with allocation at the level of the individual. Patients were recruited from general practices in England in the areas surrounding our four recruiting centres: (1) Bristol, (2) Exeter, (3) Hull/York and (4) Manchester/Keele. The primary outcome was measured at 12 weeks. The double-blinded randomised allocation was maintained for a period of 12 months, although participants could be unblinded at their request or at the request of their GP after measurement of the primary outcome at 12 weeks; outcomes were also measured at 24 weeks and 12 months.
An economic evaluation was conducted alongside the randomised controlled trial (RCT) to evaluate the cost-effectiveness of the intervention at 12 months (see Chapter 4). A nested qualitative study was conducted to explore patients’ and GPs’ views of the use of an additional antidepressant (see Chapter 5). The trial protocol has been published. 23
Ethics approval and research governance
Ethics approval for the study was given by South East Wales Research Ethics Committee (REC) Panel C (reference: 12/WA/0353). Bristol Clinical Commissioning Group (CCG) and other relevant CCGs provided research governance assurance. Clinical trial authorisation was provided by the Medicines and Healthcare products Regulatory Agency (MHRA). The trial sponsor was the University of Bristol. The trial was registered as EudraCT number 2012-000090-23 (January 2012) and ISRCTN06653773 (September 2012). A summary of the changes made to the original protocol is given in Table 1.
| Description | Submitted | Approved |
|---|---|---|
| Original submission (v8.3) | 8 November 2012 | N/A |
| Amendment 1 (protocol v8.4) | 23 January 2013 | 25 January 2013 |
| Revised submission | ||
| Not setting an upper age limit | ||
| Clarification of DMEC to review data | ||
| Defining an adequate dose of a SSRI/SNRI | ||
| Unblinding option at 12 weeks | ||
| Dementia included in exclusion criteria | ||
| Clarification of GP changes to drug regime because of failure to respond | ||
| Amendment 2 (protocol v8.5) | 3 April 2013 | N/A |
| Changes requested by REC | ||
| Addition of PCT sites | ||
| Clarification of minimisation criteria and of issue of medicine to unblinded participants | ||
| GP information sheet revised | ||
| PIS | ||
| Amendment 3 (protocol v8.6) | 16 April 2013 | 23 April 2013 |
| Response to REC-suggested changes | ||
| Change wording from ‘tablet’ to ‘capsule’ | ||
| Amendment 4 (protocol v8.7) | 4 October 2013 | 4 October 2013 |
| Dates for project milestones revised | ||
| Appendix 3: UHB’s AE SOP replaced with MIR trial-specific AE SOP | ||
| Amendment 5 (protocol v8.8) | 3 July 2015 | 11 September 2015 |
| Section 13.3 Expected Adverse Events and Reactions updated to reflect a revised SMPC | ||
| Immediate implementation because of urgent safety measures |
Participants
The trial sought to recruit participants from primary care who had depression that had not responded to at least 6 weeks of treatment with SSRI or SNRI antidepressants, prescribed at an adequate dose. We planned to recruit 470 participants over 18 months from 96 practices in four centres.
Inclusion criteria (all must apply)
Eligible patients were those who met all of the following criteria.
-
Aged ≥ 18 years and in primary care.
-
Treated for depression for at least 6 weeks with any one of the following SSRI or SNRI antidepressants at recommended BNF doses – fluoxetine, sertraline, citalopram, escitalopram, fluvoxamine, paroxetine, duloxetine or venlafaxine (see Appendix 1, Table 25 for the adequate dose table).
-
Adhered to their medication. Adherence to medication is difficult to measure. To operationalise our definition of treatment resistance, we used the Morisky four-item self-report measure of compliance,24 as adapted for CoBalT. 25 The Morisky measure has previously been validated against electronic monitoring bottles, with a score of zero (range 0–4) indicating at least 80% compliance. 18,26 Given the relatively long half-life of antidepressant medication, individuals who have forgotten to take one or two tablets were not excluded.
-
Scored ≥ 14 points on the Beck Depression Inventory-II (BDI-II). 27
-
An ICD-10 diagnosis of depression [assessed using the Clinical Interview Schedule – Revised (CIS-R)]. 28
Exclusion criteria (presence of any warrants exclusion)
General practitioners were asked to exclude patients who fulfilled any of the following exclusion criteria at the time of the record search:
-
patients currently taking combined or augmented antidepressant treatment
-
patients having their medication managed by a psychiatrist
-
patients with dementia (formal diagnosis), bipolar disorder, psychosis or alcohol or substance abuse/dependence
-
women who were pregnant, planning a pregnancy or breastfeeding
-
patients who were unable to complete the study questionnaires
-
patients who had had a previous adverse reaction to mirtazapine
-
patients currently being treated with a monoamine oxidase inhibitor (MAOI), including moclobemide, or with other medical contraindications to mirtazapine.
Recruitment of participants
We used two methods of recruitment: record search and in-consultation recruitment.
Method 1: search of general practice computerised records to identify patients being treated for depression
The search of general practice computerised records identified all patients in the appropriate age range who had been prescribed SSRI or SNRI antidepressants for ≥ 6 weeks and who were currently being prescribed an antidepressant medication at an adequate dose for depression, as recommended in the BNF. 19
The GPs then screened this list of patients and excluded those patients who fulfilled any of the exclusion criteria listed above. A letter of invitation and a brief information leaflet about the study were sent by the general practice to the remaining potential participants. This letter sought permission for the research team to contact potential participants and to send a questionnaire asking about their depressive symptoms and adherence to antidepressant medication. Potential participants replied directly to the study team indicating whether or not they agreed to be contacted. One reminder was sent to those who did not respond to the initial letter of invitation.
On the reply slip, those who did not wish to participate were asked to indicate their age, sex and reason for non-participation. In addition, non-participants were asked to indicate their willingness to take part in a brief telephone interview to discuss their reasons for non-participation.
Anonymised data on the age and sex of those patients who were mailed an invitation to participate but who did not respond (or who refused to participate when invited during the consultation) were collected to assess the generalisability of the study findings.
Method 2: in-consultation recruitment
The GPs could also invite patients to take part in the study during a consultation. In such cases, they provided patients with an information leaflet about the study and obtained permission to pass their contact details to the research team. The research team then mailed a questionnaire to potential participants asking them about their depressive symptoms and adherence to antidepressant medication.
Postal screening: assessment of depressive symptoms and adherence to antidepressants
All those who agreed to be contacted by the research team (in response to either the postal invitation or a direct invitation from their GP during a consultation) were sent a postal questionnaire. This questionnaire collected data on the following:
-
severity of depressive symptoms, using the BDI-II
-
duration of antidepressant treatment, dose of medication and adherence to medication, using the Morisky measure of compliance
-
sociodemographic variables (age, sex, marital status, ethnicity, educational qualifications, employment status, home ownership and financial difficulties).
One reminder was sent to those individuals who did not return a completed postal questionnaire within 2 weeks.
The GPs were then asked to review all of the patients who appeared to be eligible on the postal screening and to sign a form to confirm that these patients were suitable to be prescribed mirtazapine. Once GP approval was received, patients were contacted by a researcher by telephone and invited to attend a face-to-face appointment with a researcher to discuss participation in the trial and to assess their eligibility. The date, time and location of the baseline appointment were confirmed by letter. A detailed patient information sheet (PIS) and a detailed leaflet about mirtazapine were also enclosed; patients were asked to read both prior to attending the baseline appointment.
Patients who completed the screening process but were not eligible to participate received a letter informing them of this and thanking them for taking part. The letter explained that their GP had also been informed and would continue to care for them as usual. The GP received a letter which explained that the patient was ineligible as he or she had not met one or more of the eligibility criteria, but that the GP could refer the patient back to the trial if these factors changed. If the patient had given permission in the postal questionnaire, the GP also received a report that gave more detail about the inclusion criteria that were not satisfied, as well as the individual’s score on the BDI-II.
Baseline assessment
The baseline assessment was conducted in the patient’s home or at the GP’s surgery or on university premises. The researcher explained the study in detail and obtained written informed consent for the baseline assessment. If the potential participant agreed to the assessment, they completed the following questionnaires:
-
BDI-II
-
Morisky questionnaire (adherence to medication)
-
Patient Health Questionnaire-9 items (PHQ-9),29 a brief measure of depression
-
CIS-R, an in-depth psychiatric questionnaire that gives an ICD-10 diagnosis.
Patients were also asked for details of their prescribed medication, prior use of antidepressants and whether or not they were currently in receipt of psychological therapy. In addition, sociodemographic details were recorded (age, sex, ethnicity, marital status), together with information on a number of socioeconomic markers (employment status, housing situation).
Potential participants who scored ≥ 14 points on the BDI-II and who had an ICD-10 primary diagnosis of depression using the CIS-R, were told that they were potentially eligible to enter the trial [pending confirmation by the principal investigator (PI)] and were asked to provide further written consent for trial participation and indicate whether or not they were willing to be contacted about future research projects.
These potentially eligible participants were also asked to complete some further questionnaires including the:
-
General Anxiety Disorder-7 (GAD-7) questionnaire30
-
EuroQoL-5 Dimensions, five-level version (EQ-5D-5L),31 a brief measure of health-related QoL
-
Short Form questionnaire-12 items (SF-12), a brief measure of mental and physical functioning32
-
Antidepressant Side-Effect Checklist (ASEC), a measure of antidepressant side effects. 33
They were asked further questions about their history of depression, whether or not they had ever been referred to a psychiatrist and the strength of their preference for active treatment over placebo (as this could potentially affect medication adherence and outcomes). Additional information was collected on life events, financial stress, social support and use of alcohol. 34
Once the baseline assessment was complete, the local research clinician (PI) reviewed the baseline information and confirmed whether or not the patient was eligible for the MIR trial.
Informed consent
Prior to the start of the baseline assessment, patients were asked to provide written informed consent for the storage and processing of the data collected at the time of the assessment. This covered the data collected from both those who were found to be ineligible to participate in the trial as well as those who were eligible, thus, enabling the trial to be reported in line with the Consolidated Standards of Reporting Trials (CONSORT) guidelines. 35 Those patients who were identified as eligible to participate in the trial were asked to provide additional written informed consent for this purpose. The original signed and dated consent forms were held securely as part of the trial site file, with copies given to both the participants and their GPs for their records.
Randomisation, concealment of allocation and blinding
Following the baseline assessment, eligible and consenting participants were randomised using the automated randomisation service provided by the Bristol Randomised Trials Collaboration (BRTC). Randomisation was carried out by means of a computer-generated code to ensure concealment of allocation.
Randomisation was stratified by centre (n = 4) to ensure balance in terms of local differences. Minimisation was used to ensure balance in important prognostic indicators, that is, baseline BDI-II score (using approximate tertiles derived from CoBalT baseline scores: < 26 points; 26–34 points; ≥ 35 points), sex and whether or not the patient was currently receiving a psychological therapy (yes/no). We used minimisation with a probability weighing of 0.8 to reduce predictability. 36
Once this had been completed, the University Hospitals Bristol NHS Foundation Trust (UHB) pharmacy, which was the central trial pharmacy, was notified. A participant pack containing an initial 8-week supply of medication was sent by registered post from the UHB pharmacy either to participants’ GP surgery or, in exceptional circumstances, to their home.
Treatment group allocation
Participants were randomly assigned to one of two treatments: (1) one × 15-mg encapsulated mirtazapine tablet daily for 2 weeks followed by two × 15-mg encapsulated mirtazapine tablets for up to 50 weeks or (2) identical placebo tablets. Participants were free to withdraw from the medication at any time. Participants, clinicians, outcome assessors and the research team were blinded to allocation. All participants continued with their GP care and usual antidepressants as agreed by their GP. Clinicians were not restricted in their use of psychological services.
Unblinding was available through the trial pharmacy at all times in case of a medical emergency (‘emergency unblinding’). After the 12-week primary outcome had been completed, the code could also be broken at the request of a participant or his or her GP (‘elective unblinding’). Those who had not requested emergency or elective unblinding were unblinded at the end of the follow-up period or on withdrawal from the study. The trial team did not provide further supplies of the trial medication once participants had been unblinded.
Follow-up
Follow-up data collection took place at five time points: 2, 6, 12 and 24 weeks and 12 months post randomisation. Measurement of the primary outcome took place at the 12-week follow-up. The 12-month follow-up was designed to enable the investigation of any longer-term effects on study outcomes.
At 2 weeks post baseline, researchers contacted participants briefly by telephone to check that they had received and started their trial medication. At 6, 12 and 24 weeks and at 12 months, participants were asked to complete self-report outcome questionnaires. The follow-up schedule is summarised in the flow chart presented in Figure 1. Follow-up questionnaires could be completed face to face with the researcher, by telephone or by post. If a postal questionnaire was not returned, a reminder was sent.
FIGURE 1.
Follow-up schedule.
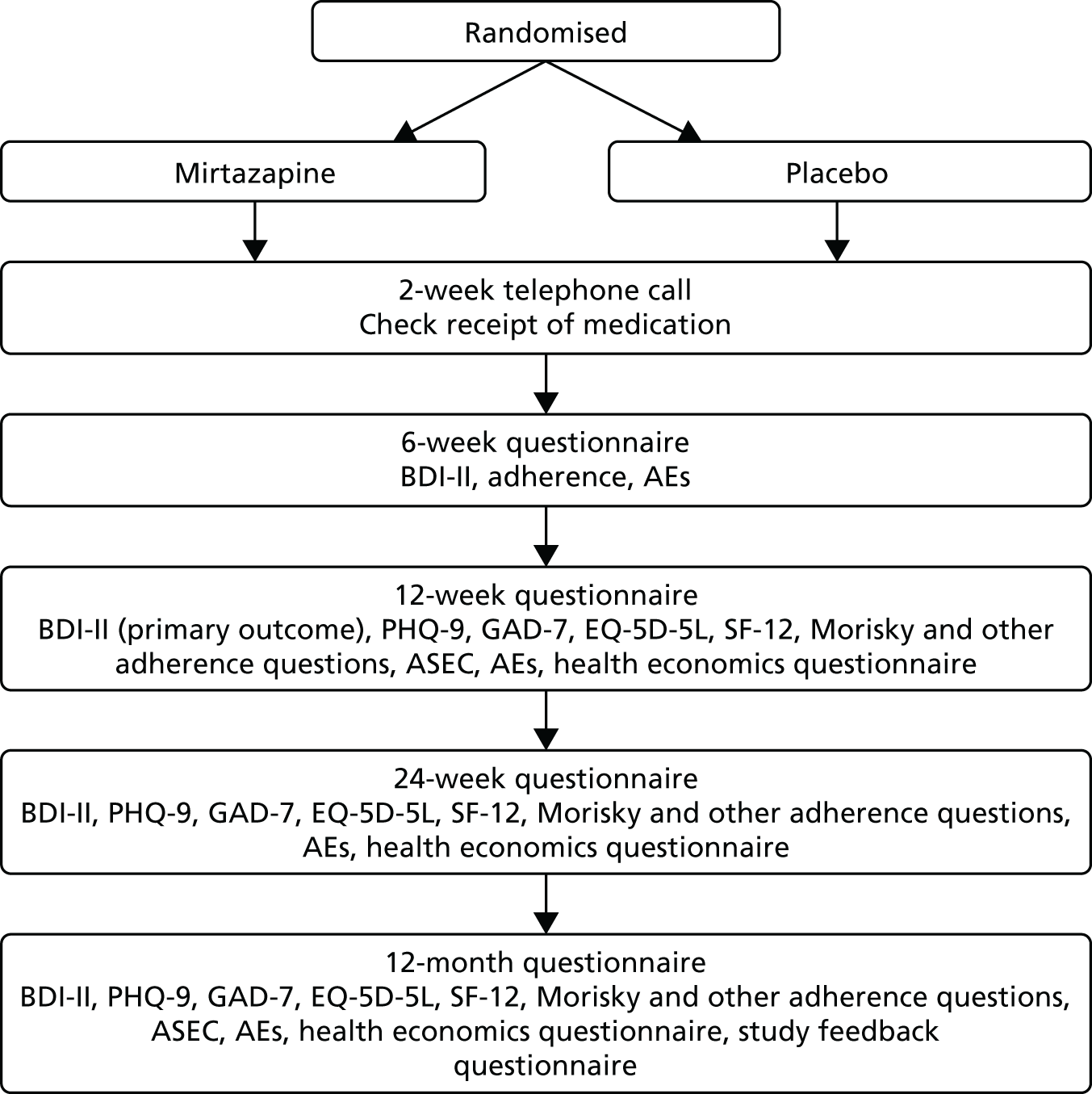
Throughout the follow-up process participants were asked about possible AEs and were advised to consult their GP about these if appropriate. Participants were sent a £5 gift voucher with the 12- and 24-week and the 12-month questionnaires, to thank them for their participation.
At the end of the 12-month follow-up period (or on withdrawal from the study), participants were advised to return to their GP to discuss their continued care.
Withdrawal of trial participants
Participants could withdraw from the trial at any time, for any reason, without their medical care being affected. When possible, data already collected would continue to be used in the trial; participants who withdrew from the trial were asked if they were still willing to provide follow-up data. If a patient withdrew, the reason for, and type of, withdrawal was documented on the case report form (CRF).
Principal investigators had the right to withdraw participants from the trial medication in the event of intercurrent illness, AEs, serious adverse events (SAEs), suspected unexpected serious adverse reactions (SUSARs) or protocol violations or for administrative reasons or other reasons; this was documented on the CRF.
Although there is no evidence that the medication is teratogenic, if a participant discovered that she was pregnant during the trial, she was instructed to stop her trial medication immediately, although she was still able to continue to participate in completion of the trial outcome measures if she wished. A longer monitoring period was put in place to establish the safe delivery of a healthy infant, at which point follow-up would stop.
Data collection and management
To standardise processes across the four centres and maximise data quality, researchers were trained to use detailed standard operating procedures (SOPs) for each stage of data collection. A number of cross-checks were routinely performed as a means of ensuring that any data inconsistencies arising from either the baseline assessment or a follow-up were identified and resolved. Trial data were entered into an OpenClinica version 3.1.4 (Waltham, MA, USA) database at each centre. A range of data validation checks were carried out in both Microsoft Access® 2013 (Microsoft Corporation, Redmond, WA, USA) and Stata® 14 (StataCorp LP, College Station, TX, USA) to minimise erroneous or missing data.
The trial sponsor took primary responsibility for ensuring that the design of the study met appropriate standards and that arrangements were in place to ensure appropriate conduct and reporting. The trial was run in accordance with Good Clinical Practice (GCP)37 and current regulatory guidance.
We employed standard strategies to ensure the quality of the data; for example, a random sample of 10% of CRFs was checked by the trial research team against entries within the database. Consistency checks on key variables were also conducted to identify unlikely or inconsistent responses. These were checked by the research team against hard-copy questionnaires and the database was updated accordingly.
Outcome measures
Primary outcome
The primary outcome was change in the BDI-II score at 12 weeks post randomisation, measured as a continuous variable.
The BDI-II is a 21-item self-report instrument used to measure the severity of depressive symptoms occurring over the previous 2 weeks; it has been widely used in depression trials and extensively validated. 27 The 21 items are rated on a four-point severity scale (0–3) and are summed to give a total score (range 0–63 points). A higher score on the BDI-II denotes more severe depression.
Secondary outcomes
The BDI-II was also completed at 24 weeks and at 12 months to assess the longer-term effect of the intervention. Other outcome measures included at the 12- and 24-week and the 12-month follow-up assessments are listed below:
-
treatment response, measured as an improvement of ≥ 50% in BDI-II score at 12 weeks compared with baseline
-
the rate of remission of symptoms, defined as a BDI-II score of < 10 points at 12 weeks
-
depression, as measured using the PHQ-9
-
change in anxiety symptoms (measured using the GAD-7) at 12 weeks
-
all of the above outcomes at 24 weeks and 12 months
-
antidepressant use and adherence (using the Morisky questionnaire and additional questions)
-
QoL (measured using the EQ-5D-5L)
-
social and physical functioning (measured using the SF-12) at 12 and 24 weeks and 12 months
-
AEs, including any new symptoms or worsening of existing symptoms, consultations for a documented deterioration in illness and SAEs (self-reported or from a review of primary care notes) and side effects (measured using the ASEC) at 12 weeks and 12 months
-
cost-effectiveness from the perspectives of the NHS, patients and society (using self-report questionnaires at 12 and 24 weeks and at 12 months and using primary care practice data on consultations, services and prescriptions over the 12-month trial period).
See Appendix 1, Table 26 for a full schedule of the questionnaires used.
Handling missing items
For the BDI-II, PHQ-9 and GAD-7, the research team dealt with any missing data at an individual item level by adopting the following rule: if ≥ 10% of the items were incomplete, the data collected on that measure for that participant were disregarded. However, if < 10% of the items on a particular measure were missing, missing item(s) were imputed using the mean of the remaining items (rounded to an integer). Therefore, when an individual had completed 19 or 20 items for the primary outcome measure (BDI-II), the remaining one or two items were imputed. For all other measures (PHQ-9 and GAD-7), the 10% rule meant that only a single item would be imputed. Data were complete for the majority of the sample. The scoring manuals for the SF-12 and EQ-5D-5L, which require the application of complex scoring algorithms, indicated that, if any item was missing, the scale score should not be calculated.
A number of sensitivity analyses were conducted to assess the impact of missing primary outcome data on the main findings.
Trial medication
The active trial drug was mirtazapine: one × 15-mg oral capsule per day for 2 weeks, followed by 2 × 15-mg oral capsules per day for up to 12 months. The mirtazapine was encapsulated and the placebo was an identical capsule filled with an inert excipient. The placebo capsule exactly matched the encapsulated mirtazapine in dimensions and appearance, so that allocation concealment and blinding of the trial were maintained.
Packaging, labelling and dispensing
The labelling of medication packs was MHRA approved and conformed to Annexe 13 of good manufacturing practice standards and Article 13.3 of Directive 2001/20/EC. 38 Each medication pack had a medicine identification number, randomly generated to ensure that mirtazapine and placebo medicine packs were indistinguishable and, thus, maintain allocation concealment. The random numbers were generated by the BRTC and provided to the manufacturer, who used them to form the identifiers.
Sharp Clinical Services (UK) Ltd (Crickhowell, UK) provided qualified person services and distribution and project management. It shipped labelled and numbered packages to the central trial pharmacy (UHB Clinical Trials Pharmacy), where the trial medication was stored under controlled conditions. Storage was secure and there were delegation logs for access, for which the trial pharmacy took responsibility. The trial pharmacy dispensed individual participant packs and oversaw the packaging and posting of those packs. Participant packs contained no more than an 8-week supply of the trial medication and were posted by recorded delivery. All deliveries were logged to ensure drug accountability. The trial medication was shipped and stored in conditions in line with the manufacturer’s stability data.
Concomitant medication
Pharmacodynamic interactions
Mirtazapine should not be administered concomitantly with MAOIs or within 2 weeks after discontinuation of MAOI therapy. Likewise, about 2 weeks should pass before patients treated with mirtazapine are treated with MAOIs. Participants in this study were not treated with MAOIs and GPs were advised to wait at least 2 weeks after stopping the trial medication before starting a MAOI.
Co-administration with other serotonergic active substances [L-tryptophan, triptans, tramadol, linezolid (Zyvox®; Pfizer, New York, NY, USA), lithium and St John’s wort (Hypericum perforatum) preparations] may lead to serotonin-associated effects; therefore, participants were advised to use these medications with caution. Mirtazapine may increase the sedating properties of benzodiazepines and other sedatives (notably most antipsychotic drugs, antihistamine H1 antagonists and opioids). Caution should be exercised when these medicinal products are prescribed together with mirtazapine.
Mirtazapine may increase the central nervous system depressant effect of alcohol. Participants were, therefore, advised to be cautious in their intake of alcohol while taking mirtazapine.
Other concomitant care (including switching, discontinuing or changing the dose of SSRI/SNRI medication and receipt of psychological therapies) was not prohibited.
Trial-stopping rules
The trial could be prematurely discontinued by the sponsor, chief investigator, regulatory authority or funder on the basis of new safety information or for other reasons given by the Data Monitoring and Ethics Committee (DMEC) or Trial Steering Committee (TSC).
The trial could be prematurely discontinued because of a lack of recruitment or on advice from the TSC. If the trial was to be prematurely discontinued, active participants would have to be informed and no further participant data would have been collected.
Justification of sample size
Original sample size calculation
The primary outcome was BDI-II score, reported as a continuous variable. It is difficult to estimate a clinically important difference in BDI-II score, although the NICE guideline panel for the first depression guideline39 suggests that this corresponds to about 3 points [standard deviation (SD) 0.35 points] on the Hamilton Depression Rating Scale (HDRS)40 for non-treatment-resistant patients and 2 points for those who are treatment resistant. The equivalent difference to 3 HDRS points for the BDI-II total score would be 3–4 points (SD 10–12 points in CoBalT), which is also a clinically important difference from the patient perspective. 41 With 200 participants in each group, we would have 91% power to detect a difference of 0.33 SDs at the 5% level. Allowing for a 15% loss to follow-up at 12 weeks, we wanted to recruit 472 patients.
For our secondary outcome, response rate, defined as a 50% reduction in symptoms using the BDI-II score, 200 patients in each group would yield 90% power to detect a difference of between 30% and 46% in response, or an odds ratio (OR) of 2, at a two-sided 5% significance level. We therefore aimed to recruit 120 patients from 24 general practices at each of the four recruiting centres.
Blinding
Participants, GPs and investigators were blinded to treatment. The effectiveness of blinding was assessed by a brief questionnaire asking participants to which arm they believed they had been allocated at the 12-week follow-up.
Statistical analysis
Baseline data analyses
Baseline sociodemographic and clinical characteristics were described by treatment group to ascertain any marked imbalances and inform any additional adjustment of the primary and secondary analyses as appropriate. Continuous variables were summarised using the mean and SD [or median and interquartile range (IQR) if the distribution was skewed] and categorical data were summarised as frequencies and proportions. All antidepressant medication use at baseline was reported by class, name, dose and duration of treatment.
Stratification, minimisation and sociodemographic variables and measures of depression and treatment preference, assessed at baseline, were explored for associations with ‘missingness’ of the primary outcome measure at 12 and 24 weeks and 12 months. Comparisons were made using chi-squared tests for categorical variables and t-tests and Mann–Whitney U-tests for continuous variables.
Primary analysis
Analysis and reporting were in line with CONSORT guidelines,35 with the primary analyses being conducted on an intention-to-treat (ITT) basis. The primary analysis was the BDI-II score at 12 weeks post randomisation, measured as a continuous variable. Linear regression modelling was used to compare the randomised groups, adjusting for stratification and minimisation baseline variables and baseline BDI-II score.
Analyses of secondary outcomes were conducted by adjusting for the baseline measure of the outcome variable and stratification and minimisation variables. Linear regression was used for continuous outcomes and logistic regression models were used for binary outcomes.
Secondary analyses
Secondary analyses of the primary and all secondary outcomes included additional adjustment for variables demonstrating a marked imbalance at baseline (ascertained using descriptive statistics).
In all analyses, we have presented regression coefficients (or ORs for binary outcomes), with 95% CIs and p-values.
Subgroup analyses
We conducted a number of prespecified analyses to investigate any differential effects in subgroups defined by a number of factors. These were carried out by introducing appropriate interaction terms in the regression models. We carried out these analyses by baseline depression severity (BDI-II) and a multilevel measure of degree of treatment resistance based on duration of symptoms and prior treatment with antidepressants.
Sensitivity analyses
In all tables, missing data are indicated in the footnotes. For our analysis of the primary outcome, we investigated the influence of missing data using sensitivity analyses that made different assumptions: ‘best’- and ‘worst’-case scenarios and multiple imputation by chained equation (MICE) to impute missing data. 42,43 When using MICE, 25 data sets were generated and 10 switching procedures undertaken. The imputation model included all variables predictive of missingness, together with all of the variables used in the primary analysis. Results from the ITT analyses of complete cases are presented alongside these analyses.
Per-protocol analyses were conducted at 12 weeks and at 12 months for the primary outcome. As these are likely to be biased, we also adopted a complier-average causal effect (CACE)44 approach.
A further sensitivity analysis using CACE methods was conducted at 24 weeks and 12 months. If we define ‘compliers’ as those who had continued taking their trial medication up until 12 weeks, we could then estimate the effect of completing a 12-week course of mirtazapine on depression outcomes at the later follow-up points (24 weeks and 12 months).
Safety reporting and disclosure
Definitions
Adverse event
Adverse events were defined as any untoward medical occurrence in a clinical trial participant. An AE did not necessarily have to have a causal relationship with the trial treatment. An AE could, therefore, be any unfavourable and unintended sign (including an abnormal finding), symptom or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product [International Conference on Harmonisation (ICH) definition]. 45 This included any occurrence that was new in onset or aggravated in severity or frequency from the baseline condition, or abnormal results of diagnostic procedures, including laboratory test abnormalities. All AEs were recorded on the CRF for the duration of participants’ direct involvement in the trial (12 months).
Serious adverse event
A SAE was defined by the ICH45 as any untoward medical occurrence that, at any dose of the trial medication, meets any of the following conditions:
-
Resulted in the death of the participant.
-
Was life-threatening. The term ‘life-threatening’ referred to an event in which the participant was at risk of death at the time of the event; it did not refer to an event that hypothetically might have caused death if it were more severe.
-
Required inpatient hospitalisation or prolongation of existing hospitalisation. For any events that were not immediately life-threatening or did not result in death or hospitalisation, but that could jeopardise the participant or required intervention to prevent one of these outcomes, the chief investigator would exercise his/her scientific and medical judgement to decide whether or not such an event required expedited reporting to UHB (who acted on behalf of the sponsor in these instances).
-
Resulted in persistent or significant disability/incapacity. This means any event that seriously disrupted the ability of the participant to lead a normal life; in other words, one that led to a persistent or permanent significant change, deterioration, injury or perturbation of the participant’s body functions or structure, physical activity and/or QoL.
-
Was a congenital anomaly/birth defect. This related to exposure to the trial drug before conception (in men or women) or during pregnancy that resulted in an adverse outcome in the child.
-
Other medical events. This related to medical events that could jeopardise the subject or required an intervention to prevent a characteristic or consequence of a SAE. Such events were referred to as ‘important medical events’ and were also considered as ‘serious’ in accordance with the definition of a SAE.
Adverse event associated with the use of the drug
An AE was considered to be associated with the use of the drug if the attribution was possible, probable or very likely, as in the following definitions:
-
Not related – an AE that was not related to the use of the drug.
-
Doubtful – an AE for which an alternative explanation was more likely, for example concomitant drug(s), concomitant disease(s) or the relationship in time suggesting that a causal relationship was unlikely.
-
Possible – an AE that might be because of the use of the drug and for which an alternative explanation, for example concomitant drug(s) or concomitant disease(s), was inconclusive. The relationship in time was reasonable and, therefore, the causal relationship could not be excluded.
-
Probable – an AE that might be the result of the use of the drug. The relationship in time was suggestive (e.g. confirmed by withdrawal from trial medication). An alternative explanation was less likely, for example concomitant drug(s) or concomitant disease(s).
-
Very likely – an AE that was listed as a possible adverse reaction and could not be reasonably explained by an alternative explanation, for example concomitant drug(s) or concomitant disease(s). The relationship in time was very suggestive (e.g. it was confirmed by withdrawal from trial medication and reintroduction).
Procedure for reporting
All AE reporting was in accordance with the MIR trial SOP for AE reporting (see Appendix 1).
All adverse events
All AEs were reported by the chief investigator from the time that a signed and dated informed consent form was obtained until completion of the last trial-related procedure (collection of follow-up data 12 months after randomisation). Those occurrences meeting the definition of SAEs were reported using the SAE form (see Appendix 1), including SAEs spontaneously reported to the investigator within 30 days after the participant had completed the trial (including post-trial follow-up). UHB, on behalf of the sponsor, would evaluate any safety information that was spontaneously reported by a chief investigator beyond the time frame specified in the protocol. All AEs, regardless of seriousness, severity or presumed relationship to the trial drug, were recorded in the source document and the CRF, together with any measures taken. All PIs recorded in the CRF their opinion concerning the relationship of the AE to the trial therapy. UHB, on behalf of the sponsor, assumed responsibility for appropriate reporting of AEs to the regulatory authorities.
Serious adverse events
All SAEs were reported to UHB (who monitored SAEs on behalf of the sponsor) and the relevant PI by a delegated member of the research team within 24 hours of their knowledge of the event. The chief investigator and trial manager were also informed. All SAEs that were not resolved by the end of the trial (i.e. by the end of the primary care notes review follow-up period), or that were not resolved on discontinuation of the participant’s participation in the trial, were followed until (1) the event resolved/stabilised/returned to baseline, (2) the event could be attributed to other factors unrelated to the trial or (3) it became unlikely that additional information could be obtained.
The death of a participant was considered a SAE, as was any event requiring hospitalisation (or prolongation of hospitalisation) that occurred during the course of a participant’s participation. Exceptions to this were hospitalisations for:
-
social reasons, in the absence of an AE
-
in-clinic protocol measures
-
surgery or procedure planned before entry into the trial (this was documented on the CRF).
Suspected unexpected serious adverse reaction
All relevant information about a SUSAR that occurred during the course of the trial was reported to the MHRA and the relevant ethics committee by UHB, on behalf of the sponsor, as soon as possible (fatal or life-threatening SUSARs were reported within 7 days; those that were not fatal or life-threatening were reported within 15 days).
The expectedness of an AE was determined by whether or not it was listed in the Summary of Medicinal Product Characteristics (SMPC), the BNF and/or the study protocol.
Expected AEs and adverse reactions are presented in Appendix 1, Box 2.
Quality assurance
Good Clinical Practice37 is an international ethical and scientific quality standard for designing, conducting, recording and reporting studies that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that originated in the Declaration of Helsinki,46 and that the clinical trial data are credible. This research trial ran in accordance with GCP.
Direct access to source data/documents
The PIs and trial site teams allowed monitors (from UHB, on behalf of the sponsor), persons responsible for the audit, representatives of the REC and representatives of the regulatory authorities to have direct access to source data/documents. This was reflected in the PIS. Trial monitoring was undertaken on behalf of the sponsor by UHB using their monitoring SOP.
Trial monitoring
Before the trial
The PIs and trial sites allowed the monitors to visit the sites and facilities where the trial took place to ensure compliance with the protocol requirements. The University of Bristol’s Green Light procedure was implemented in each of the other collaborating centres in order to document preparedness to conduct recruitment locally. A monitoring plan was agreed prior to commencement of the trial.
During the trial
The PIs allowed the monitors and/or the sponsor to:
-
inspect the sites, the facilities and the material used for the trial
-
meet all members of their team involved in the trial
-
consult all of the documents relevant to the trial
-
check that the CRFs had been filled out correctly
-
directly access source documents to compare data therein with the data in the CRFs
-
verify that the trial was carried out in compliance with the protocol and local regulatory requirements
-
carry out trial monitoring at regular intervals, depending on the recruitment rate and arranged between the chief investigator and monitor.
All information dealt with during these visits was treated as strictly confidential.
Quality assurance during the trial
We employed standard strategies to ensure the quality of the data; for example, a random sample of 20% of the CRFs was checked by the trial research team against entries in the database. Recruiting sites were asked to perform a self-audit on all entries and provide a report to the Bristol trial centre (who would report to the trial sponsor). A 10% sample audit was conducted by the UHB monitoring team.
The content of the database was validated at two stages, as follows:
-
At the data-entry stage, validation rules were set to run on submission of data in order to direct researchers to fields that required completion, should any essential fields be missed, and to flag up anomalous or incomplete entries so that researchers could correct data prior to final submission of the electronic CRF.
-
Management information regarding data quality and completeness at centre, site and patient level was generated from data within the trial database and used by the trial manager to inform the implementation and monitoring of the trial.
Standard operating procedures were developed to address each aspect of quality control and the quality assurance procedures.
Data handling
The database and randomisation system were designed to protect patient information and maintain anonymity. Data were stored securely in line with the Data Protection Act 1998. 47 The chief investigator was the custodian of the data. Access to the final data set was restricted to the MIR trial team in the first instance. The team were open to requests by other investigators to access anonymised data.
Other methodological issues
Extension to the trial in 2015
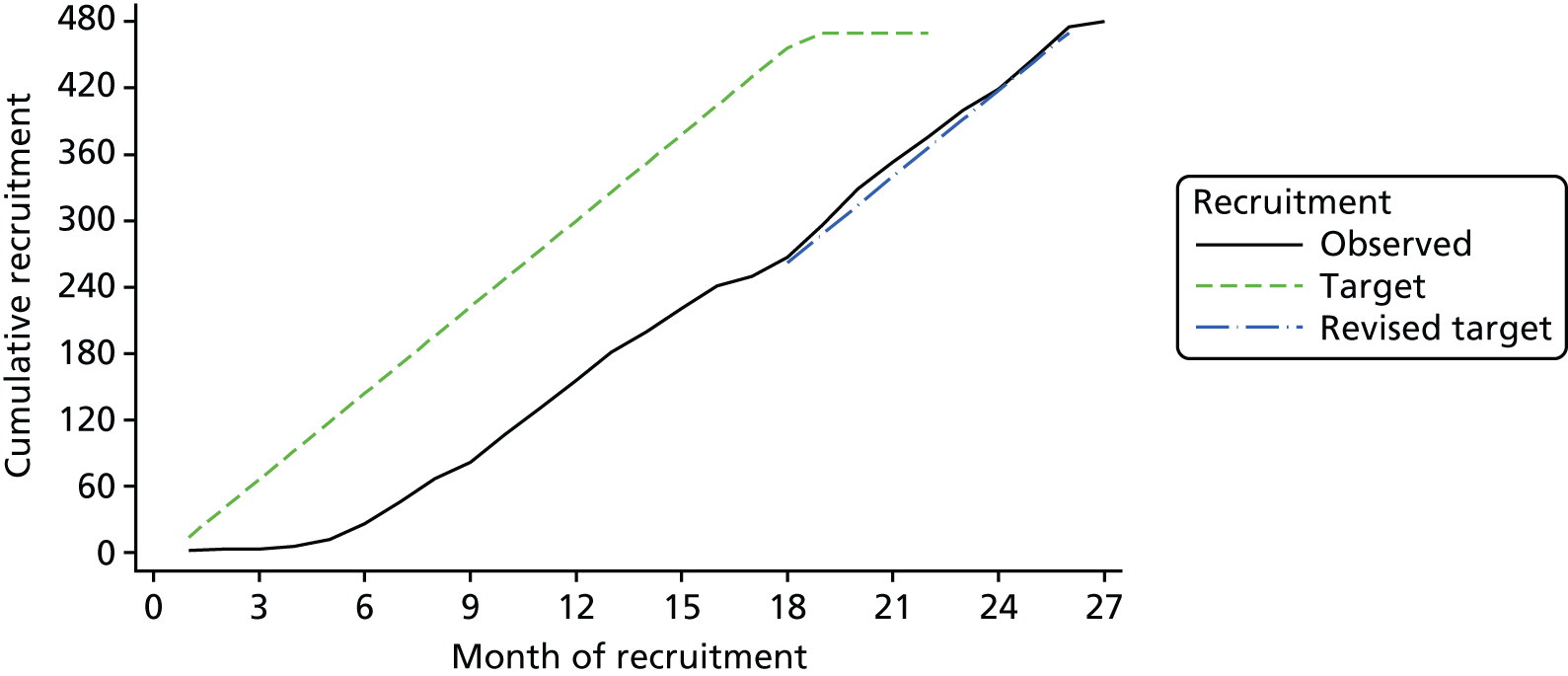
We were obliged to request additional funding for the trial to meet the recruitment target set out in the trial protocol. At the end of September 2014, we had recruited and randomised 200 participants. The recruitment period as set out in the original application was to have ended on 28 February 2015. The final target was 470 participants and it was not likely that we would achieve this within the designated time frame. Recruitment rates during the study had been good and to target. However, there were delays in set-up and in obtaining the necessary permissions from regulatory bodies and finalising contracts with the supplier of the trial medication. An extension of 7 months was awarded and we recruited 480 participants in the revised timescale.
Elective unblinding after the primary outcome at 12 weeks
In the initial study design we proposed that participants would remain blind to allocation for 52 weeks. However, we were advised by the REC that this was not ethically acceptable and that we should offer elective unblinding after the 12-week primary outcome. Therefore, after the 12-week follow-up and data collection (the primary outcome), participants who wished to continue in the study had the following two options:
-
to continue with the trial medication, blind to allocation, with the understanding that they could be unblinded at any time if they wished
-
to be unblinded to their allocation, which would mean that they would no longer receive the trial medication but would remain in the study.
The randomised blinded design was, therefore, preserved intact up until the primary outcome at 12 weeks. However, after that point, participants could discover whether they had been taking placebo or the active treatment. Those who were unblinded then had the option to discontinue their treatment or to consult their GP and request treatment with mirtazapine. Therefore, in addition to the two blinded groups who continued to receive the trial medication, there were now four unblinded groups, as follows, all of whom were included in the remaining follow-ups:
-
those who had been taking placebo up to 12 weeks and who decided to request mirtazapine from their GP
-
those who had been taking placebo up to 12 weeks and who did not request mirtazapine
-
those who had been taking mirtazapine up to 12 weeks and who decided to continue with the drug, now supplied by their GP
-
those who had been taking mirtazapine up to 12 weeks and who decided not to continue with this drug.
Chapter 3 Results
Practice details
Across four centres, 106 general practices agreed to collaborate in the MIR trial; record searches were conducted and mailings sent out. A summary of the practice characteristics of the participating general practices is presented in Table 2. The median number of patients invited to participate in the study per general practice was similar in all four centres.
| Practice details | Centre | Total | |||
|---|---|---|---|---|---|
| Bristol | Exeter | Manchester/Keele | Hull/York | ||
| Number of practices | 33 | 22 | 31 | 20 | 106 |
| Practice size (number of patients), median (IQR) | 10,100 (8442–14,019) | 9890 (4802–13,695) | 6021 (3926–9292) | 11,862 (6056–14,800) | 9200 (5869–13,353) |
| Number of full-time GPs per practice, mean (SD) | 6 (2.2) | 6 (3.9) | 5 (3.1) | 6 (2.5) | 6 (3.0) |
| Number of patients per practice | |||||
| Invited, median (IQR) | 158 (77–215) | 150 (86–261) | 118 (73–248) | 261 (143–405) | 153 (80–262) |
| Completed screening questionnaire, median (IQR) | 22 (8–28) | 23 (14–31) | 12 (6–34) | 29 (16–42) | 21 (9–32) |
| Randomised, median (IQR) | 5 (2–9) | 4 (3–8) | 2 (1–3) | 6 (4–9) | 4 (2–7) |
| Percentage of patients completing screening questionnaire who were randomised | 26 | 24 | 13 | 20 | 21 |
Flow of participants into the trial
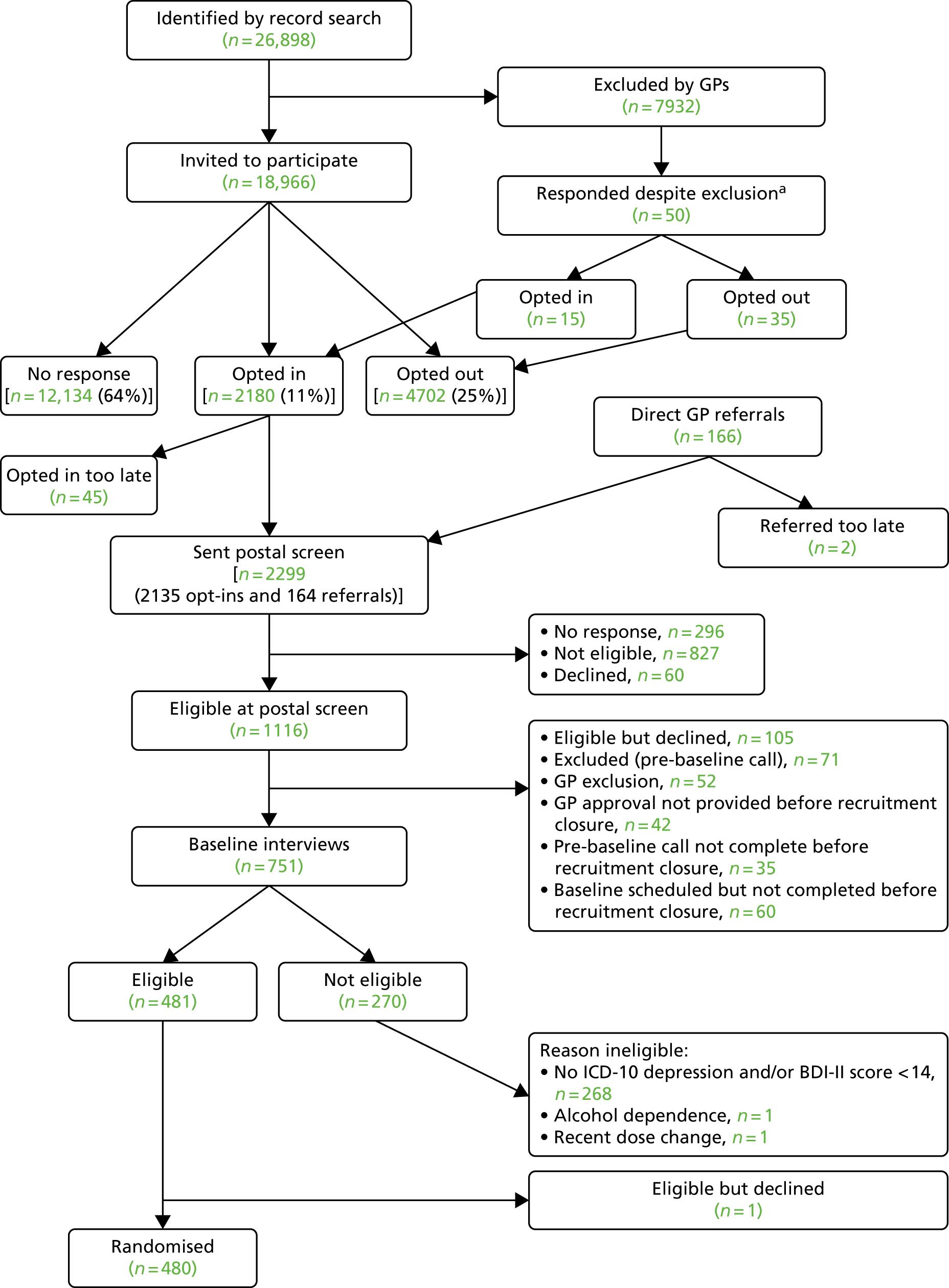
A screening process was designed first to identify those patients being treated for depression and then to assess their depressive symptoms and adherence to antidepressant medication to identify the target population who were potentially eligible to participate in the MIR trial. Potential participants were then invited to a baseline assessment with a researcher to establish their eligibility. The flow of participants through these three stages is outlined in Figure 2. The screening process commenced in August 2013 and the final patient was randomised to the trial on 6 October 2015. All follow-up data were collected between August 2015 and 31 October 2016.
FIGURE 2.
Flow of participants: recruitment pathway. a, Invited by practice despite GP exclusion. Of these, 15 patients opted in (of whom 3 were randomised). Note: Some patients may be ineligible for more than one reason.
Search of general practice computerised records to identify patients being treated for depression
There were 26,898 patients aged ≥ 18 years who were currently being prescribed antidepressants for ≥ 6 weeks at an adequate dose identified through searches of the general practice computerised records. A further 166 individuals were referred to the study directly by their GP. GPs excluded 7932 individuals who were ineligible. Fifty patients were invited as a result of an administrative error and responded, of whom 15 opted in to participate in the trial.
In total, 19,016 patients were mailed a letter of invitation to participate in the study, asking for their permission for the research team to contact them (although 50 of these were invited in error having been excluded by their GPs). GPs invited 166 patients to take part during consultation. Of these, 2180 patients (11%) responded to the letter of invitation and opted in, 4702 (25%) responded and opted out and 12,134 (64%) did not respond to the invitation (see Figure 2).
When available, information on age and sex were recorded from GP records and are summarised in Appendix 2, Tables 28 and 29.
Assessment of depressive symptoms and adherence to antidepressant medication
A screening questionnaire was mailed to 2299 potential participants, of whom 296 individuals did not complete the assessment screen and 60 declined. Of those who returned their questionnaire, 827 were not eligible; mostly this was because their BDI-II score was below the threshold of 14 points, they were not adhering to their antidepressant medication or they did not meet the criteria for an adequate dose/duration of treatment. Other reasons for ineligibility at this stage were pregnancy, breastfeeding, treatment with mirtazapine or amitriptyline and being part of another Clinical Trial of an Investigational Medicinal Product (CTIMP). In total, 1116 patients were eligible at the postal screen stage. However, 105 decided not to participate, 71 were excluded at the pre-baseline check and 52 were excluded by their GP as not suitable. A total of 137 were excluded because their checks were not completed before the closure of recruitment.
When available, information on age and sex were recorded from GP records and are summarised in Appendix 2, Table 30.
Baseline assessment of eligibility to participate in the randomised controlled trial
A total of 751 potential participants attended a face-to-face baseline appointment with a researcher to establish their eligibility to participate in the MIR trial and obtain written informed consent. In total, 270 individuals were ineligible to participate. The majority (n = 268) were ineligible because they did not meet the ICD-10 criteria for depression or failed to meet the BDI-II threshold. In total, 480 individuals were eligible to participate, gave written informed consent and were randomised.
The available information on age, sex and socioeconomic status for those who did and did not attend a baseline assessment is summarised in Table 3.
| Sociodemographic characteristics | Attendance | |
|---|---|---|
| Did not attend (N = 365) | Attended (N = 751) | |
| Age (years) | ||
| Number with available data | 365 | 751 |
| Mean (SD) | 50.22 (14.6) | 51.20 (13.4) |
| Sex | ||
| Number with available data | 365 | 751 |
| Female, n (%) | 260 (71.2) | 513 (68.3) |
| Employment status | ||
| Number with available data | 363 | 746 |
| In paid employment, n (%) | 198 (54.6) | 390 (52.3) |
| Not in employment, n (%) | 165 (45.5) | 356 (47.7) |
| Educational attainment | ||
| Number with available data | 363 | 750 |
| A level, Higher or above, n (%) | 146 (40.2) | 360 (48.0) |
| GCSE, Standard Grade or above, n (%) | 129 (35.5) | 247 (32.9) |
| No formal qualifications, n (%) | 88 (24.2) | 143 (19.1) |
| Housing | ||
| Number with available data | 364 | 750 |
| Home owner, n (%) | 208 (57.1) | 432 (57.6) |
| Tenant or living with relative/friend, n (%) | 150 (41.2) | 314 (41.9) |
| Hostel/care home, homeless or other, n (%) | 6 (1.7) | 4 (0.5) |
Summary of recruitment by centre
There were 480 participants recruited to the study from the four study centres. Bristol recruited the most participants (n = 177), Exeter recruited 122 participants, Hull/York recruited 99 participants and Manchester/Keele recruited 82 participants. A summary of recruitment by centre is presented in Appendix 2, Table 31.
A summary of the overall predicted and actual recruitment is provided in Figure 3. Owing to delays in start-up, a 7-month extension was needed to meet the final target.
FIGURE 3.
Predicted and actual recruitment.
Follow-up of participants in the trial
Of the 480 participants randomised, 241 were allocated to receive usual care and mirtazapine and the remaining 239 were allocated to receive usual care and placebo. The CONSORT flow diagram for the MIR trial is presented in Figure 4. The number of participants who withdrew from the study – as well as a breakdown of the reasons for withdrawal – is presented alongside the number who were lost to follow-up at each follow-up time point.
FIGURE 4.
Flow of participants from randomisation onwards. a, One participant allocated to usual care and placebo withdrew after 12 months of follow-up.
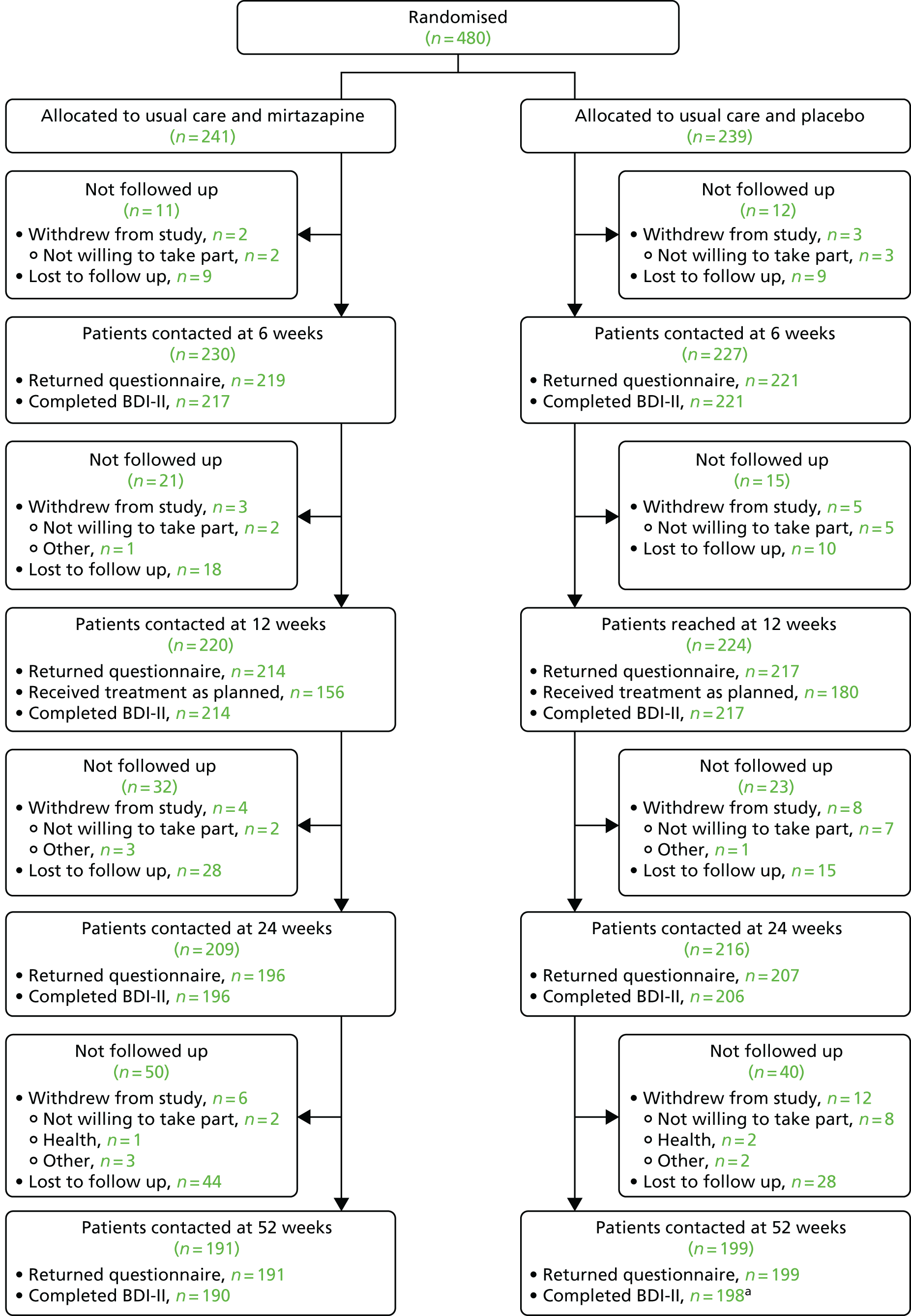
There were six withdrawals among the participants allocated to receive usual care and mirtazapine and 12 withdrawals among those allocated to receive usual care and placebo (including one participant who withdrew after 12 months of follow-up). Withdrawals tended to occur later in follow-up and the most common reason for withdrawal in both groups was a lack of willingness to take part. The timings and reasons for withdrawals are described in Figure 4 and in Appendix 2, Tables 32 and 33.
There were 19 protocol deviations affecting 20 participants (10 among those allocated to usual care and mirtazapine and 10 among those allocated to usual care and placebo). These included one instance when a participant was given the medication of another participant (both participants were allocated to the placebo arm) and one instance when one participant allocated to the placebo arm received another participant’s mirtazapine prescription and vice versa. The most common protocol deviation was participants having difficulty receiving the medication (10 participants). In one case, this led to an emergency unblinding and, in another, the GP prescribed mirtazapine. All protocol deviations are summarised in Appendix 2, Tables 34 and 35.
Baseline characteristics of randomised participants
Table 4 provides a summary of the descriptive statistics used to assess the baseline comparability of the randomised groups. Data were collected at baseline on sociodemographic characteristics, caring responsibilities, treatment preference, severity of depression and measures of depression. A number of baseline imbalances were observed, suggesting that participants in the mirtazapine group had more severe depression. Participants randomised to this group were more likely to have had a prior history of depression and their last course of antidepressants was more recent. A higher proportion of participants in the mirtazapine group than the placebo group had suicidal thoughts at baseline.
| Variable | Allocated groups | |
|---|---|---|
| Mirtazapine + usual care (N = 241), n (%) | Placebo + usual care (N = 239), n (%) | |
| Stratification variable | ||
| Centre | ||
| Bristol | 89 (36.9) | 88 (36.8) |
| Exeter | 61 (25.3) | 61 (25.5) |
| Manchester/Keele | 41 (17.0) | 41 (17.2) |
| Hull/York | 50 (20.7) | 49 (20.5) |
| Minimisation variables | ||
| Female | 168 (69.7) | 164 (68.6) |
| Baseline BDI-II score, points | ||
| 14–25 | 77 (32.0) | 79 (33.1) |
| 26–34 | 78 (32.4) | 78 (32.6) |
| ≥ 35 | 86 (35.7) | 82 (34.3) |
| Currently receiving psychological services | 33 (13.7) | 29 (12.1) |
| Sociodemographic variables | ||
| Age (years), mean (SD) | 50.4 (13.8) | 49.9 (12.5) |
| Ethnic group | ||
| White | 233 (96.7) | 235 (98.3) |
| Mixed | 6 (2.5) | 2 (0.8) |
| Asian/British Asian | 1 (0.4) | 1 (0.4) |
| Other | 1 (0.4) | 1 (0.4) |
| Marital status | ||
| Married/cohabiting | 142 (58.9) | 135 (56.5) |
| Single | 47 (19.5) | 53 (22.2) |
| Separated | 9 (3.7) | 9 (3.8) |
| Divorced | 32 (13.3) | 27 (11.3) |
| Widowed | 11 (4.6) | 15 (6.3) |
| Employment status | ||
| Employed | 109 (45.2) | 135 (56.5) |
| Unemployed | 132 (54.8) | 104 (43.5) |
| Educational attainment | ||
| Degree or equivalent | 44 (18.3) | 51 (21.3) |
| HNC, HND, SVQ (Level 4 or 5) or RSA Higher Diploma | 17 (7.1) | 28 (11.7) |
| A level, Higher or equivalent [GNVQ/NVQ Advanced, GSVQ/SVQ (Level 3) or RSA Advanced Diploma] | 54 (22.4) | 36 (15.1) |
| GSCE, Standard Grade, O level or equivalent | 72 (29.9) | 78 (32.6) |
| No formal qualification | 54 (22.4) | 46 (19.2) |
| Housing | ||
| Home owner | 129 (53.5) | 134 (56.1) |
| Tenant | 96 (39.8) | 88 (36.8) |
| Living with relative or friend | 16 (6.6) | 16 (6.7) |
| Other | 0 (0.0) | 1 (0.4) |
| Financial well-being | ||
| Comfortable/OK | 111 (46.1) | 113 (47.3) |
| Just about getting by or worse | 130 (53.9) | 126 (52.7) |
| Alcohol consumption (units), median (IQR) | 2.0 (1.0–4.0) | 2.0 (1.0–4.0) |
| Number of life events in the past 6 months, mean (SD) | 1.0 (1.0) | 1.1 (1.0) |
| Social support score, mean (SD) | 12.2 (4.1) | 12.8 (4.0) |
| Caring responsibilities | ||
| Providing care for someone who is disabled | 30 (12.4) | 37 (15.5) |
| How many children aged < 5 years live with you? | ||
| None | 213 (88.4) | 219 (91.6) |
| 1 or 2 | 27 (11.2) | 18 (7.5) |
| ≥ 3 | 1 (0.4) | 2 (0.8) |
| Treatment preference | ||
| Do you have a preference for either group? | ||
| Prefer to receive mirtazapine | 143 (59.3) | 146 (61.1) |
| Prefer to receive placebo | 2 (0.8) | 0 (0.0) |
| Do not mind either way | 96 (39.8) | 93 (38.9) |
| If you were to be allocated to the other group, how disappointed would you be?a | ||
| Very | 16 (11.0) | 25 (17.1) |
| Moderately | 54 (37.2) | 39 (26.7) |
| A little bit | 45 (31.0) | 44 (30.1) |
| Not really | 30 (20.7) | 38 (26.0) |
| Measures of depression | ||
| Suffered from depression in the past | 206 (85.5) | 190 (79.5) |
| Previous referral to a psychiatrist for depressionb | 71 (34.5) | 60 (31.6) |
| Number of prior episodes of depressionc | ||
| None | 3 (1.5) | 5 (2.6) |
| 1 | 14 (6.8) | 8 (4.2) |
| 2–4 | 82 (39.8) | 79 (41.6) |
| ≥ 5 | 107 (51.9) | 98 (51.6) |
| Duration of current course of antidepressants | ||
| < 6 months | 26 (10.8) | 20 (8.4) |
| ≥ 6 months | 215 (89.2) | 219 (91.6) |
| ICD-10 primary diagnosis | ||
| Mild | 38 (15.8) | 44 (18.4) |
| Moderate | 138 (57.3) | 144 (60.3) |
| Severe | 65 (27.0) | 51 (21.3) |
| Secondary psychiatric diagnosis according to the CIS-R | ||
| No diagnosis identified | 2 (0.8) | 5 (2.1) |
| Mixed anxiety and depressive disorder (mild) | 26 (10.8) | 31 (13.0) |
| Generalised anxiety disorder (mild) | 2 (0.8) | 3 (1.3) |
| Mixed anxiety and depressive disorder | 62 (25.7) | 52 (21.8) |
| Specific (isolated) phobia | 13 (5.4) | 12 (5.0) |
| Social phobia | 7 (2.9) | 15 (6.3) |
| Agoraphobia | 12 (5.0) | 9 (3.8) |
| Generalised anxiety disorder | 87 (36.1) | 97 (40.6) |
| Panic disorder | 30 (12.4) | 15 (6.3) |
| BDI-II score (points), mean (SD) | 31.5 (10.2) | 30.6 (9.6) |
| GAD-7 score (points), mean (SD)d | 11.3 (4.8) | 10.7 (4.8) |
| PHQ-9 score (points), mean (SD) | 16.7 (5.5) | 16.0 (5.5) |
| EQ-5D-5L score (points), mean (SD)e | 0.7 (0.3) | 0.7 (0.2) |
| SF-12 aggregate physical functioning score (points), mean (SD) | 45.7 (13.8) | 46.4 (13.1) |
| SF-12 aggregate mental functioning score (points), mean (SD) | 27.9 (9.6) | 29.2 (9.7) |
| CIS-R score, mean (SD) | 28.3 (8.2) | 27.0 (8.3) |
| Suicidal ideation (CIS-R thoughts/plans) | ||
| No suicidal thoughts | 81 (33.6) | 119 (49.8) |
| Patient feels life is not worth living | 59 (24.5) | 44 (18.4) |
| Suicidal thoughts | 101 (41.9) | 76 (31.8) |
All patients were taking a SSRI or SNRI at baseline; the most commonly prescribed treatments were citalopram (41.7%) and fluoxetine (24.6%) at doses of 20–60 mg (see Appendix 2, Table 36). There was little difference between the two groups in this respect: 40.7% of participants allocated to usual care and mirtazapine and 42.7% of participants allocated to usual care and placebo received citalopram at baseline, and 23.2% and 24.6%, respectively, were taking fluoxetine. In the mirtazapine group, 10.4% were taking a SNRI and in the placebo group 9.6% were taking a SNRI.
Losses to follow-up
Follow-up rates were slightly higher in the group allocated to usual treatment and placebo and the difference increased after 12 weeks (see Figure 4). At 12 weeks, 88.8% of participants allocated to usual care and mirtazapine and 90.8% of participants allocated to usual care and placebo were contacted for follow-up. By 24 weeks, 81.3% of participants allocated to usual care and mirtazapine were contacted compared with 86.6% of participants allocated to usual care and placebo. At 52 weeks, these proportions were 79.3% and 83.3%, respectively.
Missing data
The pattern of missing data was explored by identifying variables recorded at baseline that were associated with ‘missingness’ of the primary outcome at 12 weeks’ follow-up. Table 5 presents the frequency and proportion of missing data by baseline characteristic.
| Variable | Data, n (%) | ||
|---|---|---|---|
| Present (N = 431) | Missing (N = 49) | p-value | |
| Stratification variable | |||
| Centre | |||
| Bristol | 164 (38.1) | 13 (26.5) | 0.231 |
| Exeter | 110 (25.5) | 12 (24.5) | |
| Manchester/Keele | 73 (16.9) | 9 (18.4) | |
| Hull/York | 84 (19.5) | 15 (30.6) | |
| Minimisation variables | |||
| Female | 291 (67.5) | 41 (83.7) | 0.02 |
| Baseline BDI-II score (points) | |||
| 14–25 | 147 (34.1) | 9 (18.4) | 0.083 |
| 26–34 | 137 (31.8) | 19 (38.8) | |
| ≥ 35 | 147 (34.1) | 21 (42.9) | |
| Currently receiving psychological services | 54 (12.5) | 8 (16.3) | 0.453 |
| Sociodemographic variables | |||
| Age (years), mean (SD) | 50.7 (13.0) | 45.4 (13.8) | 0.008 |
| Ethnic group | |||
| White | 419 (97.2) | 49 (100.0) | 0.706 |
| Mixed | 8 (1.9) | 0 (0.0) | |
| Asian/British Asian | 2 (0.5) | 0 (0.0) | |
| Other | 2 (0.5) | 0 (0.0) | |
| Marital status | |||
| Married/cohabiting | 252 (58.5) | 25 (51.0) | 0.333 |
| Single | 89 (20.6) | 11 (22.4) | |
| Separated | 16 (3.7) | 2 (4.1) | |
| Divorced | 49 (11.4) | 10 (20.4) | |
| Widowed | 25 (5.8) | 1 (2.0) | |
| Employment status | |||
| Employed | 217 (50.3) | 27 (55.1) | 0.528 |
| Unemployed | 214 (49.7) | 22 (44.9) | |
| Educational attainment | |||
| Degree or equivalent | 87 (20.2) | 8 (16.3) | 0.429 |
| HNC, HND, SVQ (Level 4 or 5) or RSA Higher Diploma | 38 (8.8) | 7 (14.3) | |
| A level, Higher or equivalent [GNVQ/NVQ Advanced, GSVQ/SVQ (Level 3) or RSA Advanced Diploma] | 79 (18.3) | 11 (22.4) | |
| GSCE, Standard Grade, O level or equivalent | 139 (32.3) | 11 (22.4) | |
| No formal qualification | 88 (20.4) | 12 (24.5) | |
| Housing | |||
| Home owner | 242 (56.1) | 21 (42.9) | 0.160 |
| Tenant | 158 (36.7) | 26 (53.1) | |
| Living with relative or friend | 30 (7.0) | 2 (4.1) | |
| Other | 1 (0.2) | 0 (0.0) | |
| Financial well-being | |||
| Comfortable/OK | 208 (48.3) | 16 (32.7) | 0.038 |
| Just about getting by or worse | 223 (51.7) | 33 (67.3) | |
| Alcohol consumption, median (IQR) | 2.0 (1.0–4.0) | 2.0 (1.0–4.0) | 0.934 |
| Number of life events in the past 6 months, mean (SD) | 1.0 (1.0) | 1.3 (1.3) | 0.125 |
| Social support score, mean (SD) | 12.5 (4.1) | 12.7 (4.4) | 0.596 |
| Providing care for someone who is disabled | 60 (13.9) | 7 (14.3) | 0.944 |
| How many children aged < 5 years live with you? | |||
| None | 388 (90.0) | 44 (89.8) | 0.826 |
| 1 or 2 | 40 (9.3) | 5 (10.2) | |
| ≥ 3 | 3 (0.7) | 0 (0.0) | |
| Treatment preference | |||
| Do you have a preference for either group? | |||
| Prefer to receive mirtazapine | 255 (59.2) | 34 (69.4) | 0.358 |
| Prefer to receive placebo | 2 (0.5) | 0 (0.0) | |
| Do not mind either way | 174 (40.4) | 15 (30.6) | |
| If you were to be allocated to the other group, how disappointed would you be? | |||
| Very | 36 (14.0) | 5 (14.7) | 0.743 |
| Moderately | 82 (31.9) | 11 (32.4) | |
| A little bit | 81 (31.5) | 8 (23.5) | |
| Not really | 58 (22.6) | 10 (29.4) | |
| Measures of depression | |||
| Suffered from depression in the past | 351 (81.4) | 45 (91.8) | 0.069 |
| Family history of depression | 227 (64.7) | 36 (80.0) | 0.04 |
| Previous referral to a psychiatrist for depression | 112 (31.9) | 19 (42.2) | 0.166 |
| Number of prior episodes of depression | |||
| None | 8 (2.3) | 0 (0.0) | 0.278 |
| 1 | 20 (5.7) | 2 (4.4) | |
| 2–4 | 147 (41.9) | 14 (31.1) | |
| ≥ 5 | 176 (50.1) | 29 (64.4) | |
| Duration of current course of antidepressants, | |||
| < 6 months | 39 (9.0) | 7 (14.3) | 0.238 |
| ≥ 6 months | 392 (91.0) | 42 (85.7) | |
| ICD-10 primary diagnosis | |||
| Mild | 76 (17.6) | 6 (12.2) | 0.552 |
| Moderate | 253 (58.7) | 29 (59.2) | |
| Severe | 102 (23.7) | 14 (28.6) | |
| Secondary psychiatric diagnosis according to the CIS-R | |||
| No diagnosis identified | 7 (1.6) | 0 (0.0) | 0.569 |
| Mixed anxiety and depressive disorder (mild) | 52 (12.1) | 5 (10.2) | |
| Generalised anxiety disorder (mild) | 5 (1.2) | 0 (0.0) | |
| Mixed anxiety and depressive disorder | 100 (23.2) | 14 (28.6) | |
| Specific (isolated) phobia | 20 (4.6) | 5 (10.2) | |
| Social phobia | 19 (4.4) | 3 (6.1) | |
| Agoraphobia | 18 (4.2) | 3 (6.1) | |
| Generalised anxiety disorder | 170 (39.4) | 14 (28.6) | |
| Panic disorder | 40 (9.3) | 5 (10.2) | |
| BDI-II score (points), mean (SD) | 30.8 (9.9) | 33.8 (9.7) | 0.040 |
| GAD-7 score (points), mean (SD) | 10.9 (4.8) | 12.0 (4.7) | 0.109 |
| PHQ-9 score (points), mean (SD) | 16.2 (5.5) | 17.4 (5.4) | 0.160 |
| EQ-5D-5L score (points), mean (SD) | 0.7 (0.2) | 0.6 (0.3) | 0.064 |
| SF-12 aggregate physical functioning score (points), mean (SD) | 45.8 (13.5) | 48.4 (13.1) | 0.203 |
| SF-12 aggregate mental functioning score (points), mean (SD) | 29.0 (9.5) | 25.1 (10.2) | 0.009 |
| CIS-R score, mean (SD) | 27.5 (8.3) | 29.0 (8.0) | 0.221 |
| Suicidal ideation (CIS-R thoughts/plans) | |||
| No suicidal thoughts | 180 (41.8) | 20 (40.8) | 0.983 |
| Patient feels life is not worth living | 92 (21.3) | 11 (22.4) | |
| Suicidal thoughts | 159 (36.9) | 18 (36.7) | |
At each time point, when participants were successfully contacted, few did not complete sufficient components of the BDI-II questionnaire to calculate a score (see Figure 4).
The pattern of missing data was further explored by identifying variables recorded at baseline that were associated with ‘missingness’ of the BDI-II score at 24 weeks and 12 months, as presented in Appendix 2, Tables 37 and 38.
Primary outcome
Depressive symptoms at 12 weeks measured using the continuous Beck Depression Inventory-II score
The mean BDI-II score at 12 weeks was 18.0 points (SD 12.3 points) in participants randomised to the usual care and mirtazapine group compared with 19.7 points (SD 12.4 points) in those randomised to usual care and placebo (Table 6). Higher scores indicate more severe depression, with a maximum possible score of 63 points.
| Randomisation group | n | Mean (SD), points | Difference in meansa | 95% CI | p-value |
|---|---|---|---|---|---|
| Usual care + mirtazapine | 214 | 18.0 (12.3) | –1.83 | –3.92 to 0.27 | 0.087 |
| Usual care + placebo | 217 | 19.7 (12.4) | |||
| Total | 431 | 18.8 (12.4) |
In the primary ITT analysis, participants in the intervention group had a slightly lower BDI-II score than those in the placebo group (see Table 6). The difference – corrected for baseline BDI-II score, centre, baseline BDI-II score terciles, sex and whether or not the patient was receiving psychological therapy at baseline – was less than the minimum clinically relevant difference that we sought to detect (0.33 SDs, which in this case was 4.1 points on the BDI-II) and the confidence interval (CI) overlapped zero, which includes the possibility that there is no difference between the two treatment groups. Further adjustment for variables that suggested some imbalance between the treatment groups at baseline (history of depression, duration of current course of antidepressants and suicidal ideation) did not meaningfully change the effect estimate (difference –2.12 points, 95% CI –4.25 to 0.02 points; p = 0.052).
Subgroup analysis
We planned two a priori subgroup analyses to investigate any differential effects according to baseline depression severity (BDI-II score) and a five-level measure of degree of treatment resistance based on the duration of symptoms and prior treatment with antidepressants.
In the case of baseline depression severity, we found no evidence that this had any effect on the difference between the mirtazapine group and the placebo group (p-value for interaction with treatment group = 0.101).
The observed patterns of duration of symptoms and prior treatment with antidepressants at baseline suggested that a four-level measure of degree of treatment resistance would be more appropriate than the planned five-level measure. The four-level measure was categorised as follows: (1) no prescribed antidepressants in the past, (2) prescribed antidepressants in the past and depressed for < 1 year, (3) prescribed antidepressants in the past and depressed for 1–2 years and (4) prescribed antidepressants in the past and depressed for > 2 years. There was no evidence that this measure of treatment resistance had any effect on the difference between the treatment groups (p-value for interaction with treatment group = 0.299).
Sensitivity analyses to examine the impact of missing data
Table 7 presents the results of the ITT analysis for the 431 ‘complete cases’ with BDI-II score outcome data at 12 weeks and the results from ITT analyses in which the missing data had been estimated using three methods. In the first method, it was assumed that those with missing BDI-II score outcome data had the ‘worst’ possible score (63 points), in the second method it was assumed that those with missing BDI-II score outcome data had the ‘best’ possible score (0 points) and in the third method the missing data were imputed using the method of MICE. The ‘worst’-case scenario approach and the MICE approach yielded smaller differences than the complete-case analysis. The ‘best’-case scenario approach yielded a slightly larger estimate.
| Analysis | n | Difference in meansa | 95% CI | p-value |
|---|---|---|---|---|
| Complete case | 431 | –1.83 | –3.92 to 0.27 | 0.087 |
| ‘Best’-case scenario | 480 | –2.22 | –4.41 to –0.03 | 0.047 |
| ‘Worst’-case scenario | 480 | –1.11 | –4.11 to 1.89 | 0.469 |
| MICE | 480 | –1.78 | –3.90 to 0.34 | 0.100 |
Sensitivity analysis to examine the timing of questionnaire completion
There was evidence of a very small difference in the mean number of days between randomisation and completion of the 12-week questionnaire: 90.25 days (SD 14.15 days) in the group allocated to usual care and mirtazapine and 89.00 days (SD 10.3 days) in the group allocated to usual care and placebo. Further adjusting for this variable in a model of BDI-II scores at 12 weeks (also adjusting for the baseline BDI-II score and minimisation and stratification variables) did little to change the adjusted difference between the two treatment groups (difference –1.88 points, 95% CI –3.98 to 0.21 points).
Secondary outcomes
Beck Depression Inventory-II as a continuous score at 24 weeks’ and 12 months’ follow-up
Table 8 summarises the means and differences in mean BDI-II sores at 24 weeks’ and 12 months’ follow-up. In both groups the mean BDI-II scores declined over time. At 24 weeks’ follow-up, those in the intervention group had a BDI-II score that was 0.85 points lower than those in the placebo group after adjustment for baseline BDI-II score and stratification and minimisation variables, with a 95% CI of –3.12 to 1.43 points. At 12 months’ follow-up, the difference between the two groups was 0.17 points (95% CI –2.13 to 2.46 points). In both cases the CIs overlapped zero, suggesting no evidence of a difference between the groups. Additional adjustment for the variables that were imbalanced at baseline increased the size of the differences somewhat but the 95% CIs continued to overlap zero (24 weeks: difference –1.26 points, 95% CI –3.57 to 1.05 points, p = 0.285; 12 months: difference –0.29 points, 95% CI –2.61 to 2.03 points, p = 0.807).
| Randomisation groups | 24 weeks’ follow-up (N = 402) | 12 months’ follow-up (N = 388) | ||
|---|---|---|---|---|
| n | Mean (SD), points | n | Mean (SD), points | |
| Usual care + mirtazapine | 196 | 17.3 (12.9) | 190 | 16.8 (12.7) |
| Usual care + placebo | 206 | 18.2 (12.6) | 198 | 16.7 (12.2) |
| Regression analyses | ||||
| Follow-up time point | n | Difference in meansa | 95% CI | p-value |
| 24 weeks | 402 | –0.85 | –3.12 to 1.43 | 0.465 |
| 12 months | 388 | 0.17 | –2.13 to 2.46 | 0.886 |
Our prespecified statistical analysis plan included a repeated-measures analysis incorporating outcomes at 12 and 24 weeks and 12 months post randomisation to examine whether any treatment effects were sustained, diminished or emerged later. This analysis was not conducted, given that differences between the treatment groups at these time points showed no evidence of an important difference.
‘Response’ to treatment at 12 and 24 weeks’ and 12 months’ follow-up
Table 9 summarises the percentages and ORs of ‘response’ to treatment (defined as a reduction in depressive symptoms on the BDI-II of ≥ 50% relative to baseline) at 12 and 24 weeks’ and 12 months’ follow-up. At each of the time points, the intervention group had an increased odds of response compared with the usual care group [OR 1.39, 95% CI 0.94 to 2.07; number needed to treat (NNT) = 12]. However, the CIs at each time point overlapped 1. Additional adjustment for the variables that were imbalanced at baseline did not make any meaningful difference to the observed difference at 12 weeks’, 24 weeks’ or 12 months’ follow-up (12 weeks: OR 1.45, 95% CI 0.97 to 2.16, p = 0.074; 24 weeks: OR 1.04, 95% CI 0.69 to 1.57, p = 0.842; 12 months: OR 1.05, 95% CI 0.69 to 1.58, p = 0.834).
| Randomisation group | Length of follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 12 weeks (N = 431) | 24 weeks (N = 402) | 12 months (N = 388) | |||||||
| Total in group, N | Number (%) reporting improvement of ≥ 50% in BDI-II score vs. baseline | Regression analysis: OR,a 95% CI; p-value | Total in group, N | Number (%) reporting improvement of ≥ 50% in BDI-II score vs. baseline | Regression analysis: OR,a 95% CI; p-value | Total in group, N | Number (%) reporting improvement of ≥ 50% in BDI-II score vs. baseline | Regression analysis: OR,a 95% CI; p-value | |
| Usual care + mirtazapine | 214 | 94 (43.9) | 1.39, 0.94 to 2.07; 0.099 | 196 | 96 (49.0) | 1.01, 0.67 to 1.50; 0.977 | 190 | 97 (51.1) | 0.99, 0.66 to 1.49; 0.978 |
| Usual care + placebo | 217 | 78 (35.9) | 206 | 100 (48.5) | 198 | 101 (51.0) | |||
‘Remission’ of depression symptoms at 12 and 24 weeks’ and 12 months’ follow-up
Table 10 summarises the percentages and ORs of ‘remission’ (defined as having a BDI-II score of < 10) at 12 and 24 weeks’ and 12 months’ follow-up. Those in the intervention group had an increased odds of ‘remission’ at 12 weeks compared with those in the placebo group and the CI overlapped 1 (OR 1.29, 95% CI 0.82 to 2.02; NNT = 18). Similar results were seen at 24 weeks and by 12 months the OR approached 1. The ORs tended to be slightly increased after adjustment for those variables that were imbalanced between treatment groups at baseline, but the CIs continued to overlap 1 (12 weeks: OR 1.39, 95% CI 0.88 to 2.21, p = 0.157; 24 weeks: OR 1.31, 95% CI 0.83 to 2.09, p = 0.249; 12 months: OR 1.02, 95% CI 0.65 to 1.60, p = 0.940).
| Randomisation group | Length of follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 12 weeks (N = 431) | 24 weeks (N = 402) | 12 months (N = 388) | |||||||
| Total in group, N | Number (%) reporting BDI-II score of < 10 points | Regression analysis: OR,a 95% CI; p-value | Total in group, N | Number (%) reporting BDI-II score of < 10 points | Regression analysis: OR,a 95% CI; p-value | Total in group, N | Number (%) reporting BDI-II score of < 10 points | Regression analysis: OR,a 95% CI; p-value | |
| Usual care + mirtazapine | 214 | 63 (29.4) | 1.29, 0.82 to 2.02; 0.266 | 196 | 65 (33.2) | 1.28, 0.81 to 2.01; 0.287 | 190 | 63 (33.2) | 0.96, 0.62 to 1.50; 0.873 |
| Usual care + placebo | 217 | 53 (24.4) | 206 | 59 (28.6) | 198 | 67 (33.8) | |||
Anxiety symptoms at 12 and 24 weeks’ and 12 months’ follow-up
Anxiety symptoms were assessed at baseline and 12 and 24 weeks’ and 12 months’ follow-up using the GAD-7 questionnaire and were analysed as a continuous score. Higher scores indicate more severe symptoms of anxiety, with a maximum possible score of 21. Table 11 summarises the mean and differences in mean GAD-7 scores at 12 and 24 weeks’ and 12 months’ follow-up. At 12 weeks, after correction for baseline GAD-7 score and minimisation and stratification variables, those in the intervention group had a mean GAD-7 score that was 0.98 points lower (less anxious) than those in the placebo group, with a 95% CI ranging from a 1.93-point decrease to a 0.03-point decrease. Differences between the two treatment groups were smaller at subsequent time points and CIs overlapped zero. Additional adjustment for the variables that were imbalanced at baseline did not alter the results greatly (12 weeks: difference –1.00 points, 95% CI –1.97 to –0.02 points, p = 0.045; 24 weeks: difference –0.71 points, 95% CI –1.73 to 0.31 points, p = 0.172; 12 months: difference –0.22 points, 95% CI –1.31 to 0.86 points, p = 0.685).
| Randomisation group | Length of follow-up | |||||
|---|---|---|---|---|---|---|
| 12 weeks (N = 431) | 24 weeks (N = 401) | 12 months (N = 387) | ||||
| n | Mean (SD), points | n | Mean (SD), points | n | Mean (SD), points | |
| Usual care + mirtazapine | 214 | 7.15 (5.63) | 195 | 6.83 (5.89) | 189 | 6.81 (6.23) |
| Usual care + placebo | 217 | 7.89 (5.78) | 206 | 7.17 (5.86) | 198 | 6.80 (5.73) |
| Regression analyses | ||||||
| n | 428 | 398 | 384 | |||
| Difference in means,a 95% CI (points); p-value | –0.98, –1.93 to –0.03; 0.044 | –0.56, –1.56 to 0.44; 0.274 | –0.17, –1.23 to 0.90; 0.755 | |||
Adherence to antidepressants at 12 weeks’ follow-up
Adherence to the study medication at 12 weeks’ follow-up was assessed using information on participant-reported breaks in treatment and the Morisky questionnaire. Table 12 summarises the percentages and ORs of adherence to the study medication at 12 weeks. Although adherence was > 70% in both groups, those in the intervention group had a 0.55-fold odds of adherence to the study medication at 12 weeks compared with those in the placebo group. Adjustment for variables that were imbalanced at baseline did not alter this result (OR 0.56, 95% CI 0.34 to 0.92; p = 0.021).
| Randomisation group | Total number of participants in group | Number of participants reported as being adherent | % of participants reported as being adherent | ORa | 95% CI | p-value |
|---|---|---|---|---|---|---|
| Usual care + mirtazapine | 210 | 156 | 74.3 | 0.55 | 0.34 to 0.89 | 0.015 |
| Usual care + placebo | 214 | 180 | 84.1 | |||
| Total | 424 | 336 | 79.3 |
Although adherence at 24 weeks and 12 months formed part of our planned secondary outcome analyses, as patients were offered the chance of unblinding at 12 weeks while continuing to be followed up, the results of these analyses became impossible to interpret and, therefore, are not presented here.
Quality of life at 12 and 24 weeks’ and 12 months’ follow-up
Quality of life was measured using the EQ-5D-5L questionnaire as well as the SF-12 questionnaire, from which aggregate scores for social and physical functioning were calculated.
EuroQol-5 Dimensions, five-level version
Table 13 summarises the mean and differences in mean EQ-5D-5L scores at 12 and 24 weeks’ and 12 months’ follow-up. At 12 weeks, after correction for baseline EQ-5D-5L score and minimisation and stratification variables, participants in the intervention group had a mean score that was 0.01 points higher (better QoL) than that in the placebo group, with a 95% CI ranging from a 0.02-point decrease to a 0.05-point increase. Differences between the two treatment groups remained very small at subsequent time points and CIs overlapped zero. Additional adjustment for the variables that were imbalanced at baseline did not alter the results greatly (12 weeks: difference 0.01 points, 95% CI –0.02 to 0.05 points, p = 0.389; 24 weeks: difference 0.01 points, 95% CI –0.02 to 0.05 points, p = 0.425; 12 months: difference 0.004 points, 95% CI –0.03 to 0.04 points, p = 0.831).
| Randomisation group | Length of follow-up | |||||
|---|---|---|---|---|---|---|
| 12 weeks (N = 429) | 24 weeks (N = 403) | 12 months (N = 388) | ||||
| n | Mean (SD), points | n | Mean (SD), points | n | Mean (SD), points | |
| Usual care + mirtazapine | 213 | 0.72 (0.27) | 196 | 0.72 (0.25) | 189 | 0.72 (0.28) |
| Usual care + placebo | 216 | 0.73 (0.25) | 207 | 0.74 (0.25) | 199 | 0.75 (0.25) |
| Regression analyses | ||||||
| n | 427 | 403 | 388 | |||
| Difference in means,a 95% CI (points); p-value | 0.01, –0.02 to 0.05; 0.400 | 0.01, –0.02 to 0.05; 0.464 | 0.001, –0.04 to 0.04; 0.950 | |||
Short Form questionnaire-12 items aggregate physical functioning score
Table 14 summarises the mean and differences in mean SF-12 aggregate physical functioning scores at 12 and 24 weeks’ and 12 months’ follow-up. At 12 weeks, after correction for baseline score and minimisation and stratification variables, participants in the intervention group had a mean score that was 1.09 points lower (worse physical functioning) than that in the placebo group, with a 95% CI ranging from a 2.75-point decrease to a 0.57-point increase. Differences between the two treatment groups remained very small and approached zero at subsequent time points and CIs overlapped zero. Additional adjustment for the variables that were imbalanced at baseline did not alter the results greatly (12 weeks: difference –0.99 points, 95% CI –2.69 to 0.70 points, p = 0.250; 24 weeks: difference –1.67 points, 95% CI –3.39 to 0.06 points, p = 0.058; 12 months: difference –0.27 points, 95% CI –2.02 to 1.48 points, p = 0.760).
| Randomisation group | Length of follow-up | |||||
|---|---|---|---|---|---|---|
| 12 weeks (N = 418) | 24 weeks (N = 403) | 12 months (N = 373) | ||||
| n | Mean (SD), points | n | Mean (SD), points | n | Mean (SD), points | |
| Usual care + mirtazapine | 208 | 44.09 (12.87) | 191 | 42.88 (13.02) | 182 | 43.34 (13.42) |
| Usual care + placebo | 210 | 45.85 (12.54) | 201 | 45.37 (12.75) | 191 | 44.32 (12.49) |
| Regression analyses | ||||||
| n | 410 | 384 | 366 | |||
| Difference in means,a 95% CI (points); p-value | –1.09, –2.75 to 0.57; 0.196 | –1.54, –3.23 to 0.15; 0.075 | –0.47, –2.19 to 1.24; 0.587 | |||
Short Form questionnaire-12 items aggregate mental functioning score
Table 15 summarises the mean and differences in mean SF-12 aggregate mental functioning scores at 12 and 24 weeks’ and 12 months’ follow-up. At 12 weeks, after correction for baseline score and minimisation and stratification variables, participants in the intervention group had a mean score that was 3.91 points higher (better mental functioning) than that in the placebo group, with a 95% CI ranging from a 1.63-point increase to a 6.20-point increase. Differences between the two treatment groups became smaller at subsequent time points and CIs overlapped zero. Additional adjustment for the variables that were imbalanced at baseline made the differences slightly larger (12 weeks: difference 4.08 points, 95% CI 1.74 to 6.41 points, p = 0.001; 24 weeks: difference 2.81 points, 95% CI 0.29 to 5.34 points, p = 0.029; 12 months: difference 1.92 points, 95% CI –0.72 to 4.56 points, p = 0.154).
| Randomisation group | Length of follow-up | |||||
|---|---|---|---|---|---|---|
| 12 weeks (N = 418) | 24 weeks (N = 392) | 12 months (N = 373) | ||||
| n | Mean (SD), points | n | Mean (SD), points | n | Mean (SD), points | |
| Usual care + mirtazapine | 208 | 39.94 (12.27) | 191 | 39.89 (13.92) | 182 | 40.54 (13.80) |
| Usual care + placebo | 210 | 36.33 (12.53) | 201 | 37.91 (12.43) | 191 | 39.25 (13.09) |
| Regression analyses | ||||||
| n | 410 | 384 | 366 | |||
| Difference in means,a 95% CI (points); p-value | 3.91, 1.63 to 6.20; 0.001 | 2.32, –0.17 to 4.80; 0.068 | 1.42, –1.20 to 4.04; 0.287 | |||
Adverse events related to the trial medication at 12 weeks’ and 12 months’ follow-up
The Antidepressant Side-Effect Checklist measure
The ASEC measure of antidepressant side effects was used to assess whether or not the participant-reported AEs related to their trial medication. The questionnaire provides a list of 21 symptoms and patients are asked to report on a scale the severity of these symptoms. The ASEC score relates to the sum of the severity scores across all items and is treated as a continuous measure. The maximum score is 63 points, indicating severe side-effects.
Table 16 summarises the mean and differences in mean ASEC sores at 12 weeks’ and 12 months’ follow-up. Participants in the intervention group had a mean ASEC score at 12 weeks that was 0.35 points higher than that in the placebo group, after adjustment for baseline ASEC score as well as stratification and minimisation variables. At 12 months, the mean ASEC score in the intervention group was 0.43 points lower than that in the placebo group. For both groups, the 95% CIs overlapped zero. Additional adjustment for the variables that were imbalanced at baseline did not make a meaningful difference to the observed results (12 weeks: difference 0.26 points, 95% CI –1.15 to 1.68 points, p = 0.718; 12 months: difference –0.63 points, 95% CI –2.42 to 1.16 points, p = 0.489).
| Randomisation group | Length of follow-up | |||
|---|---|---|---|---|
| 12 weeks (N = 390) | 12 months (N = 255) | |||
| n | Mean (SD), points | n | Mean (SD), points | |
| Usual care + mirtazapine | 184 | 10.13 (7.02) | 119 | 9.50 (7.65) |
| Usual care + placebo | 206 | 9.77 (7.93) | 136 | 9.59 (8.26) |
| Regression analyses | ||||
| n | 385 | 252 | ||
| Difference in means,a 95% CI (points); p-value | 0.35, –1.04 to 1.73; 0.624 | –0.43, –2.19 to 1.33; 0.630 | ||
Adverse events and serious adverse events by 12 weeks post randomisation
By 12 weeks post randomisation, 167 patients allocated to the mirtazapine and usual care arm reported 168 AEs. The most common AEs were drowsiness (n = 41 events, reported by 41 participants) and weight increase (n = 24 events, reported by 24 participants). Fifty-three of the AEs were associated with stopping the trial medication (n = 46 participants); most of these AEs were drowsiness (n = 19). Most of the AEs were classified as mild (n = 136); only three were considered severe. Nearly half (n = 77) were considered to be either ‘probably’ or ‘definitely’ related to the trial medication and only 44 were unexpected.
This contrasts with the group allocated to placebo and usual care, in which only 91 AEs from 91 participants were reported. In this group, 11 AEs were associated with stopping the trial medication (n = 9 participants). Only five participants reported drowsiness and eight reported an increase in appetite or weight. The most common AEs were classified as ‘unpleasant dreams’ (n = 9) and musculoskeletal (n = 17). Nearly all were mild (n = 74) and none was severe.
Very few of the AEs reported by 12 weeks post randomisation were serious. In the group allocated to mirtazapine and usual care, eight participants reported eight SAEs. These were classified as follows: central nervous system/transient ischaemic attack (TIA) (n = 1), cardiovascular (n = 1), dental (n = 1), gynaecological (n = 1), pancreatitis (n = 1), psychiatric (n = 2) and respiratory (n = 1). The two psychiatric events and the pancreatitis event resulted in the participants stopping treatment.
In the group allocated to placebo and usual treatment, only three SAEs were reported by three participants [infection (n = 1) and musculoskeletal trauma (n = 2)]. In two cases, the participant stopped the trial medication. In none of the cases was the SAE thought to be related to the trial medication.
Adverse events and SAEs in the first 12 weeks and AEs between 12 and 52 weeks are described in more detail in Appendix 2, Tables 39–42.
Depression symptoms assessed using the Patient Health Questionnaire-9 items at 12 weeks’ follow-up
The primary outcome and many of the secondary outcomes were derived from the BDI-II questionnaire and, to explore the consistency of these results, responses to the PHQ-9 at 12 weeks’ follow-up were reviewed in a post hoc analysis. The PHQ-9 scores nine symptoms of depression yielding a maximum score of 27 points; higher scores indicate more severe symptoms of depression.
Table 17 summarises the mean and difference in mean PHQ-9 sores at 12 weeks. Participants in the intervention group had a mean PHQ-9 score at 12 weeks that was 1.05 points lower than that in the placebo group after adjustment for baseline PHQ-9 score and stratification and minimisation variables, with a 95% CI of –2.14 to 0.04 points. The direction of association was the same as that observed for the BDI-II score outcomes. Additional adjustment for the variables that were imbalanced at baseline made the difference slightly larger (difference –1.24 points, 95% CI –2.34 to –0.13 points; p = 0.029).
| Randomisation group | n | Mean (SD) | Difference in meansa | 95% CI | p-value |
|---|---|---|---|---|---|
| Usual care + mirtazapine | 212 | 9.74 (6.35) | –1.05 | –2.14 to 0.04 | 0.058 |
| Usual care + placebo | 217 | 10.63 (6.21) | |||
| Total | 429 | 10.19 (6.29) |
Treatment efficacy
To understand treatment efficacy, we conducted a CACE analysis of the primary outcome at 12 weeks. The results from this analysis are presented alongside the primary ITT analysis and a per-protocol analysis in Table 18. The CACE and per-protocol analyses showed slightly larger effects than the ITT analysis, but the CIs were wider.
| Analysis | Number of patients in model | Difference in meansa (points) | 95% CI (points) | p-value |
|---|---|---|---|---|
| ITT | 431 | –1.83 | –3.92 to 0.27 | 0.087 |
| Per-protocol | 327 | –2.18 | –4.60 to 0.24 | 0.077 |
| CACE | 427 | –2.39 | –5.18 to 0.40 | 0.093 |
‘As-treated’ analysis
After measurement of the primary outcome at 12 weeks’ follow-up, participants were given the option of continuing in the study blinded, being unblinded and continuing with the study follow-up or being unblinded and leaving the study. Those unblinded and continuing with the study follow-up no longer received the trial medication, but were free to obtain a prescription for mirtazapine from their GP. Because of this, each of the randomly allocated groups in the ITT analysis at 24 weeks and 52 weeks post randomisation included participants taking and not taking mirtazapine. We therefore conducted an ‘as-treated’ analysis to identify treatment differences between those taking mirtazapine and those not taking mirtazapine at 24 weeks.
This as-treated analysis compared the BDI-II scores at 24 weeks of participants receiving and adhering to mirtazapine (regardless of allocation at randomisation) with those of participants not receiving (or not adhering to) mirtazapine; all participants were adherent to their usual antidepressant treatment. These restrictions resulted in only 324 participants being considered (compared with 403 in the ITT analysis who completed a questionnaire at 24 weeks), of whom 199 (61.4%) were not receiving mirtazapine and 125 (38.6%) were receiving mirtazapine. The 12-week BDI-II scores for these groups suggest that, in those meeting our eligibility criteria, those still adherent to mirtazapine at 24 weeks had lower BDI-II scores at 12 weeks (mean 16.4 points, SD 11.6 points) than those not taking mirtazapine at 24 weeks (mean 18.6 points, SD 12.4 points).
To further understand who was taking mirtazapine at 24 weeks, we explored which groups the participants had been originally randomised to. Of the 125 participants receiving mirtazapine at 24 weeks, 117 had been randomly allocated to the mirtazapine arm of the study and eight opted to get mirtazapine from their GP. Those taking mirtazapine at 24 weeks were, therefore, composed largely of those randomised to mirtazapine and their mean 12-week BDI-II score was 15.7 points (SD 11.2 points). However, a small group of eight participants had been randomised to placebo and these participants’ BDI-II score at 12 weeks was much higher [mean 26.3 points (SD 14.5 points)]. Of the 199 participants not receiving mirtazapine at 24 weeks, 63 had been randomly allocated to the mirtazapine arm and had since stopped taking it in an adherent manner. Those never taking mirtazapine had a slightly lower BDI-II score at 12 weeks [mean 17.7 points (SD 12.0 points)] than those who were randomised to mirtazapine but who stopped [mean 20.5 points (SD 13.1 points)].
The mean 24-week BDI-II score was 14.5 points (SD 11.5 points) in the group taking mirtazapine and 18.4 points (SD 12.8 points) in the group not taking mirtazapine. After adjusting for baseline differences, those taking mirtazapine had a mean BDI-II score that was 3.9 points lower than that in those not receiving mirtazapine (95% CI –6.45 to –1.37 points).
Analysis of blinded patients at 24 weeks’ and 12 months’ follow-up
To understand outcomes in those participants who chose to remain blinded after the primary outcome, we conducted analyses of BDI-II scores at 24 weeks’ and 12 months’ follow-up in the subset of participants who chose to remain blinded at those time points, regardless of adherence to the study medication.
A 24-week questionnaire was returned by 403 participants, of whom 294 remained blinded to their treatment allocation (152 were allocated to receive mirtazapine and usual care; the remaining 142 were allocated to receive placebo and usual care). In this group of 294 participants, the mean BDI-II score at 24 weeks was slightly higher in the mirtazapine group (difference 0.26 points, 95% CI –2.41 to 2.94 points) after adjustment for baseline BDI-II score and minimisation and stratification variables. The CI overlapped zero, suggesting no evidence of a difference between the groups.
At 12 months’ follow-up, 390 participants returned their final follow-up questionnaire and only 231 remained blinded (110 were allocated to receive mirtazapine and usual care; the remaining 121 were allocated to receive placebo and usual care). Here, again, the mean BDI-II score was slightly higher at 12 months in the mirtazapine group (difference 1.48 points, 95% CI –1.57 to 4.53 points) after adjustment for baseline BDI-II scores and minimisation and stratification variables, although the CI suggests no evidence of a difference between the groups.
Chapter 4 Health economic analysis: cost-effectiveness of mirtazapine added to usual care compared with placebo added to usual care
Introduction
Aim
The aim of the economic evaluation was to assess the cost-effectiveness of mirtazapine plus usual care compared with placebo plus usual care in patients with TRD. The economic evaluation was carried out using data collected as part of the MIR RCT.
The intervention
Mirtazapine is a well-established inexpensive treatment for depression, at £0.04 per tablet (either 15-mg or 30-mg strength),48 with a typical dose being one tablet per day. If it were to show a meaningful improvement in QoL [here measured in quality-adjusted life-years (QALYs)], there is, ex ante, a high probability that it will be cost-effective, all other things being equal.
Methods
Form of analysis
The economic evaluation was a within-trial analysis, based on the data collected during the 12-month trial period. All costs that were incurred in the 12 months following randomisation were included. The perspective for the primary analysis was that of the NHS and personal social services (PSS). Personal costs to patients and productivity costs for patients and carers who missed work were included in a secondary analysis from the societal perspective.
The primary analysis was a cost–utility analysis of the cost per QALY when comparing the two groups. Results are also presented for a cost-effectiveness analysis for the primary trial outcome of BDI-II score at 12 weeks post randomisation.
Outcomes
The primary trial outcomes were:
-
change in depression symptoms measured as a continuous variable using the BDI-II score at 12 weeks compared with the baseline score
-
response in depression symptoms, a binary variable defined as a reduction of ≥ 50%.
Secondary trial outcomes of interest for the economic evaluation include:
-
QoL measured using the EQ-5D-5L
-
primary and secondary care resource use.
For the economic evaluation, the primary outcome of interest was the difference in QALYs over 12 months post randomisation compared with baseline. EQ-5D-5L data were captured at four time points (pre randomisation and 12, 24 and 52 weeks post randomisation) and an area under the curve approach was used to estimate total QALYs at 12 months, adjusting for baseline values. The EuroQol-5 Dimensions (EQ-5D) is recommended by NICE for use in cost–utility analyses. 49 Utility scores were derived using the most recent valuation tariff for the five-level version of the EQ-5D. 50 This is based on a representative sample of the UK population.
Resource-use categories
The analysis was based on the costs associated with caring for all patients from randomisation until 12 months. The following resource use data were included in the analysis.
Direct costs to the health service (NHS)
-
The cost of the treatment (mirtazapine).
-
Hospital (secondary) care related to depression or mental health including:
-
inpatient admissions
-
accident and emergency (A&E) attendances
-
outpatient appointments.
-
-
Primary and community costs including:
-
GP or nurse appointments at the surgery, by telephone or at home
-
counselling or other talking therapies (excluding CBT)
-
face-to-face or computerised CBT
-
mental health clinic attendances
-
prescribed exercise programmes
-
NHS Direct or 111
-
NHS walk-in centres.
-
Direct costs of personal social services
-
Mental health nurse home visits.
-
Occupational therapy.
-
Social worker.
-
Day centre use.
-
Self-help groups run by social services.
-
Home care worker visits.
-
Other.
Costs to patients and carers
-
Inpatient and outpatient private health care.
-
Private counselling.
-
Prescription charges.
-
Over-the-counter medicines.
-
Complementary therapies.
-
Private home care.
-
Loss of earnings from time off work.
-
Other costs.
Productivity costs
-
Time off work for patients and carers.
Data collection
Resource use data collection
Resource use data were collected primarily by patient self-report using a bespoke data collection form. The form was paper based and was incorporated into the assessment forms. Forms were mainly collected at the relevant face-to-face appointments along with other outcome measures. The form was used to collect information on public and privately paid-for health and social care, unpaid help received and employment-related data. Participants were asked about any disability payments received, but not the specific type of payment or the value of any payments.
Publicly funded health and social care resources
Data were collected in relation to patient use of NHS secondary care (inpatient, outpatient and casualty) and primary care, paid-for care, home care and informal care. Resource use data were collected at 12 and 24 weeks’ and 12 months’ follow-up. The questionnaires at 12 and 24 weeks asked participants to report all resource use in the previous 3 months, whereas the 12-month questionnaire asked participants to report resource use in the previous 6 months. Out of 480 participants, 450 (94%) completed at least one resource use data questionnaire. Complete cost data were available from 369 participants (77%) and complete QALY data from 368 participants (77%).
Secondary care resource use was included in the analysis if it related to depression or other mental illness. By contrast, all primary and community care resources were included – it was not possible from the data to make the distinction between depression-related service use and other service use. However, when completing the primary and community care component of the resource use questionnaire, participants were asked to identify only those activities that were related to their mental health.
General practice system data
Data on participant contacts with GPs, as well as prescription data, were collected from general practice electronic patient records, with participant consent. This was used as the basis for estimating GP and prescription resource use. Participants were also asked as part of the patient-reported resource use questionnaire how many GP visits they had made. To avoid probable double counting, only the GP visits recorded as part of the electronic health record were used in the analysis. Eight participants withheld their consent for data to be collected in this way. Four GP practices were unable to provide full participant data. One provided no data (n = 6 participants), one provided data on prescriptions, but not consultations (n = 5 participants) and two provided data on consultations but not prescriptions (n = 34 participants). Overall, data on GP contacts and/or prescription data were available for 436 participants from general practice record systems. Of these, 395 participants had a contact with the general practice surgery or a prescription for a depression-related medicine during the 12 months of study enrolment.
Private health and social care data
Participants were asked to report if they had used any privately funded health or social care services. This included both hospital-based care (inpatient, outpatient) and community-based care (counselling, complementary therapies, over-the-counter medications). Participants were also asked to report any prescription charges that they had paid, but not whether or not they were exempt from prescription charges or whether or not they had a pre-payment certificate.
Employment and lost productivity
To estimate the cost to the patient and society of the treatments, data were collected on employment and income, as well as whether or not participants had missed work or lost income as a result of their mental health. Participants were asked to report their usual hours worked per week, how many days they had had off owing to their mental health and the income that they had lost as a result. They were also asked to report whether or not anyone else had taken time off work to care for them as a result of their mental health and the lost work time.
Unit costs
Non-prescription resources
Unit costs have been taken when possible from NHS reference costs51 and the Personal Social Services Research Unit (PSSRU) annual publications of unit costs of health and social care. 52–56 All unit cost sources are indicated in Table 19. Primary analysis study costs have been estimated from a payer perspective (NHS and PSS). As described above, additional data were collected on patient costs, unpaid help received and lost productivity; these are included in a secondary analysis.
| Resource use | Unit cost (£) | Source |
|---|---|---|
| NHS secondary care | ||
| General inpatient ward | 303.94 | CoBalT57 |
| Low-level secure services (per bed-day) | 405.67 | PSSRU 201556 |
| A&E Mental Health Liaison Services, Adult and Elderly | 187.86 | 2015–16 reference costs51 |
| Psychotherapy, consultant led | 158.06 | 2015–16 reference costs51 |
| Community psychiatric nursing | 74.16 | CoBalT57 |
| Mental well-being clinic | 61.10 | CoBalT57 |
| Liaison psychiatry | 105.08 | 2015–16 reference costs51 |
| General practice systems data | ||
| GP consultation (in surgery) | 33.00 | PSSRU 201655 |
| GP consultation (telephone) | 26.11 | PSSRU 201655 |
| Prescriptions | – | As per drug costs in 2016 PCA data48 |
| Other GP or community services | ||
| Counselling | 52.22 | PSSRU 201453 |
| CBT (face to face) | 97.13 | PSSRU 201453 |
| CBT (computer based) | 23.37 | PSSRU 201354 |
| Mental health clinic | 61.10 | CoBalT57 |
| Exercise or physical activity scheme | 235.00 | NICE costing statement58 |
| NHS Direct or 111 | 9.13 | Evaluation report of NHS 111 pilot sites59 |
| NHS walk-in-centre | 46.79 | Evaluation report of NHS 111 pilot sites59 |
| Home visits | ||
| Mental health nurse (advanced nurse) | 60.59 | PSSRU 201556 |
| Occupational therapist | 44.00 | PSSRU 201655 |
| Social worker | 57.00 | PSSRU 201655 |
| GP | 141.50 | PSSRU 2016;55 PSSRU 201060 |
| Additional help | ||
| Home care worker | 22.35 | CoBalT57 |
| Day centre or social club | 22.35 | CoBalT57 |
| Self-help group | 15.67 | CoBalT57 |
| Informal unpaid help | ||
| Childcare | 7.20 | 2016 National Minimum Wage, 25 +63 |
| Help in the house (e.g. cooking), band 3 HCA | 25.00 | PSSRU 201655 |
| Help outside the house (e.g. shopping), band 3 HCA | 25.00 | PSSRU 201655 |
| Other help, band 3 HCA | 25.00 | PSSRU 201655 |
Some activities are not associated with a specific unit cost and it has been necessary to categorise these in a way that allows a unit cost to be assigned. For example, data extracted from general practice electronic systems provide detailed information on activity for which there is no published unit cost, such as e-mail consultations or telephone conversations with colleagues about a patient. Unit costs were assigned based on an assessment of the nearest equivalent activity in terms of time spent for which a unit cost does exist. For example, a telephone call with a colleague has no published unit cost, whereas a telephone call with a patient does. The activity is similar and so the same unit cost was applied. Activity has been costed based on whether it is most like a face-to-face appointment with a GP in the surgery or a GP telephone consultation.
Prescribed medication
Data from general practice electronic systems were used to estimate the cost of prescriptions for participants in the study, excluding the cost of study medication. However, the costs of mirtazapine were included for participants who requested unblinding and switched treatments; these are recorded in the general practice electronic systems. Only medications used for depression or other mental illnesses were included. This was defined according to whether or not the medication was listed in the BNF19 in one of the following categories:
-
antidepressants
-
antipsychotics (psychoses and related disorders)
-
hypnotics and anxiolytics.
Medication unit costs were obtained from the prescription cost analysis data published by the Department of Health and Social Care and based on August 2016 prices. 48 A detailed list of the medicines prescribed and their unit costs is provided in Appendix 3, Table 43.
The intervention
Participants were prescribed mirtazapine in line with the protocol. The cost of each prescription was based on the per-tablet cost (see Table 19). Participants could have up to eight prescriptions, with a maximum intervention cost of £28.56. Participants could stop taking trial medication for a number of reasons, discussed elsewhere in this report.
Participant direct and indirect costs
Information on personal expenditure by participants was obtained from the patient questionnaires, including lost earnings, private health and social care payments and prescription charges paid. A full list of categories and the cost to participants is presented in Table 20. No participants reported use of private inpatient care, which is not listed in Table 20. Participant costs are not included in the baseline analysis but are included in the secondary societal perspective analysis.
| Resource | Group | Difference (95% CI),a £ | |||
|---|---|---|---|---|---|
| Control | Intervention | ||||
| Participants, n | Mean cost per participant (SE), £ | Participants, n | Mean cost per participant (SE), £ | ||
| GP-extracted resource use | |||||
| GP consultation (surgery) | 61 | 135 (10) | 69 | 166 (16) | –32 (–69 to 6) |
| GP consultation (telephone) | 9 | 55 (15) | 10 | 44 (10) | 11 (–27 to 49) |
| Prescriptionsb | 163 | 44 (12) | 135 | 34 (9) | 9 (–21 to 40) |
| Other GP or community services | |||||
| Counselling | 23 | 323 (290) | 21 | 414 (537) | –90 (–350 to 169) |
| CBT (face to face) | 13 | 806 (109) | 10 | 524 (195) | 282 (–157 to 721) |
| CBT (computer based) | 2 | 46.74 (23.37) | 1 | 23.37 (–) | – (–) |
| Mental health clinic | 5 | 61.10 (0.00) | 4 | 183 (66) | – (–) |
| Exercise or physical activity scheme | 3 | 235 (0) | 1 | 235 (–) | – (–) |
| NHS Direct or NHS 111 | 4 | 9 (0) | 1 | 9 (–) | – (–) |
| NHS walk-in centre | 2 | 47 (0) | 4 | 58.75 (11.75) | – (–) |
| Any GP or community service used | 35 | 530 (89) | 31 | 454 (102) | 76 (–193 to 345) |
| Home visits | |||||
| Mental health nurse (advanced nurse) | 3 | 61 (0) | 6 | 183 (93) | – (–) |
| Occupational therapist | 0 | – | 1 | 528 (–) | – (–) |
| Social worker | 1 | 171 (–) | 1 | 684 (–) | – (–) |
| GP | 0 | – | 1 | 424.5 (–) | – (–) |
| Any home visits | 3 | 118 (57) | 4 | 546 (352) | – (–) |
| Additional help | |||||
| Home care worker | 2 | 363 (307) | 1 | 11,622 (–) | – (–) |
| Day centre or social club | 0 | – | 0 | – | – (–) |
| Self-help group | 1 | 282 (–) | 1 | 47 (–) | – (–) |
| Any additional help | 3 | 336 (179) | 2 | 5834 (5787) | – (–) |
| Informal unpaid help | |||||
| Childcare | 5 | 46.08 (19) | 4 | 36.9 (24) | – (–) |
| Help in the house (e.g. cooking) | 30 | 262 (49) | 19 | 588 (277) | –325 (–784 to 132) |
| Help outside the house (e.g. shopping) | 21 | 219 (66) | 15 | 425 (272) | –207 (–699 to 285) |
| Other help | 14 | 335 (73) | 9 | 158 (65) | 176 (–110 to 463) |
| Any informal unpaid help | 41 | 424 (93) | 30 | 638 (301) | –214 (–770 to 343) |
| Out-of-pocket costs | |||||
| Inpatient | 0 | – | 0 | – | – (–) |
| Outpatient | 1 | 5000 (–) | 0 | – | – (–) |
| Counselling | 6 | 590 (311) | 8 | 279 (114) | 311 (–337 to 959) |
| Home care | 0 | – | 1 | 30 (–) | – (–) |
| Prescription charges | 45 | 45 (6) | 42 | 44 (7) | 0.58 (–16 to 17) |
| Over-the-counter treatments | 19 | 22 (8) | 12 | 27 (9) | –5 (–30 to 20) |
| Complementary therapy | 10 | 97 (38) | 6 | 177 (66) | –80 (–230 to 70) |
| Lost income | 15 | 1392 (799) | 15 | 8748 (3799) | –7357 (–15,309 to 595) |
| Other | 6 | 287 (127) | 6 | 458 (334) | –172 (–967 to 624) |
| Any out-of-pocket costs | 48 | 252 (109) | 43 | 2680 (1422) | –2428 (–5110 to 254) |
| Productivity costs | |||||
| Patient | 35 | 3550 (1012) | 32 | 5082 (1084) | –1532 (–4491 to 1427) |
| Carer | 5 | 255 (115) | 6 | 332 (158) | –78 (–538 to 383) |
Productivity costs
Following CoBalT,57 the human capital approach was used to value time absent from work. Median hourly earnings were obtained from the Office for National Statistics (ONS) 2016 Annual Survey of Hours and Earnings. 61 A mean value across all ages and by sex was applied. Productivity costs are not included in the baseline analysis but are included in the secondary societal perspective analysis.
Unpaid help
Participants were asked to report whether or not they received any help that was not paid for either by themselves or by a third party. Such help included help with childcare, help around the house (e.g. with cooking or cleaning) and help outside the house (e.g. with shopping). The value of such help is hard to define and there is considerable debate in the literature about the most appropriate methods for valuing unpaid help. For household help, we followed the approach of the ONS and used a proxy-good method based on the wage rate of an equivalent paid helper. 62 The ONS use the wage of a care assistant to value non-continuous practical care, which we followed. 62 The hourly cost of a care assistant was obtained from the PSSRU. 55 For childcare, we used the national minimum wage for those aged ≥ 25 years. 63
Analysis
Resource use by participants in each trial arm is reported as frequencies, with means and medians as appropriate. SDs and IQRs are also reported as appropriate. Resource use data and unit cost data were combined to estimate the cost per participant for each cost category.
Missing data
When data were missing, they were assumed to be missing at random and multiple imputation (MI) was used. MI was conducted in Stata 14. The Stata MI commands were used to generate 50 imputed data sets using chained equations, which were combined for analysis using Rubin’s rule as specified as part of the -mi estimate- command. The imputation model for the analysis included the stratification and minimisation factors used for randomisation (site, baseline BDI-II score, whether or not a participant was undergoing talking therapy), age, sex, initial treatment allocation, continuous baseline EQ-5D-5L and BDI-II scores and a range of cost and quality of life variables.
Missing data were imputed for:
-
EQ-5D-5L index scores at baseline and 12, 24 and 52 weeks
-
mean patient-reported resource use costs at 12, 24 and 52 weeks
-
mean costs of GP consultations and prescriptions.
Uncertainty arising from patient variation
Uncertainty arising from individual patient variability is reported as standard errors (SEs) for the point estimates of the mean costs and QALYs. Uncertainty in the incremental cost-effectiveness ratio reported for complete-case analyses is reported as 95% CIs, based on 1000 bootstrap replications. We also report incremental net monetary benefit (NMB) with CIs for a range of willingness-to-pay (WTP) values and cost-effectiveness acceptability curves (CEACs).
Discounting
Costs and outcomes were not discounted as the study and analysis was limited to a 12-month period. All costs were estimated in 2016 prices and have been inflated as necessary using the PSSRU Pay and Prices inflation index. 55
Results
Resource use and costs
General practitioner-collected data
Results from the data collected directly from general practice electronic health records on the number of GP consultations and prescriptions are presented in Table 20. There was no significant difference in the cost of GP appointments in the surgery or GP telephone consultations between treatment groups. There was also no significant difference in the cost of prescribed medication per participant between groups. On an ITT basis, the mean cost of active treatment was £12.34 (SE £0.081).
Participant-reported resources
In addition, participants were asked to provide self-reported details of resource use. The results for all self-reported resource use categories are also presented in Table 20. Complete-case results are presented for the numbers of participants using each resource and the mean cost per participant. No difference in mean cost between the groups was observed in any resource use category.
Cost–utility
Cost-effectiveness analyses are reported, relating the costs of each strategy to change in:
-
QALYs at 12 weeks and 12 months post randomisation
-
BDI-II scores at 12 weeks and 12 months post randomisation.
The primary outcome of interest for the economic evaluation was the incremental cost per QALY at 12 months’ follow-up. In keeping with the primary trial outcome, the primary analysis for the economic evaluation was based on complete cases only. A secondary analysis based on the imputed data is also reported.
Primary analyses
12-week cost–utility
No clinically important differences in QALYs or costs were observed between the two groups at 12 weeks’ follow-up (Table 21). At the primary outcome end point of 12 weeks, participants in the active treatment arm reported QALYs of 0.163 and participants in the control arm reported QALYs of 0.162, a difference of 0.002 QALYs (95% CI –0.002 to 0.005 QALYs). This difference is not meaningful. The active treatment arm had higher costs (£65) than the control arm (£63). The difference in costs was small (£2, 95% CI –£27 to £31) and is also not meaningful.
| Primary analyses | Group | |||||
|---|---|---|---|---|---|---|
| Intervention | Control | Difference | ||||
| Costs (SE), £ | QALYs (SE) | Costs (SE), £ | QALYs (SE) | Costs (SE), £ | QALYs (SE) | |
| QALYsa | ||||||
| 12 weeks | 67 (11) | 0.163 (0.001) | 63 (10) | 0.162 (0.001) | 4 (15) | 0.002 (0.002) |
| 52 weeks | 261 (52) | 0.734 (0.009) | 192 (49) | 0.724 (0.009) | 69 (71) | 0.009 (0.013) |
| BDI-II scoreb | ||||||
| Costs (SE), £ | BDI-II score (points) (SE) | Costs (SE), £ | BDI-II score (points) (SE) | Costs (SE), £ | BDI-II score (points) (SE) | |
| 12 weeks | 67 (11) | 17.804 (0.748) | 63 (10) | 19.822 (0.743) | 4 (15) | –2.128 (1.055) |
| 52 weeks | 261 (52) | 16.7 0.824 | 192 (49) | 16.8 0.805 | 69 (71) | –0.166 (1.149) |
Assuming a WTP threshold of £20,000 per QALY, the expected NMB of the active treatment over placebo is £144, but with 95% CIs ranging from –£1413 to £1701 (Table 22).
| Analysis | WTP threshold per QALY (95% CI),a £ | |||
|---|---|---|---|---|
| £10,000 | £20,000 | £30,000 | £50,000 | |
| Complete-case analysis | ||||
| NHS and PSS perspective | ||||
| iNMB at 52 weeks | 25 (–267 to 137) | 118 (–419 to 655) | 211 (–582 to 1004) | 398 (–914 to 1709) |
| iNMB at 12 weeks | 72 (–706 to 851) | 144 (–1413 to 1701) | 216 (–2119 to 2552) | 360 (–3532 to 4253) |
| Societal perspective | ||||
| iNMB at 52 weeks | 51,926 (–160,178 to 264,030) | 103,852 (–320,356 to 528,060) | 155,778 (–480,534 to 792,090) | 259,630 (–800,890 to 1,320,149) |
| iNMB at 12 weeks | 2419 (–5475 to 10,313) | 4839 (–10,950 to 20,627) | 7258 (–16,425 to 30,940) | 12,096 (–27,374 to 51,567) |
| Imputed data analysis | ||||
| NHS and PSS perspective | ||||
| iNMB at 52 weeks | 15 (–32 to 62) | 34 (–48 to 116) | 54 (–66 to 174) | 92 (–106 to 290) |
| iNMB at 12 weeks | 25 (–270 to 321) | 118 (–417 to 654) | 212 (–575 to 999) | 398 (–898 to 1695) |
| Societal perspective | ||||
| iNMB at 52 weeks | –123 (–397 to 152) | –103 (–386 to 180) | –84 (–381 to 213) | –45 (–382 to 292) |
| iNMB at 12 weeks | –676 (–1443 to 90) | –583 (–1478 to 312) | –490 (–1560 to 581) | –303 (–1798 to 1192) |
The CEAC (see Figure 5) illustrates the proportion of times that each intervention is cost-effective based on the bootstrapped samples across a range of WTP estimates. Figure 5 illustrates that, based on the bootstrapped simulations, at 12 weeks mirtazapine was more likely to have the highest NMB. At a WTP threshold of £20,000 per QALY, mirtazapine was the more cost-effective option in 77% of the simulations, rising to 79% at a WTP threshold of £30,000 per QALY.
FIGURE 5.
Cost-effectiveness acceptability curve: probability of highest NMB at 12 weeks across a range of WTP estimates per QALY.
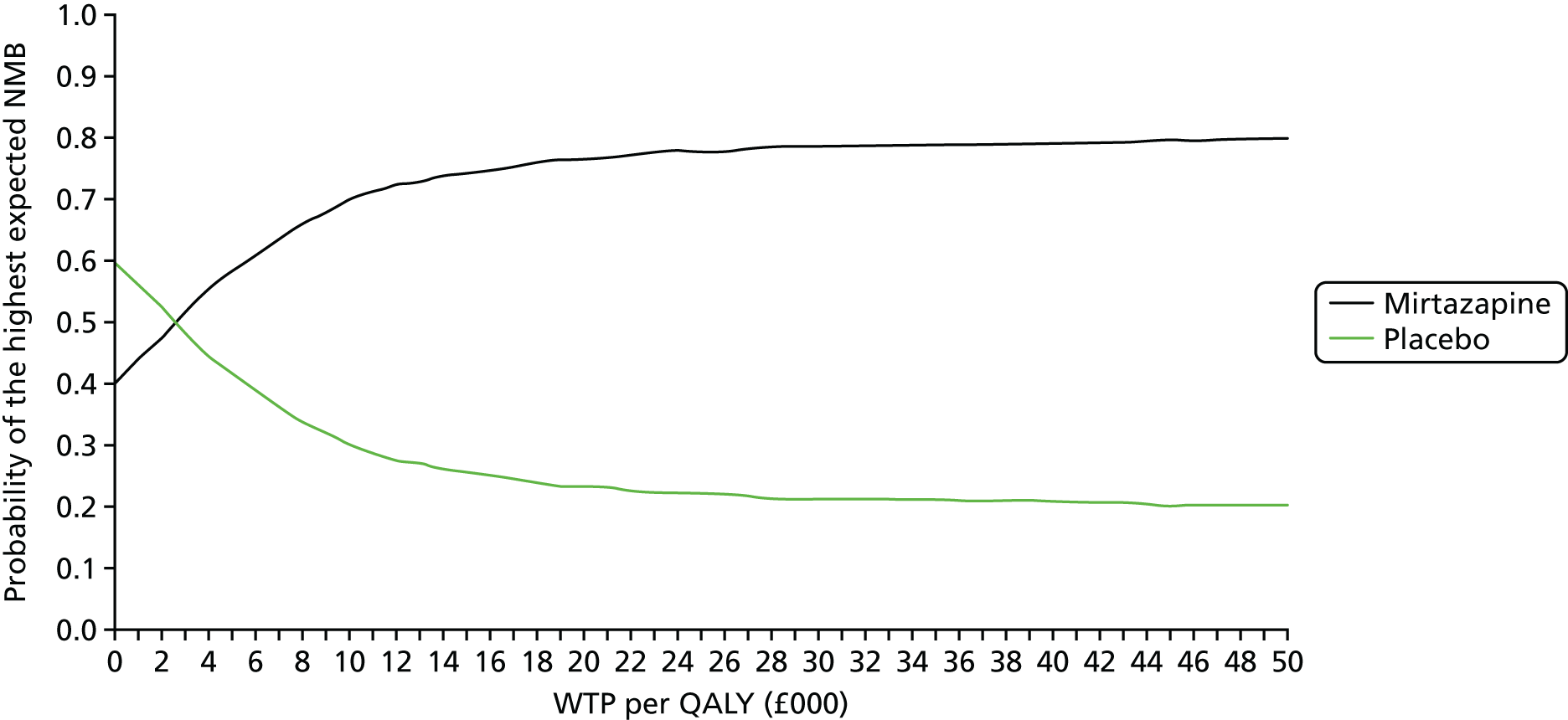
A CEAC based on the WTP for an additional 1-point improvement in BDI-II score at 12 weeks is presented in Figure 6. From this, we can see that mirtazapine is likely to have a higher expected NMB depending on the WTP for a 1-point change in BDI-II score. The shape of this curve is driven by the fact that mirtazapine provides a small improvement in BDI-II score relative to placebo. In most of the bootstrap simulations, mirtazapine is both more costly and more effective.
FIGURE 6.
Cost-effectiveness acceptability curve: probability of highest NMB at 12 weeks across a range of WTP estimates for a unit change in BDI-II score.
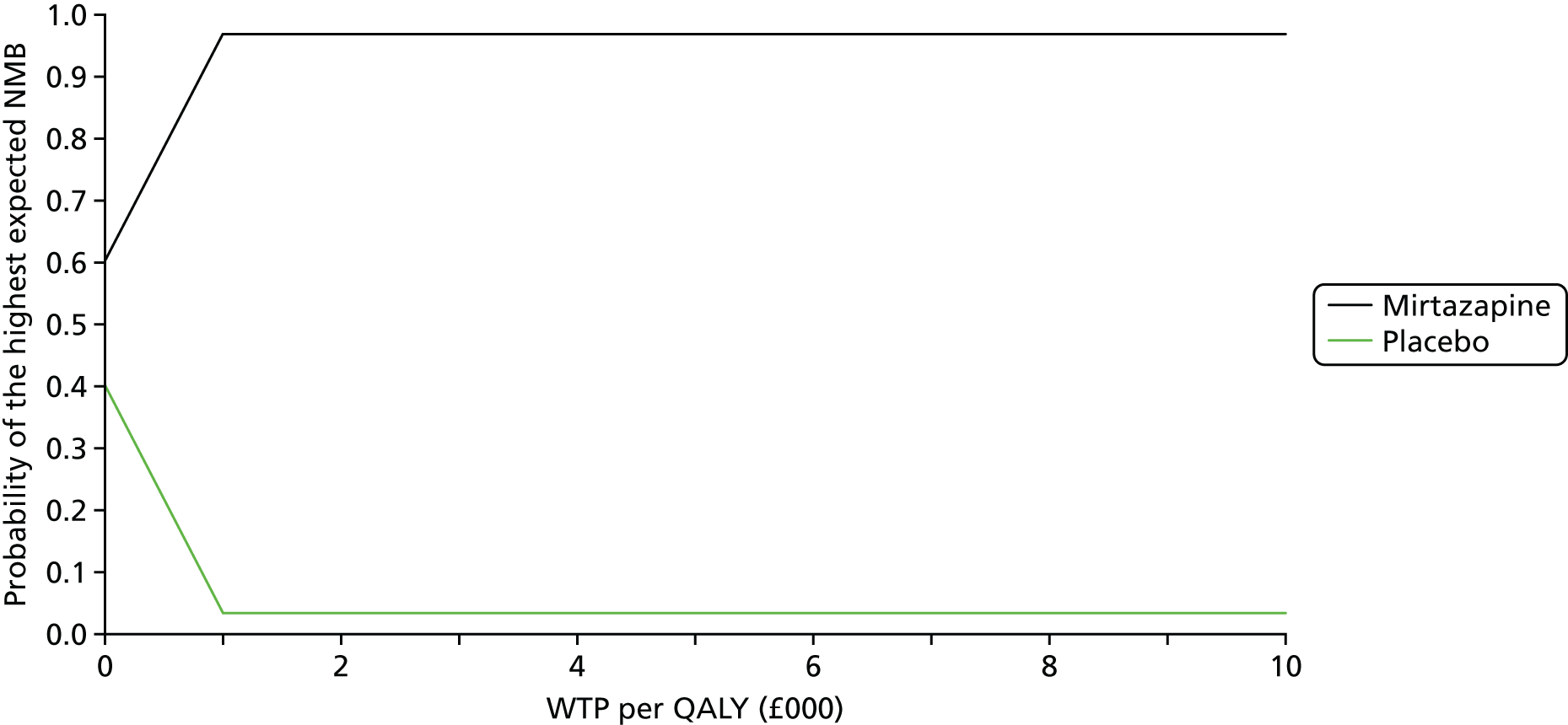
Fifty-two-week cost–utility
At 52 weeks, participants in the treatment arm had an incremental gain of 0.009 QALYs (95% CI –0.016 to 0.035 QALYs) and an incremental cost of £69 (95% CI –£74 to £206) compared with participants in the placebo arm (see Table 21). The NMB at 52 weeks is positive across the reported levels of WTP for an additional QALY – at a WTP of £20,000 per QALY, the NMB is £118 (95% CI –£419 to £655) (see Table 22). Again, consideration of the 95% CI shows a substantial degree of uncertainty around the results.
The CEAC at 52 weeks (Figure 7) shows similar results to that at 12 weeks. At a WTP of £20,000 per QALY, active treatment was the more cost-effective option in 69% of the simulations. At a WTP of £30,000 per QALY, it was more cost-effective in 71% of simulations.
FIGURE 7.
Cost-effectiveness acceptability curve: probability of highest NMB at 52 weeks across a range of WTP estimates per QALY.
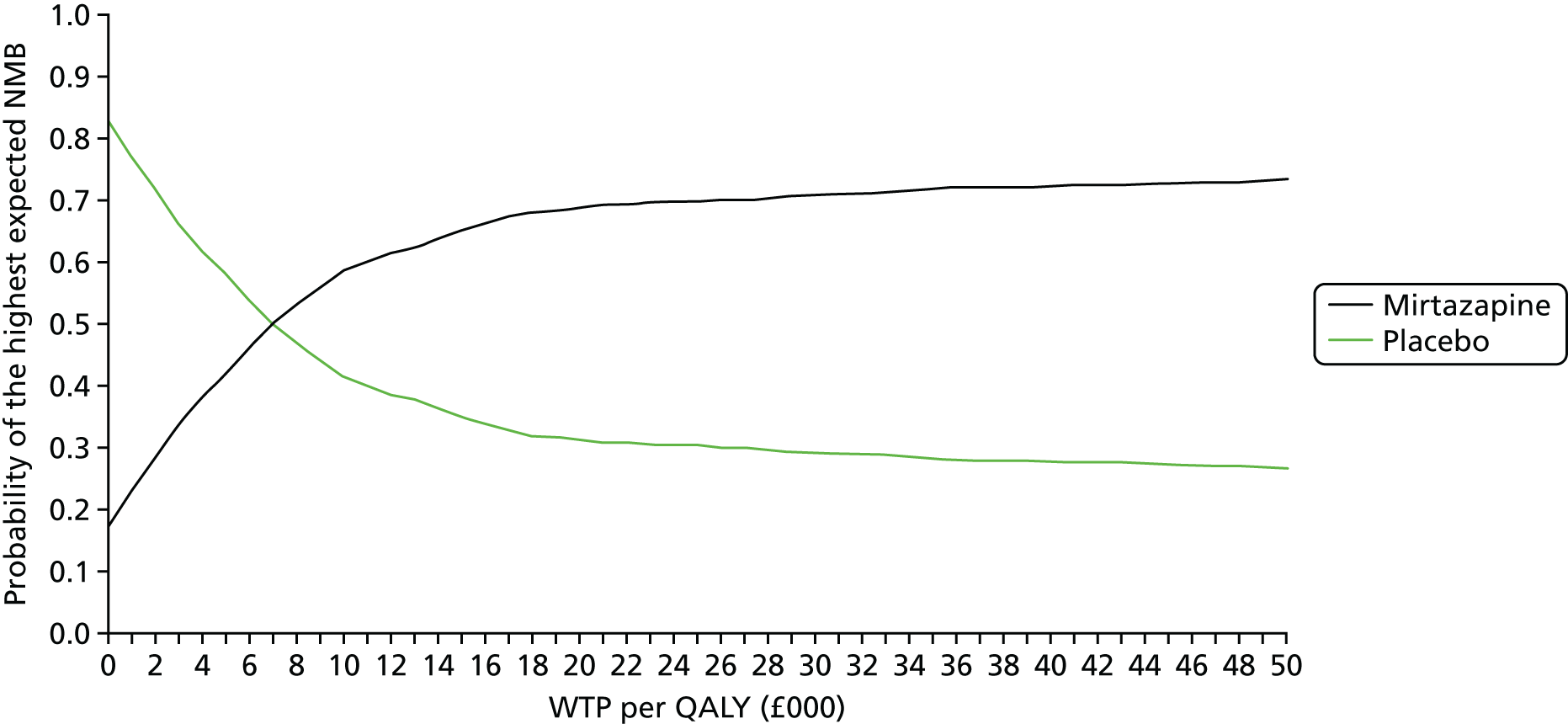
Figure 8 shows the cost-effectiveness planes for the incremental cost per QALY at 12 and 52 weeks. The overall uncertainty in the results is clear.
FIGURE 8.
Cost-effectiveness planes from a NHS and PSS perspective: incremental cost per QALY. (a) 12 weeks; and (b) 52 weeks.
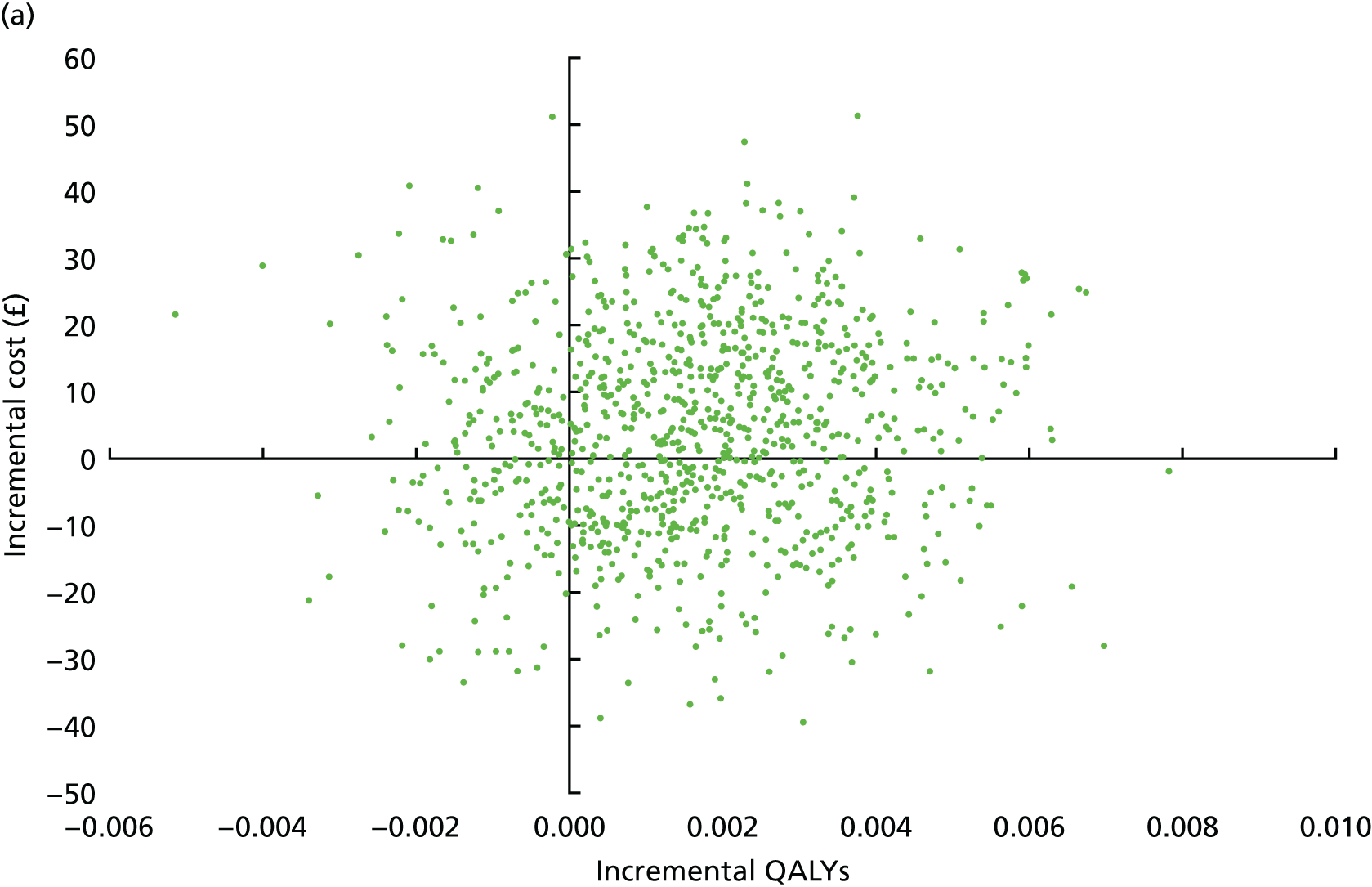
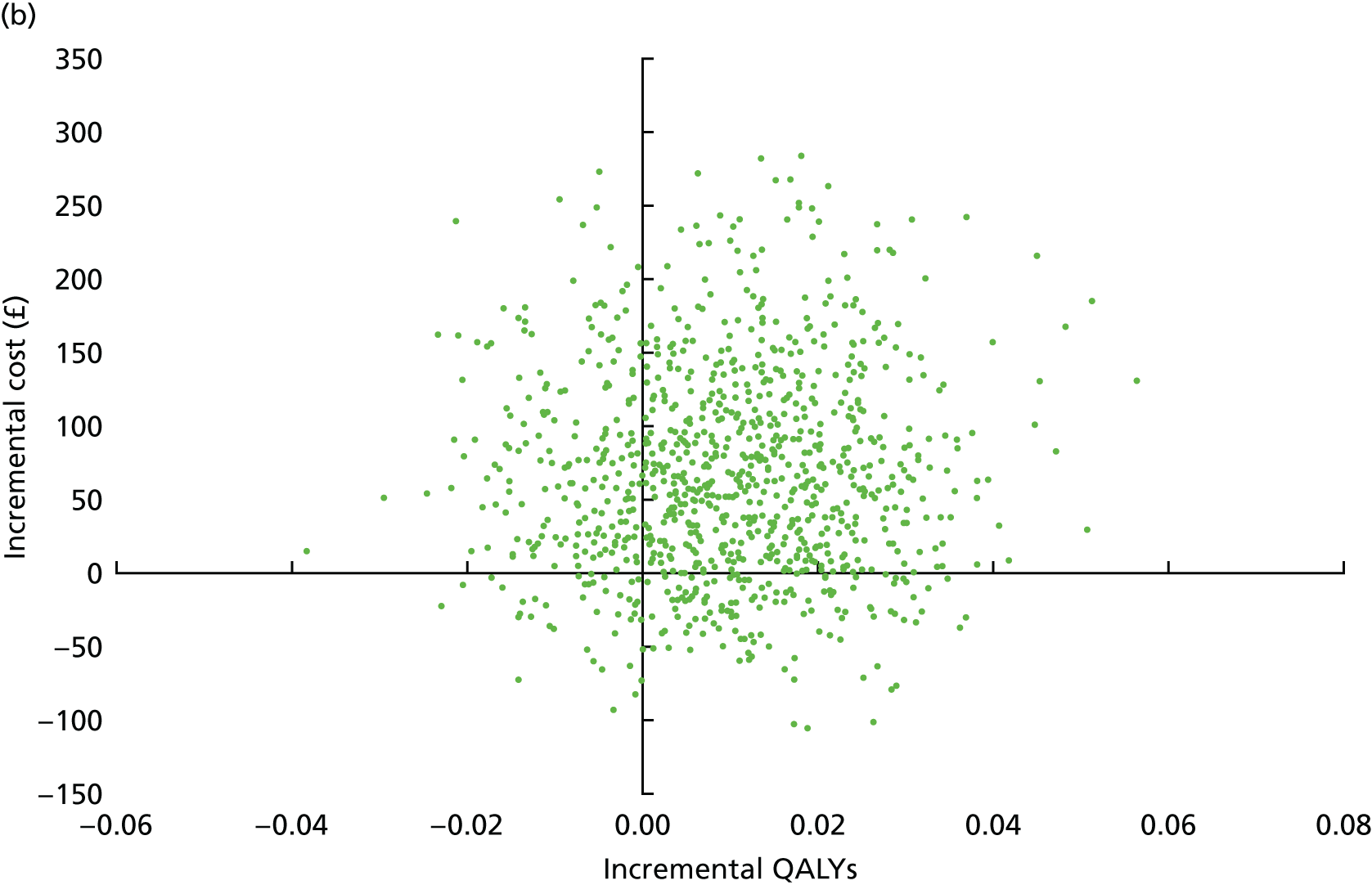
Secondary analyses
Societal perspective
In addition to the primary analysis based on the NHS and PSS perspective, we undertook an analysis based on the wider costs to society. Additional costs included in this analysis were patient out-of-pocket costs including lost income, as well as productivity losses (as described in Employment and lost productivity). Across all levels of WTP tested, mirtazapine has a negative NMB when wider societal costs are included in the analysis. At a WTP of £20,000 per QALY, the societal NMB is –£111 (95% CI –£390 to £169) at 12 weeks and –£370 (95% CI –£1199 to £459) at 52 weeks. As with all other analyses, the CIs for these estimates suggest a considerable degree of uncertainty in the results.
Imputed data
To assess the impact of missing data on the results, we also undertook analysis using MI. These results are presented in Table 22. The results also illustrate the wide variability in the NMB estimates caused by the small observed difference in QALYs at both 12 and 52 weeks. In the NHS and PSS payer perspective analysis, for a WTP threshold of £20,000, the expected NMB at 12 weeks is £118 (95% CI –£417 to £654) in the imputed data set, compared with £144 in the complete-case analysis. At 52 weeks, at a WTP threshold of £20,000, the NMB is £34 (95% CI –£48 to £116) in the imputed data set, compared with £118 in the complete-case analysis.
Subgroups
As with the analysis of the primary outcome, we undertook two a priori subgroup analyses to investigate any differential effects according to baseline depression severity (BDI-II score) and a measure of degree of treatment resistance based on the duration of symptoms and prior treatment with antidepressants. In both subgroups, we found no evidence of any effect interaction with regard to the difference between NMB (at WTP thresholds of £20,000 and £30,000) at 12 or 52 weeks.
Conclusion
The difference in QALYs at both 12 weeks and 1 year is very small and the CIs are wide and include the null. The difference in NHS and PSS costs at 12 weeks is small and not meaningful. At 52 weeks, the observed difference in NHS and PSS costs is greater, but the CIs of the difference include the null. There is no strong evidence that mirtazapine is a cost-effective use of NHS resources.
Chapter 5 Qualitative findings
Introduction
This chapter describes the methods and findings of the qualitative study embedded within the MIR trial, aiding our understanding of the acceptability of combining antidepressants in primary care, from the perspectives of people with depression and GPs.
Background
The MIR trial evaluated whether combining mirtazapine with SSRI or SNRI antidepressants resulted in better patient outcomes and more efficient NHS care than using SNRI or SSRI therapy alone. The embedded qualitative study aimed to explore the acceptability of combination treatments for depression to patients and GPs.
Patients’ health beliefs are central to an agreement about treatment being reached between patient and GP, requiring a discussion that is inclusive of, and considers the views and beliefs of, both parties. 64 Patient beliefs and attitudes influence adherence to antidepressants. 65,66 Previous literature reporting patients’ views on the management of depression suggests that some patients prefer psychological treatments to medication,67,68 but these preferences vary depending on a number of factors including age, sex and their own understanding of depression. 68,69 Negative views on antidepressants include concerns that they are addictive68 and older people may be particularly reluctant to take antidepressants. 70,71
It cannot be assumed that patients already on antidepressant medications will have a positive attitude to, or a good understanding of, the treatment or will be willing to take another antidepressant if their depressive symptoms remain. Evidence suggests that some patients using SSRI antidepressants would prefer to stop but are afraid to do so72 and that, when considering stopping the use of SSRIs, patients have a number of concerns about cessation that clinicians should attend to. 73,74 In a meta-ethnographic synthesis of qualitative evidence it was found that patients were heavily engaged in negotiating a ‘medication career’ or ‘moral career’,75 making choices about how to proceed with treatment and sense-making around their illness. This study also emphasises the vital role that GPs play in facilitating patient decision-making through discussion to aid understanding and meaning-making around depression and treatment experiences.
The views of patients who decline to take part in research can highlight salient attitudes about interventions. In their qualitative study exploring the views of people who had declined to participate in a trial of CBT for TRD, Barnes et al. 76 reported that the acceptability of the trial intervention was an important factor in determining whether or not patients chose to participate in the trial.
O’Cathain et al. 77 emphasise that the value of qualitative research to RCTs includes ‘improving the external validity of trials and facilitating interpretation of trial findings’. This chapter aims to illustrate the value of the embedded qualitative study in understanding the acceptability of a combination of antidepressants to patients and GPs and has implications for how the intervention may be received more widely by patients and GPs in primary care.
Aims
-
To explore patients’ views and experiences of taking either two antidepressant medications or an antidepressant and a placebo.
-
To identify patients’ reasons for completing or not completing the study up to 12 weeks, including stopping the study medication.
-
To explore the views of GPs on prescribing a second antidepressant in this patient group.
Methods
Ethics approval
Ethics approval was given by South East Wales Research Ethics Committee Panel C (reference: 12/WA/0352).
Design
We conducted semistructured interviews with people who were invited to participate in the trial but who declined, trial participants (who completed the trial and who withdrew) and GPs, to generate in-depth data on reasons for declining to participate, perspectives of those participating in the trial, reasons for completion of the trial or stopping the study medication and views and experiences of GPs about managing people with depression, with a focus on prescribing.
Sampling and recruitment to the qualitative study
People who declined to participate: ‘decliners’
Recruitment was from the pool of potential participants in the MIR trial. All patients invited to participate in the trial via the record search were sent a postal invitation letter (as described in Chapter 2). Respondents who declined to participate could give a reason on a form that provided closed questions to indicate their reason(s) for declining (see Figure 2 and Box 1). There was also a free-text response box for respondents to give additional details about or alternative reasons for declining. Respondents were also given the option to indicate their willingness to be contacted to be interviewed about their reasons for declining participation. If respondents indicated that they were willing to be contacted about their reasons for declining participation, these data were stored on a Microsoft Access® (Microsoft Corporation, Redmond, WA, USA) database, which could be accessed by the trial team to be used to invite a sample of willing respondents, categorised as ‘decliners’, to participate in a semistructured interview.
Purposive sampling from within the decliner population was carried out on the basis of sex, age, geographical location and reasons for declining to participate, to gain maximum variation within the sample. People who selected one or more of a, f, g and h from Box 1 were not sampled, as it was thought that these responses would yield less salient data on reasons for declining in relation to combination of antidepressants. Free-text responses were also used to direct sampling when respondents had selected ‘other’ as a closed response and when details were given in the free-text box that would indicate their suitability or unsuitability to be contacted about their reasons for declining. Examples of free-text responses can be found in Table 23, although this is not exhaustive.
-
I do not want to take part in a research study.
-
I do not want to take mirtazapine.
-
I do not want to take a placebo.
-
I only want to take one antidepressant.
-
I plan to stop taking my current antidepressant.
-
I am not taking antidepressants.
-
I am too busy.
-
I am not depressed.
-
Other.
| Free-text response examples | Deemed suitable for sampling |
|---|---|
| Fear of side effects | Yes |
| Don’t want to rock things | Yes |
| Happy with current tablets | Yes |
| I only want to take one antidepressant. I have had mirtazapine previously, hated it – I put weight on | Yes |
| I am too busy | No |
| I am on citalopram 20 mg per day to help with my bowels not for depression | No |
| I am taking sertraline for OCD not depression | No |
| Job related concerns | No |
Once identified as suitable for contact, the research assistant (Kate Dixon) made telephone contact with the respondent and gave details about the purpose of the call, the relationship of the qualitative study within the MIR trial and their rights as a potential participant of the study to confidentiality and to withdraw their consent at any time. They were then asked if they had further questions and whether or not they would be willing to participate in a telephone interview. If verbal consent was given, these interviews were usually conducted at the time of the initial telephone call or arrangements were made to call the respondent at a more convenient time.
People who participated in the MIR trial: ‘completers’ and those who stopped the study medication
At 12 weeks (once primary outcome measures were assessed), sampling of trial participants was carried out, taking sex, age and geographical location into account to gain maximum possible variation within the available sample. Our aim was to sample participants who had adhered to the trial medication up to the primary outcome at 12 weeks (hereafter referred to as ‘completers’) and those who had stopped the study medication before the primary outcome but who had remained in follow-up (hereafter referred to as ‘withdrawers’).
Participants who had reached the 12-week assessment were advised by the researcher that they may be contacted by the study team to discuss their experiences of participation in the trial. It was stressed that their participation in the qualitative study was voluntary. The research assistant received regular reports from the trial teams on participants who had reached the primary outcome measure and, following review of the participant records, would attempt to contact participants when the contact notes suggested that it was appropriate to do so. When there had been any SAEs or AEs or when a participant had declined further contact after the primary outcome measure (at 12 weeks), these individuals were not invited to participate in the qualitative study.
Participants were initially contacted by telephone and asked whether or not they would be willing to take part in an interview to discuss their experiences of being in the MIR trial. If a participant agreed, an information sheet with further details of the study was sent to the participant and a provisional date for a face-to-face interview was made (usually 1–2 weeks after the date of the telephone call to allow time for the information sheet to arrive and the participant to consider the content). The research assistant made further telephone contact 1–2 days prior to the interview to confirm that the participant was still willing to be interviewed.
If a participant withdrew from taking the tablets allocated prior to the 12-week assessment, a record of their status was noted by the research team. The research assistant received reports indicating which participants had stopped the trial medication but had consented to follow-up assessments and contact with the research team. Sampling from within the trial medication ‘withdrawer’ population was carried out on the basis of sex, age and geographical location. Following a review of the participant record and discussion with the researcher who had visited the participant, when appropriate, the research assistant attempted to contact the medication ‘withdrawer’ to gain consent for an interview. As with the ‘decliner’ participants, medication ‘withdrawers’ were offered the opportunity to discuss their reasons for withdrawing from the medication. Participants who had withdrawn from the medication gave verbal consent to discuss their reasons and interviews were conducted by telephone.
General practitioners
General practitioners were recruited from practices participating in the MIR trial. Sampling of GP practices was purposive and selection was based on the geographical location of the practice and the sex and experience of the GPs, to give maximum variation in the available population. Sampling was also contingent on whether or not the practice had patients who had been randomised to the MIR trial.
General practitioners and practice managers were initially contacted by e-mail, with the information sheet provided as an attachment, inviting them to discuss their experiences of managing people with depression and their views of using a combination of antidepressants. If there was no response to the e-mail, telephone calls were made to the practices. If a GP expressed interest in participating in an interview, the research assistant arranged a telephone interview at a time that was convenient to the GP.
Consent
Informed consent to participate in the qualitative study was obtained from all participants in the study. People invited to take part in interviews about their reasons for declining to participate in the MIR trial were provided with a short questionnaire during the trial recruitment phase on which to indicate consent to be contacted about their reasons for non-participation. When these people were contacted by telephone, they were asked to confirm that they had consented to take part in a telephone interview and for their verbal consent to record the telephone interview. Participants were reminded about their rights as a participant to confidentiality and to withdraw their consent, at the beginning and the end of the conversation, respectively. This procedure for gaining consent to conduct and record telephone interviews was also followed when contacting GPs for telephone interviews for the study.
Patients participating in the trial were sent an information sheet in the post after their 12-week outcome measure had been taken by a researcher, providing details about participation in the qualitative study. Those participants who agreed to an interview were asked to sign a written consent form to indicate their consent to participate and the researcher confirmed this consent verbally before commencing the interview. The majority of interviews with patient participants were held face-to-face, in their own homes.
Data generation
Data were generated during semistructured interviews conducted either by telephone (with patients who declined participation in the trial and GPs) or face to face (with trial participants who completed or withdrew from the trial medication).
Topic guides for the groups of interview participants (decliners, trial participants and GPs) were produced prior to the commencement of the first interviews (see Appendix 4). These topic guides were amended and adapted in response to the initial analysis of interviews and after discussion within the research team. This process was iterative throughout the period of data generation to better explore the concepts and themes that emerged as being important to the interviewees and required more in-depth examination during interviews with subsequent participants.
Data generated from each of these data sets were collected during each interview by the qualitative research assistant, using a digital audio recorder. These audio files were downloaded to, and stored securely on, a secure network at the site where the qualitative research team was based (Keele University) and handled in accordance with the research institute’s SOPs. Audio files were transcribed verbatim by an authorised transcription company and transcripts were stored securely by the research assistant in accordance with SOPs.
Each interview was checked against the audio recording, cleaned and anonymised prior to analysis.
Analysis
Analysis of the data was conducted both independently and subsequently collectively by three members of the qualitative research team: the qualitative research assistant, a research fellow (Heather Burroughs) and the co-investigator with responsibility for managing the qualitative study (Carolyn Chew-Graham). The researchers were from different professional backgrounds (psychology, health services research and anthropology, and academic primary care), which increases the trustworthiness of the analysis. 78 Transcripts were read and re-read, themes arising during analysis and data collection were discussed within the team and the topic guides were modified as data collection and analysis progressed. The qualitative research team met regularly to discuss and agree coding, revisiting the data from each data set in an iterative manner to verify coding and themes generated from these codes. This process was repeated until it was agreed that each data set had reached saturation. Analysis was initially conducted within each data set and then comparison was carried out across the data sets. NVivo 10 (QSR International, Warrington, UK) was used to store data and aid analysis.
Patient and public involvement and engagement
The qualitative research team worked with a patient and public involvement and engagement (PPIE) group to discuss transcripts from the data sets. The aim was for the lay partners to add value to the analysis by bringing their own perspectives and identifying further areas for the researchers to explore in the interviews. 79 Four meetings were held with the PPIE group over the course of data generation.
Additional perspectives provided by patients and the public on the qualitative analysis
The group initially commented on the study patient-facing documents and made valuable suggestions about how to modify phrases used. The topic guides were discussed in depth and suggestions were made to rephrase some of the prompts in the topic guides for both ‘completers’ and ‘decliners’.
At a further meeting, the PPIE group looked at two interview transcripts. The group commented on the narrative in one of the transcripts about the acceptability of medication and talking treatments. The experience reported by individuals was that antidepressants were offered initially and that there were long waiting lists for psychological therapies. The group was assured that treatment choice would be explored in interviews with MIR trial participants. The group suggested that future interviews with GPs and patients should explore what is being done in primary care while patients are awaiting psychological therapies.
The concern that GPs expressed about continuing two antidepressants was highlighted by the PPIE group and it was suggested that this should be explored in interviews with trial participants.
The principle of ‘equilibrium’ was introduced to the PPIE group as an important theme arising from the data: people who felt that they had established an equilibrium may not find participating in a trial acceptable, whereas people who were at ‘crisis point’ (not being in equilibrium) may be more likely to agree to participate in a trial (‘worth trying anything’). This perspective was, therefore, developed, in part, in collaboration with the PPIE group. The group also discussed the uncertainty about how long patients with TRD would need to take two antidepressants and felt that this should be an important focus of the analysis.
Results
Details of participants
A total of 60 interviews were conducted: 23 with ‘decliners’ (mean duration 11 minutes and 27 seconds), 23 with ‘completers’ and ‘withdrawers’ from the study medication (mean duration 47 minutes and 35 seconds) and 14 with GPs (mean duration 19 minutes and 31 seconds).
The 23 ‘decliners’ who we interviewed were distributed fairly evenly between the four centres, with seven from Bristol, six each from Exeter and Keele and four from Hull. Fifteen of the interviewees were female and eight were male, with an age range of 27–76 years. All were still blinded at the point of interview.
Illustrative data are presented with the sex and age of the participants given as identifiers in the case of trial participants (‘completers’ and ‘withdrawers’ from the study medication) and ‘decliners’. Data from GP transcripts are identified with numbers that reflect the chronological order in which the interviews were conducted.
Decliners
Table 24 lists the reasons that people gave for not wanting to participate in the MIR trial.
| Reason | Decliners (N = 4702)a n (%) |
|---|---|
| I do not want to take part in a research study | 2298 (49) |
| I plan to stop taking my current antidepressant | 1822 (39) |
| I do not want to take mirtazapine | 1692 (36) |
| I do not want to take a placebo | 1328 (28) |
| I only want to take one antidepressant | 936 (20) |
| I’m not taking antidepressants | 818 (17) |
| I am too busy | 463 (10) |
| I am not depressed | 323 (7) |
| Other reason(s) | 1321 (28) |
The most common reason for declining participation was not wanting to take part in a trial (49%). Thirty-six per cent of people invited indicated that they did not want to take mirtazapine; it is not known if this was because of prior experience of being prescribed this antidepressant. Not wanting to take more than one antidepressant was a reason given by 20% of people for declining to participate in the MIR trial. Interestingly, 17% of people suggested that they were not taking an antidepressant, even though they were being prescribed a SSRI or SNRI according to their GP records; 39% of people said that they planned to stop taking their current antidepressant.
Analysis of the ‘decliner’ data suggested that key themes were ‘the hard work of managing depression’, uncertainties about antidepressants, including questions over the value of a second antidepressant, and attainment and maintenance of a hard-won equilibrium.
The hard work of managing depression
Respondents reflected on the history of their depressive symptoms and there were rich descriptions of endeavours to manage their mental health, including self-management strategies and help-seeking.
Many respondents described delays in recognising the cause of their depression, outlining repeated investigation for physical problems until a diagnosis of depression was achieved by default:
Erm, what it went back to was – I think it was postnatal depression, it started, and I didn’t realise what it was. Erm, that was [. . .] so that was 20 years ago and I thought I’d got ME [myalgic encephalomyelitis] or, or MS [multiple sclerosis] or something, because I, I, physically, couldn’t walk. I wasn’t particularly unhappy, but I, I was physically fatigued really, really fatigued. And I struggled with that for years, going for tests, and one thing and another, and it never came to anything, because it was depression.
Female, 55 years
Once recognised as depression, respondents reported that they had tried numerous strategies and lifestyle changes to manage their own symptoms, including exercise and diet, seeking support from family and friends, taking steps to ease the pressure on themselves and pursuing psychological and alternative therapies:
Well I’d been on websites to look at the condition . . . to look at what I could do, such as the omega-3, eating well, exercising, getting as much light as you can . . . all those sort of things.
Male, 57 years
Respondents described a process of ‘trial and error’ in the search for something that works:
I took part in other things as well, like a stress-management course and an online cognitive–behavioural therapy course which, to be quite honest, I didn’t think they particularly helped me, but I did them because I was just trying everything to get out of that state.
Female, 44 years
All respondents indicated the work involved in trying to manage their symptoms and find something that helped:
. . . I’ve been doing a lot of meditation and, sort of, pursuing the mindfulness route [. . .] I think to get out of depression, you have to probably put in a lot of different things in place to be able to tackle the things that have got you there, [OK] and exercise and eating well or trying to eat well even though I didn’t have an appetite.
Female, 37 years
Medication for depression: uncertainties and fear
Some respondents described an initial reluctance to take antidepressants but, having got over this, suggested that they had come to terms with the need to use drugs to manage their mood. However, many respondents expressed concerns over the length of time that they would have to take their current antidepressant and were resistant to taking a second antidepressant:
I felt [. . .] that I didn’t wanna get involved in taking tablets for 6, 9, 12 months. I’m already 6 months into taking them now [hmm], which is longer than I thought I would be . . . I thought, ‘Oh, I’ll get rid of it. I’ll be OK. I’ll have a few months or I’ll have a couple of months off. I’ll be back to my normal self,’ but it hasn’t worked like that. Erm, and whether another antidepressant would help I really don’t know.
Male, 55 years
Such reluctance was sometimes because of a fear of (additional) side effects:
I didn’t want to start with the side effects what I got from the first one [uh-huh]. I didn’t want to start with new side effects and things.
Female, 47 years
. . . also when I read the side effects. I don’t mind the side [effects], you know, like the dry mouth, I don’t get that with these ones of mine, erm, and I have had pills in the past where I have done, and it’s not nice, is it? [. . .] And, you know, and I’ve just basically, I suppose, I’ve, you know, I’m OK with the tablets I’m on, but I don’t want to sort of erm, bring on any more symptoms, basically. [. . .] ’cos I mean when it, it was bad it was really bad and, and I’d, I’d, I don’t think I wanna go back there anymore.
Female, 48 years
Some participants, who were taking medication for other conditions, were wary of yet another drug:
I didn’t really wanna be taking another, another pill, really [. . .] again, for me it was, er, the number of pills I take is a bit of an issue.
Female, 27 years
I think it’s a good thing to have as a study, but like I say, if I was on less tablets I wouldn’t mind sort of going in for doing it. [Yeah] But as I say, I just dunno how it’ll effect, it might bugger me around with the other tablets.
Male, 60 years
None of the ‘decliners’ interviewed expressed experience of, or particular concerns about, mirtazapine.
Maintaining equilibrium
The invitation to participate in a trial was described as a difficult decision by many of the ‘decliners’ interviewed. Managing depression was seen as a series of complex decisions, with continued reflection and self-monitoring required. Respondents indicated that they were declining to take part because they feared disturbing the sense of balance they had achieved with their current antidepressant medication. The potential work involved in participation was felt to be a challenge to their current stability:
Because you know when you suddenly drop and you don’t know why? [Hmm] Erm, I’ve had a drop recently, so that, I don’t know what that was, but I had a drop recently, and I had to, sort of, look again at what I was doing [yeah]. Erm, so, adding something, changing something, is a bit scary for me [uh-huh] because I was feeling good, and I like feeling OK.
Female, 53 years
. . . but for me, taking something, erm, potentially taking something that could disrupt the way I am when I’m, you know, feel I’m on a steady level, I, I didn’t really want to, to take that risk.
Female, 27 years
I’m happy – I’m actually happy with the medication that I’m on [OK]. Erm, I, I’ve, er, I, I tried different medications to start with, and I really, it really wasn’t successful. I’m in, I’m quite content with the place that I’m in at the moment.
Female, 27 years
Other respondents, although recognising that something else may be necessary to help them manage their symptoms, expressed a preference for talking treatments:
. . . I’m on quite a low dose really, 20 mg of, erm, citalopram, and I think it was doing the job it needed to do [. . .] to get me to point where I could look at some issues.
Female, 39 years
A number of ‘decliner’ participants stated that had they been offered combination treatment at a point of crisis, when they felt out of balance and unable to cope, they might have agreed to take part in the trial because of their desperation to overcome their depression. In the following extract, the ‘decliner’ participant is responding to a question from the researcher about whether or not she would have considered taking two antidepressants if she had not felt better on one tablet. Her response suggests that if she had been ‘at rock bottom’, taking two tablets would not have been an issue if it meant getting better:
. . . I needed to do whatever it took to, you know, get well again, quite simply. So I, I would do whatever it takes. When you – I think when you really hit rock bottom you are prepared to do whatever it takes and I have absolute faith in my doctor.
Female, 42 years
The participant in the next extract is responding to the same inquiry, about how she would have felt had she been offered two antidepressants prior to feeling stable on her current antidepressant. She makes a more extreme comparison, but makes the same point about being willing to accept whatever was necessary to escape being depressed:
I think, I think if you’d caught me when I was depressed, I would have taken anything to be honest, I would have done anything to get out of it [. . .] in the first instance I struggled for a few weeks erm, thinking oh I can get out of this and it’s alright, it’s temporary and then it gets to a point where 2, 3 weeks down the line, I just thought, I can’t deal with this, I’m on a sinking ship, so I think if the study had been then, and someone had offered me, you know, I don’t know, a shot of heroin a day to get out of it, I would have done it!
Female, 38 years
The next extract is from a ‘decliner’ who, when asked about taking two antidepressants, sums up the main themes that arose from analysis of all of the ‘decliner’ transcripts. Taking mirtazapine alongside his current antidepressant may have been an option if he had had no other options, but now that he was ‘feeling well’ this was not a risk he wanted to take:
No, I mean it may well be a brilliant, you know I don’t know much about this mirtazapine and erm, you know, if I was in a more impossible situation, I probably wouldn’t mind trying it out, but erm, I was so desperate to get out of that state, I don’t want to risk it, so it is simply erm . . . now that I’m well again, not wanting to risk erm, changing that.
Male, 57 years
Why would another tablet help?
Some respondents expressed scepticism about the ‘chemical imbalance’ story that they felt had been told to them, to explain why a tablet would help their mood. Why a second antidepressant would help if the first had not was questioned:
. . . would you have had any concerns about taking two antidepressants?
Erm, no, I don’t think so . . . but I should wonder why I suppose I hadn’t had any, er, sort of beneficial reaction and maybe had to go to the doctors again and say it doesn’t seem to be working terribly well, what else can we do.
Male, 76 years
The dominance of these themes within the ‘decliner’ data set suggests that, for these people, taking part in the trial of a combination of antidepressant drugs was too great a risk to their current feelings of stability. For them, their current antidepressants were helping them maintain a level of functioning, which they wanted to protect.
Completers and withdrawers from the study medication
Analysis of data generated from interviews with participants who completed the trial or who withdrew from medication suggested four main themes, which resonated with the analysis of the ‘decliner’ data: (1) help-seeking at a point of crisis, (2) hoping for another option, (3) experiences of the trial, emphasising side effects, which led to (4) a dilemma of whether or not to continue the combination of medication.
Help-seeking at a point of crisis
People who felt that they were approached to participate in the MIR trial when they were at a point of crisis described seeing the trial as an opportunity to deal with the crisis. Thus, participants detailed the current impact of their depression on their ability to cope, to the point when they recognised they needed help:
I had a lot of work stress going on as well [uh-huh], er, and it all got on top of me [. . .].
I was massively, er, overeating, erm, oversleeping, permanent low mood, just generally unwell [. . .] So, er, at, sort of, that point, I went to the doctor and said, ‘Look, this is what’s going on. I need some help with this’.
Male, 52 years, completer
Another respondent reflected on how his feelings of desperation had led him to accept referral into the trial by his GP, in the hope that it would offer alleviation of his current condition:
Well I think I was desperate enough to take her advice that it was more likely to be helpful than anything, than a hindrance, it wasn’t going to do any harm and it might do some good.
Male, 83 years, completer
Hoping for another option
Many of the participants who agreed to take part in the trial described the hope that participation might provide them with another option to try. Thus, respondents described the protracted sequence of trying out other antidepressants, increasing the dosage of their medication, which had either been ineffective or had worked for a limited time and then seemed to stop working, and referrals to talking therapies. The hope that this trial would be another option for them to regain a sense of well-being was prevalent in accounts of these participants’ experiences:
I felt that the antidepressants, I’d already been on, you know, I was given 20 mg, and then they were increased to 40 [uh-huh], and yet I still wasn’t feeling, I wasn’t really feeling, er, 100% [uh-huh]. So I just felt that that [taking part in the trial] could be a different way of trying to get things, trying to get me a bit more better, you know.
Male, 54 years, completer
. . . I had these bouts where, maybe, I slipped back, every 6 months, 12 months, maybe even longer than that. And I found that the symptoms that I was getting were reoccurring more frequently, and I myself, was wondering, erm, if there was anything different I could try.
Female, 59 years, completer
As with the decliners, participants in the trial were keen to get back to a level of coping and well-being that they had experienced previously. Participants described that they were no longer feeling a beneficial effect from their medication and in some cases had sought out another option from their GP. The participant in the following extract gives an account of approaching her GP to ask them to ‘do something’:
I was taking sertraline erm probably from quite a while, really; about 4 months or so. And I felt they hadn’t made enough difference. [OK] Erm so I went back to the doctor and I was really worried and I said, ‘Look, I need some – either to up this medication or change it, do something’.
Female, 49 years, completer
The opportunity to participate in the MIR trial came at a time when they were seeking an alternative option to help them manage their depression.
Experiencing the trial: benefits and side effects
Feeling better: improvements in mood
A number of participants who had reached the 12-week assessment in the trial reported that they had experienced improvements in their mood and general functioning. Participants described themselves as moving towards the equilibrium that they sought:
Since taking the second tablet, I didn’t feel the need to go back to LIFT [local psychological intervention group], as well, actually. I didn’t – I felt I didn’t need to talk it through because I felt OK.
Female, 61 years, completer
I’m back to feeling more like myself, looking forward to things, planning things, generally being on top of stuff. I’m still tired because that’s because we’ve got three small children [laughs]. Bless him. Erm, but generally, yeah, like, I was picking up and, sort of, the, sort of, the weight is starting to come off as well, and it’s just, sort of, things are turning around, definitely, I think.
Male, 35 years, completer
I’m quite happy with taking these combinations particularly, particularly this one, because it seems to be working [. . .] now I wake up and I think, oh it’s quite a nice sunny day I might do such and such today you know it’s – it’s more positive definitely, er, a more positive outlook [. . .] Er, and one thing it’s done it has actually increased my appetite it’s definitely done that because I was, er, I was forgetting to eat basically.
Male, 59 years, completer
This participant was pleased both with his increased positivity and with the increase in appetite and weight gain, which contrasts with the reports of other completer participants who reported that the side effect of weight gain had caused them to withdraw from the study medication.
A couple of participants reported withdrawing from the study medication because they felt that they had regained their equilibrium:
I think the medication that I was on – got me, you know, on an acceptable level, you know. I’m not as bad as I was [. . .] And then you get to a stage where yeah, things are acceptable. You just accept things and you just carry on.
Female, 69 years, withdrawer
Side effects: increased appetite and weight
Commonly reported side effects in both completers and withdrawers were increased appetite and weight gain, which most found problematic:
. . . my sugar, craving for sugar, which I’ve always had, er, a, er, a . . . [Sweet tooth?] Yeah, and I always called it addiction, erm, and that seemed to lessen with just citalopram, and I lost a lot of weight last year; I lost over 2 stone. As soon as I started taking these tablets, I was climbing the wall, erm, immediately, for sugar [uh-huh], and I didn’t seem to have, erm, a stop button to say you’re full.
Female, 61 years, completer
The only problem I have is my weight gain issue. I’ve never been a person that’s been on a diet in my life. I’m not a person that, erm, has had to worry before about putting weight on and since I’ve been taking this, these tablets, I’ve put on 2 stone [laughs], which is quite a lot.
Female, 59 years, completer
Had my weight gain not been so severe I would’ve gone on longer taking the drug – and I told her that. I said to her that I couldn’t warrant having great thoughts and then dying of a cardiac arrest [laughs] because I’d put on so much weight. I have weight issues anyway, so it was just like the balance wasn’t right for me by the end of it.
Female, 36 years, completer – withdrew after unblinding
Two antidepressants: whether or not to continue?
Both the completers and those participants who withdrew from the study medication offered a narrative of balancing improvements in their mood and unpleasant side effects of treatment:
Yeah, I do on one hand. I do feel slower, and like I’m not getting anything done [laughs] which is a double-edged sword, isn’t it? Because like, I’m feeling obviously a lot better on one hand, and, erm, you know, is, what is the benefit of me staying on it other than it, it, you know, in case, obviously I’d take it for as long as I need to, and then I’d come off it and see if I can cope.
Female, 59 years, completer
. . . but I sometimes wonder if it’s worth it. I don’t wanna put all that weight back on.
Female, 61 years, completer
The need to balance the gain in improvement of mood with the side effects is well illustrated in this exchange:
The only problem is that I gained nearly 3 stone.
OK, er, and, er, you have mentioned that, er, you have put on weight with the medication, does that mean that you’ve actually stopped taking it?
Yes I have.
You have, OK.
Because it’s, er, believe me I’m beginning to notice that I’ve stopped taking it.
Right, OK, in terms of a decline in your positive feelings . . .
But the amount of weight that I put on was I – I, it, I couldn’t tolerate it [right] it was making me short of, it was nearly 3 stone.
And that was in the period of time you were on the trial?
It’s, well, I was alright for the first month [OK] and then it gradually crept up and this last 2 months it literally shot on and since I’ve stopped taking them I’ve actually lost 2 kilos already.
Male, 75 years, completer
General practitioner perspectives
The narratives of GPs about their management of people with depression suggested that it is complex, individualised and involves a good deal of effort when medication is used. GPs described the ‘hard work’ of managing people with depression, resonating with the patients’ perspectives. Observations about people presenting at a crisis point linked with the patient data sets.
The hard work of managing people with depression
Although GPs described managing people with depression as ‘the bread and butter of general practice’, they reflected on the complexity of the work involved in the assessment of a patient:
I mean all of it is history taking initially, finding out a little bit about the, the, the physical problems, social problems and obviously their mental health problems. There might be more significant social connotations. So they might have alcohol-related problems or drug use problems and things like that so that can make things a bit more challenging.
GP04
. . . you can’t give a global answer to that actually because it’s incredibly tailored individual . . . to individual patient circumstances and patient beliefs [. . .] so I’m – you know, I just think it’s – it’s just very individual. I think we all adopt different consultation styles for different patients, don’t we?
GP02
This multifaceted and individual assessment of patients who presented with depressive symptoms or ongoing depression, described by some GPs as a large component of their workload, was also described as one that can feel like a ‘battle’ and at times overwhelming:
Erm, so it’s, it’s inevitably a battle [yeah]. Well, not a battle, it’s a challenge to, to relate to the patient in the way that they understand. That’s what we do for a living, isn’t it? [Hmm] I suppose.
GP12
. . . we have thousands of patients who do seem to have erm, anxiety, stress, depression and mood problems [. . .] Yeah, within the practice, you know, we are seeing an increasing number of people who do present with mental health problems, or it feels like that. The reason we employ a specific nurse to help deal with those problems is a reflection on the fact that we are overwhelmed by it.
GP05
In addition, GPs described the complexity involved in negotiating management with patients:
. . . erm I think it’s – some of it’s about previous response or lack of previous depression, really. [OK] Erm the, the, I think then that’s looking at the broader context of what the patient’s like, whether it’s a personality disorder, erm and whether it’s err, circumstantial, you know, whether it’s – there’s, there’s, it – there’s quite a few things that feed into that, really.
GP10
General practitioners varied in their descriptions of TRD, with some suggesting that it was related to the patient’s lack of response to medication, while others outlined a more complex definition:
Yeah, well I mean, when I say more challenging I mean more treatment resistant I suppose so [right] if you’ve got to that point these patients are sort of, if you like, failing to respond to, um, to if you want to call it first-line treatment and so then, by definition, they’re struggling more so that, they’re sort of, so they’re more challenging in as much as, um, providing them with effective management I suppose.
GP104
For the GPs, the opportunity to participate in the MIR trial also provided them with another option to offer to patients with depression.
Help-seeking in a crisis
General practitioners’ accounts reflected the perspectives of patients – that often help-seeking was at a point of crisis:
And, and if somebody’s depressed they really – if they’re, you know, if they’re at a watershed and they’ve actually come to the doctor’s about it [. . .] because it’s often a crisis really when they’ve – or it’s been a huge step to actually come and do something about it.
GP102
. . . but a lot of the time, when people come and see us, er, it’s normally at the crisis point.
GP08
I think a lot of people by the time they’ve got to us they’re probably quite sort of . . . you know, er, they’re needing some help really.
GP03
Such help-seeking at crisis point was not just described in new presentations; GPs described how patients with long-standing mood problems would often re-present at a time of crisis:
Erm, clearly there’s a, quite an imperative to prescribe in some people, people who have been unwell for years. Some people want medication and don’t have the time or the inclination, or the enthusiasm to go for counselling, for a long period of time, and wait. They just want something done now, please.
GP114
Two antidepressants: confidence to start and whether or not to continue?
Combination of antidepressants
A number of GP respondents reported that prescribing mirtazapine in combination with SSRIs and SNRIs was something that they had already tried, although it was far from widespread across the practitioners in our sample:
. . . the advice we sort of get back [from a psychiatrist] is that you could add mirtazapine type of thing, so that sort of, as I said, has crept in to our practice a bit, almost by the back door, I think.
GPI03
. . . erm, well MIR, obviously, is asking whether adding mirtazapine in [huh-huh] is, is an option and that is sometimes done, I – you know. It’s not done very often [huh-huh] but it is occasionally done.
GP102
That’s probably, you know, I wouldn’t say it was high numbers but er, you know, probably two or three times a year or something like that, you know. Yeah.
GP10
However, most GPs expressed a degree of unfamiliarity with combining antidepressants:
I, you know, we, we manage polypharmacy all the time – if you look at some of our diabetic screens, and, erm, er, the potential for interactions, er, er, often defeat, even, the computer, and it doesn’t know quite what to do with them [uh-huh]. So, erm, er, a, a couple of antidepressants isn’t that, er, so far out of the bounds of possibility that I would worry about it, really.
GP108
Some GPs described the patients who they have offered this combination to: in order to manage outstanding symptoms, particularly sleep disturbance, when a patient has been on an antidepressant ‘for years’ or when a number of antidepressants had already been tried:
Yes I sometimes give people, er, some mirtazapine, er, because we tend to start with the SSRIs, I sometimes add in mirtazapine especially if they’re having sleep problem.
GP06
I think the time when I would tend to combine might be, er, if somebody’s already been on an antidepressant for a long time and is getting other symptoms then I might add in a different one [. . .] we’ve got some people who have been on antidepressants for years so they just might be the ones where I might prescribe two but not necessarily.
GP106
[Combining is] mainly done in, in people who have had several switches, basically.
GP10
Continuing the combination?
Although some GPs described a level of confidence in prescribing two antidepressants, all expressed caution about how long such a combination should be continued. Some speculated on how they might try to reduce one antidepressant in a combination, but none of the GPs interviewed reported experience of stopping one in the combination:
I dare say we’d reduce one to zero, first of all, and see how it goes, and then reduce the other to zero [uh-huh]. I don’t think I would stop them both at the same time [yeah]. I don’t know [uh-huh], but I think I would, I’m sure I would do that, actually. I’d, I’d choose one and say, ‘Well, how are you doing?’ And, ‘We’re gonna have to start reducing these now’ [yeah], and I would suggest that, er, ‘we get you off one first of all, for a few weeks, and see how you feel, and then withdraw the other one slowly’.
GP12
All GPs expressed concern about patients remaining on two antidepressants, suggesting that patients were commonly prescribed SSRIs without a plan to withdraw:
It’s all right initiating it, isn’t it, and – but then I think then there becomes a bit more of a long-term issue about if you – how long do they stay on them both? [huh-huh] Do you – which one do you tail off first? [huh-huh] And erm, you don’t – you wouldn’t necessary want people stuck on both antidepressants for time immemorial, which sometimes happens with SSRI, doesn’t it?
GP102
Thus, GPs described a future dilemma if antidepressants were combined.
Discussion
Summary of findings
This embedded qualitative study within the MIR trial explored reasons why people approached to participate in the MIR trial declined, the experiences of people participating in the MIR trial and the perspectives of GPs about the role of a combination of two antidepressants for people with TRD.
The narratives of people who declined to participate in the MIR trial, but who were willing to be interviewed about this decision, and the participants in the trial all described the hard work involved in managing depression and the importance of a ‘crisis point’ in precipitation of help-seeking. The people who declined to participate in the MIR trial described feeling that they were at an equilibrium and feared that participating in the trial would disturb that hard-won equilibrium. Patients may be ‘resistant to change’, rather than the depression simply being ‘treatment resistant’.
Experiencing a crisis point seemed to be a motivator to make a change and the offer of trying a second antidepressant within a trial could act as that motivator. Some ‘decliners’ expressed uncertainties about the role of medication and feared that a second tablet would not help, or could not see the logic of a second medication.
The people who participated in the trial (whether they completed the study or withdrew from medication) described the constant hope that something would help their mood, which was something that the trial offered. Withdrawal from the study medication was sometimes put down to reported side effects, particularly weight gain. Of note, at the time of the interview, those people who withdrew did not know whether they had been taking placebo or mirtazapine. People who withdrew described reaching a point at which they had to balance improvement in their mood with side effects from the second agent and, when the balance tipped against side effects, they made the decision to withdraw from the medication.
The narratives of GP respondents reflected the patient data, describing the ‘hard work’ needed to manage people with depression and how ‘treatment resistance’ is about more than a poor response to an antidepressant: it is about a complex patient presenting the GP with a challenge in management. GPs described how patients often present at a crisis point and how the MIR trial offered an option to manage such patients. Some GPs reported confidence in combining antidepressants, but others did not. All expressed concern over the dangers of leaving people on two antidepressants, reflecting that, in their experience, people did remain on a SSRI without review or monitoring and with no attempt to withdraw the drug.
Comparison with previous literature
The most common reason for people declining participation in the MIR trial was not wanting to take part in a research study, as in Barnes et al. 76 But, in contrast to the study by Barnes et al.,76 in which the ‘decliners’ expressed negative feelings about the treatment offered in the trial, in our study ‘decliners’ expressed the view that they were at an equilibrium, which would be disturbed if they chose to participate in the trial, although a second antidepressant was not thought to be logical by a number of the ‘decliners’ we interviewed.
Hughes-Morley et al. 80 described how individuals declined participation because they judged themselves ineligible or not in need of the trial therapy. Similarly, our analysis suggests that the ‘decliners’ we interviewed were not in need of the trial intervention, the second antidepressant.
Trial participants described experiencing a crisis as a motivator to participate in the trial, which resonates with Schofield et al. ,81 who reported that participants described their first course of antidepressants as typically occurring when they had ‘hit rock bottom’, having exhausted all other possibilities. Schofield et al. 81 reported that participants described a period of experimentation in which it was usual to stop and restart medication, often several times. Ultimately, these recurring cycles lead to participants becoming more expert about their condition and better able to make an informed decision about medication. This expertise resonates with our analysis.
Schofield et al. 81 also reported that, for older people, there was often an acceptance that their condition, and medication use, would be long term. This acceptance might resonate with the concept of ‘equilibrium’ that we describe in this study.
In a meta-ethnography, Malpass et al. 75 outline the role of the GP in supporting decision-making about antidepressant use and facilitating concordant relationships with patients regarding antidepressant use. Not having this discussion at entry into the trial may account for some people declining to participate.
Malpass et al. 75 recommend that GPs are aware of the competing demands that patients experience at a decision-making juncture. We identified the importance of a ‘crisis’ in influencing a decision to try something new and a sense of equilibrium in resisting change.
Strengths and limitations
Exploring the reasons for non-participation in trials is still unusual; this study describes how interviews to determine such reasons can be incorporated into a trial recruitment procedure and illuminates the reasons people declined. It should be noted that people invited to participate in the MIR trial did not necessarily have TRD – they were people being prescribed one antidepressant in primary care. That 39% of respondents indicated that they were planning on stopping their antidepressant suggests that a good proportion of those invited to participate in the MIR trial may not have been depressed. None of the ‘decliners’ interviewed expressed any experience of or concerns about mirtazapine as a particular antidepressant, even though sampling included those who had given ‘I do not want to take mirtazapine’ as a reason for declining to participate in the MIR trial.
A limitation of the study is that people who responded to the decliner questionnaire, and the invitation to participate in a telephone interview about their reasons for declining to participate in the MIR trial, were a self-selected group, which may limit the generalisability of the findings. Perhaps people with stronger opinions might be more likely to respond. In addition, the telephone interviews were relatively short (mean duration < 12 minutes).
A strength of this study was involvement of the PPIE group in the analysis,79 although some challenges arose, including difficulties in recruiting a diverse range of members of the public to carry out the role, and the research team sometimes experienced difficulties keeping the PPIE members on track.
Implications
Exploring reasons for declining to participate in the MIR trial suggests that some people who are already taking one antidepressant may be reluctant to take a second. Potential participants were wary of possible side effects, but were also unconvinced of the logic behind such a combination. They considered that, if one antidepressant is not working, it was not plausible that a second would be effective. In addition, people described being in a state of equilibrium and reluctant to make a change, reflecting that this equilibrium is ‘hard won’ and they are unwilling to risk disturbing this. Understanding a patient’s view on medication is important for GPs when discussing antidepressants. The key finding of the importance of achieving an equilibrium is vital for researchers to understand and incorporate into recruitment strategies in future trials of treatments for depression.
A crisis seems to be the motivator to try something new, including taking a combination of antidepressants, and this is the point at which people often consult their GP for help. If a patient receives an invitation to participate in a trial when they are at such a point, they may be more willing to participate. Whether or not this affected the characteristics of patients recruited to the MIR trial is not known, but we might postulate that people who felt that their mood was particularly low and who were searching for something to help were the people recruited into the MIR trial. This does not necessarily mean that people recruited were more depressed, but their perceptions of their mood might have been that things were particularly difficult at the time that referral into the MIR trial was offered by their GP or the letter of invitation to participate was received.
Varied levels of confidence were reported by GPs in initiating prescribing of two antidepressants; some GPs reported that this had ‘crept into’ their practice following advice from a psychiatrist with previous patients. Other GPs reported that combining antidepressants was not something that they were comfortable with. All GPs, however, expressed concern over the risks of continued prescribing of two antidepressants, leaving people on a combination long term, and felt that guidance would be needed to ensure safe prescribing and withdrawal of one agent.
This qualitative study highlights the concern expressed by all GP respondents that people could then be left on two antidepressants, given that many already remain on one antidepressant long term and there is a reluctance from both patient and GP to make changes. This has implications for the education and training of GPs about the management of TRD, the importance of regular review and how to facilitate continuity of care for people with depression.
Chapter 6 Discussion and conclusion
Summary of findings
This study did not find convincing evidence of a clinically important benefit for mirtazapine given in addition to a SSRI or SNRI antidepressant over placebo in primary care patients with TRD. Patients eligible for this trial had been adherent to their antidepressant treatment for ≥ 6 weeks and were still suffering from a major depressive disorder, as defined by the ICD-10. 18 Most of them had been on antidepressants for much longer than 6 weeks. Over 90% of participants had been taking antidepressants for > 6 months and 72% had been taking antidepressants for > 1 year. One in four participants were severely depressed and the majority were at least moderately depressed.
In the primary ITT analysis, both the placebo group and the intervention group showed improvements in the primary outcome at the 12-week follow-up: in the placebo group, BDI-II score improved from a baseline mean value of 30.6 points to a mean of 19.7 points and, in the intervention group, BDI-II score improved from a baseline mean value of 31.5 points to a mean of 18 points. We based our sample size calculation on detecting a between-group difference of 3–4 BDI-II points, which we argued would be clinically important. 41 The adjusted mean difference in BDI-II score between the groups at 12 weeks’ follow-up was –1.83 points (95% CI –3.92 to 0.27 points; p = 0.087) in favour of the intervention group. Although the lower limit of the 95% CI for this difference (–3.92 points) means that we cannot exclude the possibility of a clinically meaningful effect, the CI included the null, indicating that the results are also consistent with no difference between the groups.
Similar evidence of small differences between the treatment groups in favour of the mirtazapine group was observed for the secondary outcomes at 12 weeks post randomisation, but for most outcomes the 95% CIs surrounding the difference between groups included the null. At 12 weeks’ follow-up, 43.5% of participants in the treatment group met criteria for response (i.e. BDI-II score improved by ≥ 50% compared with baseline) compared with 35.9% of those in the placebo group (OR 1.39, 95% CI 0.94 to 2.07). In the intervention group, 29.4% of participants met criteria for ‘remission’ (i.e. BDI-II score of < 10 points) at 12 weeks, compared with 24.4% of participants in the placebo group (OR 1.29, 95% CI 0.82 to 2.02). This weak evidence of a small effect is supported by changes in favour of the intervention for which the CIs did not include the null, that is, in the SF-12 aggregate mental health score (between-group difference 3.91 points, 95% CI 1.63 to 6.20 points) and the GAD-7 score (between-group difference –0.98 points, 95% CI –1.93 to –0.03 points). Outcomes at later time points showed smaller between-group differences. CACE and per-protocol analyses for the primary outcome, which are designed to estimate treatment effects in those who complied with their allocated treatment, showed slightly larger between-group differences than the ITT analyses, but CIs still crossed zero and per-protocol analyses are known to be biased. Prespecified subgroup analyses, based on severity and degree of treatment resistance, did not show any evidence of effect modification.
Participants were offered elective unblinding after the primary outcome at 12 weeks. An additional sensitivity analysis at 24 and 52 weeks found no between-group differences in BDI-II score in those who remained blinded throughout the trial. An ‘as-treated’ analysis at 24 weeks comparing those who were taking mirtazapine and a SSRI or SNRI with those who were taking a SSRI or SNRI only, irrespective of whether or not they were blinded, found a mean between-group difference of 3.9 points (95% CI –6.45 to –1.37 points) on the BDI-II in favour of the intervention. However, this result is difficult to interpret as it is not a true randomised blinded comparison.
There was no evidence of a substantial increase in the burden of AEs in the mirtazapine group compared with the placebo group using the ASEC AE rating scale (between-group difference 0.26 points, 95% CI –1.15 to 1.68 points). However, mild AEs were more often reported in the mirtazapine group and were associated with stopping the medication in 46 participants in this group, compared with only nine participants in the placebo group. The most common AE reported was ‘drowsiness’. We were not able to objectively assess and quantify weight gain in the mirtazapine group.
The economic analysis found that the difference in QALYs at both 12 weeks and 1 year was very small, with wide CIs that crossed zero (between-group difference in QALYs – 12 weeks: 0.02, 95% CI –0.02 to 0.05; 52 weeks: 0.009, 95% CI –0.02 to 0.04). The difference in NHS and PSS costs at 12 weeks was small and not meaningful. At 52 weeks, the observed difference in NHS and PSS costs was greater (£69, 95% CI –£72 to £209), but the CI includes the null and this difference was unlikely to be meaningful. In the context of the results, there was no evidence that mirtazapine was a cost-effective use of NHS resources.
Qualitative findings suggest that, although many GPs are familiar with the strategy of combining two antidepressants, they are not entirely comfortable with it and have concerns about the burden of AEs for those who are on two antidepressants long term. Patients in the qualitative study who declined to join the MIR trial were often concerned about disturbing an ‘equilibrium’ that they had reached with their current treatment with a single antidepressant, whereas those who entered the study often described their motive as seeking a resolution to a ‘crisis’.
Strengths and limitations
The CONSORT statement82 and the extensions for pragmatic trials83 were followed both in the conduct and in the reporting of the trial. Randomisation was carried out by means of a computer-generated code to ensure concealment of allocation. The main analyses were conducted according to the principle of ITT and the analysis plan was agreed in advance with the TSC.
Participants, investigators and assessors were all blind to the allocation up to and including the primary outcome at 12 weeks. Voluntary unblinding of participants after the primary outcome at 12 weeks made interpretation of longer-term outcomes more difficult. The primary outcome was measured by self-report, which further reduced the possibility of observer bias. The primary outcome measure was the BDI-II score,27 and the outcomes were consistent with the other self-reported depression symptom measure used in the study, the PHQ-9. 29 All participants had an ICD-10 diagnosis of major depressive disorder using the CIS-R. 28
Follow-up rates throughout the trial were good at all sites; the overall follow-up rates were 90% at 12 weeks, 84% at 24 weeks and 81% at 52 weeks. Sensitivity analyses were conducted to assess the impact of missing data on the analysis of the primary outcome. Whether estimating the missing data assuming a ‘best’- or ‘worst’-case scenario or using MI, the observed difference in BDI-II scores between treatment groups at 12 weeks’ follow-up was small. There were some minor baseline imbalances between the two groups but adjustment for these did not materially affect the trial results.
The criteria for defining TRD that we adopted have been used elsewhere in primary care research21 and were designed to be inclusive while reflecting NICE treatment guidelines. 84 In the last few years, the idea that a definition of TRD should include at least two unsuccessful courses of treatment has been recommended. 17 Our study protocol predates this recommendation and we did not specify that participants should have had more than one course of antidepressants in the past to be eligible. However, most participants reported previous episodes of depression and were suffering from moderate to severe depression, in spite of taking antidepressants for > 6 months. Importantly, there is very little evidence to support the effectiveness of switching within or between classes of antidepressants as a strategy to manage treatment resistance85 and hence the population recruited to our trial is closely aligned to the group for whom there is uncertainty around management in primary care.
We based our view of the clinically important difference between the intervention group and the placebo group of 3–4 points on the BDI-II on previous recommendations from NICE. 84 Estimating the minimum clinically important difference (MCID) in depression is difficult and there are no universally accepted methods for establishing this. Since our protocol was written, an approach towards establishing MCID using self-rated global ratings of improvement has been developed. 41 This approach gives an estimate of a MCID in depression of a 17.5% reduction in BDI-II score for a non-TRD population, and in CoBalT,21 which studied a TRD population similar to ours, estimates that a 32% reduction would be required. These translate to differences in BDI-II score of 3.5 and 5.9 points, respectively. Based on these figures, it seems unlikely that mirtazapine would provide a clinically important benefit, although there is still considerable uncertainty around the clinically important difference in treatment outcome for this group of patients.
Adherence to medication was measured using an adapted version of a self-report measure (the Morisky measure) that had previously been validated against electronic monitoring bottles24 and that has been used in a trial of similar design in primary care. 21
Comparison with existing studies
Earlier small studies of patients who had not failed previous treatment had reported that mirtazapine in combination with a SSRI gave a greater improvement than monotherapy12 and that it was well tolerated with either a SSRI or a SNRI (venlafaxine). 13 The STAR*D study3 was a large four-stage evaluation of treatments for major depressive disorder that included assessment of the effectiveness of combinations of antidepressants for those who do not respond to single-drug treatment. Within the STAR*D study, there was a comparison of venlafaxine (a SNRI antidepressant) plus mirtazapine with tranylcypromine, a MAOI antidepressant. Participants included in this part of the STAR*D study can be characterised as having TRD in that they had not responded to either a single antidepressant or one of a number of augmentation treatments. There was a modest advantage for the combination of venlafaxine and mirtazapine over the MAOI. However, there is no placebo group in this comparison and it has been argued that the STAR*D study casts ‘limited light on the relative efficacy of combinations’. 85
The CO-MED (Combining medications to enhance depression outcomes) randomised trial14 compared the combination of venlafaxine and mirtazapine with escitalopram (a SSRI) and placebo in patients who had either recurrent depression or chronic depression lasting ≥ 2 years. There was no difference in response rates between the two groups and the burden of AEs was greater in the combined antidepressant group. There are some important differences between the CO-MED and MIR trials. Those recruited into the CO-MED trial were not necessarily taking an antidepressant at baseline and, therefore, do not conform to our definition of TRD. Those in the combined antidepressant group were treated with venlafaxine and mirtazapine rather than continuing on their established antidepressant, which in the MIR trial could be either a SSRI or a SNRI. In effect, the comparison in the CO-MED trial is between escitalopram and the mirtazapine/venlafaxine combination for a group of patients selected for chronicity and relative severity, rather than a treatment-resistant group.
Other combinations of antidepressants have also been studied. Two large RCTs examined the effect of mianserin (an analogue of mirtazapine and a drug that is infrequently used in UK practice) combined with another antidepressant compared with placebo, with inconclusive results. 86,87
We chose 30 mg of mirtazapine as being more typical of clinical practice in the primary care setting and representing a balance between efficacy and tolerability. We are not aware of clear data about a mirtazapine dose–response. Although some mirtazapine studies offer the option of increasing mirtazapine to 45 mg, the picture in combination studies is mixed. Blier et al. 13 used a fixed dose of mirtazapine of 30 mg. The CO-MED study14 allowed titration up to 45 mg but the average final dose was only 20 mg. Carpenter et al. 11 used a dose of up to 30 mg but most stayed on 15 mg, whereas the STAR*D10 trial aimed to titrate the dose of mirtazapine up to 45 mg and achieved a mean exit dose of 36 mg. Therefore, the dose used in our study was broadly in line with what has been used in previous augmentation studies, but, given the equivocal results, it is possible that an option to increase the dose to 45 mg might have increased efficacy in those not responding to 30 mg.
Lopes Rocha et al. 88 performed a systematic review and meta-analysis of studies that assessed the effect of antidepressant combinations for major depression in patients with incomplete response to an initial antidepressant. Only five studies with a total of 483 participants satisfied their inclusion criteria. Only one of these involved the addition of mirtazapine to another antidepressant. This study compared the addition of mirtazapine to a SSRI with placebo in a group of 26 patients who had not responded to ≥ 4 weeks of monotherapy. 11 Lopes Rocha et al. 88 argue that the small number of trials and methodological drawbacks of those trials preclude definitive conclusions about the effectiveness of combing antidepressants. The British Association for Psychopharmacology guidelines comment that ‘Clinical experience and open studies indicate that tolerability and safety are usually good, but there is a lack of controlled data examining the efficacy of most combinations’. 85 A search of the current literature has not found any further studies of the addition of mirtazapine to another antidepressant in a treatment-resistant group. The MIR trial, thus, represents a robust contribution to the evidence on the effectiveness of combination therapy in this group.
Comparison with non-pharmacological interventions for treatment-resistant depression
Although this is an extensive field and it is not practical to address it in a comprehensive way in this context, it is important to mention CoBalT. 21 This is a study that compared the addition of CBT to usual care for TRD, including the continuation of an antidepressant, and much of the design and recruitment strategy of the MIR trial is based on that of CoBalT. As a result, the populations in these two studies are more similar than in many TRD studies because of the wide variability in definitions of TRD in the literature, and a comparison is, therefore, of interest. In CoBalT, CBT was found to be more clinically effective and cost-effective than usual care and the OR of response in the intervention group was 3.26 (95% CI 2.10 to 5.06) at 24 weeks. However, participants in the usual care group in CoBalT were not blind to treatment allocation and were not offered any additional treatment, including placebo, and improved less than either group in the MIR trial. Although it is difficult to make a direct comparison between the two studies, it is important to note the relatively poor outcomes for TRD patients who are not offered further interventions.
Implications for current practice and suggestions for further research
Half of those who take antidepressants in an adequate dose for an adequate duration remain depressed. 3,22 This represents a substantial burden of illness and an unmet or inadequately met need. Although many can benefit from CBT, the evidence in TRD is limited to high-intensity CBT rather than briefer, less expensive therapies. High-intensity CBT is not readily available despite the IAPT programme,89 nor is it always effective in TRD. 21 In primary care, where most initial encounters between people with depression and clinicians take place, antidepressants are still very widely prescribed as a first-line treatment and the evidence suggests that more patients are taking them for longer. 90 Several pharmacological strategies have been developed to help those who do not respond to this first-line treatment but the evidence supporting them is not of very high quality. Most of the studies are small, many have not been replicated and there are multiple definitions of TRD. 85 There is, therefore, a lack of clear guidance for clinicians in an area of unmet need, which is particularly important in primary care because of the size of the TRD population. 22 As highlighted in the introduction to this study, there was reason to believe that the addition of mirtazapine to a SSRI or SNRI held therapeutic promise. The evidence of benefit from this study is not strong enough to justify the recommendation of this as a routine strategy in primary care for those who do not respond to a single antidepressant. However, the evidence does include the possibility of a clinically meaningful effect and it may be that clinicians and patients would wish to consider this combination in an informed discussion about risks and benefits.
There is a need for further research into combination and augmentation treatments for people with TRD and, in particular, the addition of drugs that can be prescribed safely and appropriately in primary care. There is modest evidence in support of augmentation of antidepressants with lithium91 and, likewise, atypical antipsychotics have been found to have some benefit. 7 Both strategies have a greater risk of AEs and, in the case of lithium, an additional administrative and cost burden of ‘near-patient testing’ in primary care.
There is also a need for research into novel treatments for depression that could be applied in TRD. For example, theta burst transcranial magnetic stimulation is a promising treatment for those who have not responded to antidepressants. 92 There is also emerging evidence of a possible causal role for cytokines in depression and that cytokine modulators may have a role as novel drugs for depression in subjects with chronic inflammation. 93
It is important that future studies in this area clearly define TRD and that this definition is appropriate to primary care. There is a need for evidence to inform the management of patients with TRD in primary care, where there is a substantial burden of severity and chronicity and where there is the greatest potential for access to care.
Acknowledgements
We are grateful to all the patients, practitioners and general practice surgery staff who took part in this research.
We would like to thank and acknowledge our sponsor and the members of our TSC [Professor John Norrie (Chairperson), Professor Clare Wilkinson, Professor Mike King and David Conlon] and DMEC [Professor Mike Crawford (Chairperson), Dr Clare Rutterford and Professor Chris Burton] for their valuable advice and support during the project.
Finally, we would also like to thank the following colleagues who have contributed to the MIR trial, through recruitment and retention of patients and provision of administrative support: Lone Gale, Marie Platt, Sue Jones and Michelle Phillips in Bristol; Caroline Jenkinson, Mary Carter and Ellie Kingsland in Exeter; Alison Lloyd, Tom Sheppard, Jan Wilson, William Cole, Alice Mackie, Kate Dixon, Heather Burroughs, Alicia Bratt and Caroline Reeves in Keele; and Helen Gibson, Carla Bratten, Michelle Platton, Alison Waring, Sarah Smith and Peter English in Hull.
Sponsorship and adoptions
The University of Bristol acted as sponsor for the study. The study was adopted by the Mental Health Research Network and the Primary Care Research Network, now collectively known as the Clinical Research Network.
Contributions of authors
David Kessler (Reader in Primary Care) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As chief investigator, he had overall responsibility for the management of the study and the Bristol site. He also provided training to the research team; drafted the introduction, main results and discussion chapters; supervised the analyses; and contributed to critical revision of the final report.
Alison Burns (Trial Manager/Research Associate; specialty: RCT management) was a trial manager, with responsibility for the day-to-day co-ordination of the trial. She also carried out data cleaning and analyses, drafted the methods chapter and contributed to drafting the results section and critical revision of the final report.
Debbie Tallon (Trial Manager; specialty: RCT management) was a trial manager, with responsibility for the day-to-day co-ordination of the trial. She also provided training to the research team and contributed to critical revision of the final report.
Glyn Lewis (Professor of Psychiatric Epidemiology; specialty: RCT design, management and analysis; psychiatry) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. He also contributed to critical revision of the final report.
Stephanie MacNeill (Research Fellow in Medical Statistics) wrote the statistical analysis plan, carried out data cleaning, conducted the analyses and contributed to drafting the results section and critical revision of the final report.
Jeff Round (Lecturer in Health Economics) conducted the cleaning of the health economic data, worked on the analyses and interpretation of the health economic data, drafted the chapter reporting the results from the health economic evaluation and contributed to critical revision of the final report.
William Hollingworth (Professor of Health Economics) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. He also worked on the analyses and interpretation of the health economic data and contributed to critical revision of the final report.
Carolyn Chew-Graham (Professor of General Practice Research; specialty: qualitative methods; primary care) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As a co-investigator, she had responsibility for the Keele site. She also led the qualitative study, drafted the qualitative chapter and contributed to critical revision of the final report.
Ian Anderson (Professor of Psychiatry) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As a co-investigator, he had responsibility for the Keele site. He also contributed to critical revision of the final report.
John Campbell (Professor of General Practice; specialty: qualitative methods; primary care) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As a co-investigator, he had responsibility for the Exeter site. He also contributed to critical revision of the final report.
Chris Dickens (Professor of Psychological Medicine) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As a co-investigator, he had responsibility for the Exeter site. He also contributed to critical revision of the final report.
Una Macleod (Professor of Primary Care Medicine) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As a co-investigator, she had responsibility for the Hull site. She also contributed to critical revision of the final report.
Simon Gilbody (Professor of Psychological Medicine and Health Services Research) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. As a co-investigator, he had responsibility for the Hull site. He also contributed to critical revision of the final report.
Simon Davies (Assistant Professor in Psychiatry) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. He also contributed to critical revision of the final report.
Tim J Peters (Professor of Primary Care Health Services Research and Head of School; specialty: RCT design, management and analysis; statistics) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. He also wrote the statistical analysis plan, supervised the data cleaning and analyses and contributed to critical revision of the final report.
Nicola Wiles (Reader in Epidemiology; specialty: RCT design, management and analysis) was responsible for the original proposal, securing funding for the trial and drafting the original proposal. She also supervised the analyses and contributed to critical revision of the final report.
All authors contributed to refinement of the trial protocol.
Publication
Kessler DS, MacNeill SJ, Tallon D, Lewis G, Peters TJ, Hollingworth W, et al. Mirtazapine added to SSRIs or SNRIs for treatment resistant depression in primary care: phase III randomised placebo controlled trial (MIR). BMJ 2018;363:k4218.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Mathers C, Loncar D. Updated Projections of Global Mortality and Burden of Disease, 2002–2030: Data Sources, Methods and Results (Working Paper). Geneva: World Health Organization; 2005.
- Prescriptions Dispensed in the Community: England 2004–14. Leeds: NHS Digital; 2015.
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163:28-40. https://doi.org/10.1176/appi.ajp.163.1.28.
- Depression in Adults: Recognition and Management. London: NICE; 2009.
- Connolly KR, Thase ME. If at first you don’t succeed: a review of the evidence for antidepressant augmentation, combination and switching strategies. Drugs 2011;71:43-64. https://doi.org/10.2165/11587620-000000000-00000.
- Nelson JC, Papakostas GI. Atypical antipsychotic augmentation in major depressive disorder: a meta-analysis of placebo-controlled randomized trials. Am J Psychiatry 2009;166:980-91. https://doi.org/10.1176/appi.ajp.2009.09030312.
- Spielmans G, Berman M, Linardatos E, Rosenlicht N, Perry A, Tsai A. Adjunctive atypical antipsychotic treatment for major depressive disorder: a meta-analysis of depression, quality of life, and safety outcomes. PLOS Med 2013;10. http://dx.doi.org/10.1371/journal.pmed.
- Zhou X, Ravindran A, Qin B, Del Giovane C, Li Q, Bauer M, et al. Comparative efficacy, acceptability, and tolerability of augmentation agents in treatment-resistant depression: systematic review and network meta-analysis. J Clin Psychiatry 2015;76:e487-98. https://doi.org/10.4088/JCP.14r09204.
- Cha DS, McIntyre RS. Treatment-emergent adverse events associated with atypical antipsychotics. Expert Opin Pharmacother 2012;13:1587-98. https://doi.org/10.1517/14656566.2012.656590.
- Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006;163:1161-72. https://doi.org/10.1176/ajp.2006.163.7.1161.
- Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry 2002;51:183-8. https://doi.org/10.1016/S0006-3223(01)01262-8.
- Blier P, Gobbi G, Turcotte JE, de Montigny C, Boucher N, Hébert C, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol 2009;19:457-65. https://doi.org/10.1016/j.euroneuro.2009.01.015.
- Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry 2010;167:281-8. https://doi.org/10.1176/appi.ajp.2009.09020186.
- Rush AJ, Trivedi MH, Stewart JW, Nierenberg AA, Fava M, Kurian BT, et al. Combining medications to enhance depression outcomes (CO-MED): acute and long-term outcomes of a single-blind randomized study. Am J Psychiatry 2011;168:689-701. https://doi.org/10.1176/appi.ajp.2011.10111645.
- Gartlehner G, Gaynes BN, Hansen RA, Thieda P, DeVeaugh-Geiss A, Krebs EE, et al. Comparative benefits and harms of second-generation antidepressants: background paper for the American College of Physicians. Ann Intern Med 2008;149:734-50. https://doi.org/10.7326/0003-4819-149-10-200811180-00008.
- World Psychiatric Association . Symposium on therapy resistant depression. Pharmacopsychiatry 1974;7.
- Conway CR, George MS, Sackeim HA. Toward an evidence-based, operational definition of treatment-resistant depression: when enough is enough. JAMA Psychiatry 2017;74:9-10. https://doi.org/10.1001/jamapsychiatry.2016.2586.
- The ICD-10 Classification of Mental and Behavioural Disorders: Diagnostic Criteria for Research. Geneva: WHO; 1992.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; n.d.
- Thase ME, Rush AJ. When at first you don't succeed: sequential strategies for antidepressant non-responders. J Clin Psychiatry 1997;58:23-9.
- Wiles N, Thomas L, Abel A, Ridgway N, Turner N, Campbell J, et al. Cognitive behavioural therapy as an adjunct to pharmacotherapy for primary care based patients with treatment resistant depression: results of the CoBalT randomised controlled trial. Lancet 2013;381:375-84. https://doi.org/10.1016/S0140-6736(12)61552-9.
- Thomas L, Kessler D, Campbell J, Morrison J, Peters TJ, Williams C, et al. Prevalence of treatment-resistant depression in primary care: cross-sectional data. Br J Gen Pract 2013;63:e852-8. https://doi.org/10.3399/bjgp13X675430.
- Tallon D, Wiles N, Campbell J, Chew-Graham C, Dickens C, Macleod U, et al. Mirtazapine added to selective serotonin reuptake inhibitors for treatment-resistant depression in primary care (MIR trial): study protocol for a randomised controlled trial. Trials 2016;17. https://doi.org/10.1186/s13063-016-1199-2.
- Morisky DE, Green LW, Levine DM. Concurrent and predictive validity of a self-reported measure of medication adherence. Med Care 1986;24:67-74. https://doi.org/10.1097/00005650-198601000-00007.
- Thomas L, Abel A, Ridgway N, Peters T, Kessler D, Hollinghurst S, et al. Cognitive behavioural therapy as an adjunct to pharmacotherapy for treatment resistant depression in primary care: the CoBalT protocol. Contemp Clin Trials 2012;33:312-19. https://doi.org/10.1016/j.cct.2011.10.016.
- George CF, Peveler RC, Heiliger S, Thompson C. Compliance with tricyclic antidepressants: the value of four different methods of assessment. Br J Clin Pharmacol 2000;50:166-71. https://doi.org/10.1046/j.1365-2125.2000.00244.x.
- Beck A, Steer R, Brown G. Manual for the Beck Depression Inventory-II. San Antonio, TX: Psychological Corporation; 1996.
- Lewis G, Pelosi AJ, Araya R, Dunn G. Measuring psychiatric disorder in the community: a standardized assessment for use by lay interviewers. Psychol Med 1992;22:465-86. https://doi.org/10.1017/S0033291700030415.
- Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13. https://doi.org/10.1046/j.1525-1497.2001.016009606.x.
- Spitzer RL, Kroenke K, Williams JB, Löwe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med 2006;166:1092-7. https://doi.org/10.1001/archinte.166.10.1092.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Ware J, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care 1996;34:220-33. https://doi.org/10.1097/00005650-199603000-00003.
- Uher R, Farmer A, Henigsberg N, Rietschel M, Mors O, Maier W, et al. Adverse reactions to antidepressants. Br J Psychiatry 2009;195:202-10. https://doi.org/10.1192/bjp.bp.108.061960.
- Saunders JB, Aasland OG, Babor TF, de la Fuente JR, Grant M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption – II. Addiction 1993;88:791-804. https://doi.org/10.1111/j.1360-0443.1993.tb02093.x.
- Schulz KF, Altman DG, Moher D. CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med 2010;152:726-32. https://doi.org/10.7326/0003-4819-152-11-201006010-00232.
- Brown S, Thorpe H, Hawkins K, Brown J. Minimization – reducing predictability for multi-centre trials whilst retaining balance within centre. Stat Med 2005;24:3715-27. https://doi.org/10.1002/sim.2391.
- National Institute for Health Research Clinical Research Network (NIHR CRN) . Good Clinical Practice (GCP): Reference Guide n.d. www.nihr.ac.uk/our-faculty/documents/GCP%20Reference%20Guide.pdf (accessed 8 February 2018).
- The Rules Governing Medicinal Products in the European Union. Brussels: European Commission, Directorate General Enterprise and Industry; 2010.
- Depression: Management of Depression in Primary and Secondary Care. London: NICE; 2004.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960;23:56-62. https://doi.org/10.1136/jnnp.23.1.56.
- Button KS, Kounali D, Thomas L, Wiles NJ, Peters TJ, Welton NJ, et al. Minimal clinically important difference on the Beck Depression Inventory-II according to the patient’s perspective. Psychol Med 2015;45:3269-79. https://doi.org/10.1017/S0033291715001270.
- Schafer JL. Multiple imputation: a primer. Stat Methods Med Res 1999;8:3-15. https://doi.org/10.1177/096228029900800102.
- van Buuren S, Boshuizen HC, Knook DL. Multiple imputation of missing blood pressure covariates in survival analysis. Stat Med 1999;18:681-94. https://doi.org/10.1002/(SICI)1097-0258(19990330)18:6<681::AID-SIM71>3.0.CO;2-R.
- Dunn G, Maracy M, Tomenson B. Estimating treatment effects from randomized clinical trials with noncompliance and loss to follow-up: the role of instrumental variable methods. Stat Methods Med Res 2005;14:369-95. https://doi.org/10.1191/0962280205sm403oa.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use . ICH Harmonised Tripartite Guideline. Clinical Safety Data Management: Definitions and Standards for Expedited Reporting 1994.
- World Medical Association . WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects n.d. www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed 8 February 2018).
- Data Protection Act 1998. London: The Stationery Office; 1998.
- Prescription Cost Analysis (PCA) Data: Monthly Data. London; 2016.
- Guide to the Methods of Technology Appraisal 2013. London: NICE; 2013.
- Devlin N, Shah K, Feng Y, Mulhern B, Van Hout B. Valuing Health-Related Quality of Life: An EQ-5D-5L Value Set for England. London: Office of Health Economics; 2016.
- National Schedule of Reference Costs: The Main Schedule. London: DHSC; 2016.
- Curtis L. Unit Costs of Health and Social Care 2011. Canterbury: Personal Social Services Research Unit, University of Kent; 2011.
- Curtis L. Unit Costs of Health and Social Care 2014. Canterbury: Personal Social Services Research Unit, University of Kent; 2014.
- Curtis L. Unit Costs of Health and Social Care 2013. Canterbury: Personal Social Services Research Unit, University of Kent; 2013.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2016. Canterbury: Personal Social Services Research Unit, University of Kent; 2016.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2015. Canterbury: Personal Social Services Research Unit, University of Kent; 2015.
- Wiles N, Thomas L, Abel A, Barnes M, Carroll F, Ridgway N, et al. Clinical effectiveness and cost-effectiveness of cognitive behavioural therapy as an adjunct to pharmacotherapy for treatment-resistant depression in primary care: the CoBalT randomised controlled trial. Health Technol Assess 2014;18. https://doi.org/10.3310/hta18310.
- Costing Statement: Exercise Referral Schemes to Promote Physical Activity: Implementing NICE Guidance PH54. London: NICE; 2014.
- Turner J, O’Cathain A, Knowles E, Nicholl J, Tosh J, Sampson F, et al. Evaluation of NHS 111 Pilot Sites: Final Report. Sheffield: University of Sheffield; 2012.
- Curtis L. Unit Costs of Health and Social Care 2010. Canterbury: Personal Social Services Research Unit, University of Kent; 2010.
- Annual Survey of Hours and Earnings: 2016 Provisional Results. London: ONS; 2016.
- Valuing Informal Adultcare in the UK. London: ONS; 2013.
- Department for Work and Pensions . National Minimum Wage and National Living Wage Rates n.d. www.gov.uk/national-minimum-wage-rates (accessed 12 March 2018).
- Snowden A, Marland G. No decision about me without me: concordance operationalised. J Clin Nurs 2013;22:1353-60. https://doi.org/10.1111/j.1365–2702.2012.04337.x.
- Moradveisi L, Huibers M, Renner F, Arntz A. The influence of patients’ preference/attitude towards psychotherapy and antidepressant medication on the treatment of major depressive disorder. J Behav Ther Exp Psychiatry 2014;45:170-7. https://doi.org/10.1016/j.jbtep.2013.10.003.
- Brown C, Battista DR, Bruehlman R, Sereika SS, Thase ME, Dunbar-Jacob J. Beliefs about antidepressant medications in primary care patients: relationship to self-reported adherence. Med Care 2005;43:1203-7. https://doi.org/10.1097/01.mlr.0000185733.30697.f6.
- Lam RW. Patients’ preferences and counselling for depression in primary care. Lancet 2001;357:575-6. https://doi.org/10.1016/S0140-6736(00)04076-9.
- van Schaik DJ, Klijn AF, van Hout HP, van Marwijk HW, Beekman AT, de Haan M, et al. Patients’ preferences in the treatment of depressive disorder in primary care. Gen Hosp Psychiatry 2004;26:184-9. https://doi.org/10.1016/j.genhosppsych.2003.12.001.
- Houle J, Villaggi B, Beaulieu MD, Lespérance F, Rondeau G, Lambert J. Treatment preferences in patients with first episode depression. J Affect Disord 2013;147:94-100. https://doi.org/10.1016/j.jad.2012.10.016.
- Gum AM, Areán PA, Hunkeler E, Tang L, Katon W, Hitchcock P, et al. Depression treatment preferences in older primary care patients. Gerontologist 2006;46:14-22. https://doi.org/10.1093/geront/46.1.14.
- Givens JL, Datto CJ, Ruckdeschel K, Knott K, Zubritsky C, Oslin DW, et al. Older patients’ aversion to antidepressants. A qualitative study. J Gen Intern Med 2006;21:146-51. https://doi.org/10.1111/j.1525-1497.2005.00296.x.
- Verbeek-Heida PM, Mathot EF. Better safe than sorry – why patients prefer to stop using selective serotonin reuptake inhibitor (SSRI) antidepressants but are afraid to do so: results of a qualitative study. Chronic Illn 2006;2:133-42.
- van Geffen EC, Hermesen JH, Heerdink ER, Egberts AC, Verbeek-Heida PM, van Hulten R. The decision to continue or discontinue treatment: experiences and beliefs of users of selective serotonin-uptake inhibitors in the initial months – a qualitative study. Res Social Adm Pharm 2011;7:134-50. https://doi.org/10.1016/j.sapharm.2010.04.001.
- Leydon GM, Rodgers L, Kendrick T. A qualitative study of patient views on discontinuing long-term selective serotonin reuptake inhibitors. Fam Pract 2007;24:570-5. https://doi.org/10.1093/fampra/cmm069.
- Malpass A, Shaw A, Sharp D, Walter F, Feder G, Ridd M, et al. ‘Medication career’ or ‘moral career’? The two sides of managing antidepressants: a meta-ethnography of patients’ experience of antidepressants. Soc Sci Med 2009;68:154-68. https://doi.org/10.1016/j.socscimed.2008.09.068.
- Barnes M, Wiles N, Morrison J, Kessler D, Williams C, Kuyken W, et al. Exploring patients’ reasons for declining contact in a cognitive behavioural therapy randomised controlled trial in primary care. Br J Gen Pract 2012;62:e371-7. https://doi.org/10.3399/bjgp12X641492.
- O’Cathain A, Thomas KJ, Drabble SJ, Rudolph A, Goode J, Hewison J. Maximising the value of combining qualitative research and randomised controlled trials in health research: the QUAlitative Research in Trials (QUART) study – a mixed methods study. Health Technol Assess 2014;18. https://doi.org/10.3310/hta18380.
- Henwood KL, Pidgeon NF. Qualitative research and psychological theorizing. Br J Psychol 1992;83:97-111. https://doi.org/10.1111/j.2044-8295.1992.tb02426.x.
- Garfield SJS, Husson F, Jackline A, Bischler A, Norton C, Franklin BD. Lay involvement in the analysis of qualitative data in health services research: a descriptive study. Res Involvement and Engagement 2016;2:1-12. https://doi.org/10.1186/s40900-016-0041-z.
- Hughes-Morley A, Young B, Hempel RJ, Russell IT, Waheed W, Bower P. What can we learn from trial decliners about improving recruitment? Qualitative study. Trials 2016;17. https://doi.org/10.1186/s13063-016-1626-4.
- Schofield P, Crosland A, Waheed W, Waquas A, Aseem S, Gask L, et al. Patients’ views of antidepressants: from first experiences to becoming expert. Br J Gen Pract 2011;61:142-8. https://doi.org/10.3399/bjgp11X567045.
- Altman DG, Schulz KF, Moher D, Egger M, Davidoff F, Elbourne D, et al. The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Ann Intern Med 2001;134:663-94. https://doi.org/10.7326/0003-4819-134-8-200104170-00012.
- Zwarenstein M, Treweek S, Gagnier JJ, Altman DG, Tunis S, Haynes B, et al. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ 2008;337. https://doi.org/10.1136/bmj.a2390.
- Depression: The NICE Guideline on the Treatment and Management of Depression in Adults. Updated Edition. London: British Psychological Society and Royal College of Psychiatrists; 2010.
- Cleare A, Pariante CM, Young AH, Anderson IM, Christmas D, Cowen PJ, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2008 British Association for Psychopharmacology guidelines. J Psychopharmacol 2015;29:459-525. https://doi.org/10.1177/0269881115581093.
- Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ. Benefits from mianserin augmentation of fluoxetine in patients with major depression non-responders to fluoxetine alone. Acta Psychiatr Scand 2001;103:66-72. https://doi.org/10.1034/j.1600-0447.2001.00148.x.
- Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertraline treatment. A randomised study of extended duration of treatment, dose increase or mianserin augmentation. Psychopharmacology (Berl) 2002;161:143-51. https://doi.org/10.1007/s00213-002-0999-0.
- Lopes Rocha F, Fuzikawa C, Riera R, Ramos MG, Hara C. Antidepressant combination for major depression in incomplete responders – a systematic review. J Affect Disord 2013;144:1-6. https://doi.org/10.1016/j.jad.2012.04.048.
- Talking Therapies: A Four-year Plan of Action. London: DHSC; 2011.
- McCrea RL, Sammon CJ, Nazareth I, Petersen I. Initiation and duration of selective serotonin reuptake inhibitor prescribing over time: UK cohort study. Br J Psychiatry 2016;209:421-6. https://doi.org/10.1192/bjp.bp.115.166975.
- Bauer M, Dell’osso L, Kasper S, Pitchot W, Dencker Vansvik E, Köhler J, et al. Extended-release quetiapine fumarate (quetiapine XR) monotherapy and quetiapine XR or lithium as add-on to antidepressants in patients with treatment-resistant major depressive disorder. J Affect Disord 2013;151:209-19. https://doi.org/10.1016/j.jad.2013.05.079.
- George MS, Lisanby SH, Avery D, McDonald WM, Durkalski V, Pavlicova M, et al. Daily left prefrontal transcranial magnetic stimulation therapy for major depressive disorder: a sham-controlled randomized trial. Arch Gen Psychiatry 2010;67:507-16. https://doi.org/10.1001/archgenpsychiatry.2010.46.
- Kappelmann N, Lewis G, Dantzer R, Jones PB, Khandaker GM. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions [published online ahead of print October 18 2016]. Mol Psychiatry 2016. https://doi.org/10.1038/mp.2016.167.
- Mirtazapine Actavis 15 mg, FCTs. Summary of Product Characteristics. Actavis; 2015.
Appendix 1 MIR trial documents
Adequate doses
A list of commonly used antidepressants with adequate doses for the MIR trial is provided in Table 25.
| Antidepressant type and name | Minimum adequate daily dose (mg) |
|---|---|
| SSRIs | |
| Citalopram | 20 |
| Escitalopram | 10 |
| Fluoxetine | 20 |
| Fluvoxamine | 100 |
| Paroxetine | 20 |
| Sertraline | 100 |
| SNRIs | |
| Duloxetine | 60 |
| Venlafaxine | 75 |
A full schedule of questionnaires used in the MIR trial is provided in Table 26.
| Questionnaire | Time point | ||||||
|---|---|---|---|---|---|---|---|
| Postal screening | Baseline | 2-week telephone call | 6 weeks | 12 weeks | 24 weeks | 12 months | |
| Consent form | Y | ||||||
| BDI | Y | Y | Y | Y | Y | Y | |
| Biographic and demographic data, including psychiatric history, life events, social support, alcohol use | Y | Y | |||||
| Views on treatment | Y | ||||||
| Medication | Y | Y | Y | Y | Y | Y | |
| Morisky questionnaire (adapted) | Y | Y | Y | Y | Y | Y | |
| CIS-R | Y | ||||||
| PHQ-9 | Y | Y | Y | Y | |||
| GAD-7 | Y | Y | Y | Y | |||
| EQ-5D-5L | Y | Y | Y | Y | |||
| SF-12 | Y | Y | Y | Y | |||
| Economics | Y | Y | Y | ||||
| Health events (SAEs) | Y | Y | Y | Y | |||
| ASEC | Y | Y | Y | ||||
| 2-week check | Y | ||||||
| Blinding questions | Y | ||||||
| Exit questionnaire | Y | ||||||
Adverse reactions of mirtazapine as listed in the SMPC (February 2015) are provided in Box 2.
-
Increase in appetite:b very common (> 1/10 patients).
-
Increase in weight:b very common (> 1/10 patients).
-
Hypnotraemia: frequency not known. a
-
Dry mouth: very common (> 1/10 patients).
-
Nausea:c common (1/100 patients to 1/10 patients).
-
Diarrhoea:d common (1/100 patients to 1/10 patients).
-
Vomiting:d common (1/100 patients to 1/10 patients).
-
Constipation:b common (1/100 patients to 1/10 patients).
-
Oral hypoaesthesia: uncommon (1/1000 patients to 1/100 patients).
-
Pancreatitis: rare (1/10,000 patients to 1/1000 patients).
-
Mouth oedema: frequency not known. a
-
Increased salivation: frequency not known. a
-
Headache:d very common (> 1/10 patients).
-
Lethargy:b common (1/100 patients to 1/10 patients).
-
Dizziness: common (1/100 patients to 1/10 patients).
-
Tremor: common (1/100 patients to 1/10 patients).
-
Paraesthesiae:d uncommon (1/1000 patients to 1/100 patients).
-
Restless legs: uncommon (1/1000 patients to 1/100 patients).
-
Syncope: uncommon (1/1000 patients to 1/100 patients).
-
Myoclonus: rare (1/10,000 patients to 1/1000 patients).
-
Convulsions: frequency not known. a
-
Serotonin syndrome: frequency not known. a
-
Oral paraesthesia: frequency not known. a
-
Dysarthria: frequency not known. a
-
Abnormal dreams: common (1/100 patients to 1/10 patients).
-
Confusion: common (1/100 patients to 1/10 patients).
-
Nightmares:d uncommon (1/1000 patients to 1/100 patients).
-
Mania: uncommon (1/1000 patients to 1/100 patients).
-
Agitation:d uncommon (1/1000 patients to 1/100 patients).
-
Hallucinations: uncommon (1/1000 patients to 1/100 patients).
-
Psychomotor restlessness (including akathisia and hyperkinesia): uncommon (1/1000 patients to 1/100 patients).
-
Aggression: rare (1/10,000 patients to 1/1000 patients).
-
Back pain:b common (1/100 patients to 1/10 patients).
-
Arthralgia: common (1/100 patients to 1/10 patients).
-
Myalgia: common (1/100 patients to 1/10 patients).
-
Rhabdomyolysis: frequency not known. a
-
Orthostatic hypotension: common (1/100 patients to 1/10 patients).
-
Hypotension:d uncommon (1/1000 patients to 1/100 patients).
-
Elevations in serum transaminase: rare (1/10,000 patients to 1/1000 patients).
-
Peripheral oedema:b common (1/100 patients to 1/10 patients).
-
Fatigue: common (1/100 patients to 1/10 patients).
-
Exanthema:d common (1/100 patients to 1/10 patients).
-
Stevens–Johnson syndrome: frequency not known. a
-
Dermatitis bullous: frequency not known. a
-
Erythema multiforme: frequency not known. a
-
Toxic epidermal necrolysis: frequency not known. a
-
Urinary retention: frequency not known. a
-
Inappropriate antidiuretic hormone secretion: frequency not known. a
The frequency of adverse reactions from spontaneous reporting, when no cases were observed in the randomised placebo-controlled patient trials, is classified as ‘not known’.
In clinical trials, these events occurred statistically significantly more frequently during treatment with mirtazapine than with placebo.
In clinical trials, these events occurred statistically more frequently during treatment with placebo than with mirtazapine.
In clinical trials, these events occurred more frequently during treatment with placebo than with mirtazapine, but not statistically significantly more frequently.
Dose reduction generally does not lead to less somnolence/sedation but can jeopardise antidepressant efficacy.
On treatment with antidepressants in general, anxiety and insomnia (which may be symptoms of depression) can develop. Under mirtazapine treatment, development or aggravation of anxiety and insomnia has been reported.
Cases of suicidal ideation and suicidal behaviours have been reported during mirtazapine therapy or early after treatment discontinuation.
Note
Any other symptom, side effect or AE listed in the SMPC or the BNF will not be regarded as unexpected.
Appendix 2 Results
| Participants | Total, N | Age | Female | ||
|---|---|---|---|---|---|
| Number with available data | Mean (SD), years | Number with available data | n (%) | ||
| Excluded | 7932 | 7750 | 53.2 (18.9) | 7749 | 5166 (66.7) |
| Potential participants | 18,966 | 18,966 | 49.8 (15.6) | 18,962 | 13,445 (70.9) |
| Participants | Total, N | Age | Female | ||
|---|---|---|---|---|---|
| Number with available data | Mean (SD), years | Number with available data | n (%) | ||
| Did not respond | 12,134 | 12,134 | 47.1 (15.3) | 12,131 | 8459 (69.7) |
| Declined | 4702 | 4702 | 56.6 (15.0) | 4702 | 3562 (75.8) |
| Accepted | 2180 | 2180 | 50.4 (14.3) | 2179 | 1456 (66.8) |
| Participants | Total, N | Age | Female | ||
|---|---|---|---|---|---|
| Number with available data | Mean (SD), years | Number with available data | n (%) | ||
| Not responded, not eligible or declined | 1183 | 1183 | 49.6 (14.8) | 1183 | 769 (65.0) |
| Eligible | 1116 | 1116 | 50.2 (13.8) | 1116 | 773 (69.3) |
| Recruitment statistics | Centre | Total | |||
|---|---|---|---|---|---|
| Bristol | Exeter | Manchester/Keele | Hull/York | ||
| Number of practices | 33 | 22 | 31 | 20 | 106 |
| Invitations/GP referrals, n | 5318 | 4042 | 5000 | 4772 | 19,132 |
| GP referrals, n | 83 | 46 | 27 | 10 | 166 |
| Total invitations, n | 5235 | 3996 | 4973 | 4762 | 18,966 |
| Number returned | 1896 | 1561 | 1957 | 1468 | 6882 |
| Number (%) accepted | 688 (36.3) | 518 (33.2) | 623 (31.8) | 517 (35.2) | 2346 (34.1) |
| Assessment screening questionnaire completed, n (%) | 573 (83.2) | 425 (82.0) | 523 (83.9) | 422 (81.6) | 1943 (82.8) |
| Eligible for baseline assessment, n (%) | 345 (60.2) | 251 (59.1) | 287 (54.9) | 233 (55.2) | 1116 (57.4) |
| Baseline assessments, n | 285 | 198 | 130 | 138 | 751 |
| Randomisations | |||||
| Number | 177 | 122 | 82 | 99 | 480 |
| Percentage of baseline assessments | 62 | 62 | 63 | 72 | 64 |
| Withdrawal and reasons | Randomised to, n (%) | Total (N = 480), n (%) | |
|---|---|---|---|
| Usual care + mirtazapine (N = 241) | Usual care + placebo (N = 239) | ||
| Any withdrawal from the trial medication | 6 (2.5) | 12 (5.0) | 18 (3.8) |
| Reason | |||
| Health | 1 (0.4) | 2 (0.8) | 3 (0.6) |
| Lack of willingness to participate | 3 (1.2) | 8 (3.3) | 11 (2.3) |
| Other | 1 (0.4) | 2 (0.8) | 3 (0.6) |
| Allocated treatment group | Days between randomisation and withdrawal from the trial medication (estimated when dates not provided) | Withdrawal decision made by | Reason | Completed further follow-up |
|---|---|---|---|---|
| Usual care + placebo | 91 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 40 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 102 | Subject | Other | No |
| Usual care + mirtazapine | 40 | Study team | Lack of willingness to participate | No |
| Usual care + placebo | 40 | Subject | Lack of willingness to participate | No |
| Usual care + mirtazapine | 176 | Subject | Health | No |
| Usual care + placebo | 52 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 41 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 471a | Subject | Lack of willingness to participate | No |
| Usual care + mirtazapine | 168 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 245 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 175 | Subject | Other | No |
| Usual care + mirtazapine | 229 | GP | Other | No |
| Usual care + mirtazapine | 29 | Subject | Lack of willingness to participate | No |
| Usual care + mirtazapine | 78 | Subject | Other | No |
| Usual care + placebo | 60 | Subject | Lack of willingness to participate | No |
| Usual care + placebo | 178 | Subject | Health | No |
| Usual care + placebo | 298 | Subject | Health | No |
| Protocol deviations | Randomised to, n (%) | Total (N = 480), n (%) | |
|---|---|---|---|
| Usual care + mirtazapine (N = 241) | Usual care + placebo (N = 239) | ||
| Any protocol deviation | 10 (4.1) | 10 (4.2) | 20 (4.2) |
| Medicine handling | 1 (0.4) | 3 (1.3) | 4 (0.8) |
| Unblinding error | 1 (0.4) | 0 (0) | 1 (0.2) |
| Delivery of medication | 5 (2.1) | 5 (2.1) | 10 (2.1) |
| Ran out of medication and GP prescribed mirtazapine | 0 (0) | 1 (0.4) | 1 (0.2) |
| SAE reporting | 2 (0.8) | 0 (0) | 2 (0.4) |
| Prescribed mirtazapine | 1 (0.4) | 1 (0.4) | 2 (0.4) |
| Allocated treatment group | Centre | Further details (exact nature dependent on type of deviation) |
|---|---|---|
| Usual care + placebo | Bristol | Medication handling error; one participant received the medication of another placebo participant |
| Usual care + mirtazapine | Exeter | Unblinding error |
| Usual care + placebo | Exeter | Unable to collect trial medication from GP |
| Usual care + placebo and usual care + mirtazapine | Exeter | Medication handling error at GP surgery; a participant allocated to receive placebo received medication of a mirtazapine participant and vice versa |
| Usual care + mirtazapine | Hull | Data protection breach |
| Usual care + mirtazapine | Keele | Delayed SAE report |
| Usual care + mirtazapine | Keele | GP prescribed mirtazapine – protocol breach |
| Usual care + placebo | Keele | GP prescribed mirtazapine |
| Usual care + mirtazapine | Hull | Participant ran out of medication |
| Usual care + mirtazapine | Hull | Participant ran out of medication |
| Usual care + placebo | Hull | Delay/difficulty contacting patient regarding medication collection |
| Usual care + placebo | Hull | Participant ran out of medication; emergency unblinding |
| Usual care + mirtazapine | Hull | Script requested but not issued. Participant almost ran out of medication |
| Usual care + placebo | Hull | Participant ran out of medication; GP prescribed mirtazapine |
| Usual care + placebo | Keele | Participant ran out of medication |
| Usual care + mirtazapine | Hull | Participant ran out of medication |
| Usual care + placebo | Hull | Participant ran out of medication |
| Usual care + placebo | Hull | Participant ran out of medication/no contact |
| Usual care + mirtazapine | Hull | Participant ran out of medication |
| Antidepressant medication | Dose (mg), median (range) | Allocated treatment group, n (%) | Total (N = 480), n (%) | |
|---|---|---|---|---|
| Mirtazapine + usual care (N = 241) | Placebo + usual care (N = 239) | |||
| Citalopram/Cipramil (SSRI) | 30 (20–60) | 98 (40.7) | 102 (42.7) | 200 (41.7) |
| Duloxetine/Cymbalta/Yentreve (SNRI) | 60 (60–60) | 1 (0.4) | 1 (0.4) | 2 (0.4) |
| Escitalopram/Cipralax (SSRI) | 20 (10–20) | 5 (2.1) | 3 (1.3) | 8 (1.7) |
| Fluoxetine/Prozac (SSRI) | 40 (20–60) | 56 (23.2) | 62 (25.9) | 118 (24.6) |
| Paroxetine/Seroxat (SSRI) | 30 (20–50) | 12 (5.0) | 6 (2.5) | 18 (3.8) |
| Sertraline/Lustral (SSRI) | 100 (100–300) | 44 (18.3) | 42 (17.6) | 86 (17.9) |
| Venlafaxine/Efexor (SNRI) | 150 (75–300) | 25 (10.4) | 23 (9.6) | 48 (10.0) |
| Variable | Data, n (%) | |
|---|---|---|
| Present (N = 402) | Missing (N = 78) | |
| Stratification variable | ||
| Centre | ||
| Bristol | 152 (37.8) | 25 (32.1) |
| Exeter | 104 (25.9) | 18 (23.1) |
| Manchester/Keele | 65 (16.2) | 17 (21.8) |
| Hull/York | 81 (20.1) | 18 (23.1) |
| Minimisation variables | ||
| Female | 275 (68.4) | 57 (73.1) |
| Baseline BDI-II score (points) | ||
| 14–25 | 140 (34.8) | 16 (20.5) |
| 26–34 | 129 (32.1) | 27 (34.6) |
| ≥ 35 | 133 (33.1) | 35 (44.9) |
| Currently receiving psychological services | 45 (11.2) | 17 (21.8) |
| Sociodemographic variables | ||
| Age (years), mean (SD) | 51.2 (12.9) | 44.6 (13.1) |
| Ethnic group | ||
| White | 391 (97.3) | 77 (98.7) |
| Mixed | 7 (1.7) | 1 (1.3) |
| Asian/British Asian | 2 (0.5) | 0 (0.0) |
| Other | 2 (0.5) | 0 (0.0) |
| Marital status | ||
| Married/cohabiting | 237 (59.0) | 40 (51.3) |
| Single | 84 (20.9) | 16 (20.5) |
| Separated | 14 (3.5) | 4 (5.1) |
| Divorced | 43 (10.7) | 16 (20.5) |
| Widowed | 24 (6.0) | 2 (2.6) |
| Employment status | ||
| Employed | 199 (49.5) | 45 (57.7) |
| Unemployed | 203 (50.5) | 33 (42.3) |
| Educational attainment | ||
| Degree or equivalent | 82 (20.4) | 13 (16.7) |
| HNC, HND, SVQ (Level 4 or 5) or RSA Higher Diploma | 34 (8.5) | 11 (14.1) |
| A level, Higher or equivalent [GNVQ/NVQ Advanced, GSVQ/SVQ (Level 3) or RSA Advanced Diploma] | 76 (18.9) | 14 (17.9) |
| GSCE, Standard Grade, O level or equivalent | 130 (32.3) | 20 (25.6) |
| No formal qualification | 80 (19.9) | 20 (25.6) |
| Housing | ||
| Home owner | 229 (57.0) | 34 (43.6) |
| Tenant | 146 (36.3) | 38 (48.7) |
| Living with relative or friend | 26 (6.5) | 6 (7.7) |
| Other | 1 (0.2) | 0 (0.0) |
| Financial well-being | ||
| Comfortable/OK | 199 (49.5) | 25 (32.1) |
| Just about getting by or worse | 203 (50.5) | 53 (67.9) |
| Alcohol consumption (units), median (IQR) | 2.0 (1.0–4.0) | 2.5 (1.0–5.0) |
| Number of life events in the previous 6 months, mean (SD) | 1.0 (1.0) | 1.2 (1.2) |
| Social support score, mean (SD) | 12.6 (4.0) | 11.6 (4.6) |
| Providing care for someone who is disabled | 56 (13.9) | 11 (14.1) |
| How many children aged < 5 years live with you? | ||
| None | 364 (90.5) | 68 (87.2) |
| 1 or 2 | 35 (8.7) | 10 (12.8) |
| ≥ 3 | 3 (0.7) | 0 (0.0) |
| Treatment preference | ||
| Do you have a preference for either group? | ||
| Prefer to receive mirtazapine | 239 (59.5) | 50 (64.1) |
| Prefer to receive the placebo | 2 (0.5) | 0 (0.0) |
| Do not mind either way | 161 (40.0) | 28 (35.9) |
| If you were to be allocated to the other group, how disappointed would you be? | ||
| Very | 32 (13.3) | 9 (18.0) |
| Moderately | 77 (32.0) | 16 (32.0) |
| A little bit | 79 (32.8) | 10 (20.0) |
| Not really | 53 (22.0) | 15 (30.0) |
| Measures of depression | ||
| Suffered from depression in the past | 331 (82.3) | 65 (83.3) |
| Family history of depression | 211 (63.7) | 52 (80.0) |
| Previous referral to a psychiatrist for depression | 105 (31.7) | 26 (40.0) |
| Number of prior episodes of depression | ||
| None | 8 (2.4) | 0 (0.0) |
| 1 | 18 (5.4) | 4 (6.2) |
| 2–4 | 139 (42.0) | 22 (33.8) |
| ≥ 5 | 166 (50.2) | 39 (60.0) |
| Duration of current course of antidepressants | ||
| < 6 months | 39 (9.7) | 7 (9.0) |
| ≥ 6 months | 363 (90.3) | 71 (91.0) |
| ICD-10 primary diagnosis | ||
| Mild | 70 (17.4) | 12 (15.4) |
| Moderate | 235 (58.5) | 47 (60.3) |
| Severe | 97 (24.1) | 19 (24.4) |
| Secondary psychiatric diagnosis according to the CIS-R | ||
| No diagnosis identified | 6 (1.5) | 1 (1.3) |
| Mixed anxiety and depressive disorder (mild) | 48 (11.9) | 9 (11.5) |
| Generalised anxiety disorder (mild) | 5 (1.2) | 0 (0.0) |
| Mixed anxiety and depressive disorder | 96 (23.9) | 18 (23.1) |
| Specific (isolated) phobia | 18 (4.5) | 7 (9.0) |
| Social phobia | 17 (4.2) | 5 (6.4) |
| Agoraphobia | 18 (4.5) | 3 (3.8) |
| Generalised anxiety disorder | 157 (39.1) | 27 (34.6) |
| Panic disorder | 37 (9.2) | 8 (10.3) |
| BDI-II score (points), mean (SD) | 30.5 (9.8) | 34.0 (10.2) |
| GAD-7 score (points), mean (SD) | 10.7 (4.8) | 12.2 (4.8) |
| PHQ-9 score (points), mean (SD) | 16.2 (5.5) | 17.4 (5.3) |
| EQ-5D-5L score (points), mean (SD) | 0.7 (0.2) | 0.7 (0.2) |
| SF-12 aggregate physical functioning score (points), mean (SD) | 29.3 (9.6) | 25.6 (10.1) |
| SF-12 aggregate mental functioning score (points), mean (SD) | 45.2 (13.8) | 49.3 (12.2) |
| CIS-R score, mean (SD) | 27.5 (8.3) | 28.8 (8.3) |
| Suicidal ideation (CIS-R thoughts/plans) | ||
| No suicidal thoughts | 173 (43.0) | 27 (34.6) |
| Patient feels life is not worth living | 86 (21.4) | 17 (21.8) |
| Suicidal thoughts | 143 (35.6) | 34 (43.6) |
| Variable | Data, n (%) | |
|---|---|---|
| Present (N = 388) | Missing (N = 92) | |
| Stratification variable | ||
| Centre | ||
| Bristol | 152 (39.2) | 25 (27.2) |
| Exeter | 95 (24.5) | 27 (29.3) |
| Manchester/Keele | 66 (17.0) | 16 (17.4) |
| Hull/York | 75 (19.3) | 24 (26.1) |
| Minimisation variables | ||
| Female | 266 (68.6) | 66 (71.7) |
| Baseline BDI-II score (points) | ||
| 14–25 | 138 (35.6) | 18 (19.6) |
| 26–34 | 124 (32.0) | 32 (34.8) |
| ≥ 35 | 126 (32.5) | 42 (45.7) |
| Currently receiving psychological services | 42 (10.8) | 20 (21.7) |
| Sociodemographic variables | ||
| Age (years), mean (SD) | 51.7 (12.5) | 43.8 (14.0) |
| Ethnic group | ||
| White | 381 (98.2) | 87 (94.6) |
| Mixed | 4 (1.0) | 4 (4.3) |
| Asian/British Asian | 1 (0.3) | 1 (1.1) |
| Other | 2 (0.5) | 0 (0.0) |
| Marital status | ||
| Married/cohabiting | 234 (60.3) | 43 (46.7) |
| Single | 77 (19.8) | 23 (25.0) |
| Separated | 10 (2.6) | 8 (8.7) |
| Divorced | 44 (11.3) | 15 (16.3) |
| Widowed | 23 (5.9) | 3 (3.3) |
| Employment status | ||
| Employed | 196 (50.5) | 48 (52.2) |
| Unemployed | 192 (49.5) | 44 (47.8) |
| Educational attainment | ||
| Degree or equivalent | 77 (19.8) | 18 (19.6) |
| HNC, HND, SVQ (Level 4 or 5) or RSA Higher Diploma | 33 (8.5) | 12 (13.0) |
| A level, Higher or equivalent [GNVQ/NVQ Advanced, GSVQ/SVQ (Level 3) or RSA Advanced Diploma] | 70 (18.0) | 20 (21.7) |
| GSCE, Standard Grade, O level or equivalent | 126 (32.5) | 24 (26.1) |
| No formal qualification | 82 (21.1) | 18 (19.6) |
| Housing | ||
| Home owner | 229 (59.0) | 34 (37.0) |
| Tenant | 136 (35.1) | 48 (52.2) |
| Living with relative or friend | 22 (5.7) | 10 (10.9) |
| Other | 1 (0.3) | 0 (0.0) |
| Financial well-being | ||
| Comfortable/OK | 196 (50.5) | 28 (30.4) |
| Just about getting by or worse | 192 (49.5) | 64 (69.6) |
| Alcohol consumption, median (IQR) | 2.0 (1.0–4.0) | 3.0 (1.0–5.0) |
| Number of life events in the past 6 months, mean (SD) | 1.0 (1.0) | 1.3 (1.2) |
| Social support score, mean (SD) | 12.6 (4.0) | 11.8 (4.5) |
| Providing care for someone who is disabled | 54 (13.9) | 13 (14.1) |
| How many children aged < 5 years live with you? | ||
| None | 353 (91.0) | 79 (85.9) |
| 1 or 2 | 32 (8.2) | 13 (14.1) |
| ≥ 3 | 3 (0.8) | 0 (0.0) |
| Treatment preference | ||
| Do you have a preference for either group? | ||
| Prefer to receive mirtazapine | 231 (59.5) | 58 (63.0) |
| Prefer to receive the placebo | 2 (0.5) | 0 (0.0) |
| Do not mind either way | 155 (39.9) | 34 (37.0) |
| If you were to be allocated to the other group, how disappointed would you be? | ||
| Very | 26 (11.2) | 15 (25.9) |
| Moderately | 75 (32.2) | 18 (31.0) |
| A little bit | 79 (33.9) | 10 (17.2) |
| Not really | 53 (22.7) | 15 (25.9) |
| Measures of depression | ||
| Suffered from depression in the past | 318 (82.0) | 78 (84.8) |
| Family history of depression | 207 (65.1) | 56 (71.8) |
| Previous referral to a psychiatrist for depression | 97 (30.5) | 34 (43.6) |
| Number of prior episodes of depression | ||
| None | 6 (1.9) | 2 (2.6) |
| 1 | 19 (6.0) | 3 (3.8) |
| 2–4 | 137 (43.1) | 24 (30.8) |
| ≥ 5 | 156 (49.1) | 49 (62.8) |
| Duration of current course of antidepressants | ||
| < 6 months | 36 (9.3) | 10 (10.9) |
| ≥ 6 months | 352 (90.7) | 82 (89.1) |
| ICD-10 primary diagnosis | ||
| Mild | 69 (17.8) | 13 (14.1) |
| Moderate | 229 (59.0) | 53 (57.6) |
| Severe | 90 (23.2) | 26 (28.3) |
| Secondary psychiatric diagnosis according to the CIS-R | ||
| No diagnosis identified | 6 (1.5) | 1 (1.1) |
| Mixed anxiety and depressive disorder (mild) | 46 (11.9) | 11 (12.0) |
| Generalised anxiety disorder (mild) | 4 (1.0) | 1 (1.1) |
| Mixed anxiety and depressive disorder | 95 (24.5) | 19 (20.7) |
| Specific (isolated) phobia | 19 (4.9) | 6 (6.5) |
| Social phobia | 16 (4.1) | 6 (6.5) |
| Agoraphobia | 18 (4.6) | 3 (3.3) |
| Generalised anxiety disorder | 152 (39.2) | 32 (34.8) |
| Panic disorder | 32 (8.2) | 13 (14.1) |
| BDI-II score (points), mean (SD) | 30.3 (9.7) | 34.4 (10.2) |
| GAD-7 score (points), mean (SD) | 10.7 (4.7) | 12.2 (4.8) |
| PHQ-9 score (points), mean (SD) | 16.1 (5.5) | 17.6 (5.2) |
| EQ-5D-5L score (points), mean (SD) | 0.7 (0.2) | 0.6 (0.2) |
| SF-12 aggregate physical functioning score (points), mean (SD) | 29.5 (9.5) | 25.3 (10.2) |
| SF-12 aggregate mental functioning score (points), mean (SD) | 45.2 (13.8) | 48.5 (12.6) |
| CIS-R score, mean (SD) | 27.2 (8.1) | 29.7 (8.8) |
| Suicidal ideation (CIS-R thoughts/plans) | ||
| No suicidal thoughts | 164 (42.3) | 36 (39.1) |
| Patient feels life is not worth living | 91 (23.5) | 12 (13.0) |
| Suicidal thoughts | 133 (34.3) | 44 (47.8) |
| Category | Number of participants | Number of events | Number of participants who stopped treatment | Intensity (n) | Relatedness (n) | Expectedness (n) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Not related/unlikely | Possibly related | Probably/definitely related | Expected | Unexpected | N/A | ||||
| Randomised to usual care + mirtazapine (N = 241) | ||||||||||||
| Allergy | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 2 | 0 | 1 | 1 | 0 |
| Anticholinergic | 16 | 16 | 3 | 14 | 2 | 0 | 1 | 5 | 10 | 13 | 3 | 0 |
| Central nervous system (drowsy) | 41 | 41 | 19 | 36 | 5 | 0 | 0 | 11 | 30 | 41 | 0 | 0 |
| Central nervous system (headache) | 4 | 4 | 1 | 4 | 0 | 0 | 2 | 1 | 1 | 3 | 1 | 0 |
| Central nervous system (other) | 4 | 4 | 1 | 3 | 1 | 0 | 0 | 3 | 1 | 4 | 0 | 0 |
| Central nervous system (TIA) | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Central nervous system (unpleasant dreams) | 11 | 11 | 2 | 9 | 2 | 0 | 0 | 4 | 7 | 9 | 2 | 0 |
| CVS | 4 | 4 | 1 | 3 | 1 | 0 | 4 | 0 | 0 | 0 | 3 | 1 |
| Dental | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Discontinuation syndrome when stopped | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| Endocrine – new diabetes | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| ENT | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Epistaxis | 2 | 2 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| Eyes | 2 | 2 | 1 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| GI (nausea) | 2 | 2 | 0 | 2 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 0 |
| GI (other) | 5 | 5 | 1 | 4 | 1 | 0 | 2 | 3 | 0 | 2 | 3 | 0 |
| GUS | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Gynaecological | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Haematological | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Infective | 2 | 2 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| Metabolic – increase in appetite | 2 | 2 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 2 | 0 | 0 |
| Metabolic – weight increased | 24 | 24 | 7 | 20 | 4 | 0 | 0 | 2 | 22 | 24 | 0 | 0 |
| Musculoskeletal (other) | 7 | 7 | 3 | 7 | 0 | 0 | 5 | 2 | 0 | 2 | 5 | 0 |
| Musculoskeletal – restless legs/cramps | 3 | 3 | 0 | 3 | 0 | 0 | 1 | 2 | 0 | 3 | 0 | 0 |
| Musculoskeletal trauma | 5 | 6 | 1 | 3 | 3 | 0 | 5 | 0 | 1 | 1 | 5 | 0 |
| Oedema | 9 | 9 | 3 | 7 | 2 | 0 | 6 | 2 | 1 | 2 | 7 | 0 |
| Pancreatitis | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| Psychiatric | 8 | 8 | 4 | 5 | 2 | 1 | 3 | 4 | 1 | 5 | 2 | 1 |
| Respiratory | 2 | 2 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 1 | 1 |
| Skin and subcutaneous tissue disorder | 3 | 3 | 2 | 3 | 0 | 0 | 2 | 1 | 0 | 2 | 1 | 0 |
| Randomised to usual care + placebo (N = 239) | ||||||||||||
| Anticholinergic | 4 | 4 | 0 | 3 | 1 | 0 | 1 | 0 | 3 | 3 | 1 | 0 |
| Central nervous system (drowsy) | 5 | 5 | 1 | 5 | 0 | 0 | 0 | 1 | 4 | 5 | 0 | 0 |
| Central nervous system (headache) | 4 | 4 | 0 | 3 | 1 | 0 | 0 | 3 | 1 | 4 | 0 | 0 |
| Central nervous system (other) | 2 | 2 | 0 | 2 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| Central nervous system (unpleasant dreams) | 9 | 9 | 1 | 7 | 2 | 0 | 0 | 6 | 3 | 8 | 1 | 0 |
| Dental | 2 | 2 | 1 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| Endocrine disorders | 2 | 2 | 1 | 1 | 1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 |
| ENT | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| GI (nausea) | 3 | 3 | 0 | 3 | 0 | 0 | 1 | 1 | 1 | 2 | 1 | 0 |
| GI (other) | 6 | 6 | 0 | 5 | 1 | 0 | 3 | 2 | 1 | 3 | 3 | 0 |
| Gynaecological | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Haematological | 3 | 3 | 1 | 3 | 0 | 0 | 3 | 0 | 0 | 0 | 3 | 0 |
| Hepatobiliary disorder | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Infective | 5 | 5 | 1 | 5 | 0 | 0 | 5 | 0 | 0 | 0 | 4 | 1 |
| Metabolic – increase in appetite | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| Metabolic – weight increased | 7 | 7 | 0 | 5 | 2 | 0 | 1 | 1 | 5 | 6 | 1 | 0 |
| Musculoskeletal (other) | 8 | 8 | 0 | 7 | 1 | 0 | 7 | 1 | 0 | 1 | 7 | 0 |
| Musculoskeletal – restless legs/cramps | 3 | 3 | 0 | 3 | 0 | 0 | 2 | 1 | 0 | 1 | 2 | 0 |
| Musculoskeletal trauma | 6 | 6 | 2 | 3 | 3 | 0 | 6 | 0 | 0 | 0 | 4 | 2 |
| Non-cardiological chest pains | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Oedema | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| Psychiatric | 5 | 5 | 0 | 4 | 1 | 0 | 0 | 4 | 1 | 5 | 0 | 0 |
| Psychosexual | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Renal and urinary disorders | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Respiratory | 3 | 3 | 2 | 3 | 0 | 0 | 3 | 0 | 0 | 0 | 3 | 0 |
| Skin and subcutaneous tissue disorders | 4 | 4 | 1 | 3 | 1 | 0 | 3 | 0 | 1 | 1 | 3 | 0 |
| Sleep disturbance | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Sweating | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Category | Number of participants | Number of events | Number of participants who stopped treatment | Intensity (n) | Relatedness (n) | Expectedness (n) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Not related/unlikely | Possibly related | Probably/definitely related | Expected | Unexpected | N/A | ||||
| Randomised to usual care + mirtazapine (N = 241) | ||||||||||||
| Central nervous system (TIA) | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| CVS | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Dental | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Gynaecological | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Pancreatitis | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| Psychiatric | 2 | 2 | 2 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 |
| Respiratory | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Randomised to usual care + placebo (N = 239) | ||||||||||||
| Infective | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Musculoskeletal trauma | 2 | 2 | 1 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 2 |
| Category | Number of participants | Number of events | Number of participants who stopped treatment | Intensity (n) | Relatedness (n) | Expectedness (n) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Not related/unlikely | Possibly related | Probably/definitely related | Expected | Unexpected | N/A | ||||
| Randomised to usual care + mirtazapine (N = 241) | ||||||||||||
| Anticholinergic | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Central nervous system (drowsy) | 3 | 3 | 1 | 3 | 0 | 0 | 0 | 3 | 0 | 3 | 0 | 0 |
| Central nervous system (other) | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Central nervous system (TIA) | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Central nervous system (unpleasant dreams) | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| CVS | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Death because of drug overdose (heroin) | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Dental | 2 | 2 | 1 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| Endocrine – new diabetes | 2 | 2 | 1 | 1 | 1 | 0 | 0 | 2 | 0 | 0 | 2 | 0 |
| ENT | 3 | 3 | 1 | 1 | 2 | 0 | 3 | 0 | 0 | 0 | 2 | 1 |
| Eyes | 2 | 2 | 1 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| GI (other) | 3 | 3 | 3 | 2 | 1 | 0 | 3 | 0 | 0 | 0 | 2 | 1 |
| GUS | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Gynaecological | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Haematological | 2 | 2 | 0 | 1 | 0 | 1 | 2 | 0 | 0 | 0 | 1 | 1 |
| Hepatobiliary disorder | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Infective | 6 | 6 | 4 | 3 | 2 | 1 | 6 | 0 | 0 | 0 | 3 | 3 |
| Metabolic – weight increased | 2 | 2 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 1 |
| Musculoskeletal (other) | 8 | 9 | 6 | 7 | 2 | 0 | 8 | 1 | 0 | 1 | 5 | 3 |
| Musculoskeletal – restless legs/cramps | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| Musculoskeletal trauma | 8 | 8 | 1 | 7 | 1 | 0 | 8 | 0 | 0 | 0 | 7 | 1 |
| Oedema | 3 | 3 | 1 | 3 | 0 | 0 | 2 | 0 | 1 | 1 | 2 | 0 |
| Psychiatric | 3 | 3 | 0 | 2 | 1 | 0 | 2 | 1 | 0 | 2 | 1 | 0 |
| Respiratory | 2 | 2 | 1 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 1 | 1 |
| Skin and subcutaneous tissue disorders | 3 | 4 | 1 | 3 | 1 | 0 | 4 | 0 | 0 | 0 | 2 | 2 |
| Randomised to usual care + placebo (N = 239) | ||||||||||||
| Vitamin B12 deficiency | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Central nervous system (headache) | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Central nervous system (other) | 2 | 3 | 1 | 1 | 2 | 0 | 3 | 0 | 0 | 0 | 0 | 3 |
| Central nervous system (unpleasant dreams) | 2 | 2 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 2 | 0 | 0 |
| Central nervous system epilepsy | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| CFS/ME | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| CVS | 3 | 3 | 1 | 2 | 1 | 0 | 3 | 0 | 0 | 0 | 2 | 1 |
| DVT | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Endocrine disorders | 2 | 2 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 2 | 0 |
| Endocrine – new diabetes | 2 | 2 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 2 | 0 | 0 |
| ENT | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Epistaxis | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| GI (other) | 4 | 5 | 4 | 3 | 2 | 0 | 5 | 0 | 0 | 0 | 2 | 3 |
| Gynaecological | 4 | 4 | 0 | 3 | 1 | 0 | 4 | 0 | 0 | 0 | 2 | 2 |
| Hepatobiliary disorder | 2 | 2 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 1 | 1 |
| Infective | 8 | 8 | 1 | 7 | 1 | 0 | 8 | 0 | 0 | 0 | 8 | 0 |
| Metabolic – weight increased | 2 | 2 | 1 | 2 | 0 | 0 | 1 | 0 | 1 | 2 | 0 | 0 |
| Musculoskeletal (other) | 5 | 5 | 2 | 5 | 0 | 0 | 5 | 0 | 0 | 0 | 5 | 0 |
| Musculoskeletal –restless legs/cramps | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| Musculoskeletal trauma | 10 | 10 | 3 | 6 | 4 | 0 | 10 | 0 | 0 | 0 | 7 | 3 |
| Oedema | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Psychiatric | 2 | 2 | 1 | 2 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| Renal and urinary disorders | 2 | 2 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| Respiratory | 2 | 2 | 1 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 1 | 1 |
| Category | Number of participants | Number of events | Number of participants who stopped treatment | Intensity (n) | Relatedness (n) | Expectedness (n) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Not related/unlikely | Possibly related | Probably/definitely related | Expected | Unexpected | N/A | ||||
| Randomised to usual care + mirtazapine (N = 241) | ||||||||||||
| Central nervous system (TIA) | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Death because of drug overdose (heroin) | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| ENT | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| GI (other) | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| GUS | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Gynaecological | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Haematological | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Hepatobiliary disorder | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Infective | 3 | 3 | 2 | 0 | 2 | 1 | 3 | 0 | 0 | 0 | 0 | 3 |
| Metabolic – weight increased | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Musculoskeletal (other) | 3 | 3 | 3 | 1 | 2 | 0 | 3 | 0 | 0 | 0 | 0 | 3 |
| Musculoskeletal trauma | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Psychiatric | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| Respiratory | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Skin and subcutaneous tissue disorders | 1 | 2 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 2 |
| Randomised to usual care + placebo (N = 239) | ||||||||||||
| Central nervous system (other) | 2 | 3 | 1 | 1 | 2 | 0 | 3 | 0 | 0 | 0 | 0 | 3 |
| Central nervous system epilepsy | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| CVS | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| ENT | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| GI (other) | 2 | 3 | 3 | 1 | 2 | 0 | 3 | 0 | 0 | 0 | 0 | 3 |
| Gynaecological | 2 | 2 | 0 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 2 |
| Hepatobiliary disorder | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Musculoskeletal trauma | 3 | 3 | 1 | 1 | 2 | 0 | 3 | 0 | 0 | 0 | 0 | 3 |
| Respiratory | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
Appendix 3 Health economic documents
All unit costs were derived from the August 2016 Prescription Cost Analysis. 48
| Drug name | Cost per quantity (£) |
|---|---|
| Amitriptyline 10-mg tablets | 0.03 |
| Amitriptyline 25-mg tablets | 0.03 |
| Amitriptyline 50-mg tablets | 0.12 |
| Amitriptyline hydrochloride 10-mg tablets | 0.03 |
| Chlordiazepoxide 10-mg capsules | 0.50 |
| Cipralex 20-mg tablets (Lundbeck Ltd) | 0.90 |
| Cipramil 20-mg tablets (Lundbeck Ltd) | 0.32 |
| Citalopram 10-mg tablets | 0.03 |
| Citalopram 20-mg tablets | 0.03 |
| Citalopram 20-mg tablets (price for 28 tablets) | 0.83 |
| Citalopram 40-mg tablets | 0.03 |
| Citalopram hydrobromide 10-mg tablets | 0.03 |
| Citalopram hydrobromide 20-mg tablets | 0.03 |
| Citalopram hydrobromide 40-mg tablets | 0.03 |
| Colestyramine 4-g oral powder sachets | 0.40 |
| Diazepam 2-mg tablets | 0.03 |
| Diazepam 5-mg tablets | 0.03 |
| Dosulepin 75-mg tablets | 0.05 |
| Duloxetine 20-mg gastro-resistant capsules | 0.20 |
| Duloxetine 30-mg gastro-resistant capsules | 0.17 |
| Duloxetine 60-mg gastro-resistant capsules | 0.16 |
| Escitalopram 10-mg tablets | 0.04 |
| Escitalopram 20-mg tablets | 0.05 |
| Escitalopram 5-mg tablets | 0.04 |
| Fluoxetine 20-mg capsules | 0.03 |
| Fluoxetine 20-mg capsules | 0.03 |
| Fluoxetine 20-mg capsules (price for 30 capsules) | 0.93 |
| Fluoxetine 60-mg capsules | 0.34 |
| Fluoxetine hydrochloride capsules 20 mg | 0.03 |
| Haloperidol 5 mg/1 ml solution for injection ampoules | 3.47 |
| Lofepramine 70-mg tablets | 0.22 |
| Lofepramine 70-mg tablets (Actavis UK Ltd) | 0.22 |
| Lorazepam 1-mg tablets | 0.08 |
| Mirtazapine 15-mg tablets | 0.04 |
| Mirtazapine 30-mg tablets | 0.04 |
| Mirtazapine 15-mg orodispersible tablets | 0.04 |
| Mirtazapine 15-mg tablets | 0.04 |
| Mirtazapine 30-mg tablets | 0.04 |
| Mirtazapine 45-mg tablets | 0.05 |
| Nitrazepam 5-mg tablets | 0.04 |
| Nortriptyline 10-mg tablets | 0.48 |
| Oxactin 20-mg capsules (Discovery Pharmaceuticals Ltd) | 0.03 |
| Oxycodone 5 mg/5 ml sugar-free oral solution | 0.04 |
| Paroxetine 10-mg tablets | 0.60 |
| Paroxetine 10 mg/5 ml sugar-free oral suspension | 0.06 |
| Paroxetine 20-mg tablets | 0.05 |
| Paroxetine 30-mg tablets | 0.06 |
| Paroxetine hydrochloride 20-mg tablets | 0.05 |
| Priadel 200-mg modified-release tablets (Sanofi) | 0.03 |
| Priadel 400-mg modified-release tablets (Sanofi) | 0.04 |
| Promethazine hydrochloride 25-mg tablets | 0.49 |
| Quetiapine 150-mg modified-release tablets | 0.04 |
| Quetiapine 50-mg modified-release tablets | 0.04 |
| Reboxetine 4-mg tablets | 0.32 |
| Sertraline 100-mg tablets | 0.05 |
| Sertraline 100-mg tablets (price for 28 tablets) | 1.49 |
| Sertraline 50-mg tablets | 0.04 |
| Sertraline hydrochloride 100-mg tablets | 0.05 |
| Sertraline hydrochloride 50-mg tablets | 0.04 |
| Sodium alginate and potassium bicarbonate oral suspension, sugar free, peppermint 500 mg + 100 mg/5 ml | 5.11 |
| Sodium picosulfate sugar-free 5 mg/5 ml oral solution | 2.37 |
| Temazepam 10-mg tablets | 0.10 |
| Temazepam 20-mg tablets | 0.10 |
| Tonpular XL 75-mg capsules (Wockhardt UK Ltd) | 0.25 |
| Trazodone 50-mg capsules | 0.36 |
| Venaxx Xl M/R 75-mg capsules | 0.37 |
| Venaxx XL 75-mg capsules (AMCo) | 0.37 |
| Venlablue XL 150-mg capsules (Bluefish Pharmaceuticals AB) | 0.36 |
| Venlablue XL 75-mg capsules (Bluefish Pharmaceuticals AB) | 0.25 |
| Venlafaxine 75-mg tablets | 0.04 |
| Venlafaxine 150-mg modified-release capsules | 1.31 |
| Venlafaxine 150-mg modified-release tablets | 1.31 |
| Venlafaxine 225-mg modified-release tablets | 0.04 |
| Venlafaxine 37.5-mg tablets | 0.04 |
| Venlafaxine 75-mg modified-release capsules | 0.04 |
| Venlafaxine 75-mg modified-release tablets | 0.04 |
| Venlafaxine 75-mg tablets | 0.04 |
| Venlalic XL 150-mg tablets (DB Ashbourne Ltd) | 0.62 |
| Venlalic XL 225-mg tablets (DB Ashbourne Ltd) | 1.12 |
| Venlalic XL 75-mg tablets (DB Ashbourne Ltd) | 0.37 |
| Vensir XL 150-mg capsules (Morningside Healthcare Ltd) | 0.14 |
| Vensir XL 75-mg capsules (Morningside Healthcare Ltd) | 0.09 |
| Zolpidem 10-mg tablets | 0.04 |
| Zolpidem 5-mg tablets | 0.04 |
| Zopiclone 7.5-mg tablets | 0.04 |
| Zopiclone 3.75-mg tablets | 0.04 |
Appendix 4 Qualitative topic guides
Glossary
- Baseline recruitment
- A meeting with the research associate in which the research associate provided a full explanation of trial participation, took consent and completed the trial baseline case report form.
- Code-break
- A record held by the University Hospitals Bristol NHS Foundation Trust of the allocation of active drug and placebo (and medicine identification number) by patient identification number.
- Index consultation
- The routine consultation between the patient and the primary care clinician responsible for the patient’s routine care, in which the patient’s general practitioner identified that the patient was suffering from depression and had had at least 6 weeks of treatment with a selective serotonin reuptake inhibitor or a serotonin–noradrenaline reuptake inhibitor antidepressant. The clinician introduced the trial, took written consent for the patient to be contacted by the study team and carried out a detailed check of eligibility, including checking for potential drug interactions on the general practice information system.
- Medicine identification number
- The unique number assigned to the investigational medicinal product at manufacture (by the investigational medicinal product manufacturer, using the randomisation data provided by the Bristol Randomised Trials Collaboration) and assigned to the patient identification number according to the randomisation schedule provided to the University Hospitals Bristol NHS Foundation Trust by the Bristol Randomised Trials Collaboration.
- Medicine pack
- The packaging containing the investigational medicinal product uniquely identified by the medicine identification number.
- Patient identification number
- The unique number assigned to a recruited patient by the research associate following receipt of the completed reply slip.
- Patient information sheet
- The information sheet given to patients by the general practitioner during the consultation or posted to potential participants by the general practice. All patients were provided with the full patient information sheet. A summary patient information sheet was also sent to participants with their baseline appointment letter.
- Randomisation data
- A list of random numbers generated by the Bristol Randomised Trials Collaboration in line with the requirements of the trial sponsor and the medicine supplier [Sharp Clinical Services (UK) Ltd, Crickhowell, UK] and provided to Sharp Clinical Services (UK) Ltd (in a manner that maintained the complete blinding of the trial team) for their use in numbering the medicine packs that were provided to the four trial centres. The bottle numbers formed the identifiers on the open code-break document sent with each delivery of medication packs.
- Randomisation schedule
- Instructions provided by the Bristol Randomised Trials Collaboration to the four trial centres regarding active compared with placebo medicine allocation.
- Responsible clinician
- The general practitioner who took responsibility for the clinical management of a patient, for the initial confirmation of the patient’s eligibility to take part in the trial (final confirmation was the responsibility of the principal investigator) and for checking for possible drug interactions on the general practice information system.
- Site
- Sites were defined as the four recruiting centres in the MIR trial (for the purposes of research and development, Research Ethics Committee applications, etc.).
- Source data
- For the MIR trial, the source data were considered to be the data recorded on the case report form by the research associate and by the patient. When data had been entered directly online, without the use of paper forms, these were taken as the source data.
- Trial Participation Card
- Trial participants were requested to carry this with them while participating in the trial. It recorded the medicine and patient identification numbers to be used for emergency unblinding.
- Trial prescription
- This was the prescription for the trial medication, issued after confirmation that a patient was eligible and randomised, and was signed by the site’s clinical leader or nominated deputy. It was then sent directly to the trial pharmacy.
List of abbreviations
- 5-HT
- 5-hydroxytryptamine
- A&E
- accident and emergency
- AE
- adverse event
- ASEC
- Antidepressant Side-Effect Checklist
- BDI-II
- Beck Depression Inventory-II
- BNF
- British National Formulary
- BRTC
- Bristol Randomised Trials Collaboration
- CACE
- complier-average causal effect
- CBT
- cognitive–behavioural therapy
- CCG
- Clinical Commissioning Group
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CIS-R
- Clinical Interview Schedule – Revised
- CO-MED
- Combining medications to enhance depression outcomes
- CoBalT
- Cognitive behavioural therapy as an adjunct to pharmacotherapy for primary care patients with treatment resistant depression: a randomised controlled trial
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- DMEC
- Data Monitoring and Ethics Committee
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- GAD-7
- Generalised Anxiety Disorder-7
- GCP
- Good Clinical Practice
- GP
- general practitioner
- HDRS
- Hamilton Depression Rating Scale
- IAPT
- Improving Access to Psychological Therapies
- ICD-10
- International Statistical Classification of Diseases and Related Health Problems, Tenth Revision
- ICH
- International Conference on Harmonisation
- IQR
- interquartile range
- ITT
- intention to treat
- MAOI
- monoamine oxidase inhibitor
- MCID
- minimum clinically important difference
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MI
- multiple imputation
- MICE
- multiple imputation by chained equation
- NA
- noradrenaline
- NICE
- National Institute for Health and Care Excellence
- NMB
- net monetary benefit
- NNT
- number needed to treat
- ONS
- Office for National Statistics
- OR
- odds ratio
- PHQ-9
- Patient Health Questionnaire-9 items
- PI
- principal investigator
- PIS
- patient information sheet
- PPIE
- patient and public involvement and engagement
- PSS
- personal social services
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SD
- standard deviation
- SE
- standard error
- SF-12
- Short Form questionnaire-12 items
- SMPC
- Summary of Medicinal Product Characteristics
- SNRI
- serotonin–noradrenaline reuptake inhibitor
- SOP
- standard operating procedure
- SSRI
- selective serotonin reuptake inhibitor
- STAR*D
- Sequenced Treatment Alternatives to Relieve Depression
- SUSAR
- suspected unexpected serious adverse reaction
- TIA
- transient ischaemic attack
- TRD
- treatment-resistant depression
- TSC
- Trial Steering Committee
- UHB
- University Hospitals Bristol NHS Foundation Trust
- WTP
- willingness to pay