

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 09/51/01. The contractual start date was in August 2011. The draft report began editorial review in December 2017 and was accepted for publication in June 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Athimalaipet V Ramanan reports grants from the National Institute for Health Research (NIHR) Health Technology Assessment (HTA) programme and Arthritis Research UK during the conduct of the study and others from AbbVie Inc. (Ludwigshafen, Germany) and the University Hospitals Bristol NHS Foundation Trust, outside the submitted work. He has received speaker fees from AbbVie Inc. and lectured in symposia, sponsored by AbbVie Inc.. He has also been on an Advisory Board organised by AbbVie Inc. and has been supported by AbbVie Inc. to attend European and American Rheumatology Society meetings. Andrew D Dick reports other from data and revenue sharing outside the submitted work for consultancy work, paid to University of Bristol by AbbVie Inc., Ashley P Jones, Andrew McKay, Anna Rosala-Hallas, Ben Hardwick, Helen Hickey, Naomi Rainford, Graeme Hickey, Ruwanthi Kolamunnage-Dona and Paula R Williamson report grants from the NIHR HTA programme and Arthritis Research UK during the conduct of the study, other from AbbVie Inc. and University Hospitals Bristol NHS Foundation Trust and personal fees from University of Liverpool, outside the submitted work. In addition, Paula R Williamson is the Director of the Clinical Trials Research Centre, which is the Clinical Trials Unit that managed the day-to-day running of this trial. Dyfrig Hughes and Patricia Woo report grants from the NIHR HTA programme and Arthritis Research UK during the conduct of the study and other from AbbVie Inc., outside the submitted work. Giovanna Culeddu and Eifiona Wood report grants from Arthritis Research UK during the conduct of the study. Sandrine Compeyrot-Lacassagne reports grants from the NIHR HTA programme and Arthritis Research UK during the conduct of the study, and grants and others from AbbVie Inc., outside the submitted work. Clive Edelsten reports grants from the NIHR HTA programme and Arthritis Research UK during the conduct of the study, and others and personal fees from AbbVie Inc., outside the submitted work. Michael W Beresford reports grants from the NIHR HTA programme and Arthritis Research UK during the conduct of the study and others from AbbVie Inc. and the University Hospitals Bristol NHS Foundation Trust, outside the submitted work.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Ramanan et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Scientific background
Children with juvenile idiopathic arthritis (JIA), the most common rheumatic disease in children, are at risk of inflammation of the uvea in the eye (uveitis). Overall, 20–25% of all paediatric uveitis is associated with JIA. 1,2 Several major risk factors are known for the development of uveitis in JIA, including oligoarticular pattern of arthritis, onset of arthritis at < 7 years of age and antinuclear antibody positivity. 3 Generally, in the initial stages of mild to moderate inflammation, the uveitis is entirely asymptomatic; therefore, current practice is to screen all children with JIA regularly for uveitis. Between 12% and 38% of patients with JIA will develop uveitis in the initial 7 years following the onset of arthritis. 4,5 Structural complications are present in 30–50% of children with JIA-associated uveitis at diagnosis. 6 Importantly, 50–75% of those children with severe uveitis will eventually develop visual impairment secondary to ocular complications including cataracts, glaucoma, band keratopathy and macular pathology. 7–9 Defining the severity of inflammation and structural complications in uveitis patients can now be more consistently described following Standardisation of Uveitis Nomenclature (SUN) guidelines. 10 These guidelines allow incorporation into randomised controlled trials and cohort studies. 10
Poor prognosticators of poor visual acuity include structural changes at presentation, need for intraocular surgery, posterior segment inflammation, abnormal intraocular pressure (IOP) and the failure to maintain long-term disease control as marked by persistent anterior chamber (AC) cell scores of ≥ 1+. 6–8,11 Despite current screening and therapeutic options (pre biologics), some 10–15% of children with JIA-associated uveitis may eventually develop bilateral visual impairment and will be certified legally blind. 12,13 It is, therefore, critical to find more effective therapeutic interventions.
Rationale for research
Methotrexate (MTX) is well established as the first-line disease-modifying agent in the management of JIA. 14,15 Topical corticosteroids are among the current approaches to treatment of mild JIA-associated uveitis. In children with moderate to severe JIA-associated uveitis, MTX is also effective. 16–18 However, there have been no prospective randomised placebo-controlled trials of MTX or corticosteroid regimens for JIA-associated uveitis.
A systematic review of the evidence of the use of MTX in JIA is restricted to joint involvement14 and does not include paediatric uveitis. Despite the scarce evidence, MTX has become the mainstay of treatment for JIA-uveitis. 19 However, up to 15–50% of children will have refractory uveitis in spite of optimal therapy with MTX. 16–18 In a small study,20 some 30% of patients started on MTX for JIA-associated uveitis did not achieve disease control during the first year of therapy and, even when remission was achieved with MTX, nearly 70% of patients will later relapse, suggesting that only 4 out of 22 (18%) patients achieved total remission. 13 In a Dutch study, only 12% were found to be in total remission 5 years after starting MTX. 20 In small, retrospective case series, other agents including ciclosporin and mycophenolate mofetil have been shown to be of partial benefit in controlling JIA-uveitis. 21,22 However, there is little evidence that they rescue MTX-refractory patients and their use is restricted because of intolerability and adverse reactions. Neither ciclosporin nor mycophenolate mofetil is very effective in controlling joint manifestations of JIA in children. 19
More recently, animal models and corroborative human evidence23 support the role of tumour necrosis factor alpha (TNF-α) in the aetiopathogenesis of uveitis and, moreover, the potential value of its inhibition as a therapeutic intervention. 24 Studies on experimental models of autoimmune uveitis have demonstrated that TNF-α plays a pivotal role in pathogenesis of intraocular inflammation,23 which has been borne out in treatment of adult uveitis. 24 In mouse models of anterior uveitis, deleting the TNF p55 receptor alone is as effective as combined tumour necrosis factor p55 and p75 knockout animal in demonstrating reduced ocular inflammation,25 equivalent to the effect of tumour necrosis factor p55 fusion protein. 26 In an animal model of uveitis, infliximab reduced disease severity,27 albeit at doses of 20 mg/kg.
Translating this to humans, several case series have been published demonstrating the efficacy of infliximab and adalimumab (Humira®; AbbVie Inc., Ludwigshafen, Germany) in the treatment of severe refractory uveitis in adults and children. 28–33 In contrast, etanercept has been reported not to halt the onset of uveitis or be more effective than placebo,34,35 and is less effective than infliximab in treating JIA-uveitis. 31,36,37 There are a number of reports of new-onset uveitis associated with etanercept use in JIA. 38 An adverse events (AEs) register-based study examining these cases determined that, although the frequency was greater for etanercept than for infliximab or adalimumab (n = 20, 4 and 2 cases, respectively), causality could not be established. 39 Etanercept is not considered to be effective in treating intraocular inflammation. 31
Intervention
Adalimumab is a fully human monoclonal antibody, engineered by gene technology that uses site-directed mutagenesis to enhance its binding efficiency to TNF-α. It does not contain non-human or artificial protein sequences. Adalimumab binds only to TNF-α and has an elimination half-life of approximately 2 weeks. The antibody has been studied extensively in vitro as well as in vivo and in animal toxicology experiments. A clinical trial of adalimumab as monotherapy or in combination with MTX in adult subjects with rheumatoid arthritis showed a significant clinical response. 40 In children with JIA, a multicentre randomised, double-blind stratified parallel-group trial has shown a significant benefit in children with active arthritis: disease flares (the primary end point) occurred in a significantly lower percentage of those receiving adalimumab than of those receiving placebo [13 of 30 (43%) vs. 20 of 28 (71%); p = 0.03]. 41
Studies in paediatric non-infectious uveitis have shown very promising results with adalimumab, with 21 out of 26 eyes from 14 children with JIA- or idiopathic-uveitis showing improvement in inflammation. 42 In another retrospective case series of 18 paediatric patients with uveitis, 88% had a substantial decrease in ocular inflammation and adalimumab showed corticosteroid-sparing potential. 28
At the time of starting the randomised controlled trial of the clinical effectiveness, SafetY and Cost-effectiveness of Adalimumab in Combination with MethOtRExate for the treatment of juvenile idiopathic arthritis associated uveitis (SYCAMORE), there were no prospective studies of efficacy and safety of anti-TNF-α agents in JIA-associated uveitis, or of their cost-effectiveness. In the randomised controlled trial of adalimumab in JIA that demonstrated efficacy and supported its safety, the most commonly reported AEs were infections and injection-site reactions. 41 Serious adverse events (SAEs) that were considered to be possibly related to the study drug by the investigator occurred in 14 patients. Seven of these included one case of bronchopneumonia, one of herpes simplex infection, one of pharyngitis and one of pneumonia, and two cases of herpes zoster infection. In this trial, there were no deaths, malignant conditions, opportunistic infections, cases of tuberculosis, demyelinating diseases or lupus-like reactions. 41 The fixed-dose model of fortnightly 20 mg of adalimumab (for 16 weeks) for children weighing < 30 kg and 40 mg for children weighing ≥ 30 kg selected for this trial is based on the data generated in the previously mentioned trial using the same dosing regimen. 41
Although there are no published economic evaluations of JIA-associated uveitis, there are a number of economic evaluations of anti-TNF-α agents (including adalimumab) in JIA. These are of interest but limited applicability, because they are not directly transferable for estimating the cost-effectiveness of treatment in the context of uveitis management. The only study to adopt a costing perspective of the NHS in the UK is Shepherd et al.,43 who constructed a cost–utility Markov model to compare abatacept, adalimumab, etanercept and tocilizumab44–47 in JIA using disease flare as the measure of efficacy. The analysis was based on four economic evaluations of biological disease-modifying antirheumatic drugs (DMARDs) in JIA. Utility values were sourced from the Prince et al. study. 47 The incremental cost-effectiveness ratios (ICERs) for adalimumab, etanercept, tocilizumab and abatacept, versus MTX, were £38,127, £32,256, £38,656 and £39,536 per quality-adjusted life-year (QALY), respectively. The model results were found to be most sensitive to changes in utility values and the differences in cost-effectiveness of the biological DMARDs were primarily due to differences in drug acquisition cost. A limitation common to economic analyses in JIA is the challenge of obtaining valid utility scores and extrapolation of effects over a longer time period, both of which can significantly influence cost-effectiveness. A recent economic evaluation of adalimumab and dexamethasone intravitreal implant (Ozurdex®; Allergan Ltd, Marlow, UK) for treating non-infectious intermediate uveitis, posterior uveitis or panuveitis in adults indicated that adalimumab was not cost-effective at £94,523 per QALY gained in active uveitis,48 but these findings may not be generalisable to children with active JIA-associated uveitis. The aim of the economic evaluation as part of the SYCAMORE trial was to assess the cost-effectiveness of adalimumab, based on utility and cost data acquired directly within the trial, and extrapolated using data on representative patients from routine care.
Objectives
The primary objective of the trial was to compare the clinical effectiveness of adalimumab in combination with MTX versus placebo with MTX alone, with regard to controlling disease activity in refractory uveitis associated with JIA.
The secondary objectives of the trial were to:
-
evaluate short-term safety and tolerability of adalimumab in combination with MTX versus MTX alone, with regard to ocular complications of treatment, AEs and laboratory assessments
-
determine quality of life and cost-effectiveness of adalimumab in combination with MTX versus MTX alone in severe uveitis associated with JIA
-
determine the clinical effectiveness of adalimumab in combination with MTX versus MTX alone, with regard to underlying JIA disease activity
-
determine the durability and magnitude of adalimumab efficacy response in sustaining inactive disease and achieving complete clinical remission
-
determine the long-term safety of adalimumab in combination with MTX versus MTX alone
-
assess the efficacy of treatment with adalimumab to permit concomitant medication reduction, in particular regional and parenteral steroids
-
develop a fully consented, trial-related tissue bank for subsequent investigation.
Chapter 2 Trial design and methods
Study design
This was a randomised, parallel-group, double-blind, placebo-controlled, multicentre clinical trial that compared the effects of adalimumab in combination with MTX versus placebo in combination with MTX in participants with active uveitis in association with JIA refractory to MTX monotherapy. Participants were randomised applying a ratio of 2 : 1 (in favour of adalimumab), stratified by centre.
Patients with persistently active JIA-associated uveitis (despite optimised MTX treatment for at least 12 weeks) were recruited from tertiary care centres throughout the UK.
A schematic of the study design can be seen in Figure 1.
FIGURE 1.
The SYCAMORE study design. OPD, outpatient department; QoL, quality of life.
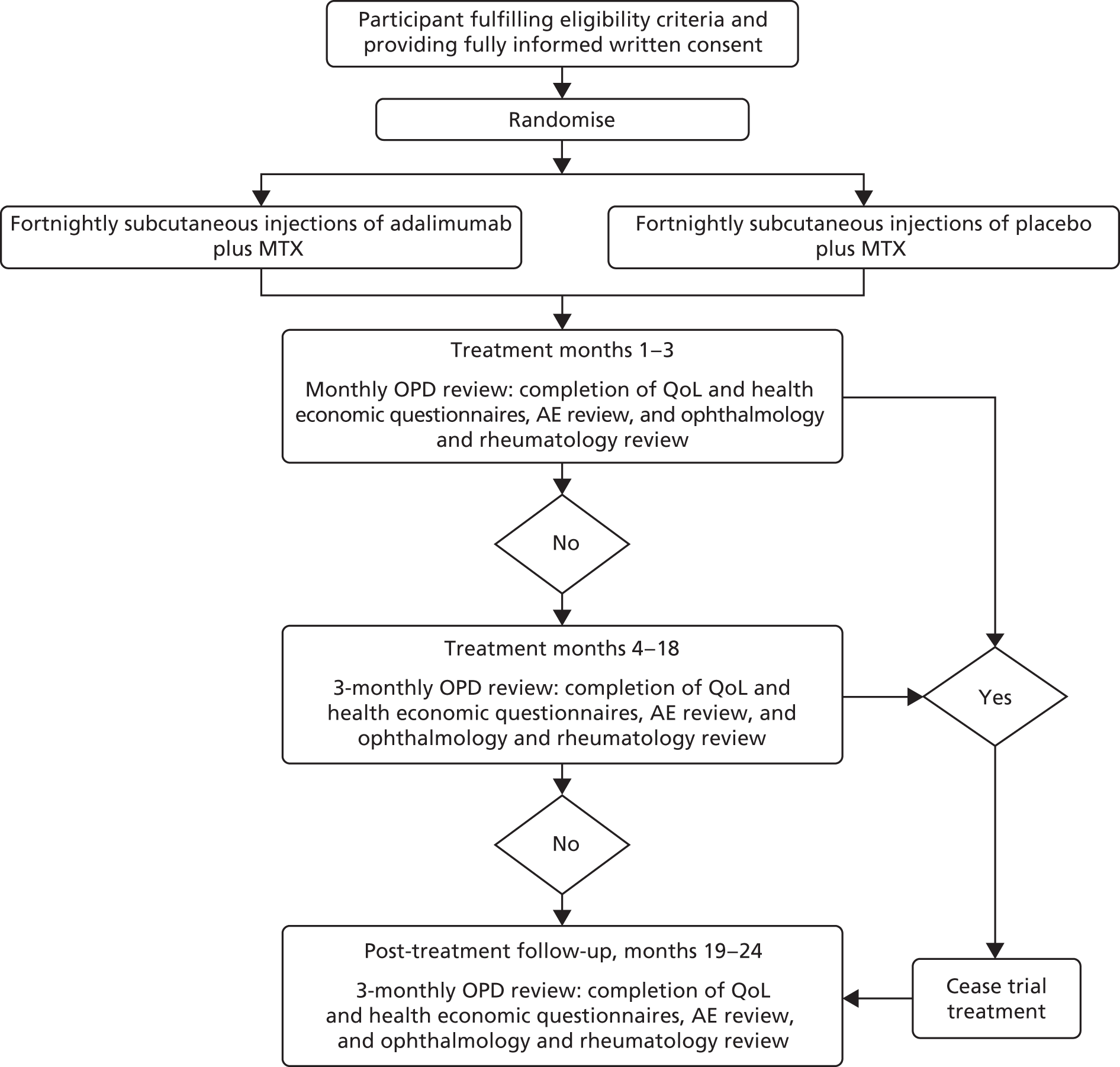
The trial protocol has previously been published in an open access journal. 49
Trial registration and ethics
The trial was registered on EudraCT on 1 June 2010 (EudraCT number 2010-021141-41) and received Clinical Trials Authorisation from The Medicines and Healthcare products Regulatory Agency on 9 May 2011 (Clinical Trials Authorisation reference number 12893/0228/001). The trial, and all subsequent protocol amendments, were reviewed and authorised by the Medicines and Healthcare products Regulatory Agency.
The trial protocol was not initiated until it had received the favourable opinion of the Main Research Ethics Committee (London – Hampstead Research Ethics Committee 11/LO/0425) on 24 June 2011. It was then reviewed at the research and development offices at participating sites.
The trial and any subsequent amendments were reviewed and approved by the National Research Ethics Service Committee London – Hampstead.
The trial was listed on the International Standard Randomised Controlled Trials Number (ISRCTN) register on the 2 September 2011 (ISRCTN10065623) and adopted onto the Medicines for Children Research Network and co-adopted onto the Ophthalmology portfolio on the 10 May 2011.
Participant inclusion and exclusion criteria
Patients who met the following eligibility criteria were considered for the trial.
Inclusion criteria
-
Children and young people aged ≥ 2 and < 18 years who fulfilled the International League of Associations for Rheumatology (ILAR) diagnostic criteria for JIA (all subgroups that had uveitis).
-
At the time of trial screening, the participant must have had active anterior uveitis, defined as a ‘sustained grade of cellular infiltrate in AC of the SUN criteria10 grade > 1+ during the preceding 12 weeks of therapy despite MTX and corticosteroid (both systemic and topical) therapy’. 10 The latest date of SUN grade score must have been the date of the screening visit.
-
Participants must have failed MTX (minimum dose of 10–20 mg/m2 once per week, with a maximum dose of 25 mg per participant). The participant must have been on MTX for at least 12 weeks and have been on a stable dose for 4 weeks prior to screening visit. (Omission of a maximum of 2 weeks’ MTX treatment within the 12 weeks was acceptable and did not render the patient ineligible, unless they were missed in the 4 weeks prior to the screening visit.)
-
No disease-modifying immunosuppressive drugs, other than MTX, in the 4 weeks prior to screening were allowed.
-
Written informed consent of adult participant or parent/legal guardian of minor, and assent, when appropriate, must have been received.
-
Participant and parent/legal guardian must have been willing and able to comply with protocol requirements.
-
Participants of reproductive potential (males and females) must have been willing to use a reliable means of contraception throughout their trial participation. Post-pubertal females must have had a negative serum pregnancy test within 10 days before the first dose of the trial drug.
-
Participant must have been able to be randomised and commence trial treatment within 2 weeks of the screening visit.
Exclusion criteria
-
Uveitis without a diagnosis of JIA.
-
Currently on adalimumab or had previously received adalimumab.
-
Have been on other biological agent within previous five half-lives of agent.
-
More than six topical steroid drops per eye per day prior to screening (this dose must have been stable for at least 4 weeks prior to screening visit).
-
For patients on prednisone or prednisone equivalent, change of dose within 30 days prior to screening.
-
For patients on prednisone or prednisone equivalent with a dose > 0.2 mg/kg per day.
-
Intra-articular joint injections within 4 weeks prior to screening.
-
Any ongoing chronic or active infection (including infective uveitis) or any major episode of infection requiring hospitalisation or treatment with intravenous antibiotics within 30 days or oral antibiotics within 14 days prior to the screening evaluation.
-
History of active tuberculosis of < 6 months treatment or untreated latent tuberculosis.
-
Participant has history of central nervous system neoplasm, active infection, demyelinating disease, or any progressive or degenerative neurological disease.
-
Poorly controlled diabetes or persistently poorly controlled severe hypertension (> 95th percentile for height/age), as deemed by the treating physician.
-
Previous history of malignancy.
-
Intraocular surgery within the 3 months prior to screening (cataract/glaucoma/vitrectomy).
-
Intraocular or periocular corticosteroids within 30 days prior to screening.
-
History of ocular herpetic disease.
-
Pregnant or nursing female.
-
Demonstrations of clinically significant deviations in any of the following laboratory parameters:
-
a platelet count of < 100,000/mm3
-
a total white cell count of < 4000 cells/mm3
-
a neutrophil count of < 1000 cells/mm3
-
aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of > 2× the upper limit of normal or serum bilirubin > 2× the upper limit of normal
-
a glomerular filtration rate (GFR) of < 90 ml/minute/1.73 m2 [GFR (ml.minimum/1.73 m2 body surface area)] = 0.55 × height (cm)/plasma creatinine (mg/dl)
-
a haematocrit of < 24%.
-
-
Having been administered a live or attenuated vaccine within 3 months prior to screening.
-
Previous randomisation into SYCAMORE to either arm of the trial.
-
An intraocular pressure of < 6 mmHg or > 25 mmHg.
-
Intraocular pressure control requiring more than one topical pressure-lowering therapy or requiring systemic acetazolamide.
Recruitment
Recruitment took place in 14 tertiary care centres throughout the UK. Participants were identified through rheumatology and ophthalmology outpatient clinics. Most tertiary care centres also set up referral links with local district general hospitals.
Informed consent
This trial recruited minors and young people aged < 16 years. Informed consent procedures reflected the legal and ethics requirements to obtain valid informed consent for this population. Prior written informed consent was required for all trial participants. In obtaining and documenting informed consent, the investigator complied with applicable regulatory requirements and adhered to good clinical practice and to the ethics principles that have their origin in the Declaration of Helsinki. 50
Information was provided to potential participants and their families verbally and in writing. All participants had the opportunity to discuss the project with the responsible investigator at site and/or a designated member of the research team. Discussions were supported with detailed written, ethics-approved patient information sheets and consent forms, provided directly to young people able to consent for themselves (defined in statutory instrument 2004 number 1031 as aged ≥ 16 years51) and parents/legal guardian of minors (aged < 16 years). Information leaflets appropriate to age and stage of development were provided to minors and their assent was obtained, when appropriate. Careful presentation was made of the known risks of the disease and trial medications and the possible benefits, as well as a detailed explanation of the trial procedures and protocol.
All participants were given the opportunity to ask any questions that may arise and to discuss the study with their surrogates and were given the time to consider the information prior to agreeing to participate.
All of the recruiting investigators were experienced rheumatologists and/or ophthalmologists familiar with imparting information to families and young people. All investigators obtaining consent had attended good clinical practice courses. When potentially eligible minors and young people were identified, they/their parent/the person with parental responsibility were approached by the investigator, or a designated member of the investigating team, and an opportunity was given to understand the objectives of the trial. The treatment schedule and trial visits were in line with standard clinical care, although they were made aware that additional travel may be needed if the trial assessments required they be reviewed at their tertiary centre rather than their local hospital. The potential risks and benefits of the anti-TNF-α agent were discussed, as were treatment failure criteria and what would happen if they chose not to enter the trial or had to withdraw from the trial for any reason. In addition, the rationale for the use of a placebo and the applied randomisation ratio was explained.
The right of the patient (non-minors) or parent/legal guardian (for minors) to refuse consent to participate in the trial without giving reasons was respected. After the patient had entered the trial, the clinician remained free to give alternative treatment to that specified in the protocol, at any stage, if they felt that it was in the best interest of the patient. However, the reason for doing so was recorded and the patient remained within the trial for the purpose of follow-up and data analysis in accordance with the treatment option to which they had been allocated. Similarly, the patient remained free to withdraw at any time from the protocol treatment and trial follow-up without giving reasons and without prejudicing their further treatment.
Adequate time to consider trial entry (generally 24 hours, although it was acknowledged that some patients/families came to a decision sooner) was allowed before written consent of the participant/parent/legal representative was obtained by the responsible clinician or other designated team member (recorded on the signature and delegation log).
Randomisation
Randomisation was undertaken during normal working hours (Monday to Friday, 09.00 to 17.00) by the pharmacy departments of participating centres on receipt of a randomisation request form and prescription from authorised clinicians. Pharmacy personnel verified that these documents were appropriately completed before proceeding.
It was the responsibility of the principal investigator or delegated research staff to:
-
notify the pharmacy of potential randomisations so that the pharmacy could ensure that adequate drug supplies were at site
-
complete the appropriate trial documents and deliver these to the pharmacy department at their centre in order that the pharmacy could proceed with a randomisation.
Participants were randomised using a secure (24-hour) web-based randomisation system, which was controlled centrally by the Clinical Trials Research Centre (CTRC). Randomisation lists were generated in a 2 : 1 ratio in favour of the active therapy. The lists were produced by an independent statistician (based at CTRC, but otherwise not involved in the trial) in Stata® version 9.2 (StataCorp LP, College Station, TX, USA) using the ralloc command. The randomisation lists were stratified by centre but in order to reduce the predictability of the randomisation sequence, the randomisation numbers were sequential across all sites (rather than within each site) to make it appear that there was no stratification by centre. For smaller sites (i.e. expected recruitment of < 10), a block size of three was used. For larger sites (i.e. expected recruitment of at least 10), random block sizes of three and six were used.
Participant treatment allocation was displayed on a secure webpage and an automated e-mail confirmation was sent to the authorised randomiser.
Description of interventions
All participants received a stable dose of MTX and either adalimumab (20 mg/0.8 ml for participants weighing < 30 kg or 40 mg/0.8 ml for participants weighing ≥ 30 kg) or placebo (0.8 ml, based on body weight) via a subcutaneous injection every 2 weeks for a maximum period of 18 months.
All participants in both arms continued to receive a stable dose of MTX at a minimum dose of 10–20 mg/m2 and a maximum dose of 25 mg as a non-IMP throughout the 18-month treatment period.
Clinical trial supplies were to be delivered to an investigator site only once the site had been initiated by the CTRC, acting on behalf of the sponsor to ensure full ethics and regulatory approvals had been granted. The size of the shipments to each site were predetermined, based on the participant recruitment target for that individual site. Recruitment was monitored centrally and drug shipment dates tailored accordingly, to ensure that pharmacies held adequate supplies of trial treatment. Pharmacies documented all shipment receipts and provided copies of this documentation to the CTRC. IMP stock was to be received by a designated member of the pharmacy department and stored at 2–8 °C, with temperature monitoring and in accordance with IMP regulations. Records of all shipments were to be kept in the pharmacy site file. All temperature excursions or damage to stock was reported to CTRC, which liaised with AbbVie Inc. for assessment.
The dose of adalimumab or placebo remained the same as at trial entry, regardless of minor fluctuations in weight that may cause a participant to cross the 30-kg threshold for the upper and lower doses.
The first dose was administered by the research/clinical team looking after the participant. Participants, or a family member, were invited to self-administer the study treatment after the first dose and taught how to do this, following the procedures in place within each participating centre. The first self-administered dose was carried out under the supervision of the clinical team, which would ensure that the participant was confident and able to carry out all parts of the procedure appropriately and accurately. The trial provided validated cooler bags for participants to transport their trial medication home. If participants did not want to self-administer the trial medication, then arrangements were put in place, on an individual basis, to ensure that trial medication was administered as prescribed.
Investigational medicinal products were labelled in accordance with regulation 46 SI2004/1031 and the detailed guidance provided in annex 13 of the EU Good Manufacturing Practice (GMP) guide. 51
Blinding
Participants, investigators, study personnel, the trial co-ordinator, statisticians and data management personnel were all blinded to the trial medication that the participant received. Pharmacy department staff were not blinded to the trial medication that the participant received.
This trial was placebo controlled and all trial assessments were carried out by health professionals, parents/carers and participants without knowledge of treatment allocation. The placebo solution for subcutaneous injection was a clear, colourless solution (matching the adalimumab vial) presented in a single-use vial for subcutaneous injection in volumes of 0.8 ml.
The packaging of the kit for adalimumab and placebo was identical. Each kit consisted of two vials of adalimumab or placebo in an outer carton. The vials of adalimumab and placebo were also identical in appearance.
Treatment allocation was concealed unless knowledge was essential for the ongoing care of the participant. If knowledge of treatment allocation was required by the responsible investigator, the process was to obtain it via the pharmacy department at the respective hospital, which would then complete an unblinding case report form (CRF) and submit this to the CTRC.
All children participating in this study during the active treatment phase of the study were immunosuppressed, in view of their concomitant MTX therapy and/or potential corticosteroid therapy, irrespective of whether they were on adalimumab or placebo. In addition, for the purpose of out-of-hours management of the patient, all participants were presumed to be on anti-TNF-α therapy and managed as such. In this way, in the event of an AE or SAE, such as an intercurrent infection, the treating clinician managed patients presuming them to be on anti-TNF-α therapy. For this reason, if unblinding was deemed necessary, this was carried out via the local pharmacy department following the procedure described below. If out-of-hours unblinding was required, this was accessed via the local pharmacy department’s on-call service.
Unblinding of individual participants during trial conduct
Procedures regarding potential unblinding were available for a number of relevant clinical scenarios.
On completion of 18 months of treatment
Although discouraged, it was acceptable to unblind participants on completion of their trial treatment (at 18 months) if this was necessary to enable appropriate ongoing treatment decisions by the participant’s clinician.
Early withdrawal from treatment
On early withdrawal from trial therapy, breaking the statistical blind was considered only when knowledge of the treatment assignment was deemed essential for the participant’s care by the participant’s physician, or a regulatory body. It was considered that it may not always be necessary to know the allocation of these participants.
Investigators were instructed that if simply ceasing trial treatment was a viable option for the participant’s care, then it was not necessary for unblinding to occur.
The procedure for unblinding during the course of the trial is set out below. The decision to unblind a single case was made when knowledge of an individual’s allocated treatment was essential to enable:
-
treatment of SAEs
-
administration of another therapy that is contraindicated by the trial treatment
-
appropriate ongoing care on cessation of allocated trial therapy.
When possible (during office hours), consent for individual unblinding was made via the trial co-ordinator at CTRC who would seek agreement of one of the lead co-chief investigators (Athimalaipet V Ramanan and Michael W Beresford).
Pharmacy departments were unblinded to the treatment allocations of participants within their centre. It was the principal investigator’s responsibility to ensure that all research personnel were aware of contact details for obtaining details of treatment allocation, if this was necessary.
The request for the allocated treatment was made to the local pharmacy department. Only the individual participant was to be unblinded and the following was documented by the pharmacy on the unblinding CRF:
-
date the information was needed
-
detailed reason for unblinding
-
identity of recipients of the unblinding information.
The local investigator ensured that all necessary CRFs to time of unblinding were completed and submitted to CTRC (if possible, completed before unblinding was performed).
All instances of unblinding were recorded and reported in writing to the CTRC by the local investigator, including the identities of all recipients of the unblinding information.
Allocation was not routinely revealed to CTRC personnel.
All instances of inadvertent unblinding were recorded and reported in writing to the CTRC by the local investigator. Reports included:
-
date of unblinding
-
detailed explanation of circumstances
-
recipients of the unblinding information
-
action to prevent further occurrence.
Data collection and management
For SYCAMORE, a paper CRF was used to collect participant data at each study visit. The paper CRF was designed by the Trial Management Group (TMG) and CTRC specifically for the study, in line with the trial protocol. Completed paper CRFs were transferred from the trial sites to the CTRC; data were then entered in to a Good Clinical Practice-compliant database (MACRO; Elsevier, Amsterdam, the Netherlands) by trial staff.
The configuration of the database was specific to SYCAMORE: there were built-in validations on certain aspects of the trial data. Any missing or inconsistent data were queried with the site using paper data query forms: query responses were completed by site staff and returned to the CTRC for entry into the database. A full audit trail of changes to the data was maintained.
Outcome measures
Primary outcome
The primary outcome for the trial was ‘treatment failure’. This was assessed at each scheduled or unscheduled visit and was defined by one or more of the following:
-
Anterior segment inflammatory score grade (SUN criteria10). Following at least 3 months of therapy –
-
two-step increase from baseline in SUN cell activity score (AC cells) over two consecutive readings
-
sustained non-improvement with entry grade of ≥ 3 for two consecutive readings
-
only partial improvement (1 grade) or no improvement, from baseline, with development of other ocular comorbidity that is sustained
-
worsening of existing (on enrolment) ocular comorbidity after 3 months
-
sustained scores, as recorded at entry grade, measured over two consecutive readings (grades 1 or 2) still present after 6 months of therapy.
-
-
Use of ineligible concomitant medications: these include medications that are not listed in the prespecified acceptable criteria or those that were not allowed.
-
Intermittent or continuous suspension of study treatment (adalimumab/placebo) for a cumulative period of no longer than 4 weeks.
Ocular comorbidities were defined as:
-
disc swelling and/or cystoid macular oedema as gauged clinically, and when possible, by optical coherence tomography evidence
-
a raised IOP (of > 25 mmHg) sustained over two consecutive visits, not responding to single ocular hypotensive agent
-
hypotony (of < 6 mmHg) sustained over two consecutive visits
-
development of unexplained reduction in vision [logarithm of the minimum angle of resolution (logMAR)] over two consecutive visits of 0.3 logMAR units (in the event of cataracts, participants remained in the trial, also if cataract surgery is required. Failure will still remain as described in end points above).
Note that an IOP of ≥ 25 mmHg or < 6 mmHg was an exclusion criterion at baseline and ocular comorbidities (i)–(iv) could be developed during follow-up only; (i) may worsen based on the existing (on enrolment) ocular comorbidity.
When a reading was required to be sustained over two consecutive visits to define treatment failure, the time of treatment failure was taken as the second of these readings.
Secondary outcomes
The following secondary outcomes were also recorded during the course of the trial:
-
Number of participants failing treatment.
-
Incremental cost-effectiveness of adalimumab added to MTX compared with MTX alone, based on:
-
health status according to the multiattribute Health Utility Index Mark 3 (HUI3)
-
resource use, estimated from participant diaries, questionnaires and routine data from patient-level information and costing systems (PLICS).
-
-
Safety, tolerability and compliance, defined as follows:
-
AEs and SAEs
-
laboratory parameters (haematological and biochemical analysis and urinalysis)
-
participant diaries and dosing records determined tolerability and compliance throughout the trial treatment period.
-
-
Use of corticosteroids over the duration of the study period and throughout follow-up, including the following:
-
total oral corticosteroid dose
-
reduction and reduction rate of systemic corticosteroid dose from entry dose
-
topical corticosteroid use (frequency) compared with use at time of entry
-
need for pulsed corticosteroid.
-
-
Optic and ocular outcomes, defined as follows:
-
number of participants with disease flares (defined by worsening based on SUN criteria) following a minimum of 3 months of disease control
-
number of participants with disease flares within the first 3 months of the study
-
visual acuity as measured by age-appropriate logMAR assessment
-
number of participants with resolution of associated optic nerve or macular oedema (as assessed by slit-lamp biomicroscopy or optical coherence tomography, where available)
-
number of participants with disease control (defined as zero cells with topical treatment for 3 and 6 months)
-
number of participants entering disease remission (defined as zero cells without topical treatment for 3 and 6 months)
-
duration of sustaining inactive disease (zero cells with or without topical treatment).
-
-
Quality-of-life assessments [as assessed by the Childhood Health Questionnaire (CHQ)52 and Childhood Health Assessment Questionnaire (CHAQ)53].
-
American College of Rheumatology (ACR) Pedi core set criteria54 at ACR 30, 50, 70, 90 and 100 levels
-
Number of participants with disease flares,55 in remission on and/or off medication,56 related to their JIA and with minimum disease activity. 57
-
Number of participants requiring change in biological and/or DMARD therapy for arthritis because of failure to respond.
-
Participants’ score on the Juvenile Arthritis Disease Activity Score (JADAS). 58 The JADAS comprises four components: a physician’s global assessment of disease activity, parent/patient global assessment of well-being, active joint count (in 27, 71 or 10 joints) and erythrocyte sedimentation rate.
The outcome ‘development of human antihuman antibody to adalimumab determined with samples collected at 1, 6 and 18 months’ was removed in version 4.0 of the trial protocol (in substantial amendment 10), because during the trial it was not possible to collect human antihuman antibody samples.
Data collection tools
Quality of life
Quality of life was measured by the use of CHQ52 and CHAQ. 53 Data collection took place on a monthly basis for the first 3 months, and then 3-monthly until withdrawal from the active phase of trial treatment.
Child Health Assessment Questionnaire
The CHAQ53 is the most widely used functional measure of disability in JIA, both in routine clinical practice throughout the UK and in clinical trials. Translated into many languages and validated in respective cultures and countries, it is easily completed and scored. It consists of eight domains, enquiring about the child/young person’s ability to manage a range of activities of daily living on a 5-point scale. Completion of the questionnaire was checked by staff.
Childhood Health Questionnaire
The CHQ52 is a generic measure of quality of life used in JIA. It explores a number of important domains including self-esteem, emotional and behavioural difficulties, and family impact. Completion of the questionnaire was checked by staff.
Sample size
Sample size calculations were undertaken using nQuery Advisor software version 4.0 (Statistical Solutions, Saugus, MA, USA).
Details of the original and revised sample size calculations are given below. Sample size revisions were necessary as a result of lower patient availability than expected.
Original trial sample size calculation
The original sample size calculation was based on data on failure rates from 62 patients on MTX in a comparable population provided by Clive Edelsten from Great Ormond Street Hospital for Children NHS Trust (2008). After 3 months, 11 patients had disease control based on grade 0 SUN criteria (18%) and, therefore, based on the trial inclusion criteria, would not be eligible for the trial. At 15 months following the start of treatment with MTX, 23 out of the 51 patients who had failed at 3 months had achieved disease control (45%), leaving 28 patients (55%) who had not. A total of 140 participants (adalimumab, n = 93; placebo, n = 47) were required to detect a relative reduction of 50% between a failure rate of 60% and 30% with 90% power (to optimise the detection of a significant difference between treatment regimens if one truly exists) at a 5% significance level, using a 2 : 1 randomisation.
The advent of biological therapies in JIA has led international investigators to a paradigm shift in the treatment of JIA and related complications, leading to significantly more ambitious outcomes in clinical trials, including elimination of inflammation and normalisation of short- and long-term function. 15,59 To this end, in JIA, instead of previously accepted clinical outcomes of 30% absolute difference in outcome between active agent and placebo,60 increasingly significant differences are being expected, with new definitions of response being established for use in clinical trials, such as clinical remission and minimal disease activity. 56,57 Indeed, 40% of patients in the adalimumab-JIA trial were reported as showing an ACR Pedi 100% response (100% response rate) at 2 years. 41
The clinically relevant outcomes of JIA-uveitis may take years to develop and the relationship between isolated measures of clinical activity and long-term outcomes remains ill-defined. Recent studies do suggest that the length of continuously controlled activity is likely to be of more clinical relevance than short-term improvements in levels of activity.
In view of these factors, as well as the expectation expressed unanimously through consultation with parent representatives in the development of the trial protocol, a minimum of 50% relative difference in failure rate between interventions was set. Based on the nature of the disease (potentially resulting in loss of vision) and a meeting of investigators representing participating centres as well as consumer representatives, and their experience of compliance from current usage of biological therapies in JIA-uveitis, it was estimated that loss to follow-up would be approximately 10%. Therefore, the sample size was increased by approximately 10% to allow for this, giving a total number of 154 participants (adalimumab, n = 102; placebo, n = 52).
The null hypothesis underlying this trial was that there is no significant difference between adalimumab and placebo in controlling disease activity of JIA-associated uveitis that is unresponsive to MTX therapy.
Revised sample size calculation for the primary outcome
The original sample size calculation had a power of 90%, but given a series of challenges arising, including those that were faced during recruitment, it was proposed by the TMG, and agreed on by the Trial Steering Committee (TSC) and Independent Data and Safety Monitoring Committee (IDSMC), as well as the sponsor and trial funders, to revise this. Reducing power to what is a universally accepted convention of 80% power maintained the status of this trial as an internationally relevant and robust contribution to the evidence base for the safety of this intervention, and was felt to be clinically acceptable. This was both acceptable to patients/families and clinicians, and would enable the sample size to be markedly reduced, and, therefore, more feasible within a reasonable period.
Furthermore, as of September 2013, there had only been one participant who had withdrawn and refused to provide primary outcome data. Therefore, it was reasonable to assume that the original assumption of adding 10% to the sample size calculation to account for missing data could be reduced to 5%.
The total sample size (including 5% drop out) that was required to detect the difference between a placebo proportion of 60% and treatment group proportion of 30% (with 0.05 two-sided significance level) was 114 participants.
Tissue bank
A blood sample collection system was developed, in accordance with Arthritis Research UK’s guidelines61 on detailed clinical and related material banks.
Written information was provided to families for this part of the study and written informed consent (with assent when appropriate) obtained for those who wished to provide blood samples. Participants who did not give consent to provide samples were eligible to take part in the main part of the trial.
Samples have been collected for future analysis and could be used as a resource to investigate the pharmacogenetics, aetiopathogenesis and identification of biomarkers of JIA-associated uveitis, for example. Understanding the genetic basis of age-specific disease processes allows consideration of the unique and rapid period of human development through to adulthood. Pharmacologic modulation of developing gene networks may have unintended and unanticipated consequences that do not become apparent or relevant until later in life. Early predictors of response allow future personalised treatment prescription in children.
Blood samples were collected pre treatment (0 months) and at two time points post treatment (at 3 months and 18 months).
Patient and public involvement
Patient and public involvement (PPI) representatives have been an integral part of SYCAMORE since the initial prioritisation, design stage and funding applications. PPI representatives provided detailed input into all aspects of the protocol design and all subsequent amendments and patient information sheets and consent forms and amendments. The patient information sheets, consent forms and assent forms were reviewed and feedback was given by the Medicine for Children Research Network young person’s advisory group. PPI representatives also provided input into any information sent to patients, such as letters to explain the closure of recruitment and the frequently asked questions section on the SYCAMORE website.
Changes to the protocol
Over the course of the whole trial, eight amendments were made to the protocol. These consisted of six non-substantial and 15 substantial amendments. A summary of amendments follows and the full list is reported in the trial protocol.
Version 1.0 (25 February 2011) amended to version 1.1 (8 September 2011)
The first amendment to the protocol made corrections to typographical errors in the original protocol.
Version 1.1 (8 September 2011) amended to version 2.0 (30 September 2011)
The second amendment added clarification to the tissue bank section; added clarification to the primary end point section and introduced an end point for the intermittent or continuous suspension of adalimumab/placebo; added clarification that patients cannot have previously received adalimumab; added two further exclusion criteria points relating to IOP; removed the limit on how many times patients can be screened; added a window for adalimumab/placebo injections; added clarification of topical treatment after 3 months of trial treatment; changed the dose range of allowed MTX to 10–20 mg/m2; and added clarification of treatment timelines and visit windows.
Version 2.0 (30 September 2011) amended to version 3.0 (25 April 2013)
The third amendment to the protocol made changes to the monthly visit windows to allow a window of 7 days; clarified in the table of assessments that the Clinical Service Receipt Inventory (CSRI) questionnaire is completed at baseline only; changed the timeline for tuberculosis assessment from 4 weeks to 12 weeks prior to baseline; and clarified that haematological and biochemical samples taken at screening can be used for the baseline visit.
Version 3.0 (25 April 2013) amended to version 4.0 (25 September 2013)
The fourth amendment to the protocol reduced the sample size from 154 to 114 participants and the duration of follow-up post treatment from 18 months to 6 months; changed the assessment of reduction of vision from number of letters to logMAR units; added clarification to inclusion and exclusion criteria; added systemic acetazolamide to the list of medication not permitted; removed the requirement for collection of human antihuman antibody samples; window for MTX administration added change to the collection of routine PLICS data; and added clarification on the definition of ‘end of trial’.
Version 4.0 (25 September 2013) amended to version 4.1 (11 August 2014)
The fifth amendment to the protocol added text to say that the IDSMC may request an interim analysis of the primary outcome.
Version 4.1 (11 August 2014) amended to version 5.0 (11 August 2014)
The sixth change to the protocol clarified that patients are classed as withdrawals and not treatment failures if they miss > 4 weeks of MTX treatment; added further clarification that haematological and biochemical blood results can be used for baseline (if taken at screening) only if assessment was completed within the previous 15 days; added clarification to tissue bank samples to state that the 3-month samples should be taken at the very next opportunity if not taken at 3 months and that the 18-month samples should be taken if patient ends treatment early.
Version 5.0 (11 August 2014) amended to version 6.0 (17 April 2015)
The seventh change to the protocol stated that the blinded phase of the trial had been stopped and that all patients on adalimumab would continue to be treated but that patients on placebo would stop treatment and proceed to follow-up.
Version 6.0 (17 April 2015) amended to version 6.1 (14 July 2016)
The eighth change to the protocol clarified that JADAS was a secondary outcome and clarified SAE reporting procedures.
Compliance with intervention
Participant diaries and dosing records determined tolerability and compliance throughout the trial period. The parent/guardian of a participant maintained a diary for all trial and other medications that were administered outside the trial visit (i.e. at home). In the diary, the date and time that the drug was administered were recorded. The dosing records were reviewed and verified for compliance at each visit by the research personnel at the trial centre.
Trial management and oversight
Trial Management Group
The TMG was a multidisciplinary team comprising the co-chief investigators, several co-investigators, PPI representatives, a sponsor representative, health economists and members of the CTRC (see Appendix 1). The TMG was responsible for the day-to-day clinical and practical aspects of the trial.
Independent Data and Safety Monitoring Committee
The IDSMC comprised two independent ophthalmologists, a statistician and a paediatric rheumatologist (see Appendix 1). The main responsibilities of the IDSMC were to safeguard the interests of the SYCAMORE participants, assess the safety and efficacy of the interventions during the course of the trial and monitor the overall progress and conduct of the trial. The IDSMC met at least annually during the course of the trial and provided recommendations to the TSC. The statistical team at the CTRC produced reports for the IDSMC.
Trial Steering Committee
The membership of the TSC included an independent rheumatologist, an independent ophthalmologist and an independent statistician, as well as representatives from the TMG (see Appendix 1). An observer from the sponsor and from the funder(s) were also invited to meetings. The TSC met at least annually, shortly after the IDSMC meeting. Its main role was to provide overall oversight of the trial.
Chapter 3 Statistical methods
Interim analysis
Interim monitoring reports of the accumulating data were performed at regular intervals (at least annually) for review by the IDSMC. In addition to the interim monitoring reports, the IDSMC requested that an interim analysis of the primary outcome was to be undertaken. The Peto–Heybittle stopping rule was applied to both interim analyses of the primary outcome. This required an extreme p-value of p < 0.001 as evidence to indicate potentially stopping for benefit. This approach was used to allow the IDSMC flexibility with the number and timing of further analyses, based on current safety and efficacy data, as it has the added benefit of preserving an overall two-sided type I error of 0.05 for the final analysis62 (see Chapter 4).
Final analysis
The results of this report are based on the data collected during three phases of the trial. Chapter 5 presents the results from the double-blind phase of the trial, Chapter 6 presents the results from the integrated analysis of the double-blind phase and the open-label phase, and Chapter 7 presents the results of the integrated analysis of the double-blind phase, open-label phase and the follow-up phase of the trial.
The data presented in this report are based on data at the final database lock that occurred on 2 August 2017.
The results from the blinded phase of the trial that were presented in the published manuscript of SYCAMORE were based on data snapshots taken on 11 September 2015 (primary outcome and AE data) and 24 May 2016 (secondary outcomes).
Because data were still being received from sites during this period, there were minor ongoing updates and changes to the database. The differences between the results in the New England Journal of Medicine manuscript63 and Chapter 5 in this report are documented in Appendix 2.
There were three statistical analysis plans (SAPs) written for the final analysis of study results. The first SAP was written by the trial statistician and contained detail only of the analyses for the primary outcome and the safety data of the blinded phase of the trial. The second and third SAPs were written after the completion of the primary analysis (after the blind had been broken to treatment allocation) and, therefore, written by independent statisticians who were blinded to the allocation of the trial. The second SAP described the analysis that was conducted on the secondary outcomes for the blinded phase of the trial and the third SAP described the analyses that were conducted for the open-label and follow-up phases of the trial. All three SAPs are available at www.journalslibrary.nihr.ac.uk/programmes/hta/095101 (accessed 4 February 2019).
General statistical considerations
The primary and secondary outcomes were all analysed using the (two-sided) 5% level of significance and 95% confidence intervals (CIs) are presented throughout. There were no adjustments for multiple testing; rather, all secondary analyses were treated as hypothesis generating.
The primary and secondary analyses used the principle of intention to treat (ITT) based on all the randomised participants, meaning that participants who consented and were randomised were analysed on the basis of the treatment they were randomised to, regardless of whether or not they received it. If consent for treatment was withdrawn but the participant was happy to remain in the study for follow-up, they were followed up until completion. However, if they decided to withdraw consent completely, then the reasons for withdrawal of consent were collected (if possible) and reported for both groups.
All statistical analyses were conducted using SAS® V9.3 (SAS Institute, Cary, NC, USA), except for the joint modelling and competing risk analyses, which were conducted in R (R Foundation for Statistical Computing, Vienna, Austria).
Analysis of baseline data
Demographic and baseline characteristics were summarised for each treatment group and overall, using descriptive statistics. No formal statistical testing was performed on these data. Descriptive statistics, including the number of observations; mean; standard deviation (SD); median, minimum and maximum for continuous variables; and counts and percentages for discrete variables, are presented as appropriate.
Analysis of the primary outcome
The primary outcome of ‘treatment failure’ was a time-to-event outcome. For those patients who entered the trial and whose eyes (both) met the entry criteria, the time to the first eye to fail treatment was used. This was not observed in all participants; those participants who did not experience an event were classed as censored. The event time or censoring was calculated by subtracting the randomisation date from one of the following scenarios:
-
Participants who failed treatment – date of the visit at which they failed treatment.
-
Participants who completed the trial treatment phase without failing treatment – censored at date of 18-month treatment visit.
-
Participants who discontinued treatment early and agreed to follow-up –
-
if they were assessed to be a treatment failure during a follow-up visit within 18 months following randomisation: date of follow-up visit
-
if they were not assessed to be a treatment failure during their follow-up visits: censored at date of last follow-up visit within 18 months following randomisation.
-
-
Participants that were lost to follow-up – censored date of last treatment visit.
Survival estimates were calculated using the method of Kaplan and Meier, with curves for each treatment group presented graphically with numbers at risk.
The p-value obtained from the log-rank test and the hazard ratios (HRs) with 95% CIs were used to assess differences in failure estimates across treatment groups. The statistical test for the primary end point was performed at a two-sided significance level of 0.05.
Participants who withdrew from trial treatment (providing they did not withdraw from the entire study or withdraw consent) moved to the follow-up phase of the trial, were assessed for the primary outcome and could still contribute to the ITT analysis.
Any participants who withdrew from follow-up contributed primary outcome data until the point at which they withdrew from the trial.
Missing data were monitored and strategies developed to minimise this occurrence. Missing data were handled by considering the robustness of the complete-case analysis to sensitivity analyses using various imputation assumptions; this was informed by data collected on the reasons for missing data.
Nine sensitivity analyses were carried out to determine the robustness of the results from the primary analysis:
-
Best case – all participants who withdrew from treatment were treated as censored at time of treatment withdrawal.
-
Worst case – all participants who withdrew from treatment were treated as treatment failures (i.e. events at time of treatment withdrawal).
-
Methotrexate – any participants who withdrew from treatment because of MTX intolerance were classified as treatment failures at the time of treatment withdrawal.
-
Component 1 of primary outcome – all participants who failed for component 1 at a treatment failure assessment had their event date as the mid-point between this visit and the previous visit instead of the date of this visit.
-
Component 2 of primary outcome – all participants who failed for component 2 at a treatment failure assessment had their event date as the date that they commenced concomitant medications (a) used against predefined acceptable criteria (see SYCAMORE protocol49) or (b) any of the concomitant medications not allowed. The event date was determined by the co-chief investigators making a clinical decision following review of the participants’ concomitant medications taken since their previous visit.
-
Component 3 – all participants who failed for component 3 at a treatment failure assessment had their event date as the exact date that they qualified as ‘intermittent or continuous suspension of study treatment (adalimumab/placebo) for a cumulative period longer than 4 weeks’. 28 The event date was determined by the chief investigator making a clinical decision following review of a participant’s trial treatment dose recordings in the treatment diaries.
-
Any missing primary outcome data – any cases of missing data for any of the primary outcome components (except for unscheduled visits) had data imputed on a worst-case basis, because the missing data could have meant that a participant failed earlier than recorded. All participants were treated the same, regardless of whether or not they had a treatment failure.
-
Loss to follow-up – in the primary analysis of the primary outcome, participants who were lost to follow-up were treated as withdrawals, assuming that they were non-informative. The reasons for loss to follow-up, when available, were blindly reviewed by Michael W Beresford (co-chief investigator) and Andrew Dick (ophthalmology expert on the TMG) to see whether or not any might be related to the prognosis. If any were deemed to be related, a sensitivity analysis would be undertaken, assuming these participants to be a treatment failure at the time of last recorded visit.
-
Incorrectly identified to be a treatment failure – once a participant was deemed to have failed treatment, treatment was stopped and they entered the follow-up phase of the study, providing that they still wished to be followed up. Any participants wrongly identified as treatment failures by the assessing physician would be classed as a withdrawal at their time of ‘treatment failure’.
Analysis of secondary outcomes
The secondary continuous outcomes were analysed using the following methods:
-
Chi-squared test, relative risk (RR) and 95% CI for the number of participants –
-
failing treatment
-
needing pulsed corticosteroids
-
having uveitis disease flares
-
having resolution of associated optic nerve or macular oedema
-
with uveitis disease control
-
entering disease remission for uveitis
-
undergoing JIA disease flare
-
with minimum disease activity of JIA
-
having remission of their JIA on and off medication
-
requiring a change in biologics due to failure to respond from arthritis.
-
-
Change from baseline –
-
laboratory parameters (haematological and biochemical assessments).
-
-
Poisson regression –
-
total oral corticosteroid dose
-
systemic corticosteroid dose.
-
-
Competing risks –
-
reduction in systemic corticosteroid dose
-
topical corticosteroid use.
-
-
Joint modelling of longitudinal and time-to-treatment failure data –
-
visual acuity
-
CHAQ and CHQ
-
ACR 30, 50, 70, 90 and 100
-
JADAS.
-
-
Random intercept model –
-
duration of sustaining inactive disease.
-
No between-group statistical analyses were conducted for compliance data or urinalysis data; instead, summary data are presented for these outcomes.
Adverse events were tabulated by the Medical Dictionary for Regulatory Activities (MedDRA) version 18.0 system by organ class and preferred term. In addition, summaries by severity and relationship to study drug were completed. SAEs and events that led to premature withdrawal were listed in detail. No formal testing of AE or SAE data was planned.
Chapter 4 Interim analysis results
Initial meeting
The first meeting of the IDSMC took place on 2 August 2011. During this meeting, the IDSMC reviewed the study protocol, was updated on study progress and agreed the IDSMC charter and also the expedited safety reports that it wished to receive.
Future meetings
The IDSMC met at regular intervals following the initial meeting (11 April 2012, 18 December 2012, 3 July 2013 and 18 February 2014). During these meetings, the committee was updated on the progress of the trial and was also presented with a report that contained data on recruitment, data completeness and safety.
There were two sessions held during the meetings, an open and closed session. Present at the open session were the independent members of the IDSMC and also relevant members of the TMG (i.e. co-chief investigators, lead trial ophthalmologist, trial co-ordinator, statistical team). During the open session, data were presented overall and not split by treatment group. The open session was then followed by a closed session, which was attended only by the independent members of the IDSMC and the statistical team responsible for the production of the report.
On 18 February 2014, the IDSMC decided that the recruited sample was approaching a size at which an interim analysis was needed in order to allow them to protect participants from potential harm or to stop the trial early if there was a clear benefit. This allowed for avoidance of delay by bringing the benefits to patients once the question that had been posed in the trial had been answered with sufficient statistical certainty.
The IDSMC, therefore, requested that an interim analysis be conducted and the results be presented at the next IDSMC meeting, which was to be held on 29 September 2015.
Statistical methods
A full SAP was written prior to conducting the interim analysis.
The Peto–Haybittle stopping rule was applied to the interim analysis of the primary outcome. This required an extreme p-value of < 0.001 as evidence to stop for benefit. This approach was used to allow the IDSMC flexibility with the number and timings of further analyses, based on current safety and efficacy data, as it had the added benefit of preserving an overall two-sided type I error of 0.05 for the final analysis.
Survival estimates were calculated using the method of Kaplan and Meier, with curves for each treatment group presented graphically along with numbers at risk.
The log-rank test was used to assess differences in failure estimates across treatment groups.
Interim analysis 1
The first interim analysis report was based on follow-up data from 71 participants who had been randomised (adalimumab, n = 48; placebo, n = 23). Safety data (SAE/AE) were based on data from 64 participants (adalimumab, n = 43; placebo, n = 21).
There was a total of nine treatment failures recorded in 48 participants in the adalimumab group (19%) and 13 treatment failures recorded in 21 participants in the placebo group (62%), and five withdrawals from 48 participants recorded in the adalimumab group (10%) and four withdrawals from 23 participants recorded in the placebo group (17%).
The Kaplan–Meier plot is shown in Figure 2. The log-rank chi-squared statistic was 15.77 and the associated log-rank p-value was < 0.0001.
FIGURE 2.
Interim analysis 1: Kaplan–Meier plot.
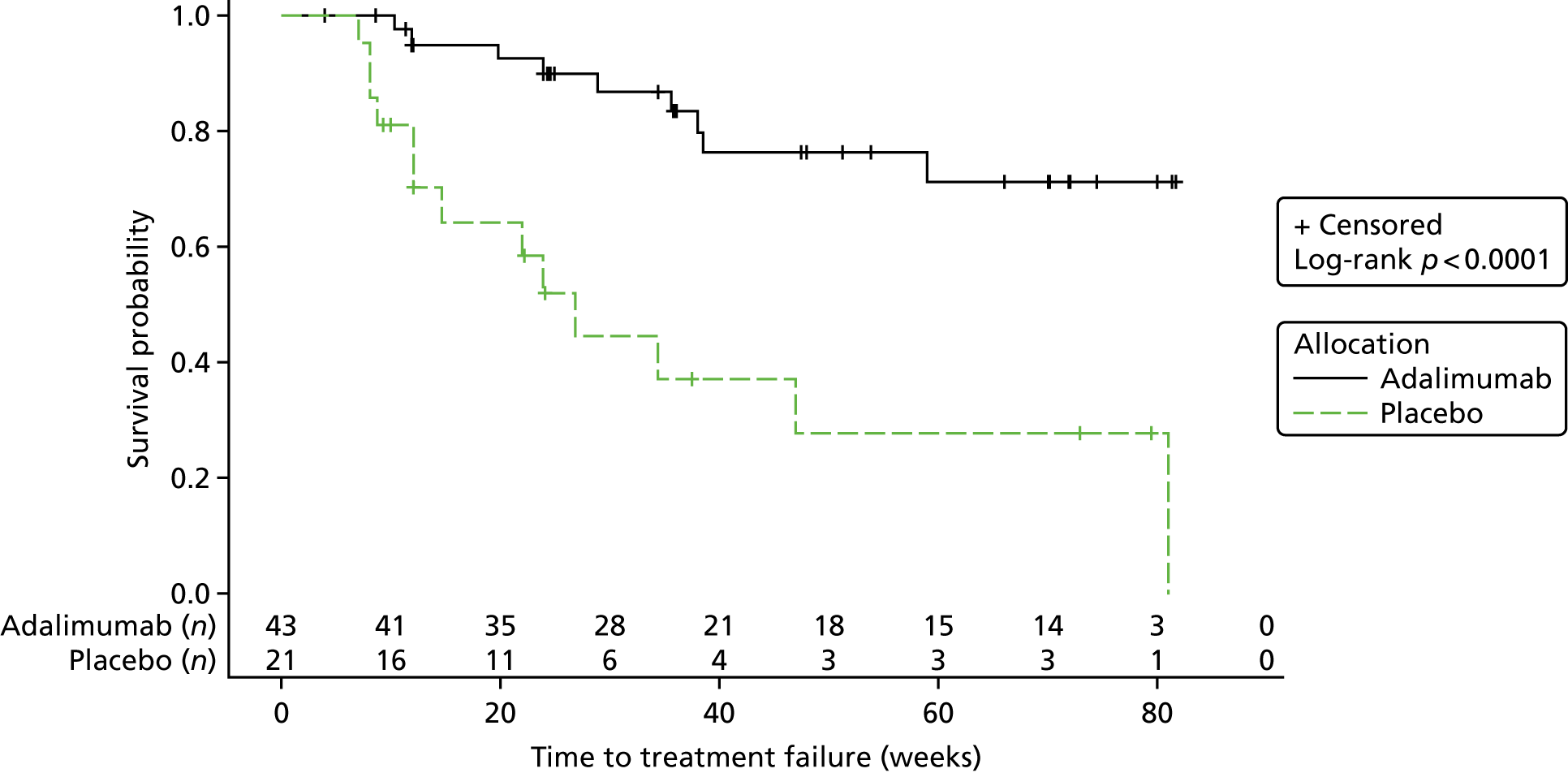
At the time of the first interim analysis, 353 AEs had been reported in 34 of the 43 participants in the adalimumab group who qualified for the safety analysis set (i.e. received at least one dose of treatment). There had been 89 AEs reported in 14 out of the 21 participants in the placebo group who qualified for the safety analysis set (i.e. received at least one dose of treatment).
There had been 10 SAEs reported in 9 participants from a total of 43 participants in the adalimumab group (21%). There had been one SAE reported from a total of 21 participants in the placebo group (5%).
The IDSMC considered the report and made the recommendation that the trial should continue with recruitment. However, it did make three observations with regard to the trial based on the report that it received:
-
We (the IDSMC) have been much reassured over recent months, and in the current report, that the recruitment rate is now running just ahead of the amended estimated rate. We would like to commend the TMG on overcoming significant difficulties to achieve this favourable situation.
-
We do, however, remain concerned that some centres are performing exceptionally badly; there is, for example, a marked contrast between the performance of Bristol, where ≈50% of those screened are recruited, and Birmingham, where < 1% of those screened are being recruited. While these two are at the extreme ends, there are several other poorly recruiting centres – this appears as a weakness with potential implications for the representativeness of the trial sample. An investigation into the problems in these centres, with a view to resolving their issues, would be time well spent.
-
We are also concerned about the levels of missing data, which seem high for certain variables. For example, in table 10.2 [referring to the meeting report], in 14% the relationship between the AE and the trial medication is missing and in 5% the severity of the AE is missing. Whilst we appreciate that you are taking steps to reduce these rates of missing data, we would like to register our concern in regard to this problem of missing data, which is evident in other parts of the report. Please take all steps to recover missing data and reduce the levels going forward.
The TMG provided further information with regards to missing data for both the primary outcome and the safety data. The response contained a more detailed breakdown of missing and unobtainable data for each component of the primary outcome and safety data, by site and overall.
This thorough investigation of the missing and unobtainable data reassured the IDSMC that the monitoring procedures being implemented by the TMG and the engagement of sites with the data manager were ensuring that these figures were kept to a minimum.
The IDSMC requested that a further interim analysis be conducted at their next meeting in March 2015.
Interim analysis 2
The IDSMC met on the 25 March 2015 to discuss the results of the second interim analysis of the SYCAMORE data.
At the time of the report, there were a total of 85 participants who had been randomised (adalimumab, n = 57; placebo, n = 28). From these 85 participants:
-
80 were included in the analysis (adalimumab, n = 54; placebo, n = 26).
-
Five were excluded –
-
four had no randomisation CRF input on the trial database
-
one had their randomisation CRF input on the trial database but had not had any further follow-up visits input to the trial database.
-
There were a total of 10 withdrawals from the trial: five withdrawals from the adalimumab group (n = 57) (9%) and five withdrawals from the placebo group (n = 28) (18%). There were a total of 27 treatment failures in the trial: 12 treatment failures on adalimumab (21%) and 15 on placebo (54%).
The Kaplan–Meier plot is shown in Figure 3. The log-rank chi-square statistic was 14.63 and the associated log-rank p-value was 0.0001.
FIGURE 3.
Interim analysis 2: Kaplan–Meier plot.
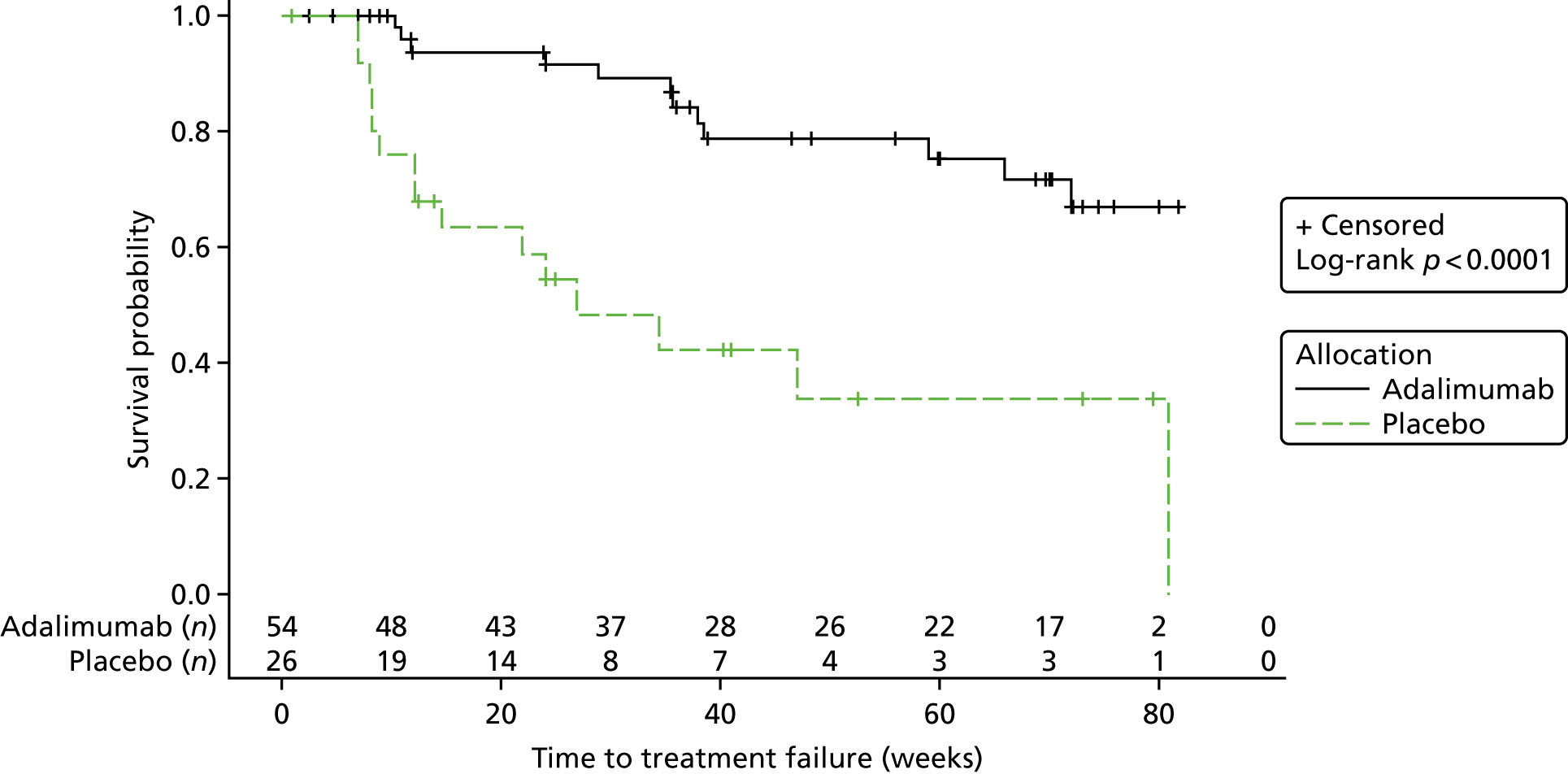
The safety data set was based on 80 participants (adalimumab, n = 54; placebo, n = 26).
There were a total of 10 withdrawals from the trial: five withdrawals from the adalimumab (n = 57) (9%) and five withdrawals from the placebo group (n = 28) (18%). There were a total of 27 treatment failures in the trial: 12 treatment failures on adalimumab (21%) and 15 on placebo (54%).
Following the review of the second interim analysis, the IDSMC carefully considered evidence from the first interim analysis, the subsequent satisfactory conclusion of the vast majority of missing data, and also the continued presence of such a strong treatment effect. The IDSMC subsequently agreed unanimously that:
-
The levels of missing data were acceptable with all reasonable efforts being made to collect outstanding missing data items where feasible, or to confirm the fact that the data were unobtainable.
-
The AEs and SAEs were in keeping with expectations for the medications in use, based on clinical experience and previous published reports.
-
The statistical significance of the beneficial effect of the investigational medicinal product (IMP) in the interim analysis substantially exceeded the predetermined requirement for consideration of stopping the trial on the basis of a powerful positive treatment effect.
-
The IDSMC should obtain guidance on procedure prior to making any stopping recommendation. Options for stopping included either to immediately stop and fully close the trial or to stop recruitment and continue collecting data until all current participants had completed their passage through the protocol.
It was decided by the IDSMC that no immediate recommendation should be made, but that the IDSMC chairperson should take advice in a timely manner, following which the IDSMC would communicate further among themselves before arriving at a final recommendation.
Summary of Independent Data and Safety Monitoring Committee and Trial Steering Committee meetings following interim analysis 2
The IDSMC and TSC met on 8 April 2015 in a combined meeting that involved all of the independent members of both committees, as well as both co-chief investigators and the lead statistician (non-independent members). The IDSMC advised the TSC that the trial should stop recruiting with immediate effect. They further recommended that all participants currently in the trial should continue in their randomly allocated treatment regimen, blinded, and follow treatment scheduling as per protocol. The IDSMC did not unblind the TSC (or co-chief investigators) to the results of the trial during the meeting. The TSC decided to consider the recommendations of the IDSMC overnight and meet again on 9 April 2015.
The independent members of the TSC met with the co-chief investigators and lead statistician (non-independent members of the TSC) again on 9 April 2015. The chairperson of the TSC contacted the chairperson of the IDSMC, requesting that the IDSMC make formal recommendations to the TSC in a formal document; they agreed to meet again on 10 April 2015.
The same independent members of the TSC, the co-chief investigators and lead statistician met on 10 April 2015 and discussed the formal recommendations that had been made by the IDSMC.
The voting members of the TSC (the three independent members and one non-independent member) then voted unanimously in favour of the following specific IDSMC recommendations:
-
Recruitment to SYCAMORE should not be reinstated. This was based on a positive signal of efficacy of the IMP (adalimumab) versus placebo, which exceeded the prespecified level.
-
All participants in the trial should be invited to attend a final blinded assessment visit. On completion of assessments, their treatment allocation should then be unblinded.
-
All participants in the active arm of the trial (adalimumab) should continue follow-up in an open-label fashion, as this long-term follow-up would contribute important additional data on quality of life, utilities and long-term efficacy. Subsequent changes in therapy would be at the discretion of the treating clinical team.
Chapter 5 Clinical effectiveness results: blinded phase of study
The results presented in this chapter are from the blinded period of the trial only.
Participant recruitment
The first participant was randomised into SYCAMORE on 27 October 2011 and the last participant was randomised on 31 March 2015. The last blinded visit took place on 16 June 2015.
Fourteen of the 17 sites randomised at least one participant and five centres randomised five or more participants. The flow of participants through the trial is represented in the Consolidated Standards of Reporting Trials (CONSORT)64 flow diagram in Figure 4.
FIGURE 4.
The CONSORT flow diagram for all trial participants.
A total of 332 patients were assessed for eligibility from 519 screenings (patients could be screened on multiple occasions).
The number of participants enrolled overall by month is provided in Appendix 3 (see Figure 13); the screening overview by centre is also given in Appendix 3 (see Table 40). The main reasons for ineligibility included the following: the patient did not have active uveitis (49%), the patient had not failed on MTX or been on MTX for at least 12 weeks with a stable dose (19%), the patient had been on another biological agent within the previous five half-lives of the agent (6%), the patient had more than six topical steroid eye drops per day at randomisation (6%) or other reasons (11%). For five patients, it was not clear whether or not they were eligible.
A total of 130 patients were eligible to participate in the trial from 139 screening visits; 90 patients were randomised. The reasons for the 45 patients (49 screening visits) not providing consent are given in Appendix 3 (see Table 41).
Recruitment rates
The original target sample size of 154 participants was expected to be achieved within 30 months of recruitment. This original target was based on feasibility data provided by each of the centres that took part in the trial. The actual rates of recruitment during the trial were lower than anticipated (see Appendix 3, Figure 13) and, therefore, the recruitment period of the trial was extended by 36 months on 16 June 2014, to allow for additional time to recruit the necessary number of participants.
The sample size of the trial was also revised, which meant that the target sample size was reduced from 154 to 114 participants. Other strategies were used to improve recruitment, including the aligning of the rheumatology and ophthalmology clinics in any sites that did not already have this service and holding a series of regional investigator meetings and local investigator meetings each month via teleconference, which included members of the CTRC and the co-chief investigators and lead SYCAMORE ophthalmologists. Regular newsletters were sent to sites to keep sites aware of recruitment and to maintain interest in the trial. All sites were also encouraged to build referral links with district general hospitals to allow referrals of potential patients for screening.
Comparison of interventions
The ITT analysis population included all 90 participants who were randomised (see Figure 4). There were no exclusions from either the safety or the ITT population.
All participants who withdrew consent for trial continuation contributed outcome data up until the point of withdrawal.
The memberships of the analysis set for the primary outcome and safety data set were determined and documented prior to the blinding being broken and the treatment allocations being requested.
Trial completion and trial exit
There were 19 participants who withdrew prematurely during the blinded treatment phase of the trial (Table 1). There were a total of 11 (18%) withdrawals in the adalimumab group (nine continued into the follow-up phase of the trial and two withdrew complete consent) and eight (23%) in the placebo group (seven continued into the follow-up phase of the trial and one withdrew complete consent). The majority of premature withdrawals were for non-safety reasons (see Table 1). The most common reason for withdrawal in the adalimumab group was because of MTX intolerance and in the placebo group it was the worsening of uveitis (that did not meet the exit criteria).
| Reasons | Treatment group, n (%) | Total, n (%) | |
|---|---|---|---|
| Adalimumab | Placebo | ||
| Safety | 2 (3.3) | 1 (3.3) | 3 (3.3) |
| AE | 2 (3.3) | 0 (0) | 2 (2.2) |
| SAE | 0 (0) | 1 (3.3) | 1 (1.1) |
| Non-safety | 9 (15) | 7 (23.3) | 16 (17.8) |
| Consultant discretion owing to disease activity | 0 (0) | 1 (3.3) | 1 (1.1) |
| Family circumstances | 0 (0) | 1 (3.3) | 1 (1.1) |
| Flare of JIA | 0 (0) | 1 (3.3) | 1 (1.1) |
| MTX intolerance | 4 (6.7) | 0 (0) | 4 (4.4) |
| Needle phobia | 1 (1.7) | 0 (0) | 1 (1.1) |
| Participant felt no benefit | 0 (0) | 1 (3.3) | 1 (1.1) |
| Recurrent infections | 1 (1.7) | 0 (0) | 1 (1.1) |
| Refused injections | 1 (1.7) | 0 (0) | 1 (1.1) |
| Unable to tolerate adalimumab/placebo | 0 (0) | 1 (3.3) | 1 (1.1) |
| Use of medication that was not permitted | 1 (1.7) | 0 (0) | 1 (1.1) |
| Withdrawal of consent – no reason | 1 (1.7) | 0 (0) | 1 (1.1) |
| Worsening of uveitis (not meeting exit criteria) | 0 (0) | 2 (6.7) | 2 (2.2) |
| Overall | 11 (18.3) | 8 (26.7) | 19 (21.1) |
Baseline characteristics
The demographic baseline data of the 90 participants randomised across all centres were comparable between the two groups (Table 2). The mean age in the placebo group was slightly lower than that in the adalimumab group and the proportion of females and males was approximately the same in the two treatment groups, with more females than males.
| Variable | Treatment group | Total (N = 90) | |
|---|---|---|---|
| Adalimumab (N = 60) | Placebo (N = 30) | ||
| Number of study eyes, n (%) | |||
| Unilateral | 43 (72) | 22 (73) | 65 (72) |
| Bilateral | 17 (28) | 8 (27) | 25 (28) |
| Age at randomisation (years) | |||
| Mean (SD) | 9.07 (3.94) | 8.56 (3.79) | 8.90 (3.88) |
| Sex, n (%) | |||
| Female | 47 (78) | 23 (77) | 70 (78) |
| Male | 13 (22) | 7 (23) | 20 (22) |
| Weighta (kg), n (%) | |||
| < 30kg | 33 (60) | 17 (57) | 50 (56) |
| ≥ 30kg | 26 (44) | 13 (43) | 39 (44) |
The distribution in weight was similar in both groups, as was physical global assessment of disease activity, antinuclear antibody and double-stranded deoxyribonucleic acid.
A total of 65 (72%) participants entered the trial with one eye that was eligible for evaluation (i.e. they met the inclusion criteria for active uveitis in one eye only) and 25 (28%) participants entered the study with two eyes that were eligible (i.e. they met the inclusion criteria for active uveitis in both eyes). Therefore, a total of 115 eligible eyes entered into the study.
Table 3 shows ocular data collected at baseline, presented at the eye level rather than the individual level for ease of reading. The overall mean for logMAR score was 0.05; this was slightly higher in the placebo group than in the treatment group. Of the eligible eyes, 76 (66%) had a score of 1+ for the AC score; the numbers in each of these categories were similar in both groups, as were the flare score and vitreous haze grading. The mean IOP in each group was similar, with an overall mean of 14.54 mmHg.
| Variable | Treatment group | Total (N = 115) | |
|---|---|---|---|
| Adalimumab (N = 77) | Placebo (N = 38) | ||
| Topical steroid drops scorea | |||
| Mean (SD) | 2.04 (1.38) | 2.20 (1.57) | 2.09 (1.44) |
| LogMAR scoreb | |||
| Mean (SD) | 0.04 (0.15) | 0.07 (0.12) | 0.05 (0.14) |
| AC cells (SUN) (%) | |||
| 1+ | 52 (68) | 24 (63) | 76 (66) |
| 2+ | 18 (23) | 11 (29) | 29 (25) |
| 3+ | 6 (8) | 3 (8) | 9 (8) |
| 4+ | 1 (1) | 0 (0) | 1 (1) |
| Flare score (SUN), n (%) | |||
| 0 | 18 (23) | 12 (32) | 30 (26) |
| 1+ | 49 (64) | 23 (61) | 72 (63) |
| 2+ | 10 (13) | 3 (8) | 13 (11) |
| LOCS III grading: pseudophakic, n (%) | |||
| No | 77 (100) | 38 (100) | 115 (100) |
| LOCS III grading: nuclear,c n (%) | |||
| N0 | 69 (96) | 36 (97) | 105 (96) |
| NI | 3 (4) | 1 (3) | 4 (4) |
| LOCS III grading: cortical,d n (%) | |||
| No cortical cataract | 56 (88) | 29 (94) | 85 (90) |
| Control | 3 (5) | 0 (0) | 3 (3) |
| CI | 4 (6) | 2 (7) | 6 (6) |
| CII | 1 (1) | 0 (0) | 1 (1) |
| LOCS III grading: posterior,e n (%) | |||
| 0 | 68 (92) | 29 (81) | 97 (88) |
| PI | 5 (7) | 4 (11) | 9 (8) |
| PII | 1 (1) | 3 (8) | 4 (4) |
| Other structural changes: central band keratopathy covering visual axis, n (%) | |||
| No | 75 (97) | 38 (100) | 113 (98) |
| Yes | 2 (3) | 0 (0) | 2 (2) |
| Other structural changes: synchiae, n (%) | |||
| No | 59 (77) | 32 (84) | 91 (79) |
| Yes | 18 (23) | 6 (16) | 24 (21) |
| Other structural changes: iris bombe, n (%) | |||
| No | 77 (100) | 38 (100) | 115 (100) |
| Other structural changes: membrane formation, n (%) | |||
| No | 75 (97) | 38 (100) | 113 (98) |
| Yes | 2 (3) | 0 (0) | 2 (2) |
| Other structural changes: neovascularisation, n (%) | |||
| No | 77 (100) | 38 (100) | 115 (100) |
| IOP (mmHg) | |||
| Mean (SD) | 14.76 (3.85) | 14.11 (4.27) | 14.54 (3.99) |
| Vitreous haze grading, n (%) | |||
| 0 | 65 (84) | 32 (84) | 97 (84) |
| 0.5+ | 8 (10) | 4 (11) | 12 (10) |
| 1+ | 3 (4) | 2 (5) | 5 (4) |
| 2+ | 1 (1) | 0 (0) | 1 (1) |
When the best and worst scores were entered for those with two eligible eyes, the baseline results were very similar to those when the eyes were looked at independently.
Table 4 shows the baseline data describing the classification by disease subtype of the participants’ JIA status. A total of 53 (59%) participants had persistent oligoarticular JIA and 21 (23%) had extended oligoarticular JIA. The proportions for each of the subcategories were similar for both groups. The mean overall JIA disease duration was 5.33 years, with the duration being slightly longer in the treatment group. A total of 66 (73%) participants had a negative rheumatoid factor (RF); this was slightly higher in the adalimumab group.
| Variable | Treatment group | Total (N = 90) | |
|---|---|---|---|
| Adalimumab (N = 60) | Placebo (N = 30) | ||
| Type of JIA (ILAR classification), n (%) | |||
| Extended oligoarthritis | 14 (23) | 7 (23) | 21 (23) |
| Persistent oligoarthritis | 36 (60) | 17 (57) | 53 (59) |
| Polyarthritis RF negative | 8 (13) | 4 (13) | 12 (13) |
| Polyarthritis RF positive | 1 (2) | 1 (3) | 2 (2) |
| Psoriatic arthritis | 1 (2) | 1 (3) | 2 (2) |
| Disease durationa (years) | |||
| Mean (SD) | 5.58 (3.69) | 4.81 (3.19) | 5.33 (3.53) |
| Physician global assessment of disease activityb | |||
| Mean (SD) | 0.76 (1.48) | 0.83 (1.09) | 0.78 (1.36) |
| Active joint count (all joints) | |||
| Mean (SD) | 0.57 (2.03) | 1.1 (2.23) | 0.74 (2.10) |
| Swollen joint count (all joints) | |||
| Mean (SD) | 0.55 (1.66) | 1.0 (1.55) | 0.70 (1.63) |
| Antinuclear antibody,c n (%) | |||
| Negative | 24 (42) | 10 (40) | 34 (42) |
| Positive | 33 (58) | 15 (60) | 48 (58) |
| Double-stranded deoxyribonucleic acid,d n (%) | |||
| Negative | 47 (94) | 22 (92) | 69 (93) |
| Positive | 3 (6) | 2 (8) | 5 (7) |
| RF,e n (%) | |||
| Negative | 46 (98) | 20 (87) | 66 (94) |
| Positive | 1 (2) | 3 (13) | 4 (6) |
Unblinding of randomised treatment
There were 57 (63%) participants who were unblinded during the course of the trial: 38 (67%) in the adalimumab group and 19 (33%) in the placebo group. A breakdown of the stage of the trial that the participants were in at the time of unblinding is given in Table 5.
| Reason | Treatment group, n (%) | |
|---|---|---|
| Adalimumab | Placebo | |
| Completed 18 months of treatment | 12 (32) | 1 (5) |
| Based on recommendation of IDSMC | 10 (26) | 4 (21) |
| Treatment failure | 11 (29) | 8 (42) |
| Withdrawal | 5 (13) | 6 (32) |
| Total | 38 | 19 |
Protocol deviations
Protocol deviations were monitored centrally via evaluation of inclusion/exclusion criteria at trial entry and throughout the course of the trial. During the course of the blinded phase of the trial, a total of 74 (82%) participants had at least one major protocol deviation (Table 6). All protocol deviations were agreed with the co-chief investigators before the final treatment allocations were requested on 11 September 2015.
| Institution | Total, n | Any protocol deviation, n (%) | At least one protocol deviation related to, n (%) | |||
|---|---|---|---|---|---|---|
| Inclusion criteria | Exclusion criteria | Treatment regimen | Study assessment | |||
| The Leeds Teaching Hospitals NHS Trust | 2 | 2 (100) | 0 (0) | 0 (0) | 1 (50) | 2 (100) |
| University Hospitals of Leicester NHS Trust | 3 | 3 (100) | 2 (67) | 0 (0) | 2 (67) | 2 (67) |
| Norfolk and Norwich University Hospitals NHS Foundation Trust | 5 | 5 (100) | 0 (0) | 0 (0) | 3 (60) | 4 (80) |
| The Newcastle upon Tyne Hospitals NHS Foundation Trust | 5 | 4 (80) | 1 (20) | 0 (0) | 3 (60) | 4 (80) |
| Hull and East Yorkshire Hospitals NHS Trust | 1 | 1 (100) | 0 (0) | 0 (0) | 1 (100) | 1 (100) |
| University Hospital Southampton NHS Foundation Trust | 4 | 4 (100) | 2 (50) | 1 (25) | 2 (50) | 4 (100) |
| University Hospitals Bristol NHS Foundation Trust | 28 | 23 (82) | 2 (7) | 1 (4) | 15 (54) | 20 (71) |
| Birmingham Children’s Hospital NHS Foundation Trust | 1 | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Alder Hey Children’s NHS Foundation Trust Hospital | 7 | 6 (85) | 1 (14) | 0 (0) | 4 (57) | 6 (856) |
| Central Manchester University Hospitals NHS Foundation Trust | 4 | 4 (100) | 1 (25) | 0 (0) | 3 (75) | 4 (100) |
| Sheffield Children’s NHS Foundation Trust | 4 | 4 (100) | 0 (0) | 0 (0) | 2 (50) | 4 (100) |
| Great Ormond Street Hospital for Children NHS Trust | 22 | 13 (59) | 2 (9) | 0 (0) | 9 (41) | 12 (55) |
| Royal Hospital for Sick Children Edinburgh – NHS Lothian | 1 | 1 (100) | 0 (0) | 0 (0) | 1 (100) | 1 (100) |
| Royal Belfast Hospital for Sick Children | 3 | 3 (100) | 0 (0) | 0 (0) | 2 (67) | 3 (100) |
| Total | 90 | 74 (82) | 11 (12) | 2 (2) | 48 (53) | 68 (76) |
Primary outcome
Adalimumab plus MTX significantly delayed the time to treatment failure compared with placebo and MTX. There were a total of 14 (23%) treatment failures for the 60 participants in the adalimumab group and 17 (57%) failures for the 30 participants in the placebo group. The median time to treatment failure was 24.10 weeks (95% CI 14.70 weeks to 81.00 weeks) in the placebo group and was not reached in the adalimumab group within the 18-month treatment period because fewer than half of the subjects experienced treatment failure at the conclusion of the trial (Figure 5).
FIGURE 5.
Primary outcome ITT Kaplan–Meier plot.
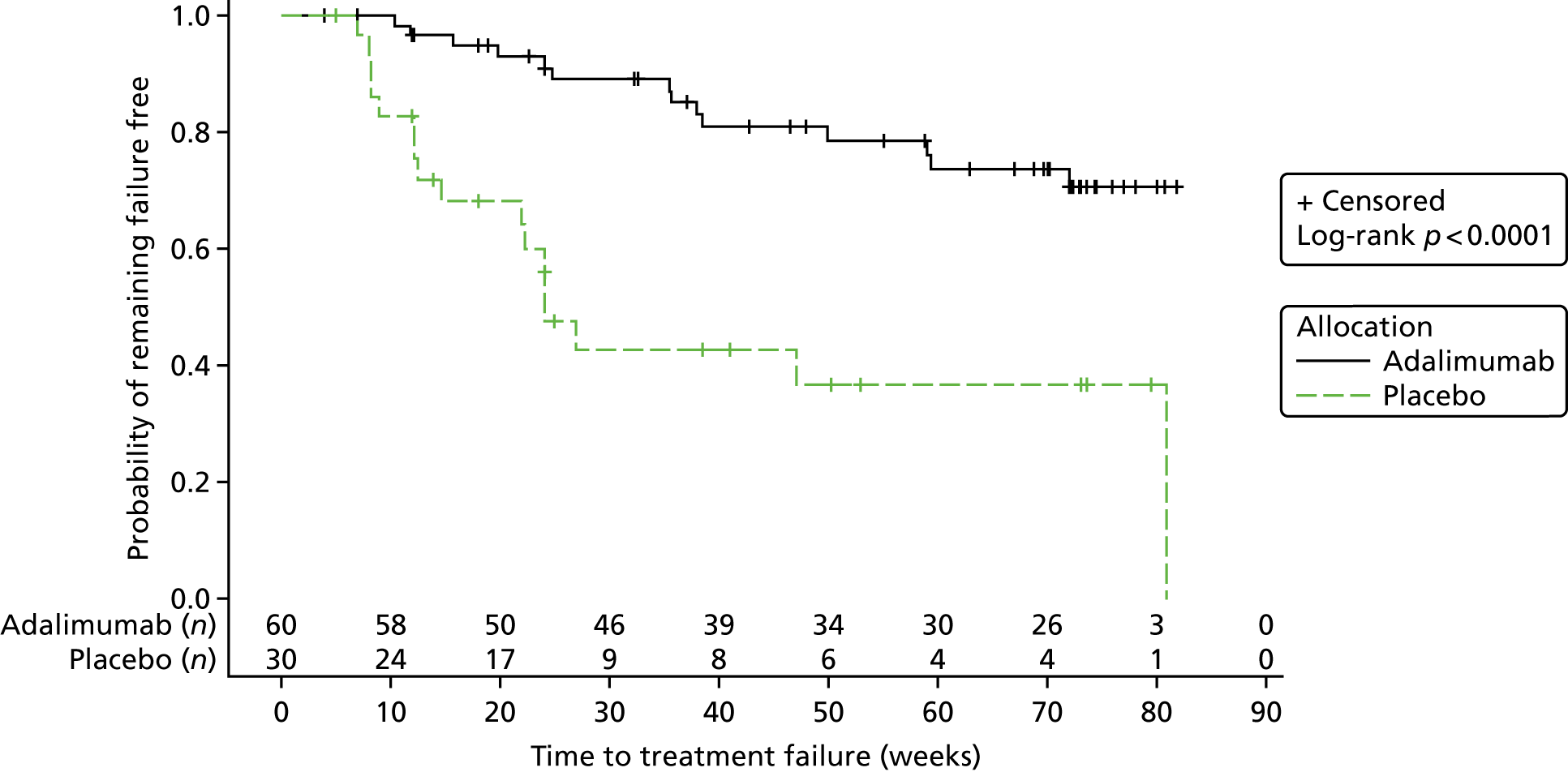
Reasons for the treatment failure of each participant can be found in Appendix 3 (see Table 42).
In the adalimumab group, five participants (trial numbers 0114011, 0116006, 0116026, 0249024 and 0030073) were classified as treatment failures because they had taken permitted concomitant medications against the acceptable criteria, three participants (trial numbers 0243023, 0249043 and 0133058) were given non-permitted concomitant medications, three participants (trial numbers 0069039, 0116062 and 0116066) had missed doses that met the failure criteria, two participants (trial numbers 0246055 and 0069076) had sustained SUN scores (as recorded at entry grade) that were still present after 6 months of therapy and one participant (trial number 0030035) had both taken permitted concomitant medications against the acceptable criteria and missed doses that met the failure criteria.
In the placebo group, two participants (trial numbers 0114033 and 0116067) received permitted concomitant medications against the acceptable criteria, seven participants (trial numbers 0249019, 0249025, 0246030, 0243032, 0540037, 0393075 and 0249086) were given non-permitted concomitant medications, one participant (trial number 0116003) missed doses that met the failure criteria and seven participants (trial numbers 0116008, 0036018, 0116005, 0249029, 0249047, 0116051 and 0249059) had sustained SUN scores (as recorded at entry grade) that were still present after 6 months of therapy.
Four participants who were classified as treatment failures did not subsequently enter the follow-up phase of the trial and withdrew completely from the trial with no further follow-up. One of these was in the adalimumab group (2%) and three were in the placebo group (10%).
The HR indicated that treatment with adalimumab significantly decreased the hazard of treatment failure by 75% (HR 0.25, 95% CI 0.12 to 0.51). The results of the log-rank test offered strong statistical evidence that the placebo group and adalimumab group differed with respect to time to treatment failure (p < 0.0001), relative to placebo. The HR was derived using Statistical Analysis Systems Procedure (SAS PROC) proportional hazards regression methods with no stratification factors.
Test of proportional hazards assumption
The assumption of proportional hazards was tested by including an interaction between time and treatment group in the Cox proportional hazards model. There was no evidence (p = 0.15) that the interaction was not zero and, therefore, no evidence that the HR was not constant over time.
Sensitivity analyses
The results of the nine sensitivity analyses can be seen in Table 7, which contains information on the number of participants analysed in each group, the number of treatment failures, the number of participants censored, the log-rank chi-squared statistic, the log-rank p-value, the HR and the 95% CIs.
| Analysis | Participants, N | Treatment group | Log-rank chi-squared statistic | Log-rank p-value | HR | 95% CI | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | ||||||||||
| Participants, n | Treatment failures, n | Censored, n | Participants, n | Treatment failures, n | Censored, n | ||||||
| ITT | 90 | 60 | 14 | 46 | 30 | 17 | 13 | 16.72 | < 0.0001 | 0.25 | 0.12 to 0.51 |
| 1: best case | 90 | 60 | 13 | 47 | 30 | 15 | 15 | 19.98 | < 0.0001 | 0.21 | 0.10 to 0.44 |
| 2: worst case | 90 | 60 | 24 | 36 | 30 | 23 | 7 | 24.17 | < 0.0001 | 0.25 | 0.14 to 0.46 |
| 3: MTX | 90 | 60 | 19 | 41 | 30 | 17 | 13 | 10.45 | 0.001 | 0.34 | 0.18 to 0.68 |
| 4: component 1 | 90 | 60 | 14 | 46 | 30 | 17 | 13 | 17.67 | < 0.0001 | 0.24 | 0.12 to 0.49 |
| 5: component 2 | 90 | 60 | 14 | 46 | 30 | 17 | 13 | 16.93 | < 0.0001 | 0.24 | 0.12 to 0.50 |
| 6: component 3 | 90 | 60 | 14 | 46 | 30 | 17 | 13 | 16.77 | < 0.0001 | 0.25 | 0.12 to 0.51 |
| 7: missing PO | 90 | 60 | 14 | 46 | 30 | 17 | 13 | 16.72 | < 0.0001 | 0.25 | 0.12 to 0.51 |
| 8: loss to follow-upa | – | – | – | – | – | – | – | – | – | – | – |
| 9: incorrect TFb | – | – | – | – | – | – | – | – | – | – | – |
There were no losses to follow-up and no incorrect treatment failures; therefore, sensitivity analyses eight and nine were not conducted. The results of the other sensitivity analyses indicate that the original conclusion from the primary analysis was robust with regard to the assumptions that were made. The overall statistical significance in the sensitivity analyses did not change.
Additional analyses
Development of uveitis in non-study eye
There were 43 (72%) participants who had unilateral vision in the adalimumab group and 22 (73%) participants who had unilateral vision in the placebo group. Those participants who had bilateral vision [17 (28%) in the adalimumab group and 8 (27%) in the placebo group] were not eligible for this analysis as they had uveitis in both eyes at baseline.
There were five (17%) participants in the placebo group who developed uveitis (defined as sustained AC cell scores of ≥ 1+ over two consecutive visits) in the non-study eye and one (2%) participant in the adalimumab group who developed uveitis in the non-study eye [the participant in the adalimumab group had a baseline AC cell score of 1+ in their non-eligible eye, but they were taking too many drops in this eye (left) for it to be eligible].
There were two participants (one in the adalimumab group and one in the placebo group) who had a single AC cell score of ≥ 1+ and had a treatment failure in their study eye at the same visit.
Time-to-treatment failure in both eyes
This analysis was not possible because only one participant (in the placebo group) failed in both eyes at different times.
Development of comorbidity on treatment failure
One participant developed cataract in the adalimumab group; none in the placebo group developed cataract. Three participants developed IOP in the adalimumab group, whereas none in the placebo group developed IOP.
There were so few participants in either of the two treatment groups who developed a comorbidity that any modelling, including the development of a comorbidity, was not possible.
Post hoc analyses
Time to treatment response
There were 44 participants in the adalimumab group and eight participants in the placebo group who were classified as having a treatment response; the difference between the two groups was statistically significant (log-rank p-value = 0.002). The HR indicated that those participants on adalimumab were over three times more likely to achieve a treatment response than those on placebo (HR 3.01, 95% CI 1.41 to 6.41).
Proportion of responders/failures/no change
Proportion of responders/failures/no change at 3 months
There were 20 (35%) participants in the adalimumab group and three (10%) participants in the placebo group who were classified as having a treatment response before 3 months. The Cochran–Armitage trend test showed a significant difference between the treatment groups at 3 months (p = 0.004).
There were three patients excluded from the analyses because they had not reached the 3-month time point.
Proportion of responders/failures/no change at 6 months
There was a total of 20 (37%) patients in the adalimumab group and three (11%) patients in the placebo group who were classified as having a treatment response prior to 6 months. The Cochran–Armitage trend test showed a significant difference between the treatment groups at 6 months (p = 0.004).
Nine participants were excluded from the analyses because they had not reached the 6-month time point.
Area under the curve of anterior chamber cells in eligible eye
There was a significant difference in the median number of AC cells between the two groups (median number of AC cells –0.79, 95% CI –0.96 to –0.63; p < 0.0001) in favour of the adalimumab group. Similar results were obtained when the best or worst score was used for participants with two eligible eyes.
Secondary outcomes
Number of participants failing treatment
Fourteen (23%) participants in the adalimumab group and 17 (57%) participants in the placebo group were classified as having treatment failures. The risk of having a treatment failure was statistically significantly reduced by 60% (RR 0.40, 95% CI 0.23 to 0.72; p = 0.002) in the adalimumab group compared with placebo.
Safety, tolerability and compliance
Adverse events and serious adverse events
Throughout the course of the trial, 733 (non-serious, n = 713; serious, n = 20) AEs were recorded. A total of 85 participants (out of 90) experienced at least one AE. There were 619 AEs reported in 59 (98%) participants in the adalimumab group and 114 AEs reported in 26 (87%) participants in the placebo group. The rate of AEs in the adalimumab group (10.60 per patient-year, 95% CI 9.77 per patient-year to 11.44 per patient-year) was greater than that in the placebo group (7.21 per patient-year, 95% CI 5.89 per patient-year to 8.53 per patient-year).
The commonest AEs in the adalimumab group were classified as infections and infestations (83%), respiratory, thoracic and mediastinal disorders (55%), general disorders and administration-site conditions (52%), gastrointestinal disorders (47%), investigations (32%), musculoskeletal and connective tissue disorders (27%), nervous system disorders (27%) and eye disorders (25%). Aside from eye disorders (27%), common AEs reported in the placebo group were consistently lower [infections and infestations (47%), respiratory (20%), thoracic and mediastinal disorders (27%), general disorders and administration-site conditions (30%), gastrointestinal disorders (13%), investigations (23%), musculoskeletal and connective tissue disorders (13%) and nervous system disorders (27%) (Table 8)].
| System Organ Class | Treatment group | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | ||||||||
| Events (n) | Patients (n) | % of patients | Events (n) | Patients (n) | % of patients | Events (n) | Patients (n) | % of patients | |
| Blood and lymphatic system disorders | 5 | 5 | 8 | 0 | 0 | 0 | 5 | 5 | 6 |
| Eye disorders | 19 | 15 | 25 | 9 | 8 | 27 | 28 | 23 | 26 |
| Gastrointestinal disorders | 79 | 28 | 47 | 14 | 9 | 30 | 93 | 37 | 41 |
| General disorders and administration-site conditions | 130 | 31 | 52 | 15 | 8 | 27 | 145 | 39 | 43 |
| Immune system disorders | 3 | 3 | 5 | 1 | 1 | 3 | 4 | 4 | 4 |
| Infections and infestations | 149 | 50 | 83 | 30 | 14 | 47 | 179 | 64 | 71 |
| Injury, poisoning and procedural complications | 15 | 12 | 20 | 5 | 3 | 10 | 20 | 15 | 17 |
| Investigations | 39 | 19 | 32 | 6 | 4 | 13 | 45 | 23 | 26 |
| Metabolism and nutrition disorders | 3 | 3 | 5 | 0 | 0 | 0 | 3 | 3 | 3 |
| Musculoskeletal and connective tissue disorders | 31 | 16 | 27 | 8 | 7 | 23 | 39 | 23 | 26 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 5 | 5 | 8 | 0 | 0 | 0 | 5 | 5 | 6 |
| Nervous system disorders | 31 | 16 | 27 | 10 | 4 | 13 | 41 | 20 | 22 |
| Psychiatric disorders | 5 | 2 | 3 | 2 | 1 | 3 | 7 | 3 | 3 |
| Reproductive system and breast disorders | 6 | 2 | 3 | 0 | 0 | 0 | 6 | 2 | 2 |
| Respiratory, thoracic and mediastinal disorders | 84 | 33 | 55 | 9 | 6 | 20 | 93 | 39 | 43 |
| Skin and subcutaneous tissue disorders | 13 | 8 | 13 | 5 | 4 | 13 | 18 | 12 | 13 |
| Surgical and medical procedures | 2 | 2 | 3 | 0 | 0 | 0 | 2 | 2 | 2 |
The majority of AEs in both treatment groups were deemed to be mild or moderate in intensity. Overall, 8% (five events in five participants) of the adalimumab group had at least one severe AE and 7% (three events in two participants) of the placebo group had one severe AE. The five severe AEs in the adalimumab group were cataract, injection-site reaction, glaucoma, arthralgia and arthritis; the three severe AEs in the placebo group were AC flare (two events in the same participant) and uveitis.
A total of 17 SAEs were reported in 13 (22%) participants in the adalimumab group and three SAEs in two (7%) participants in the placebo group during the course of the blinded phase of the trial. The rate of SAEs was greater in the adalimumab group (0.29 per patient-year) than in the placebo group (0.19 per patient-year).
A participant listing of SAEs for adalimumab and placebo can be found in Appendix 3 (see Table 43). All but one of the SAEs were classified as mild to moderate. This SAE was reported in a participant on placebo whose vision had deteriorated while on medication; the participant’s treatment allocation was revealed and, consequently, they commenced on dexamethasone drops and anti-TNF-α.
Laboratory parameters
Haematological
The data relating to haematological parameters are summarised below.
Table 9 gives the mean difference [standard error (SE)] of change in haematological laboratory parameters from baseline to each treatment visit of adalimumab compared with placebo. Differences marked by an asterisk were significant at the 5% level. None of the mean changes in haematological assessments was considered to be clinically significant.
| Variable | Month, mean difference (SE) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 6 | 9 | 12 | 15 | 18 | |
| Haematocrit (%) | 0.44 (0.60) | 0.27 (0.56) | –0.73 (0.67) | –1.23 (0.76) | –0.81 (1.24) | –1.93 (1.54) | 0.12 (1.69) | 1.62 (1.88) |
| Haemoglobin (g/dl) | 0.08 (0.17) | 0.02 (0.17) | –0.12 (0.18) | –0.41 (0.25) | –0.38 (0.37) | –0.52 (0.41) | –0.37 (0.44) | –0.09 (0.68) |
| Red blood cell count ( × 1012/l) | 0.00 (0.06) | –0.00 (0.06) | –0.06 (0.06) | –0.20 (0.08)a* | –0.18 (0.11) | –0.18 (0.14) | –0.25 (0.14) | –0.17 (0.17) |
| White blood cell count ( × 1012/l) | 0.33 (0.53) | 0.09 (0.66) | –0.22 (1.17) | 1.17 (0.69) | 0.06 (0.99) | 0.90 (1.36) | 1.36 (1.32) | 0.75 (1.63) |
| Neutrophils ( × 109/l) | –0.07 (0.45) | –0.66 (0.52) | –0.83 (1.10) | 0.64 (0.58) | –0.45 (0.70) | 0.04 (0.95) | 0.40 (1.08) | 0.03 (1.29) |
| Lymphocytes ( × 109/l) | 0.39 (0.15)* | 0.52 (0.20)* | 0.42 (0.23) | 0.32 (0.19) | 0.53 (0.38) | 0.72 (0.42) | 0.61 (0.63) | 0.34 (0.32) |
| Monocytes ( × 109/l) | 0.04 (0.04) | 0.07 (0.05) | 0.07 (0.06) | 0.12 (0.06) | 0.09 (0.09) | 0.10 (0.10) | 0.20 (0.11) | 0.10 (0.13) |
| Basophils ( × 109/l) | 0.01 (0.01) | 0.00 (0.01) | –0.00 (0.01) | –0.00 (0.01) | –0.01 (0.01) | –0.01 (0.01) | 0.01 (0.01) | –0.01 (0.01) |
| Eosinophils ( × 109/l) | 0.04 (0.06) | 0.06 (0.05) | 0.14 (0.05)* | 0.11 (0.08) | –0.02 (0.06) | –0.02 (0.26) | 0.06 (0.10) | 0.17 (0.13) |
| Platelet count ( × 109/l) | –3.34 (12.80) | –6.79 (11.60) | –21.86 (13.48) | –17.27 (13.30) | –14.41 (20.23) | –6.78 (30.89) | –15.56 (37.23) | –15.05 (29.94) |
| ESR (mm/hour) | 0.09 (3.49) | –0.85 (2.71) | –0.03 (2.43) | –1.84 (4.25) | –2.49 (3.40) | 3.40 (5.34) | –1.50 (6.25) | 5.39 (8.31) |
| Plasma viscositya | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
There was evidence of a greater change in lymphocytes from baseline to months 1 and 2 and in eosinophils from baseline to 3 months in the adalimumab group than in the placebo group. The placebo group experienced a greater change in mean red blood cell count from baseline to 6 months than the adalimumab group.
Biochemical
The data relating to biochemical parameters are summarised below.
Table 10 gives the mean difference (SE) of change in biochemical laboratory parameters from baseline to each treatment visit of adalimumab compared with placebo. Differences marked by an asterisk were significant at the 5% level.
| Variable | Month, mean difference (SE) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 6 | 9 | 12 | 15 | 18 | |
| CRP (mg/l) | 1.15 (2.43) | 0.19 (1.43) | 0.06 (3.09) | –0.99 (1.78) | –1.82 (2.16) | –0.25 (2.22) | 1.26 (3.69) | 6.06 (12.90) |
| Urea (mmol/l) | –0.43 (0.21)* | –0.55 (0.25)* | –0.57 (0.27)* | –0.38 (0.36) | –0.69 (0.49) | –1.02 (0.54) | –1.45 (0.86) | –1.57 (0.72)* |
| Creatinine (mmol/l) | –1.19 (1.86) | –2.22 (2.27) | 0.51 (2.29) | 0.45 (1.86) | –0.94 (5.36) | 0.88 (3.09) | 0.15 (3.89) | –23.64 (8.68)* |
| Sodium (mmol/lL) | 1.07 (0.61) | 1.14 (0.64) | –0.05 (0.55) | 0.19 (0.96) | 0.05 (1.11) | 0.39 (1.29) | –0.59 (1.88) | 0.57 (2.04) |
| Potassium (mmol/l) | –0.05 (0.10) | 0.01 (0.11) | 0.00 (0.13) | –0.29 (0.14)* | 0.15 (0.33) | 0.18 (0.32) | 0.07 (0.21) | –0.09 (0.19) |
| Calcium (mmol/l) | 0.02 (0.02) | 0.01 (0.03) | 0.04 (0.03) | 0.01 (0.03) | –0.03 (0.05) | 0.03 (0.05) | 0.02 (0.06) | –0.06 (0.08) |
| Inorganic phosphate (mmol/l) | 0.05 (0.06) | 0.05 (0.06) | –0.03 (0.06) | 0.02 (0.07) | 0.08 (0.12) | –0.11 (0.10) | –0.06 (0.14) | –0.09 (0.14) |
| Glucose (mmol/l) | –0.06 (0.30) | –0.33 (0.30) | 0.15 (0.30) | 0.43 (0.40) | 0.11 (0.77) | 0.10 (0.77) | 0.69 (0.74) | 0.17 (0.84) |
| Chloride (mmol/l) | 0.86 (0.63) | 0.31 (0.68) | 1.19 (0.76) | 1.24 (0.98) | 0.41 (1.23) | 0.45 (1.33) | –1.25 (1.78) | –0.38 (1.96) |
| Bicarbonate (mmol/l) | 0.28 (1.08) | –0.41 (1.00) | –1.34 (1.01) | 0.02 (1.53) | –3.70 (2.42) | 1.52 (2.00) | 0.39 (3.36) | 0.20 (2.90) |
| Total bilirubin (mmol/l) | 1.08 (0.89) | 1.25 (0.93) | –0.35 (0.96) | 0.84 (1.50) | 2.11 (1.69) | 2.74 (1.75) | 0.06 (3.43) | –3.40 (2.13) |
| ALT (IU/l) | 2.67 (8.05) | 6.80 (7.31) | –0.86 (6.11) | 1.51 (9.53) | 25.83 (41.90) | 0.66 (10.92) | 9.19 (14.94) | 4.29 (13.68) |
| AST (IU/l) | –3.32 (2.47) | –3.63 (2.72) | –1.02 (4.31) | –4.41 (4.89) | 6.03 (26.32) | –11.75 (6.85) | –12.00 (13.41) | 8.76 (13.63) |
There was evidence of a greater change in urea from baseline to months 1, 2, 3 and 18; in potassium from baseline to 6 months; and in creatinine from baseline to 18 months in the placebo group than in the adalimumab group. None of the mean changes in biochemical assessments was considered to be clinically significant.
Urinalysis
Table 11 shows the number of abnormal urinalysis assessments in each treatment group over time. Details on the microscopic analysis are presented in Table 12. Overall, the results from the urinalysis were not clinically significant.
| Visit | Allocation | Number of abnormal assessments (number of participants) |
|---|---|---|
| Baseline | Adalimumab | 19 (17) |
| Placebo | 15 (13) | |
| Month 1 | Adalimumab | 20 (15) |
| Placebo | 12 (8) | |
| Month 2 | Adalimumab | 23 (18) |
| Placebo | 9 (7) | |
| Month 3 | Adalimumab | 15 (13) |
| Placebo | 9 (7) | |
| Month 6 | Adalimumab | 17 (11) |
| Placebo | 4 (4) | |
| Month 9 | Adalimumab | 15 (12) |
| Placebo | 5 (4) | |
| Month 12 | Adalimumab | 12 (11) |
| Placebo | 4 (3) | |
| Month 15 | Adalimumab | 8 (8) |
| Placebo | 1 (1) | |
| Month 18 | Adalimumab | 6 (6) |
| Placebo | 1 (1) |
| Time point | Allocation | Assessments, n | Normal assessments, n (%) | Abnormal assessments, n (%) | If abnormal, clinically significant, n (%) | Assessment recorded as not applicable, n (%) | Missing, n (%) |
|---|---|---|---|---|---|---|---|
| Baseline | Adalimumab | 17 | 9 (53) | 7 (41) | 1 (14) | 1 (6) | 0 (0) |
| Placebo | 13 | 7 (54) | 4 (31) | 0 (0) | 2 (15) | 0 (0) | |
| 1 month | Adalimumab | 15 | 8 (53) | 3 (20) | 0 (0) | 4 (27) | 0 (0) |
| Placebo | 8 | 4 (50) | 3 (38) | 0 (0) | 1 (13) | 0 (0) | |
| 2 months | Adalimumab | 18 | 11 (61) | 3 (17) | 1 (33) | 3 (17) | 1 (6) |
| Placebo | 7 | 3 (43) | 3 (43) | 0 (0) | 0 (0) | 1 (14) | |
| 3 months | Adalimumab | 13 | 9 (69) | 3 (23) | 0 (0) | 1 (8) | 0 (0) |
| Placebo | 7 | 4 (57) | 2 (29) | 0 (0) | 0 (0) | 1 (14) | |
| 6 months | Adalimumab | 11 | 6 (55) | 3 (28) | 2 (67) | 2 (18) | 0 (0) |
| Placebo | 4 | 4 (100) | 0 (0) | N/A | 0 (0) | 0 (0) | |
| 9 months | Adalimumab | 12 | 6 (50) | 6 (50) | 2 (33) | 0 (0) | 0 (0) |
| Placebo | 4 | 3 (75) | 0 (0) | N/A | 0 (0) | 1 (25) | |
| 12 months | Adalimumab | 11 | 7 (64) | 3 (27) | 2 (67) | 1 (9) | 0 (0) |
| Placebo | 3 | 1 (33) | 2 (67) | 0 (0) | 0 (0) | 0 (0) | |
| 15 months | Adalimumab | 8 | 5 (63) | 2 (25) | 0 (0) | 1 (13) | 0 (0) |
| Placebo | 1 | 0 (0) | 0 (0) | N/A | 1 (100) | 0 (0) | |
| 18 months | Adalimumab | 6 | 2 (40) | 1 (20) | 1 (100) | 2 (40) | 0 (0) |
| Placebo | 1 | 0 (0) | 1 (100) | 0 (0) | 0 (0) | 0 (0) |
Participant diaries and dosing records
Participant diaries and dosing records determined tolerability and compliance throughout the trial treatment period. Treatment compliance was estimated using participant treatment diaries and accountability logs.
Treatment diaries were used to estimate participant compliance by dividing the number of doses recorded as taken in the treatment diary by the expected number of doses the participant should have taken (on the basis of the time the participant was on treatment). According to the treatment diaries, compliance of adalimumab was 84% and compliance of placebo was 74%. MTX compliance was estimated to be 62% for the adalimumab group and 50% for the placebo group (see Appendix 3, Tables 44 and 45, respectively).
The accountability logs were used to provide another estimate of adalimumab and placebo compliance by dividing the sum of the number of used vials returned (and the number of missing vials) by the number of vials issued. Estimated compliance for the adalimumab group was 94% and for the placebo group was 90%.
Four participants in the adalimumab group (0069039, 0030035, 0116062 and 0116066) had their medication stopped. Participant 0030035 failed treatment because of missed doses that met failure criteria and taking permitted concomitant medications against acceptable criteria, and one participant (0116003) in the placebo group failed treatment because they missed more than the required number of doses while on treatment.
Four participants had their study medication stopped because of missed doses of MTX: 0116052, 0248064, 0540060 and 0246055. These participants were all receiving adalimumab at the time that they met the threshold for missed doses of MTX.
Use of corticosteroids over duration of study period
Total oral corticosteroid dose
One participant in the placebo group and five participants in the adalimumab group received oral corticosteroids during the course of the blinded treatment phase. The five participants in the adalimumab group were on study treatment for a total of 5.28 years and the placebo participant was on study treatment for 0.17 years.
The total oral dose for the placebo group was 640 mg (standardised per patient-year, was 3767.74 mg) and 4248.5 mg for the adalimumab group (standardised per patient-year 804.31 mg). A rate ratio of 0.21 (95% CI 0.20 to 0.23; p < 0.0001) indicated that participants on placebo required more oral corticosteroids per patient-year than those randomised to adalimumab and there was evidence at the 5% level of a statistically significant difference between the two groups.
Reduction in and rate of systemic corticosteroid dose from entry dose
Reduction in systemic corticosteroid dose from entry dose
Reduction in systemic corticosteroid dose from entry dose to 0 mg
At the beginning of the study, there were six participants (adalimumab group, n = 5; placebo group, n = 1) who were prescribed systemic corticosteroids (permitted dose < 0.2 mg/kg/day; median dose 0.14 mg/kg/day). Three adalimumab-treated participants stopped systemic corticosteroids (median duration 18.14 weeks). The placebo group participant stopped systemic corticosteroids after 5.57 weeks.
No comparative analysis could be performed because the statistical algorithm did not converge.
Reduction in systemic corticosteroid dose from entry dose to < 5 mg
At the beginning of the study, there were three participants (adalimumab group, n = 2; placebo group, n = 1) who were on ≥ 5 mg of systemic corticosteroids.
No comparative analysis could be performed because the statistical algorithm did not converge.
One participant on adalimumab had a reduction to < 5 mg of systemic corticosteroids and one participant ended treatment before having a reduction to < 5 mg. The participant on placebo had a reduction to < 5 mg.
Rate of systemic corticosteroid dose from entry dose
The result of this analysis was the same as that of the total oral corticosteroid analysis.
Topical corticosteroid use (frequency) compared with entry use
Time to reduction to fewer than two drops in topical corticosteroid
The outcome was time to reduction to fewer than two drops per day for those participants already at more than two drops per day at baseline. There were 63 participants who were on more than two drops per day at baseline [18 (60%) in the placebo group and 45 (75%) in the adalimumab group] and who were, therefore, included in the analysis.
Twenty-four (53.3%) of the 45 participants on adalimumab and three (16.70%) of the 18 participants on placebo reached fewer than two drops per day before treatment failure (or the 18-month treatment visit). Five participants (11.10%) on adalimumab and one (5.60%) participants on placebo reached the 18-month visit before reaching fewer than two drops per day and 16 (35.6%) of the adalimumab group and 14 (77.8%) of the placebo group had a treatment failure/withdrawal before reaching fewer than two drops per day.
The time to reduction to fewer than two drops per day was statistically significant in favour of adalimumab (HR 3.99, 95% CI 1.18 to 13.48; p = 0.03); the incidence plot is shown in Figure 6.
FIGURE 6.
Incidence plot of time to reduction to fewer than two drops per day.
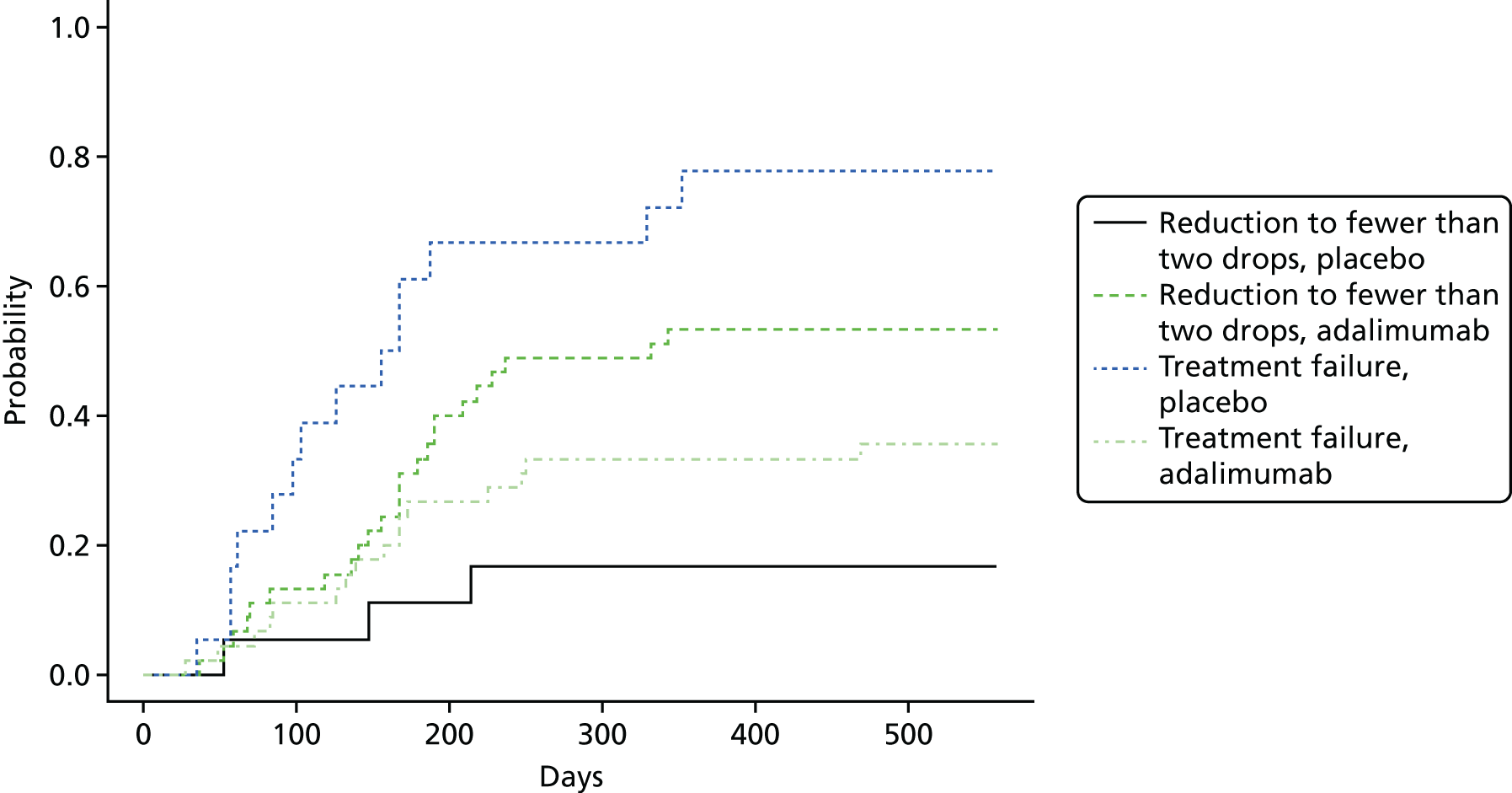
Time to reduction to zero drops in topical steroid (post hoc analysis)
The outcome was time to reduction to zero drops for those participants already at more than zero drops at randomisation. There were 74 participants [25 (34%) on placebo and 49 (66%) on adalimumab] who were on more than zero drops at randomisation and who were, therefore, included in the analysis.
Twenty-five (51%) of the 49 participants on adalimumab and four (16%) of the 25 participants on placebo reached zero drops before treatment failure (or the 18-month treatment visit). Six participants (12%) on adalimumab and two participants (8%) on placebo reached the 18-month visit before reaching zero drops and 18 (37%) of the adalimumab group and 19 (76%) of the placebo group had a treatment failure/withdrawal before reaching zero drops.
The time to reduction to zero drops was statistically significant in favour of adalimumab (HR 4.01, 95% CI 1.40 to 11.51; p = 0.01); the incidence plot is shown in Figure 7.
FIGURE 7.
Incidence plot for time to reduction to zero drops.
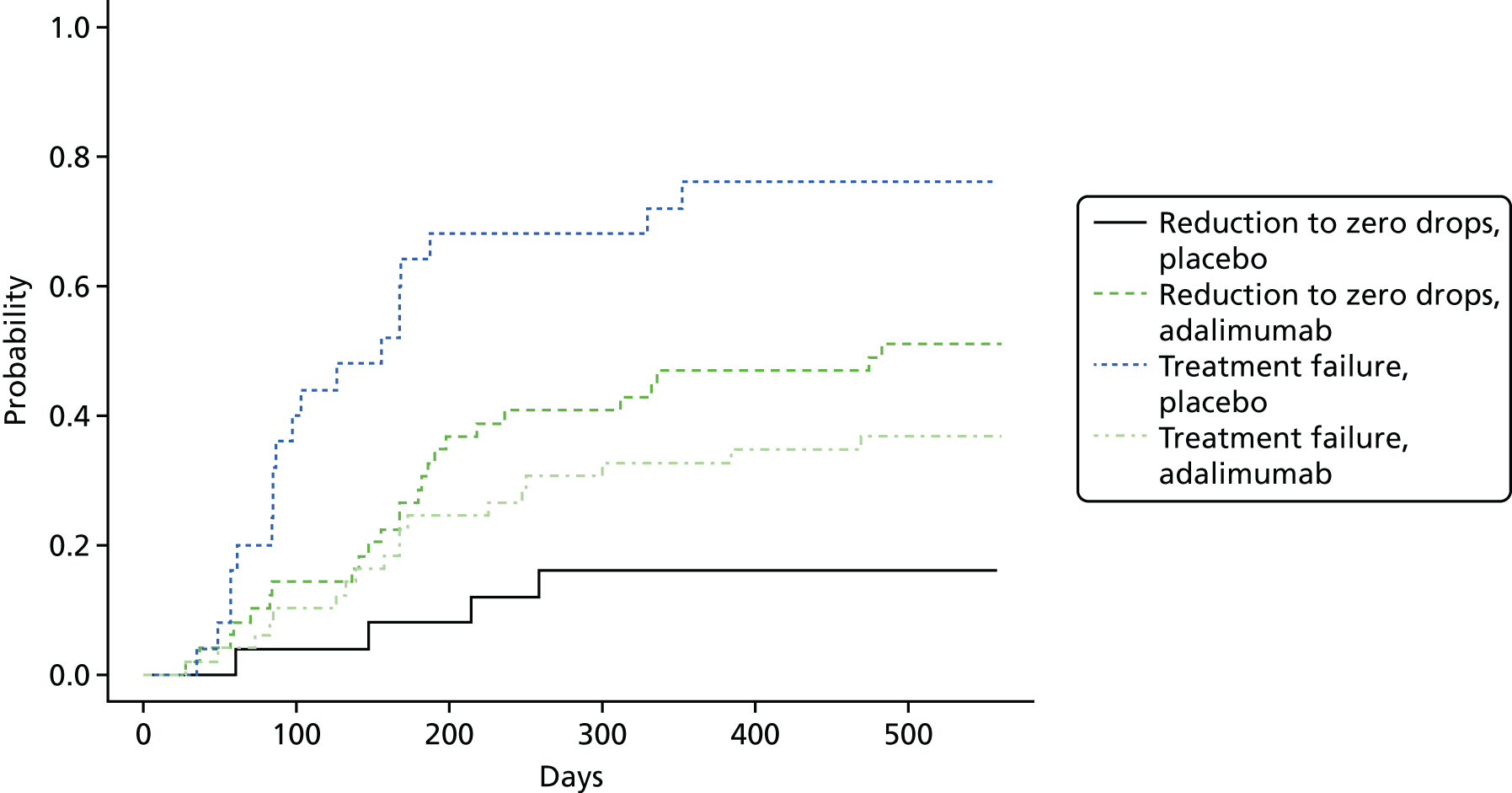
Need for pulsed corticosteroid
One participant in the placebo group (3%) and two participants in the adalimumab group (3%) required pulsed corticosteroids during the course of the blinded phase. There was no evidence of a difference in the risk of requiring pulsed corticosteroids between the two treatment groups (RR 1.00, 95% CI 0.09 to 10.59; p > 0.99).
Optic and ocular
Number of participants having disease flares (as defined by worsening on standardised uveitis nomenclature criteria) following 3 months of disease control
One of the 30 participants in the placebo group (3%) and 5 of the 60 participants in the adalimumab group (8%) experienced 3 months of disease control with a subsequent disease flare; there was no statistically significant difference between the two groups (RR 2.50, 95% CI 0.31 to 20.45; p = 0.66).
The inference from the analysis of participants who had disease control in both eligible eyes followed by a flare in at least one eye was the same as the analysis of disease control in one eye.
Number of participants having disease flare within the first 3 months
Three participants in the placebo group (10%) and no participants in the adalimumab group had a disease flare in the first 3 months of treatment. There was statistically significant evidence at the 5% level (p = 0.03) of a difference (RR 0.07, 95% CI 0.004 to 1.36) between the two groups.
No participants in the placebo group who had a flare within the first 3 months entered the study with two eligible eyes; therefore, the analysis on both eyes was not possible.
Visual acuity measured by age-appropriate logarithm of the minimum angle of resolution assessment
Two analyses were conducted using joint modelling. In each analysis, when only one eye was involved, the single logMAR value was used. When there were two eyes involved, the two analyses were:
-
analysis 1 – taking the best logMAR measurement (the minimum of the two values)
-
analysis 2 – taking the worst logMAR measurement (the maximum of the two values).
The parameter estimates for analyses 1 and 2 are presented in Table 13. For analyses 1 and 2, the results of the joint modelling showed that the treatment effects (adalimumab) on the longitudinal logMAR are –0.01 (95% CI –0.07 to 0.02) and –0.02 (95% CI –0.07 to 0.02), respectively, implying that there is no significant difference between the treatments on logMAR. These estimates are adjusted for failure because of treatment dropout from the trial.
| Analysis | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| 1 | Longitudinal | Intercept | 0.01 | –0.02 to 0.04 | 0.76 |
| Baseline | 0.71 | 0.51 to 0.93 | < 0.0001 | ||
| Time | –0.001 | –0.01 to 0.001 | 0.22 | ||
| Adalimumab | –0.01 | –0.07 to 0.02 | 0.51 | ||
| Survival | Adalimumab | –1.37 | –2.26 to –0.70 | 0.001 | |
| HR | 0.25 | 0.10 to 0.50 | 0.001 | ||
| Association | γ0 | 3.29 | –2.42 to 12.15 | 0.39 | |
| 2 | Longitudinal | Intercept | 0.02 | –0.02 to 0.05 | 0.36 |
| Baseline | 0.82 | 0.61 to 1.07 | < 0.0001 | ||
| Time | –0.002 | –0.01 to 0.0004 | 0.21 | ||
| Adalimumab | –0.02 | –0.07 to 0.02 | 0.36 | ||
| Survival | Adalimumab | –1.36 | –2.35 to –0.69 | 0.001 | |
| HR | 0.26 | 0.10 to 0.50 | 0.001 | ||
| Association | γ0 | 3.52 | –3.09 to 9.44 | 0.29 |
Appendix 3 presents data on the logMAR score for participants in the trial, split by treatment group and time point (see Table 46, and Figures 14 and 15).
Sensitivity analysis
The residuals from the separate fitted linear missed models (LMMs) for the logMAR indicated slight departures from the normality assumption. In general, fixed-effects estimates are robust to non-normal errors in LMMs. 65 The histograms of baseline logMAR scores (for analyses 1 and 2) appeared approximately normal (not shown) and, therefore, no further analysis from log-transformed data was considered.
In the primary analysis, a random-intercepts model for the longitudinal submodel was fitted. The second sensitivity analysis investigated fitting a random-intercepts and random-slopes model. This showed that inferences remained similar. For analyses 1 and 2, the treatment effect on the longitudinal outcome was –0.01 (95% CI –0.05 to 0.03) and for the random-intercepts and random-slopes model the treatment effect was –0.01 (95% CI –0.05 to 0.03); these were not statistically significant.
Number of participants with resolution of associated optic nerve or macular oedema (as assessed by slit lamp biomicroscopy or optical coherence tomography, where available)
Four participants in the adalimumab group (6.67%) had associated optic nerve at baseline or developed it at some point during the study, two (50%) of these cases were resolved during the study. There were no participants who had associated optic nerve at baseline or developed this during the course of the study in the placebo group. It was, therefore, not possible to carry out the planned statistical test of these data.
Two participants in the placebo group (7%) had macular oedema at baseline or developed it during the course of the study, compared with four participants in the adalimumab group (7%).
Three participants in the adalimumab group (75%) and no participants in the placebo group had resolution of the macular oedema (RR 5.00, 95% CI 0.34 to 74.52). This was based on the assumption that, if the macular oedema occurred in both eligible study eyes, then resolution only had to occur in at least one of these eyes.
Two of the three participants who had resolution of macular oedema were eligible in both eyes. Because both of these participants experienced resolution in both eyes, there was no difference in results when considering the assumption that the resolution must occur in both eligible eyes.
Number of participants with disease control (defined as zero cells, with topical treatment for 3 and 6 months)
Three months
Two participants in the placebo group (7%) and 23 in the adalimumab group (38%) had disease control for at least 3 months (RR 5.75, 95% CI 1.45 to 22.78; p = 0.001).
One (50%) of the two participants with disease control in the placebo group had both eyes eligible at baseline and did not have disease control in both eyes. Of the 23 participants in the adalimumab group, five participants (22%) had both eyes eligible at baseline and all five participants had disease control in both study eyes for at least 3 months. The inference of the analysis when both eligible eyes had to have disease control was the same as that for at least one eligible eye.
Six months
At 6 months, one participant (3%) in the placebo group and 17 participants in the adalimumab group (28%) had disease control in at least one of their eligible eyes (RR 8.50, 95% CI 1.19 to 60.87; p = 0.005). Four of the 17 participants (24%) had two eyes eligible at the beginning of the study.
All four participants in the adalimumab group, who were eligible in both eyes at the beginning of the study, had disease control in both eyes for at least 6 months. The inference of the analysis when both eligible eyes had to have disease control was the same as that for at least one eligible eye.
Number of participants entering disease remission (defined as zero cells, without topical treatment for 3 and 6 months)
Three months
At 3 months, one participant in the placebo group (3.33%) and 15 in the adalimumab group (25%) had entered disease remission in any of their eligible eyes (RR 7.50, 95% CI 1.04 to 54.12; p = 0.02).
One participant in the placebo group was eligible in both eyes at the beginning of the study, but had disease remission in only one eye; four participants in the adalimumab group were eligible in both eyes at the beginning of the study and all four had disease remission in both eyes. The inference of the analysis when both eligible eyes had to be in remission was the same as that for at least one eligible eye.
Six months
At 6 months, no participants in the placebo group and 13 in the adalimumab group (23%) had entered disease remission in both of their eligible eyes (RR 13.72, 95% CI 0.84 to 223.26, p = 0.004).
Four participants in the adalimumab group (29%) were eligible in both eyes at the beginning of the study. After 6 months, three of these four participants had entered remission in both of their eligible eyes and one had not. The inference of the analysis when both eligible eyes had to be in remission was the same as that for at least one eligible eye.
Duration of sustaining inactive disease (zero cells in the anterior chamber, with or without topical treatment)
The difference in the total amount of time that participants sustained inactive disease was statistically significant between the two treatment groups. The estimated mean days of sustained inactive disease was 16.36 days (SE 23.79 days) for the placebo group and 180.91 days (SE 16.81 days) for the adalimumab group, with participants in the adalimumab group spending 164.55 more days (95% CI 104.41 days to 224.69 days; p < 0.0001) with inactive disease than those in the placebo group.
Quality-of-life assessment
Childhood Health Questionnaire
Overall, the mean scores for the CHQ psychosocial subscale (PsS) were very similar in both treatment groups, with the adalimumab group having slightly higher scores (see Appendix 3, Table 47, and Figure 16). The treatment effect on the longitudinal CHQ PsS score was 2.31 (95% CI –0.44 to 5.40), implying that there is no difference between the treatments on the score; however, the p-value (0.06) is close to the margin of statistical significance (Table 14).
| Outcome | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| PsS | Longitudinal | Intercept | 15.84 | 7.49 to 23.13 | 0.0002 |
| Baseline | 0.68 | 0.54 to 0.83 | < 0.0001 | ||
| Time | 0.05 | –0.17 to 0.23 | 0.66 | ||
| Adalimumab | 2.31 | –0.44 to 5.40 | 0.15 | ||
| Survival | Adalimumab | –1.66 | –2.55 to –0.89 | 0.0002 | |
| HR | 0.19 | 0.08 to 0.41 | 0.0002 | ||
| Association | γ0 | –0.14 | –0.28 to –0.01 | 0.04 | |
| PhS | Longitudinal | Intercept | 20.85 | 12.32 to 30.54 | < 0.0001 |
| Baseline | 0.57 | 0.39 to 0.75 | < 0.0001 | ||
| Time | –0.02 | –0.21 to 0.12 | 0.83 | ||
| Adalimumab | 1.16 | –2.41 to 5.05 | 0.55 | ||
| Survival | Adalimumab | –1.40 | –2.35 to –0.67 | 0.001 | |
| HR | 0.25 | 0.10 to 0.51 | 0.001 | ||
| Association | γ0 | –0.06 | –0.17 to 0.03 | 0.18 |
The summary statistics for each time point for CHQ physical subscale (PhS) and the mean profile plots are presented in Appendix 3 (see Table 48 and Figure 17). The treatment effect on the longitudinal CHQ-PhS score was 1.16 (95% CI –2.41 to 5.05), which implies that there is no difference between the treatments on this score (see Table 14). This estimate is adjusted for failure because of dropout from the trial.
Sensitivity analysis
The normality assumption was considered for each CHQ score. The log-transform of baseline CHQ scores resulted in distributions that were more normal (not shown), although they still remained somewhat skewed. The residuals from the separate fitted LMMs for the CHQ scores indicated departures from the normality assumption. The log-transformations did not lead to an improvement in model fit according to the Q–Q plot of the residuals (not shown). In general, fixed-effects estimates are robust to non-normal errors in LMMs; therefore, the inferences from the untransformed (raw) CHQ scores are used.
The log-likelihood values for the primary analysis (random-intercepts only) and the sensitivity analysis model (random-intercepts and random-slopes) were calculated. Although the likelihood is increased for the random-intercepts and random-slopes model, it estimates two additional parameters: a variance component for the random slope and a correlation term. Trading off the improvement in goodness of fit against model complexity, only marginal model improvement for PhS was found. However, for PsS, there was greater evidence in favour of the random-intercepts and random-slopes models. The inference on the PhS score remained the same: 1.49 (95% CI –1.87 to 5.45). The treatment effect on the longitudinal PsS score was 2.09 (95% CI –0.74 to 5.21), implying that there was no difference between the treatments on this score too.
As a sensitivity analysis, the missing baseline values (n = 15) were imputed with the mean observed values for the other participants and the 57 intermediate missing measurements; the approach was used as per the predefined methodology in the SAP. After imputing the data, the joint model was refitted. The treatment effect on PhS is halved for the imputation analysis; however, both the primary and the imputation analysis treatment effects remain statistically non-significant. The treatment effect on the CHQ-PsS also remained non-significant.
Childhood Health Assessment Questionnaire
The treatment effect on the longitudinal CHAQ is –0.14 (95% CI –0.31 to 0.02) and implies that there is no difference between the treatments on CHAQ (Table 15). This estimate is adjusted for failure because of dropout from the trial. However, the p-value (0.08) is close to the margin of statistical significance.
| Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|
| Longitudinal | Intercept | 0.20 | 0.05 to 0.35 | 0.01 |
| Baseline | 0.65 | 0.49 to 0.75 | < 0.0001 | |
| Time | –0.01 | –0.01 to 0.001 | 0.06 | |
| Adalimumab | –0.14 | –0.31 to 0.02 | 0.09 | |
| Survival | Adalimumab | –1.46 | –2.23 to –0.80 | 0.0001 |
| HR | 0.23 | 0.11 to 0.45 | 0.0001 | |
| Association | γ0 | 0.64 | –0.89 to 2.04 | 0.35 |
The summary statistics for the CHAQ are shown for each treatment group for each scheduled study visit. The mean profile plots for each treatment group are given in Appendix 3 (see Table 49 and Figure 18).
Sensitivity analysis
The distribution of baseline CHAQ scores is non-normal. This is predominantly because of the large proportion of zero baseline scores (n = 12 out of 87 remaining participants). However, the effect of the log-transformation had little effect on the normality assumption of the residuals; therefore, no further analysis was carried out.
In the first sensitivity analysis that considered the model specification, the model included a treatment-to-time interaction term, as the treatment effect in the longitudinal submodel in the primary analysis, it was significant at a 10% level. The interaction term in the fitted model was statistically significant. Furthermore, the fixed effects for time and treatment were both significant at the 5% level under this model. The joint log-likelihood values for the model with and without the interaction term indicated an improvement in model fit.
The treatment effects on the longitudinal outcome CHAQ were –0.20 (95% CI –0.37 to –0.06) and implied that CHAQ was significantly lower in the adalimumab group.
The second sensitivity analysis considered a random-intercepts and random-slopes model. There was no apparent increase in likelihood for the random-intercepts and random-slopes model. In addition, the random-intercepts and random-slopes model estimates two additional parameters: a variance component for the random slope and a correlation term. Trading off the improvement in goodness of fit against model complexity, there was no model improvement. The sensitivity analysis that examined missing data used imputation; the joint model was refitted and the inferences remained statistically significant.
American College of Rheumatology Pedi core set criteria at ACR 30, 50, 70, 90 and 100
The results for the joint model of the ACR Pedi and time-to-treatment failure can be seen in Table 16. None of the improvements on ACRs is significantly different between treatments. The treatment effect on ACR30 is 0.04 (95% CI –1.37 to 1.59), ACR50 is –0.70 (95% CI –2.15 to 0.77), ACR70 is –1.08 (95% CI –2.70 to 0.46), ACR90 is –0.33 (95% CI –2.22 to 1.39) and ACR100 is –0.32 (95% CI –1.85 to 1.17). These estimates are adjusted for an informatively missing outcome because of treatment failure.
| Outcome | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| ACR30 | Longitudinal | Intercept | –1.35 | –2.65 to –0.23 | 0.02 |
| Time | 0.03 | –0.03 to 0.09 | 0.36 | ||
| Adalimumab | 0.04 | –1.37 to 1.59 | 0.98 | ||
| Survival | Adalimumab | –2.13 | –3.10 to –1.22 | < 0.001 | |
| HR | 0.12 | 0.05 to 0.29 | < 0.001 | ||
| Association | γ0 | –0.23 | –0.51 to 0.01 | 0.07 | |
| ACR50 | Longitudinal | Intercept | –1.68 | –2.98 to –0.53 | 0.003 |
| Time | 0.06 | –0.003 to 0.13 | 0.06 | ||
| Adalimumab | –0.70 | –2.15 to 0.77 | 0.37 | ||
| Survival | Adalimumab | –2.22 | –3.30 to –1.23 | < 0.001 | |
| HR | 0.11 | 0.037 to 0.29 | < 0.001 | ||
| Association | γ0 | –0.27 | –0.58 to –0.02 | 0.04 | |
| ACR70 | Longitudinal | Intercept | –2.67 | –3.93 to –1.46 | < 0.001 |
| Time | 0.07 | –0.001 to 0.14 | 0.06 | ||
| Adalimumab | –1.08 | –2.70 to 0.46 | 0.16 | ||
| Survival | Adalimumab | –2.29 | –3.58 to –1.28 | < 0.001 | |
| HR | 0.10 | 0.03 to 0.28 | < 0.001 | ||
| Association | γ0 | –0.32 | –0.79 to 0.003 | 0.05 | |
| ACR90 | Longitudinal | Intercept | –4.48 | –6.03 to –3.06 | < 0.001 |
| Time | 0.09 | 0.01 to 0.17 | 0.04 | ||
| Adalimumab | –0.33 | –2.22 to 1.39 | 0.72 | ||
| Survival | Adalimumab | –2.59 | –4.40 to –1.37 | < 0.001 | |
| HR | 0.07 | 0.01 to 0.26 | < 0.001 | ||
| Association | γ0 | –0.41 | –0.93 to –0.04 | 0.03 | |
| ACR100 | Longitudinal | Intercept | –4.98 | –6.25 to –3.88 | < 0.001 |
| Time | 0.05 | –0.05 to 0.15 | 0.30 | ||
| Adalimumab | –0.32 | –1.85 to 1.17 | 0.65 | ||
| Survival | Adalimumab | –2.17 | –3.55 to –1.22 | < 0.001 | |
| HR | 0.11 | 0.03 to 0.29 | < 0.001 | ||
| Association | γ0 | –0.28 | –1.08 to 0.22 | 0.34 |
Sensitivity analysis
The time effect was significant at the 10% level for all ACRs except for ACR 30 and 100. Models (estimates not shown in this report) were assessed with time-by-treatment interaction term, but resulted in non-significant effects for time, treatment and time–treatment.
For each model, the deviance information criterion (DIC) statistic was extracted, which can be thought of as the Bayesian analogue of the Akaike information criterion (AIC).
For ACR 90 and 100, the separate longitudinal submodels did not fit or appear to converge. Model fit and convergence problems were not unexpected, because the event (ACR = 1) rates were relatively low for ACR 90 and 100. The event rates were:
-
ACR30 – 35.5% (n = 125)
-
ACR50 – 28.7% (n = 101)
-
ACR70 – 18.5% (n = 65)
-
ACR90 – 13.4% (n = 47)
-
ACR100 – 5.4% (n = 19).
For ACR 70, 90 and 100, the event rates had decreased immensely, from 35.5% for ACR30 to 18.5% for ACR70, 13.4% for ACR90 and 5.4% for ACR100. The primary models for ACR 30, 50 and 70 (compared with the model in sensitivity analysis 1) had overwhelmingly smaller DIC values; therefore, no further models were considered. Note that owing to the problems outlined above, it was not possible to assess whether or not there was an improvement between the primary model and all additional models fitted for ACR 70, 90 and 100.
Number of participants undergoing disease flare, in remission on and/or off medication for their juvenile idiopathic arthritis, and with minimum disease activity
Number of participants undergoing disease flare
Three participants (10%) in the placebo group and no participants in the adalimumab group had at least one case of disease flare of their JIA. In total, there were three episodes of disease flare in the three participants in the placebo group.
The RR for disease flare was 0.07 (95% CI 0.004 to 1.36; p = 0.03). The inferences drawn from the 95% CI and p-value are different with respect to showing statistical significance; this may be because there were low numbers of participants who had a disease flare in each group.
Number of participants in remission on and/or off medication for their juvenile idiopathic arthritis
These outcomes will be reported in Chapter 6 because this outcome can be reported only during the follow-up period.
Number of participants with minimum disease activity
There were 74 participants (82.2%) who had oligoarticular JIA, of whom 21 (23.3%) had extended oligoarthritis and 53 (58.9%) had persistent oligoarthritis. Fourteen participants (15.6%) had polyarticular JIA, of whom 12 (13.3%) had RF-positive polyarthritis and two (2.2%) had RF-negative polyarthritis. Two participants (2.2%) had psoriatic arthritis.
Of the 74 participants who had oligoarticular JIA, 50 (68%) received adalimumab [14 (28%) out of 50 participants had extended oligoarthritis and 36 (72%) had persistent oligoarthritis] and 24 (32%) received placebo [seven (29%) out of 24 participants had extended oligoarthritis and 17 (71%) had persistent oligoarthritis].
Of the 14 participants who had polyarticular JIA, nine (64%) received adalimumab [eight (89%) had RF-negative polyarthritis and one (11%) had RF-positive polyarthritis] and five (36%) received placebo [four (80%) out of five had RF-negative polyarthritis and one (20%) had RF-positive polyarthritis).
At baseline, one (3%) participant in the placebo group and four (7%) participants in the adalimumab group had minimum disease activity. The number of participants with minimum disease activity at each time point is reported in Table 17. A total of 19 (32%) participants in the adalimumab group and four participants in the placebo group had at least one case of minimum disease activity during the course of the trial. The RR was 2.33 (95% CI 0.87 to 6.24) and the associated p-value from the chi-squared test was 0.08, indicating that there was no statistically significant difference between the two groups.
| Treatment visit | Treatment group | Total | ||||
|---|---|---|---|---|---|---|
| Adalimumab | Placebo | |||||
| Number with oligoarticular JIA or polyarticular JIA | Number (%) with minimum disease activity | Number with oligoarticular JIA or polyarticular JIA | Number (%) with minimum disease activity | Number with oligoarticular JIA or polyarticular JIA | Number (%) with minimum disease activity | |
| Baseline | 59 | 4 (7) | 29 | 1 (3) | 88 | 5 (6) |
| 1 month | 59 | 5 (8) | 29 | 0 (0) | 88 | 5 (6) |
| 2 months | 57 | 1 (2) | 24 | 1 (4) | 81 | 2 (3) |
| 3 months | 55 | 8 (15) | 18 | 3 (17) | 73 | 11 (15) |
| 6 months | 47 | 5 (10) | 12 | 0 (0) | 59 | 5 (8) |
| 9 months | 42 | 4 (10) | 7 | 0 (0) | 49 | 4 (8) |
| 12 months | 34 | 3 (9) | 5 | 1 (20) | 39 | 4 (10) |
| 15 months | 27 | 3 (11) | 3 | 0 (0) | 30 | 3 (10) |
| 18 months | 23 | 2 (9) | 3 | 0 (0) | 26 | 2 (8) |
Number of participants requiring change in biological or disease-modifying antirheumatic drugs therapy because of a failure to respond from arthritis
One (3%) participant in the placebo group and two (3%) participants in the adalimumab group required a change in their biological drug therapy or DMARD therapy because of failure to respond from their arthritis. This result was not statistically significant (RR 1.00, 95% CI 0.20 to 5.09; p = 0.99).
Juvenile Arthritis Disease Activity Score
The parameter estimates for each of the joint models of JADAS 10, 27 and 71 can be seen in Table 18.
| Outcome | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| JADAS10 | Longitudinal | Intercept | 0.62 | 0.24 to 1.07 | 0.003 |
| Baseline | 0.42 | 0.25 to 0.56 | < 0.0001 | ||
| Time | –0.01 | –0.03 to 0.01 | 0.27 | ||
| Adalimumab | –0.35 | –0.78 to 0.01 | 0.07 | ||
| Survival | Adalimumab | –2.38 | –3.92 to –1.48 | 0.25 | |
| HR | 0.09 | 0.02 to 0.23 | 0.25 | ||
| Association | γ0 | 1.11 | 0.11 to 2.85 | 0.10 | |
| JADAS27 | Longitudinal | Intercept | 0.62 | 0.25 to 1.06 | 0.003 |
| Baseline | 0.42 | 0.24 to 0.57 | < 0.0001 | ||
| Time | –0.01 | –0.03 to 0.01 | 0.27 | ||
| Adalimumab | –0.34 | –0.76 to 0.03 | 0.08 | ||
| Survival | Adalimumab | –2.37 | –3.97 to –1.47 | 0.25 | |
| HR | 0.09 | 0.02 to 0.23 | 0.25 | ||
| Association | γ0 | 1.10 | 0.08 to 2.88 | 0.11 | |
| JADAS71 | Longitudinal | Intercept | 0.63 | 0.24 to 1.08 | 0.003 |
| Baseline | 0.42 | 0.25 to 0.56 | < 0.0001 | ||
| Time | –0.01 | –0.03 to 0.01 | 0.26 | ||
| Adalimumab | –0.36 | –0.78 to 0.004 | 0.07 | ||
| Survival | Adalimumab | –2.38 | –3.88 to –1.48 | 0.25 | |
| HR | 0.09 | 0.02 to 0.23 | 0.25 | ||
| Association | γ0 | 1.11 | 0.11 to 2.81 | 0.0981 |
The distribution of baseline JADAS scores showed that a log-transformation led to a reduction in skewness. Therefore, the primary analysis joint models are fitted under this transformation.
The treatment effects on the longitudinal JADAS 10, 27 and 71 were all non-significant at the 5% level, implying that there is no difference in scores between the treatments. However, all three p-values are close to the margin of statistical significance.
Sensitivity analysis
For each outcome, a treatment–time interaction term was fitted because the treatment effect in the longitudinal submodel was significant at the 10% level. In all cases, there was some negligible increase in likelihood, and the corresponding AICs were slightly higher for the models with interaction, which did not support selection of the models with interaction terms. The estimated treatment effect on the longitudinal JADAS 10 and 71 also had p-values slightly below the 5% level.
The second model-specification sensitivity analysis considered a random-intercepts and random-slopes model. The log-likelihood values for the primary analysis (random-intercepts only) and the sensitivity analysis model (random intercepts and random slopes) showed that the likelihood is increased for the random-intercepts and random-slopes model. Trading off the improvement in goodness of fit against model complexity, the AIC confers evidence of model fit improvement. The treatment effects on all three longitudinal JADAS scores are marginally significant, with p-values just above 5%.
When the sensitivity analysis was conducted examining the effects of missing data, the treatment effects for each outcome in the longitudinal submodel were statistically significant (p < 0.05). Moreover, the association parameters are also significant (p < 0.05), implying that high JADAS values lead to a significantly high risk of treatment failure. This would suggest that the additional data have led to an increased power to detect the treatment effects and latent association parameters.
Chapter 6 Clinical effectiveness results: open-label phase
The results reported in this chapter are based on the integrated analysis of the blinded and open-label phase for participants in the adalimumab group compared with the results from the blinded phase for participants in the placebo group. The last participant visit in the open-label phase took place on 29 June 2016.
Primary outcome
During the course of the open-label phase of the trial, there were three additional treatment failures that occurred in the adalimumab arm. One participant was classified as having a treatment failure because they had taken permitted concomitant medications against the acceptable criteria and two participants had sustained scores (as recorded at entry grade) that were still present after 6 months of therapy (see Appendix 3, Table 50).
There were a total of 17 (28.3%) treatment failures for the 60 participants in the adalimumab group and 17 (56.7%) failures for the 30 participants in the placebo group. Median time to treatment failure was 24.1 weeks (95% CI 14.7 weeks to 81 weeks) in the placebo group and not reached in the adalimumab group within the 18-month treatment period because fewer than half of the subjects experienced treatment failure at the conclusion of the study (Figure 8).
FIGURE 8.
Primary outcome Kaplan–Meier plot for blinded and open-label phase.
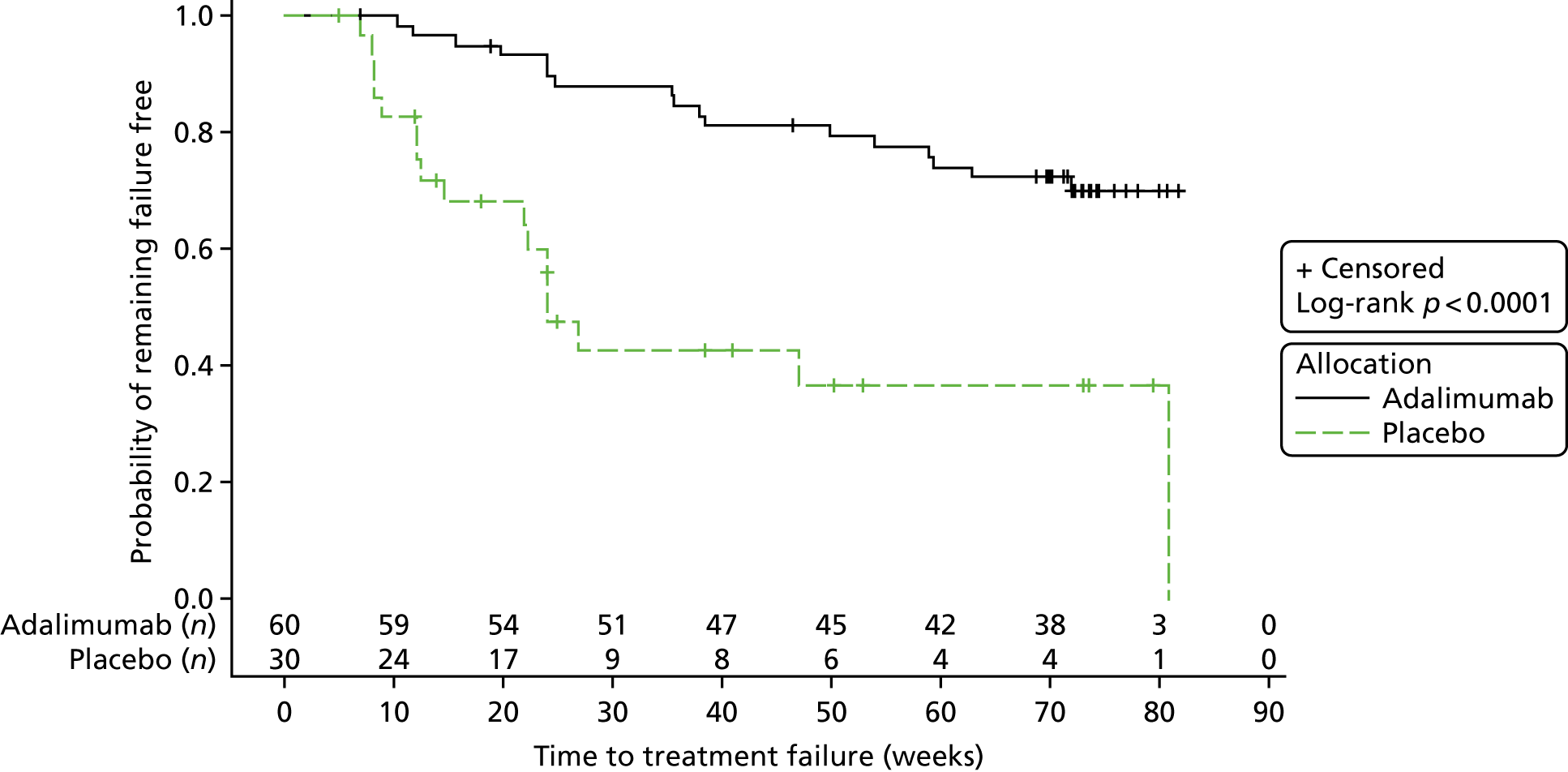
The results of the log-rank test from SAS PROC LIFETEST (SAS Institute In, Cary, NC, USA) offered strong statistical evidence that the placebo and adalimumab groups differed with respect to time to treatment failure.
The HR indicated that treatment with adalimumab significantly decreased the hazard of treatment failure by 74% (HR 0.26, 95% CI 0.13 to 0.51; p < 0.0001), relative to placebo.
Test of proportional hazards assumption
The assumption of proportional hazards was tested by including an interaction between time and treatment group in the Cox proportional hazards model. There was no evidence (p = 0.1371) that the interaction was not zero and, therefore, no evidence that the HR was not constant over time.
Sensitivity analyses
There were no losses to follow-up during the course of the trial and, therefore, sensitivity analysis 8 was not conducted. The results of the other sensitivity analyses indicate that the original conclusion from the primary analysis was robust with regard to the changes that were made. The overall statistical significance of the sensitivity analyses did not change.
Additional analyses
Development of uveitis in non-study eye
There were no additional occurrences of this outcome during the open-label phase of the trial.
Time to treatment failure in both eyes
This analysis was not possible because only one participant (in the placebo group) failed in both eyes at different times.
Development of comorbidity from treatment failure
There were no additional occurrences of this outcome during the open-label phase of the trial. There were so few numbers in the two treatment groups of those who developed a comorbidity that any modelling, including the development of a comorbidity, was not possible.
Post hoc analyses
Time-to-treatment response
During the open-label phase of the trial, there were three participants in the adalimumab group who achieved treatment response, meaning that, overall, during the blinded and open-label phases, a total of 47 participants in the adalimumab group were classified as having a treatment response. The difference between the two groups was statistically significant (log-rank p-value = 0.003). The HR indicated that participants on adalimumab were just under three times more likely to achieve a treatment response than those on placebo (HR 2.96, 95% CI 1.40 to 6.27).
Proportion of responders/failures/no change
Proportion of responders/failures/no change at 3 months
During the open-label phase of the trial, there were no further occurrences of response at 3 months and the overall conclusion showed a significant difference between the treatment groups at 6 months.
Proportion of responders/failures/no change at 6 months
During the open-label phase of the trial, there were no further occurrences of response at 6 months and the overall conclusion showed a significant difference between the treatment groups at 6 months.
Area under the curve of anterior chamber cells in eligible eye
There was a significant difference in the median number of AC cells between the two groups from the overall data of –0.81 (95% CI –0.99 to –0.64; p < 0.0001) (results from eye level favouring the adalimumab group), with similar results obtained when the best or worst score was used for participants with two eligible eyes.
Secondary outcomes
Number of participants failing treatment
Seventeen participants in the adalimumab group (28.33%) and 17 participants in the placebo group (56.67%) were classified as having treatment failures. The risk of having a treatment failure was statistically significantly reduced by 54% (RR 0.46, 95% CI 0.26 to 0.83; p = 0.01) in the adalimumab group compared with placebo.
Safety, tolerability and compliance
Adverse events and serious adverse events
During the open-label phase of the trial, 63 AEs occurred in 12 participants and two SAEs occurred in two participants (one SAE was reported outside the 30-day window of treatment cessation). For completeness, the SAE that was reported outside the reporting time window has been reported in Appendix 3 (see Table 43) only and is not included in any of the total numbers. The SAE that was reported during follow-up was in relation to joint swelling of the right knee and the severity was classified as mild and judged ‘unlikely’ to be related to the study drug.
A total of 86 participants (out of 90) experienced at least one AE. A total of 682 AEs were reported in 60 participants (100%) in the adalimumab group and 114 AEs reported in 26 participants (86.7%) in the placebo group. The rate of AEs in the adalimumab group (9.86 per patient-year) was greater than that in the placebo group (7.21 per patient-year).
The most common AEs in the adalimumab group were classified as infections and infestations (85.0%); general disorders and administration-site conditions (55%); respiratory, thoracic and mediastinal disorders (55.0%); gastrointestinal disorders (48.3%); investigations (35.0%); nervous system disorders (31.7%); eye disorders (28.3%); musculoskeletal and connective tissue disorders (26.7%); and injury, poisoning and procedural complications (23.3%) (Table 19).
| System Organ Class | Treatment group | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | ||||||||
| Events (n) | Patients (n) | % of patients | Events (n) | Patients (n) | % of patients | Events (n) | Patients (n) | % of patients | |
| Blood and lymphatic system disorders | 5 | 5 | 8.3 | 0 | 0 | 0.0 | 5 | 5 | 5.6 |
| Ear and labyrinth disorders | 1 | 1 | 1.7 | 0 | 0 | 0.0 | 1 | 1 | 1.1 |
| Eye disorders | 21 | 17 | 28.3 | 9 | 8 | 26.7 | 30 | 25 | 27.8 |
| Gastrointestinal disorders | 87 | 29 | 48.3 | 14 | 9 | 30.0 | 101 | 38 | 42.2 |
| General disorders and administration-site conditions | 135 | 33 | 55.0 | 15 | 8 | 26.7 | 150 | 41 | 45.6 |
| Immune system disorders | 4 | 4 | 6.7 | 1 | 1 | 3.3 | 5 | 5 | 5.6 |
| Infections and infestations | 164 | 51 | 85.0 | 30 | 14 | 46.7 | 194 | 65 | 72.2 |
| Injury, poisoning and procedural complications | 18 | 14 | 23.3 | 5 | 3 | 10.0 | 23 | 17 | 18.9 |
| Investigations | 43 | 21 | 35.0 | 6 | 4 | 13.3 | 49 | 25 | 27.8 |
| Metabolism and nutrition disorders | 3 | 3 | 5.0 | 0 | 0 | 0.0 | 3 | 3 | 3.3 |
| Musculoskeletal and connective tissue disorders | 32 | 16 | 26.7 | 8 | 7 | 23.3 | 40 | 23 | 25.6 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 5 | 5 | 8.3 | 0 | 0 | 0.0 | 5 | 5 | 5.6 |
| Nervous system disorders | 36 | 19 | 31.7 | 10 | 4 | 13.3 | 46 | 23 | 25.6 |
| Psychiatric disorders | 5 | 2 | 3.3 | 2 | 1 | 3.3 | 7 | 3 | 3.3 |
| Reproductive system and breast disorders | 11 | 3 | 5.0 | 0 | 0 | 0.0 | 11 | 3 | 3.3 |
| Respiratory, thoracic and mediastinal disorders | 95 | 33 | 55.0 | 9 | 6 | 20.0 | 104 | 39 | 43.3 |
| Skin and subcutaneous tissue disorders | 14 | 9 | 15.0 | 5 | 4 | 13.3 | 19 | 13 | 14.4 |
| Surgical and medical procedures | 3 | 3 | 5.0 | 0 | 0 | 0.0 | 3 | 3 | 3.3 |
Laboratory parameters (haematological, biochemical analysis and urinalysis)
When the data from the open-label period were combined with the data from the blinded phase of the trial, there were no changes to the clinical conclusions of the analyses of the haematological, biochemical and urinalysis data.
Participant diaries and dosing records
On average, treatment compliance for the adalimumab group during the open-label phase of the study, was 84%, according to the participant diaries, and 87%, according to the accountability logs. Overall, treatment compliance for the adalimumab group during the blinded phase of the trial and the open-label phase of the study combined was 83%, according to the participant diaries, and 94%, according to the accountability logs.
The average compliance with MTX for the adalimumab group during the open-label phase of the trial (according to participant diaries) was 74%; the average compliance with MTX for the adalimumab group overall for both phases of the trial was 61%.
Use of corticosteroids over duration of study period
Total oral corticosteroid dose
One participant in the placebo group and five participants in the adalimumab group received oral corticosteroids during the course of the study. The five participants in the adalimumab group were on study treatment for a total of 6.03 years and the placebo participant was on study treatment for 0.17 years.
The total oral dose for the placebo group was 640 mg (which was 3767.74 mg standardised per patient-year) and 4267.5 mg for the adalimumab group (which was 707.70 mg standardised per patient-year). A rate ratio of 0.19 (95% CI 0.17 to 0.20) indicated that participants on placebo required more oral corticosteroids per patient-year than those on adalimumab and there was evidence at the 5% level of a statistically significant difference between the two groups (p < 0.0001).
Reduction in and rate of systemic corticosteroid dose from entry dose
Reduction in systemic corticosteroid dose from entry dose
This analysis was not able to be performed because the statistical algorithm did not converge.
This analysis was not able to be performed because the statistical algorithm did not converge.
Rate of systemic corticosteroid dose from entry dose
The result of this analysis was the same as that of the total oral corticosteroid analysis.
Topical corticosteroid use (frequency) compared with entry use
Time to reduction to fewer than two drops in topical corticosteroid
The time to reduction to fewer than two drops per day was statistically significant in favour of adalimumab (HR 4.25, 95% CI 1.26 to 14.31; p = 0.02).
Time to reduction to zero drops in topical steroid (post hoc analysis)
The time to reduction to zero drops per day was statistically significant in favour of adalimumab (HR 4.26, 95% CI 1.49 to 12.2; p = 0.01).
Need for pulsed corticosteroid
During the open-label phase of the trial, there were no additional participants who required pulsed steroids. In total, one participant in the placebo group (3.33%) and two participants in the adalimumab group (3.33%) required pulsed corticosteroids during the course of the study. The RR showed that there was no evidence of a difference in the risk of requiring pulsed corticosteroids between the two treatment groups (RR 1.00, 95% CI 0.09 to 10.59; p > 0.99).
Optic and ocular
Number of participants having disease flares (as defined by worsening on Standardisation of the Uveitis Nomenclature criteria) following 3 months’ disease control
All events of disease flare following disease control took place in the blinded phase of the trial (i.e. there were no further events within the open-label phase).
Number of participants having disease flares within the first 3 months
One participant in the adalimumab arm failed treatment within the open-label phase of the study.
Overall, three participants in the placebo group (10%) and one participant in the adalimumab group (3%) had a disease flare in the first 3 months of treatment. There was no statistically significant evidence at the 5% level (p = 0.11) of a difference (RR 0.17, 95% CI 0.02 to 1.54) between the two groups.
Visual acuity measured by age-appropriate logarithm of the minimum angle of resolution assessment
The integrated analysis for the data on the logMAR score for participants in the trial split by treatment group and time point can be found in Table 20.
| Visit | Treatment group | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | Total | ||||||||||||||||
| Best score | Worst score | Best score | Worst score | Best score | Worst score | |||||||||||||
| n | Mean (SD) | Median (Range) | n | Mean (SD) | Median (Range) | n | Mean (SD) | Median (Range) | n | Mean (SD) | Median (Range) | n | Mean (SD) | Median (Range) | n | Mean (SD) | Median (Range) | |
| Baseline | 60 | 0.04 (0.15) | 0.00 (–0.23 to 0.56) | 60 | 0.05 (0.16) | 0.00 (–0.23 to 0.56) | 30 | 0.06 (0.12) | 0.05 (–0.13 to 0.40) | 30 | 0.08 (0.12) | 0.06 (–0.10 to 0.40) | 90 | 0.04 (0.14) | 0.00 (–0.23 to 0.56) | 90 | 0.06 (0.14) | 0.03 (–0.23 to 0.56) |
| 1 month | 60 | 0.03 (0.17) | 0.00 (–0.30 to 0.80) | 60 | 0.04 (0.18) | 0.00 (–0.30 to 0.80) | 30 | 0.02 (0.16) | 0.00 (–0.28 to 0.38) | 30 | 0.06 (0.17) | 0.04 (–0.28 to 0.38) | 90 | 0.02 (0.17) | 0.00 (–0.30 to 0.80) | 90 | 0.05 (0.18) | 0.00 (–0.30 to 0.80) |
| 2 months | 58 | 0.02 (0.17) | 0.00 (–0.20 to 0.56) | 58 | 0.04 (0.19) | 0.00 (–0.20 to 0.75) | 25 | 0.05 (0.18) | 0.00 (–0.15 to 0.76) | 25 | 0.06 (0.18) | 0.02 (–0.15 to 0.76) | 83 | 0.03 (0.17) | 0.00 (–0.20 to 0.76) | 83 | 0.04 (0.19) | 0.00 (–0.20 to 0.76) |
| 3 months | 57 | 0.00 (0.16) | 0.00 (–0.20 to 0.80) | 57 | 0.02 (0.19) | 0.00 (–0.20 to 0.88) | 19 | 0.01 (0.11) | 0.00 (–0.13 to 0.24) | 19 | 0.03 (0.12) | 0.00 (–0.13 to 0.28) | 76 | 0.00 (0.14) | 0.00 (–0.20 to 0.80) | 76 | 0.02 (0.18) | 0.00 (–0.20 to 0.88) |
| 6 months | 51 | 0.02 (0.19) | –0.02 (–0.20 to 0.88) | 51 | 0.02 (0.19) | 0.00 (–0.20 to 0.88) | 12 | 0.05 (0.16) | 0.02 (–0.18 to 0.30) | 12 | 0.07 (0.19) | 0.02 (–0.18 to 0.38) | 63 | 0.02 (0.18) | 0.00 (–0.20 to 0.88) | 63 | 0.03 (0.19) | 0.00 (–0.20 to 0.88) |
| 9 months | 48 | –0.01 (0.14) | 0.00 (–0.25 to 0.40) | 48 | –0.01 (0.14) | 0.00 (–0.25 to 0.40) | 7 | 0.00 (0.17) | –0.08 (–0.10 to 0.36) | 7 | 0.04 (0.20) | –0.08 (–0.10 to 0.36) | 55 | –0.01 (0.14) | 0.00 (–0.25 to 0.40) | 55 | 0.00 (0.14) | 0.00 (–0.25 to 0.40) |
| 12 months | 43 | –0.02 (0.14) | –0.02 (–0.23 to 0.34) | 43 | –0.01 (0.14) | 0.00 (–0.23 to 0.34) | 5 | 0.03 (0.14) | 0.02 (–0.10 to 0.26) | 5 | 0.08 (0.17) | 0.03 (–0.10 to 0.26) | 48 | –0.02 (0.14) | –0.01 (–0.23 to 0.34) | 48 | 0.00 (0.14) | 0.00 (–0.23 to 0.34) |
| 15 months | 38 | –0.01 (0.12) | 0.00 (–0.25 to 0.40) | 38 | 0.00 (0.12) | 0.00 (–0.25 to 0.40) | 3 | 0.00 (0.26) | –0.10 (–0.20 to 0.30) | 3 | 0.00 (0.26) | –0.10 (–0.20 to 0.30) | 41 | –0.01 (0.13) | 0.00 (–0.25 to 0.40) | 41 | 0.00 (0.13) | 0.00 (–0.25 to 0.40) |
| 18 months | 34 | 0.00 (0.13) | 0.00 (–0.22 to 0.28) | 34 | 0.01 (0.12) | 0.00 (–0.22 to 0.28) | 3 | 0.02 (0.21) | –0.10 (–0.10 to 0.26) | 3 | 0.02 (0.21) | –0.10 (–0.10 to 0.26) | 37 | 0.00 (0.13) | 0.00 (–0.22 to 0.28) | 37 | 0.01 (0.12) | 0.00 (–0.22 to 0.28) |
The parameter estimates from the joint modelling for analyses 1 and 2 (for the blinded and open-label phase of the trial combined) are shown in Table 21.
| Analysis | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| 1 | Longitudinal | Intercept | 0.01 | –0.03 to 0.04 | 0.76 |
| Baseline | 0.70 | 0.51 to 0.92 | < 0.0001 | ||
| Time | –0.002 | –0.004 to 4 × 10–4 | 0.17 | ||
| Adalimumab | –0.01 | –0.06 to 0.02 | 0.53 | ||
| Survival | Adalimumab | –1.33 | –2.25 to –0.73 | 0.001 | |
| HR | 0.26 | 0.11 to 0.48 | 0.001 | ||
| Association | γ0 | 2.86 | –2.41 to 10.37 | 0.41 | |
| 2 | Longitudinal | Intercept | 0.02 | –0.02 to 0.05 | 0.37 |
| Baseline | 0.81 | 0.60 to 1.06 | < 0.0001 | ||
| Time | –0.002 | –0.004 to 3 × 10-4 | 0.17 | ||
| Adalimumab | –0.02 | –0.07 to 0.02 | 0.37 | ||
| Survival | Adalimumab | –1.32 | –2.23 to –0.70 | 0.001 | |
| HR | 0.27 | 0.11 to 0.50 | 0.001 | ||
| Association | γ0 | 3.31 | –2.14 to 8.43 | 0.27 |
The results for integrated analyses 1 and 2 for the treatment effects (adalimumab) on the longitudinal logMAR are 0.01 (95% CI –0.06 to 0.02) and –0.02 (95% CI –0.07 to 0.02), respectively, implying that there is no significant difference between the treatments on logMAR.
These estimates are adjusted for the failure caused by treatment dropout from the trial.
Sensitivity analysis
The inferences of the sensitivity analyses for the data from the blinded phase of the trial combined with the open-label data were the same as those from the blinded phase alone.
Number of participants with resolution of associated optic nerve or macular oedema (as assessed by slit lamp biomicroscopy or optical coherence tomography, where available)
There were no further occurrences of optic nerve resolution or macular oedema in the open-label phase of the study.
Number of participants with disease control (defined as zero cells, with topical treatment for 3 and 6 months)
Three months
There were four participants in the adalimumab group who achieved disease control for 3 months during the open-label phase in at least one eligible eye. This meant that, overall, two participants in the placebo group (7%) and 27 in the adalimumab group (45%) had disease control for at least 3 months (RR 6.75, 95% CI 1.72 to 26.51; p = 0.0002).
The inferences for disease control in all eligible eyes were the same as those for disease control in one eligible eye.
Six months
There were five participants in the adalimumab group who achieved disease control for 3 months during the open-label phase in at least one eligible eye meaning that, overall, one participant (3%) in the placebo group and 22 participants in the adalimumab group (36.67%) had disease control in at least one of their eligible eyes (RR 11.00, 95% CI 1.56 to 77.74; p = 0.0003).
The inferences for disease control in all eligible eyes were the same as those for disease control in one eligible eye.
Number of participants entering disease remission (defined as zero cells, without topical treatment for 3 and 6 months)
Three months
There were no further cases of disease remission in the open-label phase.
Six months
There were no further cases of disease remission in the open-label phase.
Duration of sustaining inactive disease (zero cells in the anterior chamber, with or without topical treatment)
The difference in the total number of days that participants sustained inactive disease was statistically significant between the two treatment groups [16.31 days (SE 25.69 days) for the placebo group and 225.43 days (SE 18.15 days) for the adalimumab group], with participants in the adalimumab group spending approximately 209 (mean difference 208.80, 95% CI 143.91 to 273.69) more days with inactive disease than those in the placebo group (p < 0.0001).
Quality-of-life assessment
Childhood Health Questionnaire
The treatment effect on the longitudinal CHQ-PsS score (blinded and open-label phases) was 2.37 (95% CI –0.41 to 5.47), which implies that there is no difference between the treatments on the score (Table 22).
| Outcome | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| PsS | Longitudinal | Intercept | 15.37 | 6.54 to 22.59 | 0.0004 |
| Baseline | 0.69 | 0.55 to 0.85 | < 0.0001 | ||
| Time | 0.08 | –0.10 to 0.23 | 0.34 | ||
| Adalimumab | 2.37 | –0.41 to 5.47 | 0.14 | ||
| Survival | Adalimumab | –1.56 | –2.45 to –0.85 | 0.0002 | |
| HR | 0.21 | 0.09 to 0.43 | 0.0002 | ||
| Association | γ0 | –0.11 | –0.24 to –0.01 | 0.07 | |
| PhS | Longitudinal | Intercept | 20.68 | 12.17 to 30.70 | < 0.0001 |
| Baseline | 0.58 | 0.38 to 0.75 | < 0.0001 | ||
| Time | 0.04 | –0.11 to 0.16 | 0.56 | ||
| Adalimumab | 1.23 | –2.31 to 5.28 | 0.53 | ||
| Survival | Adalimumab | –1.34 | –2.30 to –0.67 | 0.001 | |
| HR | 0.26 | 0.10 to 0.51 | 0.001 | ||
| Association | γ0 | –0.05 | –0.15 to 0.03 | 0.24 |
The treatment effect on the longitudinal CHQ-PhS score (blinded and open-label phases) was 1.23 (95% CI –2.31 to 5.28), which implies that there is no difference between the treatments on this score (see Table 22). This estimate is adjusted for the failure due to dropout from the trial.
Sensitivity analysis
The inferences of the sensitivity analyses for the data from the blinded phase of the trial combined with the open-label data were the same as those from the blinded phase alone.
Childhood Health Assessment Questionnaire
The overall treatment effect (blinded and open-label phases) on the longitudinal CHAQ was –0.14 (95% CI –0.32 to 0.003), which implies that there is no difference between the treatments on CHAQ (Table 23). This estimate is adjusted for the failure due to dropout from the trial. However, the p-value (0.08) is close to the margin of statistical significance.
| Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|
| Longitudinal | Intercept | 0.20 | 0.05 to 0.35 | 0.01 |
| Baseline | 0.65 | 0.49 to 0.76 | < 0.0001 | |
| Time | –0.01 | –0.01 to 4 × 10–4 | 0.01 | |
| Adalimumab | –0.14 | –0.32 to 0.01 | 0.08 | |
| Survival | Adalimumab | –1.42 | –2.19 to –0.73 | 0.0001 |
| HR | 0.24 | 0.11 to 0.48 | 0.0001 | |
| Association | γ0 | 0.43 | –1.29 to 1.70 | 0.54 |
Sensitivity analysis
The inferences of the sensitivity analyses for the data from the blinded phase of the trial combined with the open-label data were the same as those from the blinded phase alone.
American College of Rheumatology Pedi core set criteria at ACR 30, 50, 70, 90 and 100
The results for the joint modelling of ACR Pedi and time to treatment failure can be seen in Table 24. None of the improvements on ACRs is significantly different between treatments. All estimates are adjusted for the failure caused by dropout from the trial.
| Outcome | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| ACR30 | Longitudinal | Intercept | –1.35 | –2.54 to –0.25 | 0.02 |
| Time | 0.04 | –0.01 to 0.09 | 0.11 | ||
| Adalimumab | 0.01 | –1.37 to 1.49 | 0.99 | ||
| Survival | Adalimumab | –1.98 | –2.97 to –1.07 | < 0.001 | |
| HR | 0.14 | 0.05 to 0.34 | < 0.001 | ||
| Association | γ0 | –0.21 | –0.50 to 0.03 | 0.10 | |
| ACR50 | Longitudinal | Intercept | –1.69 | –2.97 to –0.50 | 0.01 |
| Time | 0.07 | 0.01 to 0.13 | 0.02 | ||
| Adalimumab | –0.71 | –2.20 to 0.78 | 0.34 | ||
| Survival | Adalimumab | –2.12 | –3.15 to –1.24 | < 0.001 | |
| HR | 0.12 | 0.04 to 0.29 | < 0.001 | ||
| Association | γ0 | –0.26 | –0.58 to –0.02 | 0.04 | |
| ACR70 | Longitudinal | Intercept | –2.72 | –4.10 to –1.54 | < 0.001 |
| Time | 0.08 | 0.01 to 0.14 | 0.02 | ||
| Adalimumab | –1.11 | –2.68 to 0.42 | 0.16 | ||
| Survival | Adalimumab | –2.34 | –4.06 to –1.18 | < 0.001 | |
| HR | 0.10 | 0.02 to 0.31 | < 0.001 | ||
| Association | γ0 | –0.38 | –0.99 to –0.04 | 0.02 | |
| ACR90 | Longitudinal | Intercept | –4.74 | –6.48 to –3.21 | < 0.001 |
| Time | 0.11 | 0.04 to 0.18 | 0.004 | ||
| Adalimumab | –0.48 | –2.47 to 1.44 | 0.62 | ||
| Survival | Adalimumab | –2.55 | –4.21 to –1.30 | < 0.001 | |
| HR | 0.08 | 0.015 to 0.27 | < 0.001 | ||
| Association | γ0 | –0.38 | –0.85 to –0.07 | 0.01 | |
| ACR100 | Longitudinal | Intercept | –5.13 | –6.38 to –3.92 | < 0.001 |
| Time | 0.10 | 0.01 to 0.18 | 0.03 | ||
| Adalimumab | –0.43 | –1.91 to 1.04 | 0.57 | ||
| Survival | Adalimumab | –2.37 | –4.52 to –1.15 | < 0.001 | |
| HR | 0.09 | 0.01 to 0.32 | < 0.001 | ||
| Association | γ0 | –0.44 | –1.18 to 0.15 | 0.18 |
Sensitivity analysis
The inferences of the sensitivity analyses for the data from the blinded phase of the trial combined with the open-label data were the same as those from the blinded phase alone.
Number of participants undergoing disease flare, in remission on and/or off medication for their juvenile idiopathic arthritis, and with minimum disease activity
Number of participants undergoing disease flare
During the open-label phase of the trial, there were no additional occurrences of participants undergoing a disease flare; therefore, the results of the combined analyses are the same as those reported during the blinded phase.
Number of participants in remission on and/or off medication for their juvenile idiopathic arthritis
Number of participants in remission on medication for their juvenile idiopathic arthritis
Seven participants (12%) in the adalimumab group and 17 (57%) participants in the placebo group could not be included in the analysis because they had not been on medication for the required amount of time (6 months).
Ten (19%) of those in the adalimumab analysis population achieved remission; none of the placebo participants did. The risk of having remission while on medication in the adalimumab group was greater than for those on placebo but was not statistically significant (RR 5.40, 95% CI 0.30 to 87.40; p = 0.19).
Number of participants in remission off medication for their juvenile idiopathic arthritis
There were 45 (75%) participants in the adalimumab group and 21 (70%) in the placebo group who could not be included in the analysis because they had not been off medication for the required amount of time (12 months).
No participants in either the adalimumab group or the placebo group achieved remission off medication for their JIA.
Number of participants with minimum disease activity
During the open-label phase of the trial, 22 (37%) participants in the adalimumab group (this was three more than the result from the blinded phase alone) and four participants in the placebo group had at least one case of minimum disease activity during the course of the trial. The RR was 2.70 (95% CI 1.03 to 7.12) and the associated p-value from the chi-squared test was 0.03, indicating that there was a statistically significant difference between the two groups.
Number of participants requiring change in biological or disease-modifying antirheumatic drug therapy as a result of failure to respond from arthritis
There were no further cases of participants requiring a change in biological or DMARD therapy as a result of failure to respond from arthritis in the open-label phase.
Juvenile Arthritis Disease Activity Score
The parameter estimates for each of the joint models of JADAS 10, 27 and 71 can be seen in Table 25. The treatment effect on the longitudinal JADAS 10, 27 and 71 was non-significant at the 5% level, implying that there is no difference between the treatments on the scores. However, all three p-values are close to the margin of statistical significance.
| Outcome | Component | Parameter | Estimate | 95% CI | p-value |
|---|---|---|---|---|---|
| JADAS10 | Longitudinal | Intercept | 0.61 | 0.22 to 1.07 | 0.004 |
| Baseline | 0.43 | 0.24 to 0.57 | < 0.0001 | ||
| Time | –0.01 | –0.02 to 0.007 | 0.26 | ||
| Adalimumab | –0.35 | –0.76 to 0.02 | 0.08 | ||
| Survival | Adalimumab | –2.25 | –3.65 to –1.37 | 0.15 | |
| HR | 0.11 | 0.03 to 0.25 | 0.15 | ||
| Association | γ0 | 1.07 | –0.02 to 2.56 | 0.09 | |
| JADAS27 | Longitudinal | Intercept | 0.60 | 0.21 to 1.04 | 0.004 |
| Baseline | 0.42 | 0.24 to 0.57 | < 0.0001 | ||
| Time | –0.01 | –0.02 to 0.01 | 0.28 | ||
| Adalimumab | –0.33 | –0.75 to 0.04 | 0.09 | ||
| Survival | Adalimumab | –2.24 | –3.67 to –1.35 | 0.15 | |
| HR | 0.11 | 0.03 to 0.26 | 0.15 | ||
| Association | γ0 | 1.05 | –0.01 to 2.54 | 0.09 | |
| JADAS71 | Longitudinal | Intercept | 0.62 | 0.22 to 1.08 | 0.004 |
| Baseline | 0.43 | 0.24 to 0.57 | < 0.0001 | ||
| Time | –0.01 | –0.02 to 0.01 | 0.26 | ||
| Adalimumab | –0.35 | –0.77 to 0.01 | 0.07 | ||
| Survival | Adalimumab | –2.25 | –3.63 to –1.37 | 0.15 | |
| HR | 0.11 | 0.03 to 0.25 | 0.15 | ||
| Association | γ0 | 1.07 | –0.03 to 2.49 | 0.09 |
Sensitivity analysis
The inferences of the sensitivity analyses for the data from the blinded phase of the trial combined with the open-label data were the same as those from the blinded phase alone.
Chapter 7 Clinical effectiveness results: follow-up phase
The corticosteroid data results reported in this chapter are based on the integrated analysis of the blinded, open-label and follow-up phases for participants in the adalimumab group compared with the results from the blinded and follow-up phases for participants in the placebo group.
The laboratory data results reported in this chapter are from the follow-up period of the trial only.
The follow-up period for the trial was reduced from 18 months (six follow-up visits) to 6 months (two follow-up visits) in the revision to version 3.0 of the protocol, following the advice of the funders (HTA and Arthritis Research UK; 1 March 2013). The last participant visit took place on 14 December 2016.
Laboratory parameters (haematological, biochemical analysis and urinalysis)
The reduced follow-up period meant that only a small proportion of participants had data at six time points. Therefore, only data from the first two follow-up visits have been presented in this report.
Haematological
Overall, statistically significant changes from baseline to follow-up visits 1 and 2 were not observed in haematological assessments. None of the mean changes to either follow-up visit in haematological assessments was considered to be clinically significant.
Biochemical
Overall, statistically significant changes from baseline to follow-up visits 1 and 2 were not observed in biochemical assessments. None of the mean changes to either follow-up visit in biochemical assessments was considered to be clinically significant.
Urinalysis
-
Follow-up visit 1:
-
There were nine abnormal assessments from five participants (the results were greater than a trace, and so they needed microscopic urinalysis) in the placebo group. The results of the microscopic urinalysis showed that one was normal and four were abnormal (one of these four was clinically significant).
-
There were 19 abnormal assessments from 12 participants (the results were greater than a trace, and so they needed microscopic urinalysis) in the adalimumab group. The results of the microscopic urinalysis showed that five were normal and five were abnormal (two of these five were clinically significant); two were classified as not applicable.
-
-
Follow-up visit 2:
-
There were 10 abnormal assessments from seven participants (the results were greater than a trace, and so they needed microscopic urinalysis) in the placebo group. The results of the microscopic urinalysis showed that four were normal and three were abnormal (one of these three was clinically significant).
-
There were seven abnormal assessments from six participants (the results were greater than a trace, and so they needed microscopic urinalysis) in the adalimumab group. The results of the microscopic urinalysis showed that four were normal and two were abnormal (none of these two was clinically significant).
-
Use of corticosteroids over duration of study period (blinded, open-label and follow-up phases)
Total oral corticosteroid dose
One participant in the placebo group and five participants in the adalimumab group were receiving oral corticosteroids at the start of the study. The five participants in the adalimumab group were in the study for a total of 8.65 years and the placebo participant was in the study for 0.17 years.
The total oral dose for the placebo group was 640 mg (standardised per patient-year, 3767.74 mg) and 6837.5 mg in the adalimumab group (standardised per patient-year, 790.27 mg). A rate ratio of 0.21 (95% CI 0.19 to 0.23) indicated that participants on placebo required more oral corticosteroids per patient-year than those on adalimumab; there was evidence at the 5% level of a statistically significant difference between the two groups.
Reduction in and rate of systemic corticosteroid dose from entry dose
Reduction in systemic corticosteroid dose from entry dose
Reduction in systemic corticosteroid dose from entry dose to 0 mg
This analysis was not able to be performed because the statistical algorithm did not converge.
Reduction in systemic corticosteroid dose from entry dose to < 5 mg
This analysis was not able to be performed because the statistical algorithm did not converge.
Rate of systemic corticosteroid dose from entry dose
The result of this analysis was the same as that of the total oral corticosteroid analysis.
Topical corticosteroid use (frequency) compared with entry use
Time to reduction to fewer than two drops in topical corticosteroid
The time to reduction to fewer than two drops per day was statistically significant, in favour of adalimumab (HR 4.74, 95% CI 1.41 to 16; p = 0.01).
Time to reduction to zero drops in topical steroid (post hoc analysis)
The time to reduction to zero drops per day was statistically significant, in favour of adalimumab (HR 5.24, 95% CI 1.82 to 15.1; p = 0.002).
Need for pulsed corticosteroid
During the follow-up period of the trial, two participants required pulsed corticosteroids. One participant in the placebo group (3%) and four participants in the adalimumab group (7%) required pulsed corticosteroids during the course of the study. There was no evidence (p = 0.66) of a difference in the risk of requiring pulsed corticosteroids between the two treatment groups (RR 2.00, 95% CI 0.23 to 17.12).
Chapter 8 Economic evaluation
Methods
The economic analysis adopted the perspective of the NHS and Personal Social Services providers in England. A trial-based evaluation was extrapolated by 10 years using a Markov model in order to assess the costs and consequences of adalimumab treatment over an appropriate analytical time horizon. The primary outcome of the economic evaluation is the incremental cost per QALY with adalimumab in addition to MTX versus MTX alone.
Resource use and costs
Within-trial costs were estimated by measuring health-care resource use associated with both arms of the trial during the study period, including (1) adalimumab, MTX and other concomitant medication costs; (2) outpatient and accident and emergency (A&E) visits and contact with health-care professionals, including general practitioners (GPs) and school nurses; (3) hospitalisations; and (4) management of AEs.
The measurement of resource use required complementary approaches using data collected as part of the trial and as part of routine care. Trial participants’ use of health-care services was obtained from:
-
Medication forms. All medication use from 3 months before randomisation was recorded by trial physicians at each trial visit and was supplemented by participant diary records.
-
Baseline forms. Research nurses completed the relevant sections of the baseline forms to identify participant contact with hospital and health-care professionals in the 3 months before randomisation.
-
Three-monthly patient questionnaires. Research nurses completed the relevant sections of the patient questionnaires during face-to-face interviews with trial participants to identify overnight hospital stays, number of nights, reason for admission and type of ward. The patient questionnaires were also used to identify contacts with health-care professionals including GPs, consultants, nurses, psychologists and rheumatologists, as well as the places and/or means of contact (i.e. A&E, outpatient, GP practice, home visits, telephone, text and e-mail).
-
Electronic PLICS data and/or patient administration systems (PAS) data. These were accessed via the participating hospitals’ finance departments to identify inpatient stays, use of intensive care or high-dependency units, and outpatient visits.
-
AE or SAE forms. Research nurses completed the AE and SAE forms when trial participants were admitted to hospital for events considered possibly related to the study drug, which included bronchopneumonia, herpes simplex infection, pharyngitis and pneumonia.
-
Participant diaries recorded GP, social worker, district nurse and hospital visits.
In addition, for the estimation of long-term costs, longitudinal data for patients with JIA-associated and idiopathic uveitis were obtained from the Bristol Regional Tertiary Paediatric Uveitis clinic. This cohort provided data on the number and nature of surgeries performed from diagnosis with follow-up at 1, 3, 5 and 10 years.
Unit costs
All resource use was valued in monetary terms using appropriate UK unit costs estimated at the time of analysis (cost year: 2016). The costs of adalimumab, MTX and all other concomitant medications were based on the cost of items dispensed by Pharmacy and Appliance Contractors in England,66 supplemented by the British National Formulary67 and retail pharmacy prices when necessary (Table 26). The cost of adalimumab, which is not dose sensitive, was based on a once-per-fortnight subcutaneous injection. The formulation of MTX used, which is available in injectable, oral tablet or oral solution form, was recorded in the concomitant medication forms during follow-up. The cost of MTX was based on a once-weekly schedule; oral tablets were costed following best practice of all doses being made up as a multiple of a 2.5-mg dose tablet to avoid potential errors with combining different tablet strengths. All other medication costs were based on duration of use as recorded in the concomitant medication forms and participant diaries. All tablets and oral liquids were costed on a unit dose basis, whereas eye drops, creams, lotions and inhalers were costed on a per pack basis.
| Medication | Formulation | Cost per unit (£) | Source |
|---|---|---|---|
| Adalimumab | Humira® 40 mg or 80 mg pre-filled syringea | 352.14 | BNF67 |
| MTX | Metoject® PEN (medac GmbH, Stirling, UK) 7.5 mg/0.15 mlb | 14.85 | PCA66 |
| MTX | Metoject PEN 10 mg/0.2 ml | 15.29 | PCA66 |
| MTX | Metoject PEN 12.5 mg/0.25 ml | 16.50 | PCA66 |
| MTX | Metoject PEN 15 mg/0.3 ml | 16.57 | PCA66 |
| MTX | Metoject PEN 17.5 mg/0.35 ml | 17.50 | PCA66 |
| MTX | Metoject PEN 20 mg/0.4 ml | 17.84 | PCA66 |
| MTX | Metoject PEN 22.5 mg/0.45 ml | 18.45 | PCA66 |
| MTX | Metoject PEN 25 mg/0.5 ml | 18.48 | PCA66 |
| MTX | MTX tablet 2.5 mg | 0.06 | PCA66 |
| MTX | MTX oral solution 2 mg/ml, S/F | 2.65 | PCA66 |
Healthcare Resource Groups (HRGs) were used as the main currency of the economic analysis for hospital episodes (Tables 27 and 28). These most closely reflect actual payments, with cost codes allocated based on the latest available National Tariff68 (these are bundled care packages, reimbursed at a national level on the basis of the NHS Payment by Results Scheme)69 and, for unbundled care packages, the latest National Schedule,70 including A&E and outpatient contacts with time spent on consultations estimated through discussions with research nurses. Personal Social Services Research Unit71 unit costs were applied to all other primary health-care resource use items (Table 29).
| Service code | HRG code | HRG name | Attendance | Cost per episode (£) | Source |
|---|---|---|---|---|---|
| 130 | BZ22Z | Intermediate Vitreous Retinal Procedures | OP procedure | 142 | National Tariff68 |
| 130 | BZ23Z | Minor Vitreous Retinal Procedures | OP procedure | 109 | National Tariff68 |
| 130 | WF01B | Ophthalmology | OP first attendance – single professional | 113 | National Tariff68 |
| 130 | WF02B | Ophthalmology | OP first attendance – multi professional | 125 | National Tariff68 |
| 130 | WF01A | Ophthalmology | OP follow-up attendance – single professional | 64 | National Tariff68 |
| 130 | WF02A | Ophthalmology | OP follow-up attendance – multiprofessional | 94 | National Tariff68 |
| 216 | WF01B | Paediatric Ophthalmology | OP first attendance – single professional | 136 | National Tariff68 |
| 216 | WF01A | Paediatric Ophthalmology | OP follow-up attendance – single professional | 82 | National Tariff68 |
| 262 | WF01A | Paediatric Rheumatology | OP attendance | 203 | NHS Reference Costs 2015 to 2016 70 |
| 410 | WF01A | Rheumatology | OP follow-up attendance – single professional | 103 | National Tariff68 |
| 410 | WF01B | Rheumatology | OP first attendance – single professional | 225 | National Tariff68 |
| 410 | WF02B | Rheumatology | OP first attendance – multi professional | 246 | National Tariff68 |
| 410 | WF02A | Rheumatology | OP follow-up attendance – multiprofessional | 165 | National Tariff68 |
| 420 | WF01B | Paediatrics | OP first attendance – single professional | 222 | National Tariff68 |
| 420 | WF01A | Paediatrics | OP follow-up attendance – single professional | 135 | National Tariff68 |
| 420 | WF02A | Paediatrics | OP follow-up attendance – multiprofessional | 156 | National Tariff68 |
| 650 | WF01A/WF01B | Physiotherapy | OP attendance | 48 | NHS Reference Costs 2015 to 2016 70 |
| Service code | HRG code | HRG name | Attendance | Cost per episode (£) | Source |
|---|---|---|---|---|---|
| 262/410 | – | Rheumatology | Day case | 246 | National Tariff68 |
| 262/410 | HB29Z | Minimal knee procedures for non-trauma, with length of stay 1 day or less | Day case | 356 | National Tariff68 |
| 262/410 | PA64A | Non-surgical ophthalmology, with length of stay 0 days | Day case | 552 | National Tariff68 |
| 262/410 | PH34D | Paediatric, musculoskeletal or connective tissue disorders, with CC score 0 | Day case | 590 | NHS Reference Costs 2015 to 2016 70 |
| 262/410 | HB39Z | Minimal foot procedures for non-trauma, with length of stay 1 day or less | Day case | 672 | National Tariff68 |
| 262/410 | PA34B | Musculoskeletal or connective tissue disorders, without CC | Day case | 688 | National Tariff68 |
| 262/410 | PH34C | Paediatric, musculoskeletal or connective tissue disorders, with CC score 1–2 | Day case | 696 | NHS Reference Costs 2015 to 2016 70 |
| 262/410 | PA34A | Musculoskeletal or connective tissue disorders, with CC | Day case | 988 | National Tariff68 |
| Profession | Unit cost (£) | |||||||
|---|---|---|---|---|---|---|---|---|
| Surgery | Home | Telephone | Text message(s) | A&E | Outpatient visits | Additional visits | ||
| GP | 44.00 | 65.00 | 26.98 | 19.00 | 7.60 | 44.00 | 44.00 | 44.00 |
| Nurse | 14.42 | 14.42 | 8.82 | 7.35 | 2.94 | 29.40 | 44.10 | 44.10 |
| Consultant | 36.34 | 69.00 | 16.33 | 11.50 | 4.60 | 46.00 | 69.00 | 69.00 |
| Optometrist | 79.19 | 79.19 | 16.33 | 11.50 | 4.60 | 79.19 | 79.19 | 79.19 |
| Psychologist | 26.10 | 26.10 | 6.18 | 4.35 | 1.74 | 26.10 | 26.10 | 26.10 |
The costs of surgery were based on the NHS National Schedule of Reference Costs70 paediatric ophthalmology and outpatient procedures (Table 30). Clinical opinion was used to assign the most relevant HRG code for each type of surgery.
| Recorded surgery | Code | Description in schedule | Unit cost (£) | Source |
|---|---|---|---|---|
| Cataract | BZ32B | Intermediate, cataract or lens procedures, with CC score 0–1 | 208 | NHS Reference Costs 2015 to 2016 70 |
| Vitrectomy | BZ85Z | Very major or major, vitreous retinal procedures, 18 years and under | 334 | NHS Reference Costs 2015 to 2016 70 |
| Trabeculectomy | BZ94B | Intermediate, glaucoma or iris procedures, with CC score 0 | 401 | NHS Reference Costs 2015 to 2016 70 |
| Iridectomy | BZ94B | Intermediate, glaucoma or iris procedures, with CC score 0 | 401 | NHS Reference Costs 2015 to 2016 70 |
| Capsulotomy | BZ33Z | Minor, cataract or lens procedures | 140 | NHS Reference Costs 2015 to 2016 70 |
| Glaucoma tube | BZ93B | Major, glaucoma or iris procedures, with CC score 0–1 | 106 | NHS Reference Costs 2015 to 2016 70 |
Cost analysis
All medication, patient questionnaire and baseline form-reported hospital stays were costed irrespective of whether or not they were condition related or not.
Bundled National Tariff costs were based on the hospital spell and incorporated excess ward days and whether the case was elective or an emergency. 72 Tariff codes were obtained primarily from PLICS and PAS data, but if unavailable, were assigned an appropriate HRG code based on reason for admission, condition and any complications, by referring to AEs, SAEs, baseline forms and patient questionnaires. Locally negotiated unbundled costs were similarly identified and costs were assigned directly from the National Schedule of Reference Costs. 70 Reported health-care professional contacts in the patient questionnaires and baseline forms were multiplied by unit costs to estimate total costs.
Medication and hospitalisation use were costed for the trial-based analysis period of baseline to 18 months. If a medication administration spanned the period preceding randomisation, or beyond the 18-month time horizon, an adjustment was made to apportion costs to only those administered during the 0- to 18-month time horizon. Participants admitted to hospital were included if the hospital episode start date commenced within the 0- to 18-month time horizon.
Outcomes
The health outcome for the economic evaluation was the QALY, calculated from utilities measured from responses to the Health Utilities Index (HUI) questionnaire administered at baseline and at 3, 6, 9, 12 and 18 months. This was selected in preference to the EuroQol-5 Dimensions (EQ-5D) for its validity in paediatric populations. The HUI is a 40-item questionnaire that assesses health-related quality of life on eight single attributes: vision, hearing, speech, ambulation, dexterity, emotion, cognition and pain. Each HUI level code is estimated from responses to single questions or from a pattern of responses to a specific series of questions. Single attribute utility functions supply scores that express the morbidity for a person for each attribute. The health-related quality of life for a subject was determined by applying a multiattribute utility function to estimate HUI3 scores. 73
Modelled extrapolation
The Markov model was informed by a patient-level longitudinal data set of patients with idiopathic and JIA-associated uveitis. The only outcome recorded in both SYCAMORE and the Bristol data set74 (see Resource use and costs), and which is expected to directly affect patients’ health-related quality of life, was visual acuity, based on logMAR scores. To align with the model framework, the Bristol data74 were stratified as no visual impairment (VI) (logMAR < 0.3) and VI (logMAR ≥ 0.3). Health states were defined by patients’ vision in the worst eye as this was deemed the most clinically relevant. For trial participants, visual logMAR scores were recorded during all protocol-based visits and at unscheduled clinic visits. Data were ordered chronologically for each participant and time in each visual state (no VI, VI) was interpolated. The proportion of time in each state (no VI, VI) for each arm of the trial was used to determine the initial distribution of participants across the states of the Markov model (Figure 9). The cycle length was specified as 1 year, and a half-cycle correction applied.
FIGURE 9.
Markov model structure.
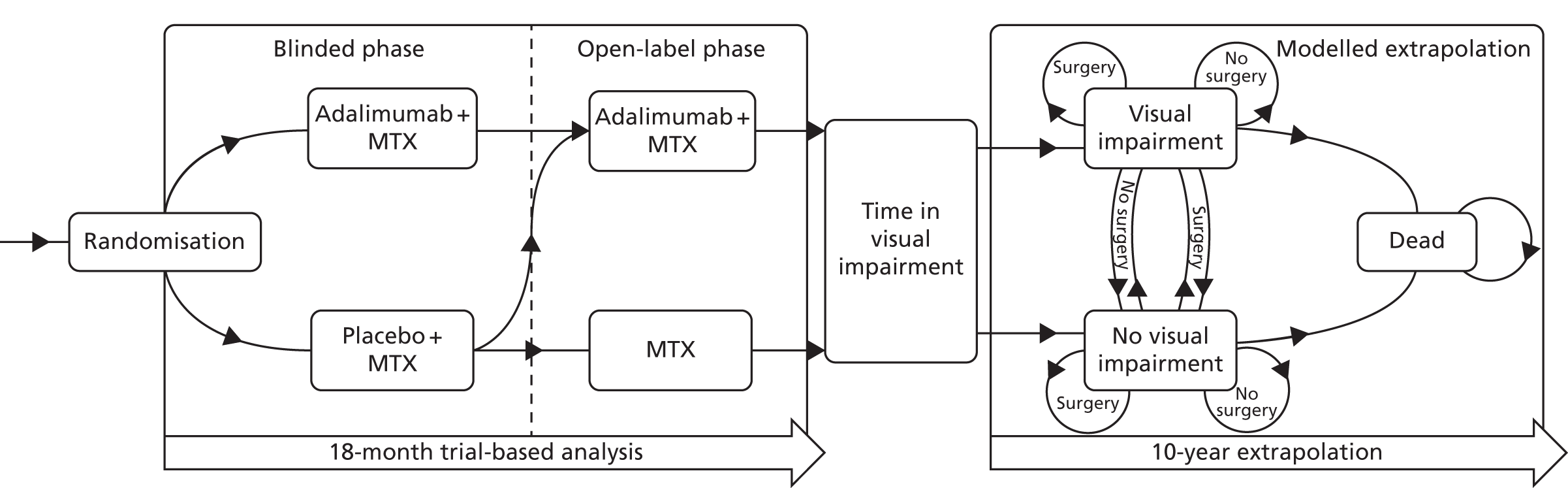
The Bristol cohort provided logMAR scores at diagnosis and at 1, 3, 5 and 10 years. This provided four time periods over which transitions among states may occur. Transitions between visual states, or to the same visual state, were defined as being either with or without surgery, which, along with transitions to the death state, resulted in 11 possible transitions. Annual transition probabilities were estimated by converting pooled transition probabilities over the 10-year time horizon into rates and back into transition probabilities.
The cost of surgery was taken as being the mean cost of a transition with surgery according to the longitudinal data set, as some transitions were associated with multiple surgery costs.
Published all-cause mortality data75 were adjusted for age but not sex because of the mixed cohort. A standardised mortality ratio of 3.9 (95% CI 0.8 to 11.3) for non-systematic JIA was applied. 76
Incremental analysis
Base-case analysis
The cost-effectiveness of adalimumab plus MTX compared with MTX alone was evaluated by its ICER, calculated by the formula:
where ΔCosts is the difference in mean total costs between intervention groups and ΔQALY is the difference in mean QALYs between intervention groups.
The base-case analysis was defined as pertaining to the 18-month trial period plus the 10-year extrapolation, based on an imputed data set to account for missing data and adjusting for the crossover of participants in the placebo arm who had access to adalimumab after the end of the blinded phase.
The base-case assumptions were that participants in the adalimumab arm of the trial had access to adalimumab for a further 3 years beyond the 18-month trial and then continued with MTX monotherapy [Michael Beresford (University of Liverpool), Andrew Dick (Bristol Eye Hospital), Athimalaipet Ramanan (Bristol Royal Hospital for Children), Eifiona Wood (Bangor University), Giovanna Culeddu (Bangor University) and Dyfrig Hughes (Bangor University), personal communication, 2017]. Trial drug costs are based on full adherence to adalimumab and MTX, in accordance with doses as defined in the protocol. Costs and QALYs beyond the first year were discounted at an annual rate of 3.5%.
Missing data and crossover
There were missing utility values at baseline and at 3, 6, 9, 12 and 18 months, and missing data for assessing the duration of time in visual impairment. Missing values were imputed using multiple imputation by chained equations. 77 Ten imputed data sets were created using predictive mean matching from a set of imputation models constructed from all potential prognostic factors (trial arm, age, sex, baseline visual impairment) and outcome variables (cost and exposure to adalimumab during the open-label phase).
The instrumental variable method was used to limit the bias that would result from participants randomised to placebo having access to adalimumab during post-trial closure follow-up. 78 Instrumental variable regressions for total costs and QALYs were calculated over the 18-month time horizon, considering received treatment (adalimumab), sex and age as covariates. For the 10-year modelled extrapolation, state-specific costs (excluding trial drug costs, which were added into the Markov model separately) and QALYs were derived using instrumental variable regressions, with treatment (adalimumab) and time in visual state as covariates.
Sensitivity analyses
Univariate sensitivity analysis
A number of sensitivity analyses were conducted to assess the robustness of the analysis. These included exploration of the impact of different time horizons of analysis and a series of sensitivity analyses relating to medication adherence, which tested assumptions based on the number of vials issued, accountability logs and recordings in participant diaries.
Separately, sensitivity analyses were conducted to assess the impact of participants having access to adalimumab for only the trial period (18 months) or for the duration of the model (18 months plus 10 years), based on the proportion of participants in the adalimumab arm who had access to adalimumab beyond the 18-month trial period in the open-label phase of the trial.
Owing to uncertainty in the proportion of participants who entered the Markov model with visual impairment, sensitivity analyses were conducted based on the CIs of these proportions. The Markov model was estimated with the proportions generated from upper to lower (H : L) and lower to upper (L : H) values of the CIs for both arms of the trial.
Finally, a sensitivity analysis was conducted without the discounting of future costs and QALYs.
Probabilistic sensitivity analysis
Mean costs and QALYs and differences between intervention groups in costs and QALYs were based on a bootstrapped analysis using 10,000 replicates. The 95% central range was based on the 2.5 and 97.5 percentiles of the bootstrap values.
A probabilistic sensitivity analysis of the base case was performed using a Monte Carlo simulation with 10,000 replicates, sampling each parameter simultaneously within its distribution. Probability distributions for regression-based analyses were generated using Cholesky decomposition.
Uncertainty in the ICER was represented as a cost-effectiveness plane and cost-effectiveness acceptability curves, which present the probability of adalimumab being cost-effective for given ceiling thresholds of costs per QALY. 79 Estimates of ICERs were compared with the £20,000- to £30,000-per-QALY threshold of cost-effectiveness set by NICE. 80
Scenario analyses
Complete-case data
Scenario analyses were conducted for complete-case data over 18 months. This was also extended to the 10-year extrapolation by specifying an ordinary least squares regression, using complete-case resource use, non-trial drug costs and QALYs to determine state-specific costs and utilities for both the adalimumab and placebo arms, based on time in visual state.
Disregarding crossover
A further analysis was conducted without regard for post-trial closure crossover from placebo to adalimumab. This was based on the imputed data set over 18 months and the 10-year extrapolation phase. Seemingly unrelated regressions for total costs and QALYs predicted by trial arm were conducted for imputed data over the 18-month time horizon. For the corresponding extrapolation, resource use, non-trial drug costs and QALYs were generated by seemingly unrelated regressions with trial arm and time in visual state as covariates. Trial drug costs were added afterwards.
All summary data and regression-based analyses were conducted using Stata version 13. The Markov model was analysed in Microsoft Excel® 2013 (Microsoft Corporation, Redmond, WA, USA) and reported in accordance with the Consolidated Health Economic Evaluation Reporting Standards. 81
Results
Resource use and cost analysis
Participants’ use of health-care resources and the corresponding NHS costs were comparable at baseline in both intervention groups for the 3 months prior to randomisation (Table 31). Only resource use associated with concomitant medications (excluding MTX) was statistically different between groups. However, the costs of the concomitant medications were not major cost drivers and accounted for only 3% of the total costs for the adalimumab arm and 1% of total costs for the placebo arm. The main cost drivers for the 3 months prior to randomisation were inpatient admissions (52% of the total) and outpatient visits (35% of total costs).
| Item of resource use | Treatment group, mean, (£) (95% CI) | Difference in mean, (£) (95% CI) | |
|---|---|---|---|
| Adalimumab (n = 60) | Placebo (n = 30) | ||
| MTX | 153 (127 to 179) | 168 (131 to 200) | –14 (–56 to 31) |
| Other concomitant medications | 51 (31 to 78) | 19 (12 to 26) | 32 (11 to 61) |
| Inpatient admissions | 867 (542 to 1239) | 768 (549 to 973) | 100 (–299 to 525) |
| Outpatient visits | 570 (450 to 705) | 534 (345 to 759) | 37 (–221 to 272) |
| GP visits | 100 (66 to 143) | 97 (56 to 147) | 3 (–59 to 64) |
| Nurse visits | 80 (54 to 109) | 87 (49 to 126) | –7 (–53 to 40) |
| Other | 166 (103 to 238) | 184 (103 to 277) | –19 (–129 to 91) |
| Total costs | 1614 (1312 to 1946) | 1526 (1072 to 2047) | 88 (–510 to 652) |
Each trial participant had available a level of resource use data that was costed and treated as complete from baseline to 18 months’ follow-up. Tables 32 and 33 present the disaggregated health-care resource use and costs, respectively, over this period. Mean total costs were £15,980 (95% CI £14,213 to £17,943; n = 60) for adalimumab plus MTX and £6248 (95% CI £3922 to £8889; n = 30) for placebo plus MTX. Adalimumab use was the main driver of the differences in costs between groups, contributing to 88% of the difference in total costs. The cost of concomitant medications and optician visits differed between arms, but these were not major cost drivers, accounting for 3% and 0.2% of the difference in total costs, respectively. There was no statistical difference in resource use between the other items. The annualised cost of the trial medications (adalimumab and MTX) differed by £8720 between groups (£11,118 adalimumab plus MTX vs. £2398 MTX alone).
| Item of resource use | Treatment group, mean count (range if > 0) [number of participants] | Difference in means | |
|---|---|---|---|
| Adalimumab | Placebo | ||
| GP visits | 3.5 (1–14) [35] | 2.75 (1–6) [12] | 0.75 |
| Nurse visits | 3.3 (1–12) [22] | 3.5 (1–10) [4] | –0.20 |
| Physiotherapist | 2.9 (1–7) [10] | 3.7 (3–4) [3] | –0.80 |
| Optician | 2.0 (1–3) [8] | 0.0 (0) [0] | 2.00 |
| Psychologist | 1.4 (1–3) [5] | 1.75 (1–4) [4] | –0.35 |
| OP – HRG BZ22B | 2.0 (1–3) [4] | 1.0 (1) [1] | 1.00 |
| OP – HRG BZ23Z | 1.25 (1–2) [4] | 1.0 (1) [1] | 0.25 |
| OP – HRG WF01A | 6.07 (1–18) [26] | 5.7 (1–15) [10] | 0.37 |
| OP – HRG WF01B | 1.4 (1–2) [10] | 1.16 (1–2) [6] | 0.24 |
| OP – HRG WF02A | 3.6 (1–10) [5] | 3.0 (1–5) [3] | 0.60 |
| IP – HRG PA34A | 8.5 (1–31) [11] | 5.25 (1–19) [8] | 3.25 |
| IP – HRG PA34B | 10 (4–18) [3] | 1.5 (1–3) [4] | 8.50 |
| IP – HRG PA64A | 2.0 (1–3) [2] | 2.0 (2–2) [1] | 0.00 |
| IP – HRG PH34C | 1.2 (1–2) [5] | 4.0 (3–5) [3] | –2.80 |
| IP – HRG PH34D | 3.4 (1–7) [5] | 1.4 (1–3) [5] | 2.00 |
| Item of resource use | Treatment group, mean, (£) (95% CI) | Difference in means, (£) (95% CI) | |
|---|---|---|---|
| Adalimumab (n = 60) | Placebo (n = 30) | ||
| Adalimumab | 10340 (9392 to 11,245) | 1761 (722 to 2951) | 8579 (7065 to 9978) |
| MTX | 778 (638 to 910) | 637 (462 to 816) | 141 (–80 to 364) |
| Concomitant medications | 540 (379 to 743) | 249 (92 to 471) | 291 (11 to 549) |
| Inpatient HRGs | 2522 (1195 to 4135) | 2549 (1166 to 4267) | –27 (–2198 to 2158) |
| Outpatient HRGs | 700 (434 to 1011) | 692 (294 to 1191) | 8 (–559 to 510) |
| GP visits | 91 (64 to 122) | 48 (23 to 79) | 43 (2 to 84) |
| Optician | 21 (8 to 37) | 0 | 21 (8 to 37) |
| Nurse visits | 18 (9 to 27) | 7 (0 to 18) | 11 (–3 to 23) |
| Physiotherapist | 12 (4 to 21) | 9 (0 to 20) | 3 (–10 to 15) |
| Psychologist | 3 (0 to 6) | 6 (1 to 14) | –3 (–12 to 4) |
| Total cost | 15,980 (14,213 to 17,943) | 6248 (3922 to 8889) | 9732 (6562 to 12,793) |
The Bristol cohort provided data from 91 patients with JIA-associated uveitis and 66 with idiopathic uveitis (Table 34). The mean age of the patients was 8 years (SD 3.8 years) and 60% were female, which is comparable with the SYCAMORE cohort. Thirty-seven surgeries in 25 patients were recorded in the Bristol data set,74 corresponding to 7.87 per 100 patient-years of follow-up. The frequency of surgeries per patient in a single time period are summarised in Table 35. Drug data were not dated and were, therefore, not used to stratify the data, as it was assumed that, following a standard care pathway, only patients whose disease had progressed furthest would be prescribed biologics, which would bias the model against adalimumab. For the Bristol cohort, the mean cost of surgeries between any two points of follow-up was £419.
| Characteristics | Patients |
|---|---|
| Age at diagnosis (years) | |
| Mean (SD) [range] | 7.9 (3.8) [1–15] |
| Sex, n (%) | |
| Male | 60 (38.2) |
| Ethnicity, n (%) | |
| Caucasian | 122 (78.2) |
| Asian | 6 (3.9) |
| African | 1 (0.6) |
| Other | 6 (3.9) |
| Unknown | 22 (14.1) |
| Aetiology, n (%) | |
| JIA | 91 (58.3) |
| Idiopathic | 66 (42.3) |
| Type of uveitis, n | |
| Anterior | 120 |
| Intermediate | 28 |
| Panuveitis | 8 |
| Posterior | 1 |
| Year of diagnosis, n (%) | |
| 1997–2000 | 10 (6.4) |
| 2001–5 | 37 (23.6) |
| 2005–10 | 61 (38.9) |
| 2011–15 | 48 (30.6) |
| Biologics received, n (%) (abatacept, adalimumab, etanercept, infliximab, tocilizumab) | |
| None | 104 (66.2) |
| One | 41 (26.1) |
| Two | 9 (5.7) |
| Three | 2 (1.3) |
| Five | 1 (0.6) |
| Adalimumab | 47 (30.0) |
| LogMAR > 0.3 at diagnosis, n (%)a | |
| Best eye | 12 (9.5) |
| Worst eye | 47 (37.3) |
| Surgical procedures, nb | |
| Capsulotomy | 3 |
| Cataract | 15 |
| Glaucoma tube | 1 |
| Iridectomy | 2 |
| Trabeculotomy | 10 |
| Vitrectomy | 6 |
| Surgeries in a single time period (n) | Number recorded | Cost per person (£) |
|---|---|---|
| None | 243 | 0 |
| Cataract | 8 | 208 |
| Glaucoma tube | 1 | 106 |
| Trabeculotomy | 5 | 401 |
| Vitrectomy | 3 | 334 |
| Capsulotomy; cataract | 1 | 348 |
| Capsulotomy (2), trabeculectomy (2) | 1 | 1082 |
| Cataract, iridectomy | 1 | 610 |
| Cataract (2), iridectomy | 1 | 817 |
| Cataract, trabeculectomy | 1 | 610 |
| Cataract, trabeculectomy (2) | 1 | 1011 |
| Cataract, vitrectomy | 1 | 542 |
| Vitrectomy (2) | 1 | 667 |
Outcomes
Utility and quality-adjusted life-years
Health Utility Index questionnaires were not completed for all participants, meaning that baseline utilities were missing for 12 participants in the adalimumab group and nine participants in the placebo group. For participants with complete baseline responses to the HUI questionnaire, mean utility values were 0.83 (95% CI 0.76 to 0.89; n = 48) for the adalimumab group and 0.87 (95% CI 0.78 to 0.96; n = 21) for the placebo group (Table 36). No significant differences in utility scores were noted at baseline; however, differences were reported for the 18-month utility scores, with a mean difference of 0.06 (95% CI 0.01 to 0.11) in favour of placebo.
| Analysis | Health outcomes | Treatment group, mean (95% CI) | Difference in means (95% CI) | |
|---|---|---|---|---|
| Adalimumab | Placebo | |||
| Complete casea | Baseline utility | 0.83 (0.76 to 0.89) | 0.87 (0.78 to 0.96) | –0.042 (–0.15 to 0.07) |
| 18-month analysis, time in VI (proportion) | 0.03 (0.01 to 0.07) | 0.02 (0.00 to 0.07) | 0.01 (–0.04 to 0.06) | |
| Utility at 18 months | 0.94 (0.88 to 0.98) | 0.99 (0.97 to 1.00) | –0.06 (–0.11 to –0.01) | |
| QALYs over 18 months | 1.40 (1.35 to 1.45) | 1.45 (1.41 to 1.50) | –0.05 (–0.12 to 0.02) | |
| Imputed datab | Baseline utility | 0.84 (0.77 to 0.90) | 0.86 (0.78 to 0.95) | –0.03 (–0.13 to 0.08) |
| 18-month analysis, time in VI (proportion) | 0.05 (0.02 to 0.08) | 0.11 (0.06 to 0.17) | –0.06 (–0.12 to –0.01) | |
| Utility at 18 months | 0.93 (0.87 to 0.99) | 0.92 (0.50 to 1.33) | 0.01 (–0.18 to 0.20) | |
| QALYs over 18 months | 1.35 (1.30 to 1.41) | 1.28 (1.15 to 1.41) | 0.07 (–0.04 to 0.18) | |
Visual acuity
Quality-adjusted life-year scores were calculated for both the complete-case and imputed analyses. Owing to missing data, QALY values were calculated only for three participants randomised to placebo and 25 participants randomised to adalimumab. In the complete-case analysis, QALY scores were lower in the adalimumab group [1.40 (95% CI 1.35 to 1.45)] than in the placebo group [1.45 (95% CI 1.41 to 1.5)], although the difference was not significant. After imputation, the QALY scores were higher for adalimumab than placebo [1.35 (95% CI 1.30 to 1.41) and 1.28 (95% CI 1.15 to 1.41), respectively] but, again, not significantly different.
Figure 10 illustrates the distribution of HUI3 level scores by treatment arm, attributes and time. Fewer participants in the placebo arm than in the adalimumab arm completed the HUI3; completion rates further reduced in both arms over the trial period. At baseline, 82% of participants in the adalimumab group and 72% of participants in the placebo group reported level 1 vision (no visual impairment); by 18 months, 76% of participants in the adalimumab group and 75% of participants in the placebo group were in the level 1 vision category. However, at 18 months, responses to this attribute were reported for only four participants in the placebo group, compared with 34 participants in the adalimumab group. For pain, at baseline, 49% (29/59) of participants in the adalimumab group and 58% (17/29) of participants in the placebo group reported level 1 pain score (no pain). At 18 months, 82% (27/33) of participants in the adalimumab group and 100% (4/4) of participants in the placebo group reported level 1 pain score, but, again, interpretation is hampered by low reporting rates at 18 months, especially in the placebo arm. No participants reported hearing-related problems for the duration of the trial.
FIGURE 10.
Distribution of participants’ responses to each HUI3 attribute, by treatment allocated and time. Levels range from 1 to 6, with 6 representing the most severe problem. The numbers of completed responses are reported by treatment arm. (a) Vision; (b) hearing; (c) speech; (d) ambulation; (e) dexterity; (f) emotions; (g) cognition; and (h) pain.
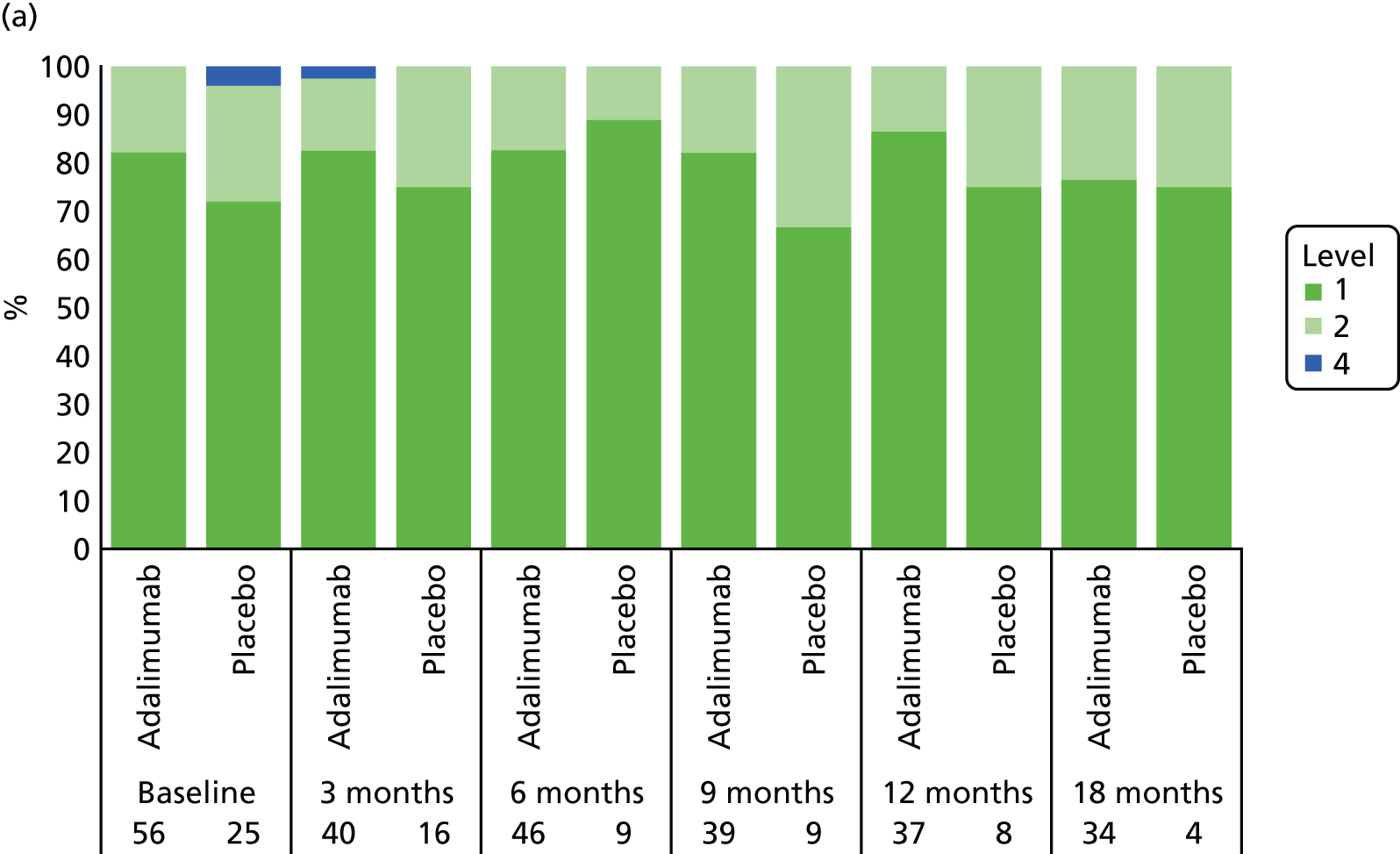
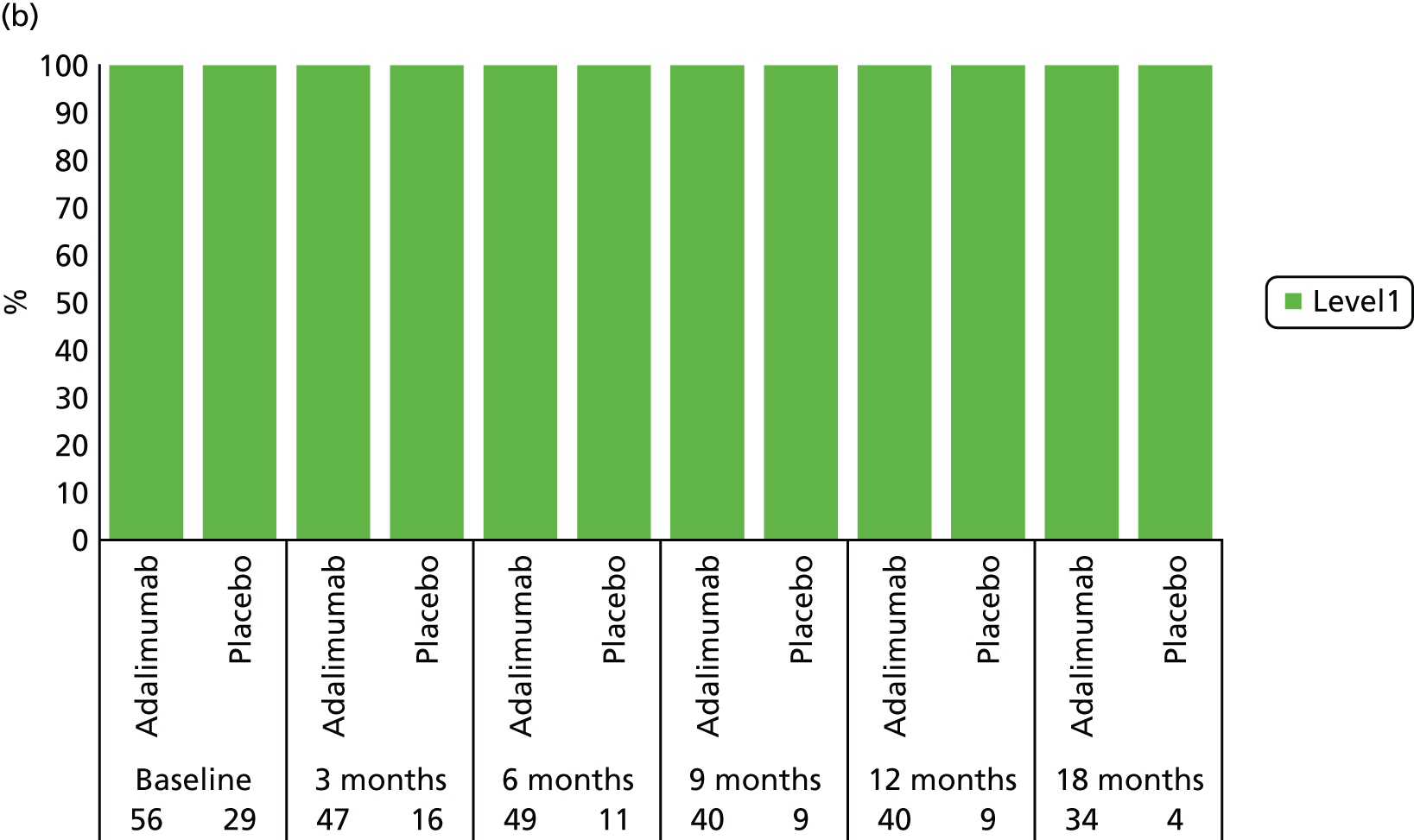
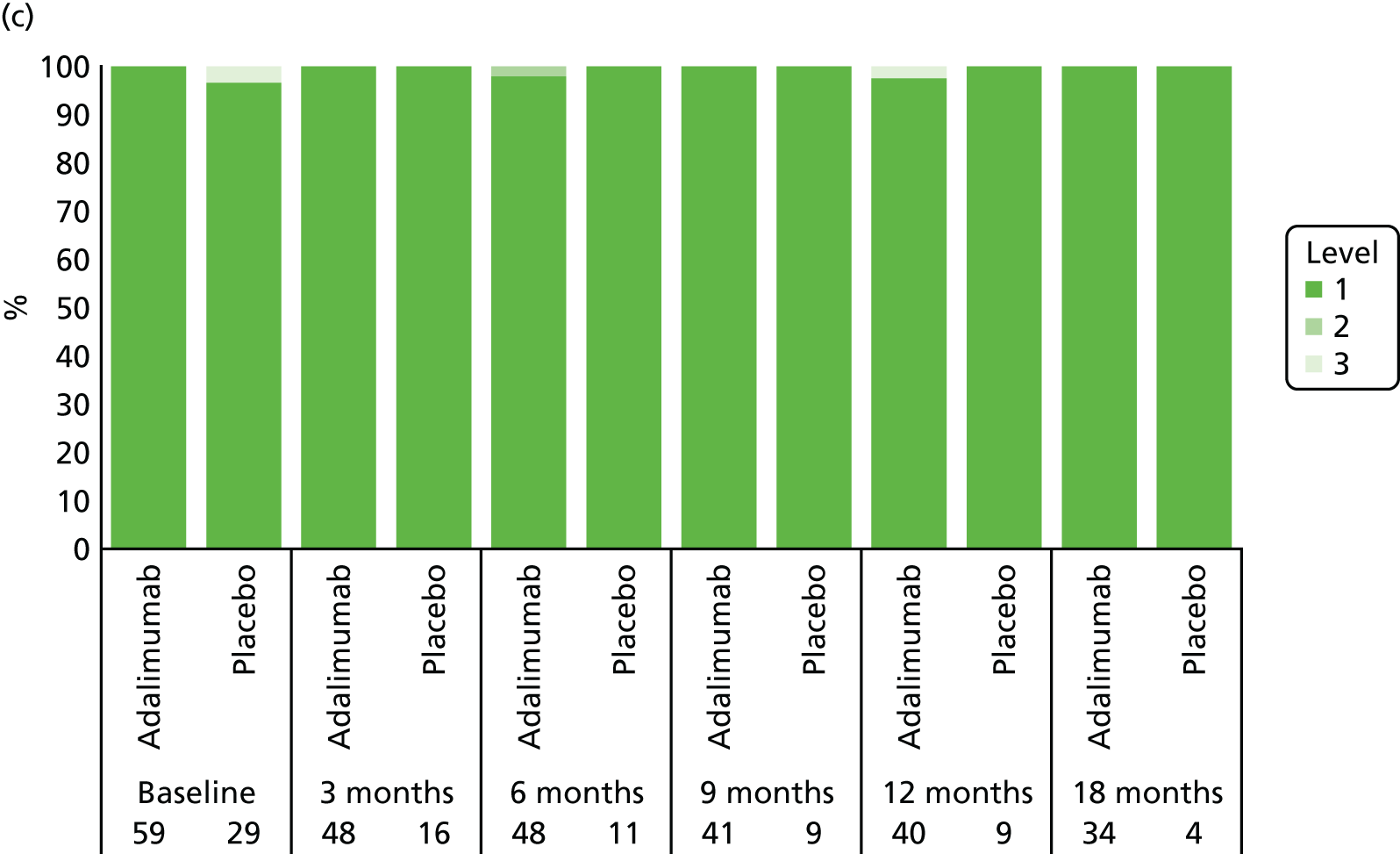
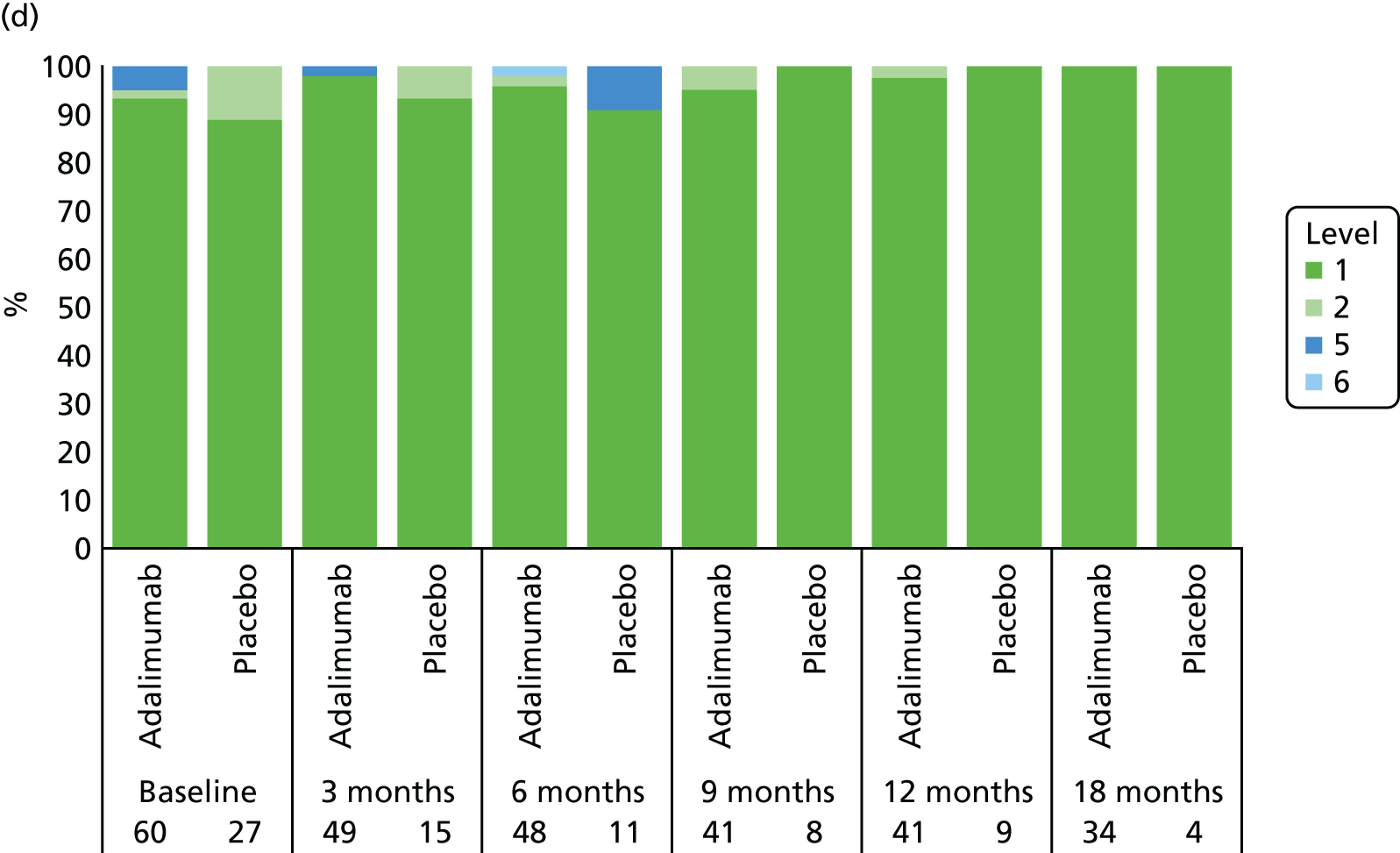
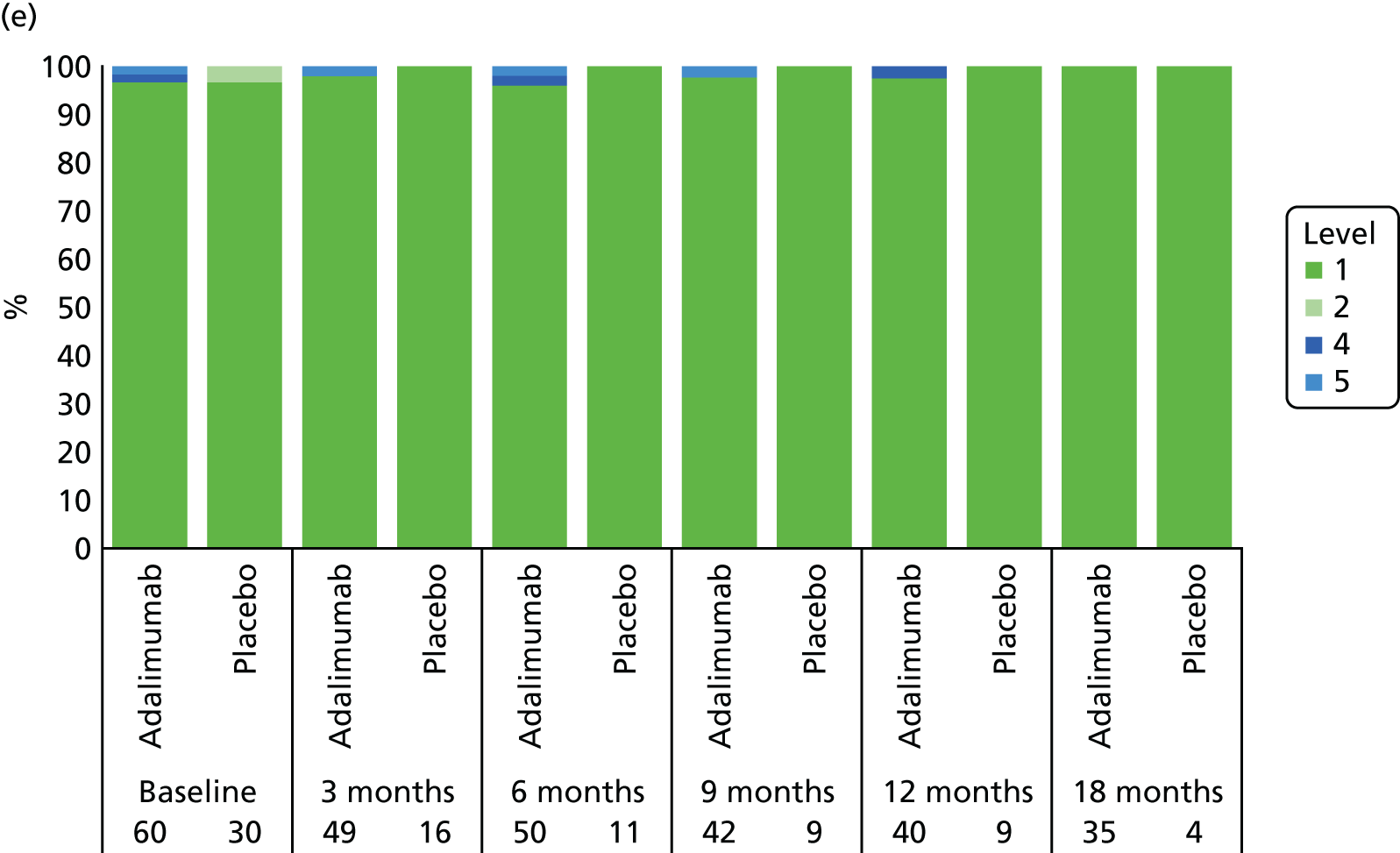
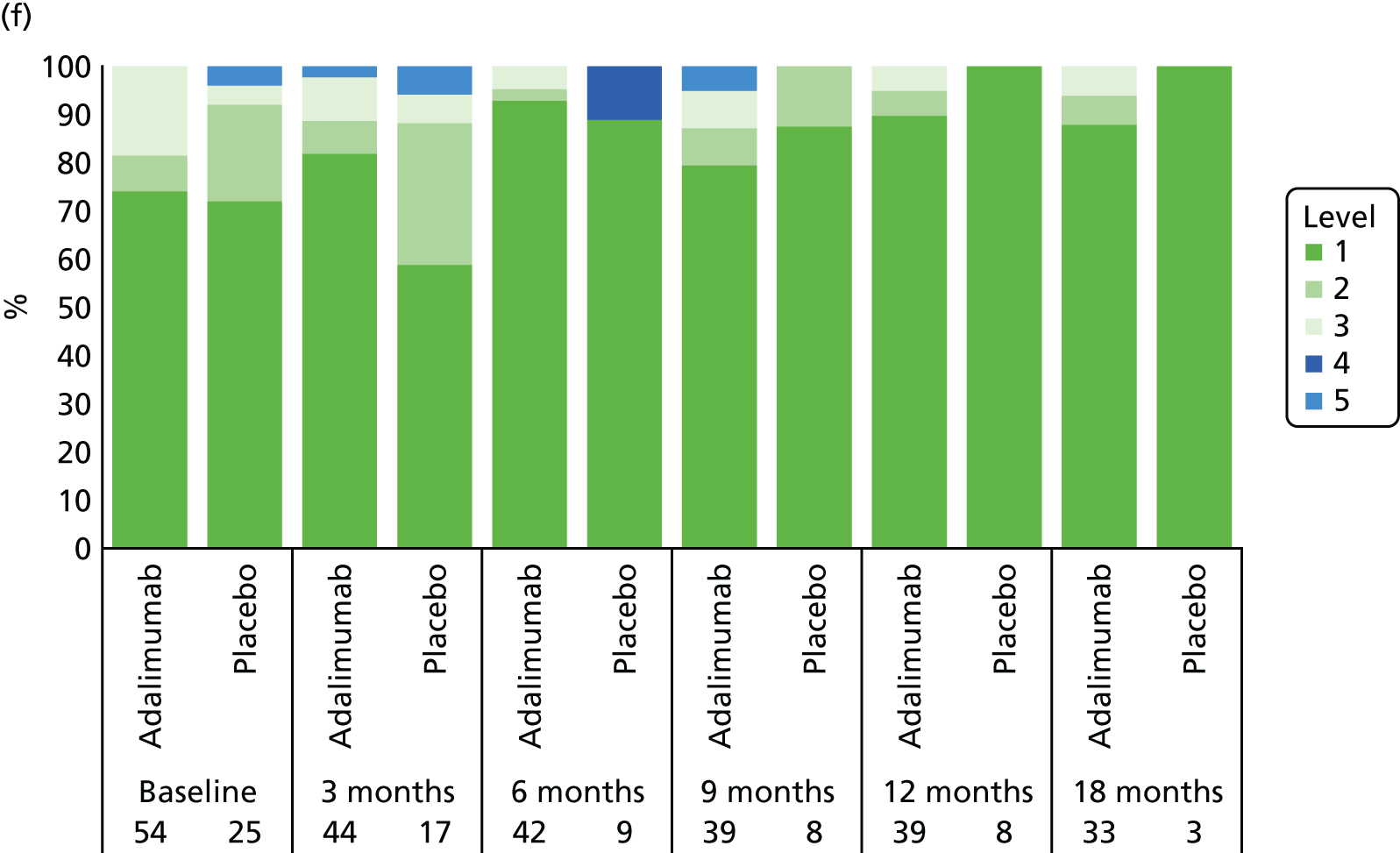
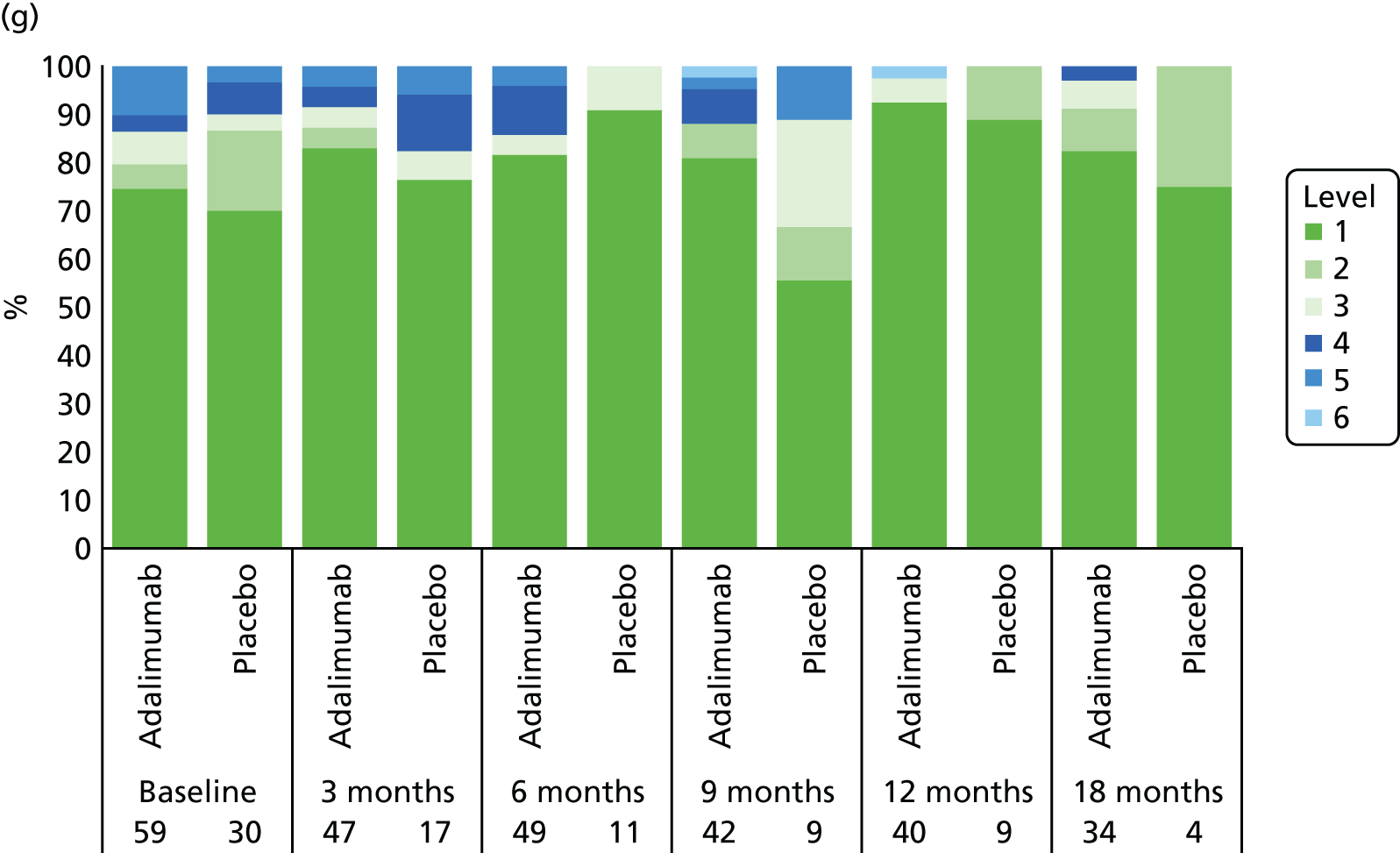
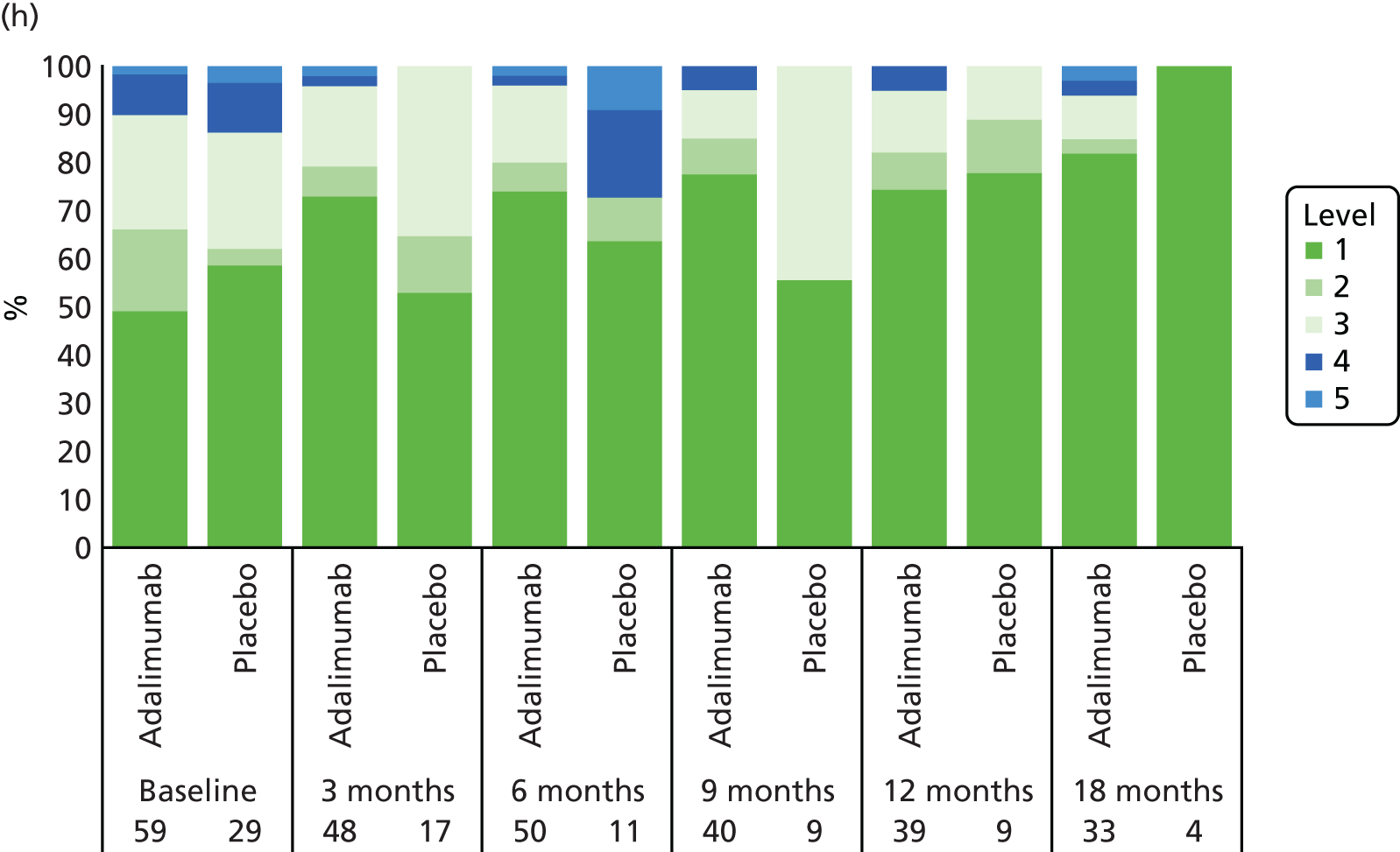
Visual acuity (logMAR) scores were available for 52 participants with complete-case data: 43 participants in the adalimumab group and nine participants in the placebo group. Baseline logMAR scores were complete and indicated the proportions of participants in the VI health state at baseline as 11.67% for adalimumab and 6.67% for placebo, which was anticipated to introduce bias in the ‘time in visual impairment’ outcome. At 18 months, 36 participants in the adalimumab arm of the trial and eight participants in the placebo arm had no time in the VI state. Mean proportion of time in the VI health state was 0.03 for participants in the adalimumab group and 0.02 for participants in the placebo group (difference 0.01, 95% CI –0.04 to 0.06) (see Table 36).
Following imputation, participants randomised to adalimumab spent 5.3% of time in VI during the 18-month analysis, whereas participants randomised to placebo spent 11.1% of time in VI. By including VI at baseline and time in VI, alongside demographics, costs and utility outcomes, the imputation model corrected for the imbalance in VI at baseline. In the imputed analyses, the rate of VI is higher in the placebo arm than in the adalimumab arm.
Data on logMAR from the Bristol cohort were available for 126, 117, 93, 75 and 22 patients at baseline and at 1, 3, 5 and 10 years, respectively. Based on the stratification of logMAR, 69 patients were recorded as having VI at one or more time point, whereas 88 patients were never recorded as having VI. There were 268 observed transitions over the four periods when transition was possible.
Analysis
Base-case analysis
All Markov model inputs and their distributions are presented in Table 37.
| Parameter | Point estimate | Distribution (distribution parameters) | Source |
|---|---|---|---|
| 18-month data (trial based) | |||
| Cost coefficient | |||
| Adalimumab | 14,374.01 | Cholesky decomposition | Trial data (SYCAMORE) |
| Age | –257.72 | ||
| Sex | –445.89 | ||
| Constant | 3765.78 | ||
| QALY coefficient | |||
| Adalimumab | 0.11 | Cholesky decomposition | Trial data (SYCAMORE) |
| Age | –0.00 | ||
| Sex | –0.02 | ||
| Constant | 1.26 | ||
| Month 19–138 (Markov model): base-case model assumptions | |||
| Cost coefficients | |||
| Adalimumab arm (excluding trial drug costs) | 1437.13 | Cholesky decomposition | Trial data (SYCAMORE) |
| Time in visual impairment | 2662.57 | ||
| Constant | 1603.05 | ||
| Drug cost | |||
| Adalimumab | 7411.73 | Gamma∼(8.11, 1370.33) | Trial data (SYCAMORE) |
| MTX | 1598.17 | Gamma∼(0.56, 4315.70) | Trial data (SYCAMORE) |
| Surgery cost (per surgery transition) | 418.71 | None (fixed) | Bristol data74 (see Resource use and costs) |
| Discount rate: cost (per annum) | 0.035 | None (fixed) | NICE80 |
| QALY coefficients | |||
| Adalimumab arm (excluding trial drug costs) | 0.07 | Cholesky decomposition | Trial data (SYCAMORE) |
| Time in visual impairment | –0.00 | ||
| Constant | 0.83 | ||
| Discount rate: QALY (per annum) | 0.035 | None (fixed) | NICE80 |
| Probabilities | |||
| Proportion of VI | |||
| Adalimumab arm | 0.05 | Beta∼(4.75, 85.25) | Trial data (SYCAMORE) |
| Placebo arm | 0.11 | Beta∼(10.04, 79.96) | Trial data (SYCAMORE) |
| Transition probability from | |||
| No VI to no VI (no surgery) | 0.95 | Dirichlet∼(162, 4, 14, 2) approximated by standardised series of gamma distributions | Bristol data74 (see Resource use and costs) |
| No VI to no VI (surgery) | 0.01 | ||
| No VI to VI (no surgery) | 0.04 | ||
| No VI to VI (surgery) | 0.01 | ||
| VI to no VI (no surgery) | 0.33 | Dirichlet∼(29, 6, 38, 13) approximated by standardised series of gamma distributions | Bristol data74 (see Resource use and costs) |
| VI to no VI (surgery) | 0.06 | ||
| VI to VI (no surgery) | 0.47 | ||
| VI to VI (surgery) | 0.14 | ||
| Mortality ratea | 0.000071 | None (fixed) | Human Mortality Database75 |
| Standardised mortality ratio | 3.9 | Log-normal∼(3.9, 2.6785) | Davies et al.76 |
Over the time horizon of the 18-month trial plus 10-year extrapolation, the total costs of the adalimumab arm were £70,719. The total costs of the placebo arm were £31,403. Total QALYs were 8.60 for the adalimumab arm and 8.29 for the placebo arm.
The incremental costs and QALYs for adalimumab were £39,316 and 0.30, respectively, resulting in an ICER of £129,025 per QALY gained.
Sensitivity analyses
Univariate sensitivity analysis
The results of the sensitivity analyses are presented in Table 38. These demonstrate that the model is most sensitive to adalimumab usage. The most cost-effective scenario is based on the assumption of adherence to MTX and adalimumab as recorded in the participant diaries over the 18-month time horizon (incremental cost of £117,514 per QALY gained) and the 18-month plus 10-year time horizon (incremental cost of £115,708 per QALY gained). However, participant diary recording of doses can be subject to unreliability and does not reflect the costs to the NHS of prescriptions issued. The least cost-effective analysis relates to adalimumab use being based on the number of vials issued, which was greater than the base-case assumption based on time in the study (110.55%), resulting in ICERs of £149,040 and £140,576 per QALY gained over 18 months and 18-month plus 10-year time horizons, respectively.
| Sensitivity analysis | Costs (£) | QALY | ICER (£) | ||||
|---|---|---|---|---|---|---|---|
| Treatment group | Incremental | Treatment group | Incremental | ||||
| Adalimumab | Placebo | Adalimumab | Placebo | ||||
| Base casea | 70,719 | 31,403 | 39,316 | 8.60 | 8.29 | 0.30 | 129,025 |
| Time horizon of analysis | |||||||
| 18 months | |||||||
| Adalimumab adherence based on vials issued | 17,399 | 1734 | 15,666 | 1.36 | 1.25 | 0.11 | 149,040 |
| Adalimumab adherence based on accountability logs | 15,716 | 2095 | 13,621 | 1.36 | 1.25 | 0.11 | 129,587 |
| Adalimumab and MTX adherence based on participant diaries | 14,345 | 1993 | 12,352 | 1.36 | 1.25 | 0.11 | 117,514 |
| 18 months + 10 years | |||||||
| Adalimumab treatment for 18 months only | 45,504 | 31,403 | 14,101 | 8.40 | 8.29 | 0.11 | 133,656 |
| Adalimumab treatment for 18 months + 10 years | 120,262 | 31,403 | 88,858 | 8.99 | 8.29 | 0.70 | 127,646 |
| Adalimumab adherence based on vials issued | 74,011 | 31,175 | 42,835 | 8.60 | 8.29 | 0.30 | 140,576 |
| Adalimumab adherence based on accountability logs | 68,800 | 31,536 | 37,264 | 8.60 | 8.29 | 0.30 | 122,291 |
| Adalimumab and MTX adherence based on participant diaries | 59,896 | 24,638 | 36,258 | 8.60 | 8.29 | 0.30 | 115,708 |
| No discounting | 77,634 | 36,743 | 40,621 | 9.88 | 9.57 | 0.32 | 128,886 |
| 23% of participants who were administered adalimumab beyond 18 monthsb | 51,304 | 31,403 | 19,900 | 8.44 | 8.29 | 0.15 | 131,511 |
| Adalimumab : placebo VI proportions H : L | 70,864 | 31,147 | 39,716 | 8.60 | 8.29 | 0.30 | 130,586 |
| Adalimumab : placebo VI proportions L : H | 70,574 | 31,659 | 38,915 | 8.60 | 8.29 | 0.31 | 127,471 |
The analysis in which the duration of adalimumab treatment was increased resulted in a lower ICER (£127,646 per QALY gained). Conversely, a shorter duration of treatment of 18 months, compared with 3 years in the base case, reduces the incremental QALY gain and raises the ICER to £133,656 per QALY gained.
Probabilistic sensitivity analysis
Based on the results of the probabilistic sensitivity analysis applied to the base case, the mean total costs of the adalimumab arm were £70,951 [95% credible range (CR) £45,204 to £123,764]. The mean total costs of the placebo arm were £31,587 (95% CR £5308 to £83,320). Mean QALYs were 8.60 (95% CR 8.00 to 9.19) for the adalimumab arm and 8.29 (95% CR 7.42 to 9.17) for the placebo arm.
The mean incremental costs and QALYs for adalimumab were £39,364 (95% CR £24,728 to £58,235) and 0.31 (95% CR –0.04 to 0.66), respectively.
The cost-effectiveness plane (Figure 11) and the cost-effectiveness acceptability curve (Figure 12) indicate that adalimumab is highly unlikely to be cost-effective in the £20,000- to £30,000-per-QALY threshold range. The probability of being cost-effective at the £30,000-per-QALY threshold is < 1%. In 96% of simulations, adalimumab was both more costly and more effective; however, in 4% of simulations, adalimumab was seen to be less effective than placebo and remained more costly.
FIGURE 11.
Cost-effectiveness plane.
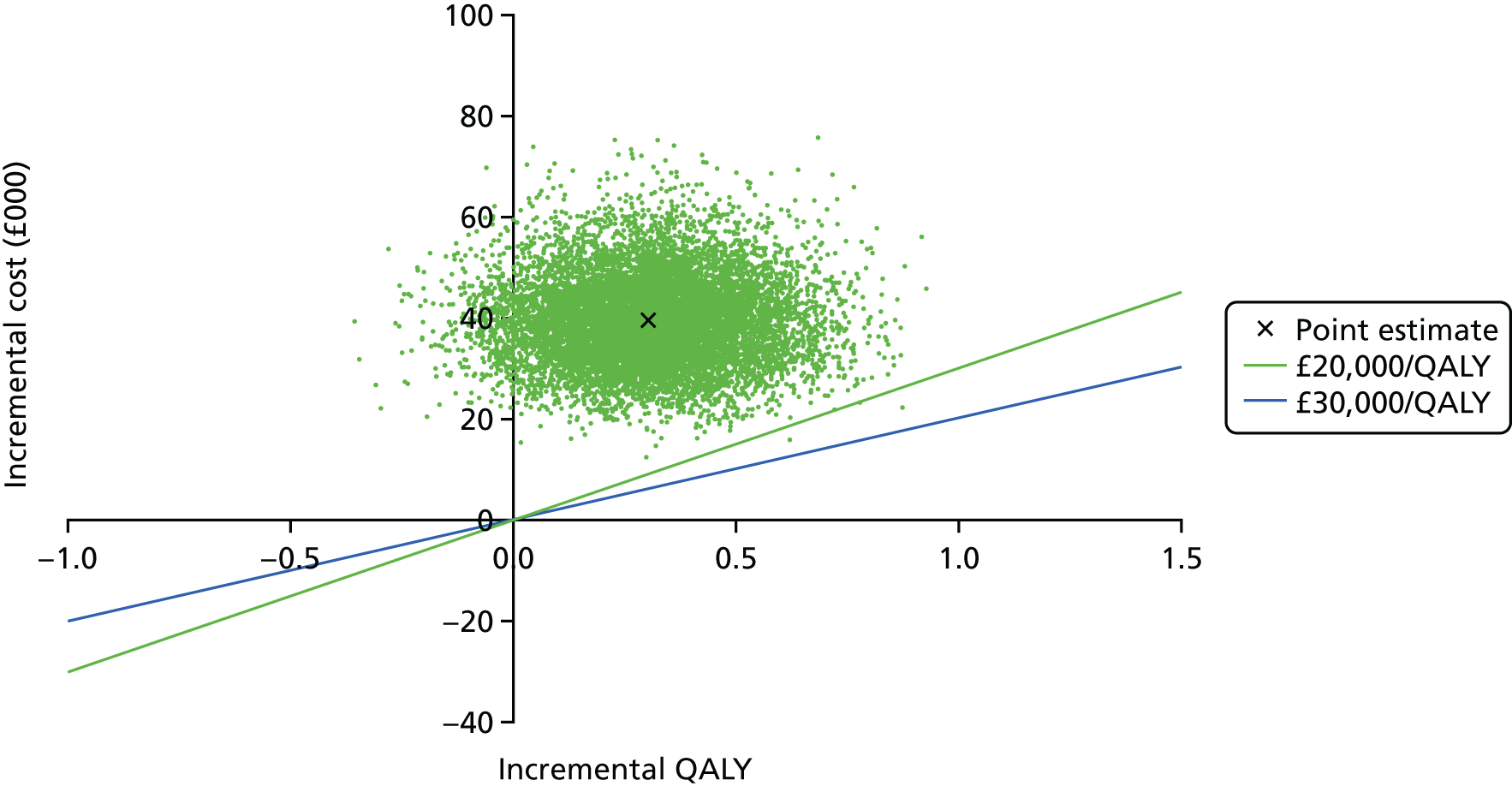
FIGURE 12.
Cost-effectiveness acceptability curve.
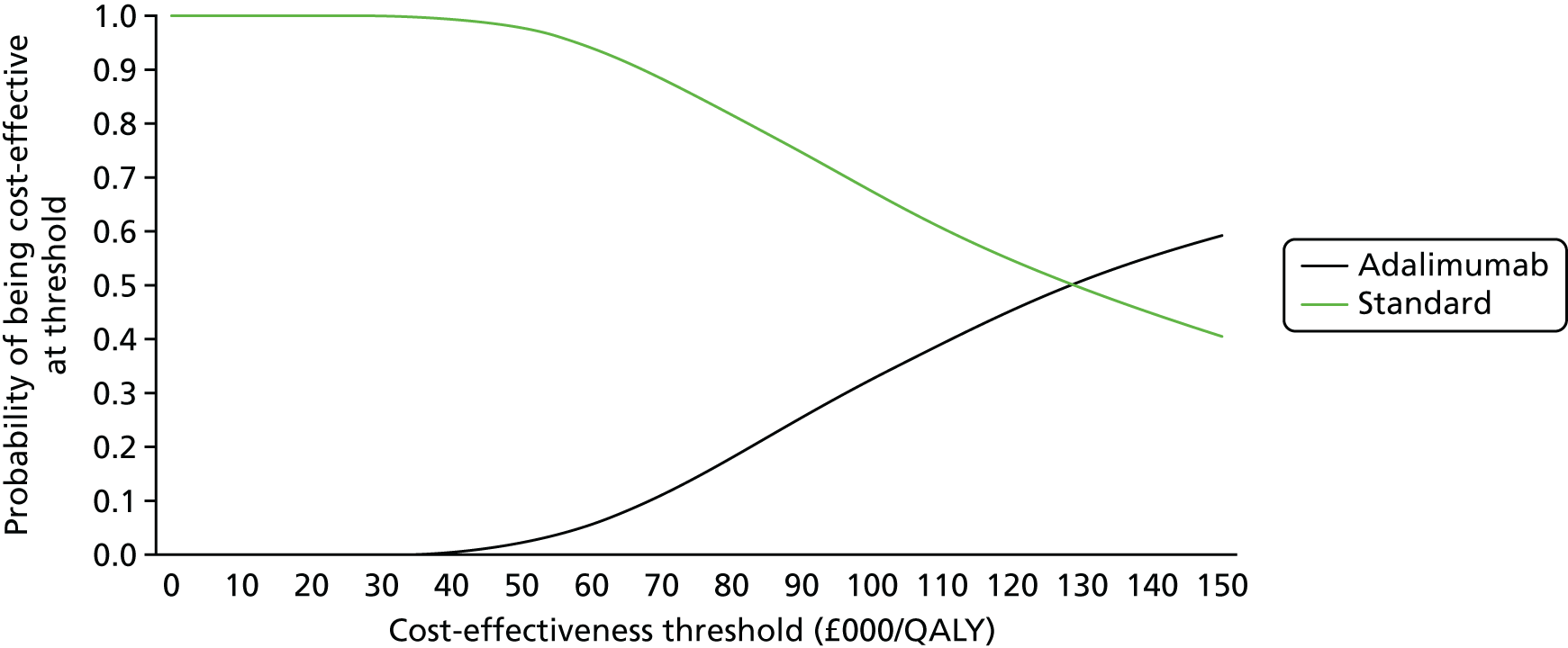
Scenario analyses
The complete-case analysis was limited by sparse QALY data for participants in the placebo group (n = 3) and should be interpreted with caution. For the imputed data, the ICER is robust to a variety of assumptions (Table 39). Taking into account the post-trial closure crossover from placebo to adalimumab has little impact on the ICER during the 18-month time horizon, but it has greater effect in the extrapolated phase, as patient benefit associated with adalimumab administered to participants in the placebo group in follow-up is adjusted away from the placebo arm to the adalimumab arm.
| Scenario | Costs (£) | QALYs | ICER (£) | ||||
|---|---|---|---|---|---|---|---|
| Treatment group | Incremental | Treatment group | Incremental | ||||
| Adalimumab | Placebo | Adalimumab | Placebo | ||||
| Complete-case analysis | |||||||
| 18-month time horizon | 15,891 | 5904 | 9988 | 1.40 | 1.45 | –0.05 | Dominated |
| + 10-year time horizon | 78,331 | 45,622 | 32,710 | 9.55 | 9.60 | –0.05 | Dominated |
| Imputed data not accounting for crossover | |||||||
| 18-month time horizon | 15,891 | 5904 | 9988 | 1.35 | 1.28 | 0.07 | 135,431 |
| + 10-year time horizon | 72,685 | 39,000 | 33,685 | 8.71 | 8.50 | 0.21 | 158,259 |
| Imputed data accounting for crossover | |||||||
| 18-month time horizon | 16,336 | 1962 | 14,374 | 1.36 | 1.25 | 0.11 | 136,751 |
| + 10-year time horizon | 70,719 | 31,403 | 39,316 | 8.60 | 8.29 | 0.30 | 129,025 |
Chapter 9 Discussion
The objectives of SYCAMORE, a randomised, double-blind, placebo-controlled trial were to investigate the clinical efficacy, safety and cost-effectiveness of adalimumab in combination with MTX for the treatment of JIA-associated uveitis in patients who had active uveitis despite having been on MTX for at least 12 weeks (with a stable dose for 4 weeks prior to the screening visit).
A total of 90 participants with active uveitis were randomised from 14 sites in the UK. No participants were excluded from the primary analysis and, therefore, the ITT data set contained all 90 participants. All participants received at least one dose of their allocated treatment and the safety data set also contained all 90 participants.
The majority of participants were female (77.8%) with a mean age of 8.9 years and had one eligible eye (72.2%). All participants had JIA-associated uveitis, with the majority of eligible eyes having mild or moderate uveitis (66.1% had activity of 1+ and 25.2% had activity of 2+); the type of JIA that was most common was persistent oligoarthritis (58.9%). There was no statistical testing of the baseline characteristics but numerically the two groups were very similar.
The primary efficacy outcome was time to treatment failure between the adalimumab and placebo groups. During the blinded phase of the study, the risk of treatment failure was significantly reduced by 75% for participants in the adalimumab group compared with participants in the placebo group (HR 0.25, 95% CI 0.12 to 0.51; p < 0.0001 from the log-rank test).
The results of the sensitivity analyses showed that the conclusions of the primary analysis were robust to changes that were made. These results all remained highly statistically significant.
The clinical secondary outcome variables that were both clinically and statistically significant in favour of adalimumab included greater disease control (uveitis) at 3 months and 6 months in at least one eligible eye and in all eligible eyes; greater disease control (uveitis) at 6 months; increased number of participants entering disease (uveitis) remission at 3 months and 6 months in at least one eligible eye and in all eligible eyes; increased duration of inactive disease (uveitis); ability to reduce to fewer than two corticosteroid drops and zero drops; and reduced total oral corticosteroid use.
As expected, the greatest proportion of uveitis patients have the oligoarticular subtype form of JIA. Therefore, measures of disease activity that look at articular disease did not show statistically significant differences.
The AE profile was consistent with the safety profile established across the approved indications of adalimumab. There were no deaths during the course of the trial. There were two AEs in the adalimumab group and one SAE in the placebo group, which led to the withdrawal of the participant.
As adalimumab is a potent immunosuppressive agent, as noted in previous literature,82 children treated with adalimumab in combination with MTX in this trial (integrated analysis of double-blind and open-label data) had a greater rate of AEs per year than placebo-treated children (9.86 vs. 7.21, respectively). The majority of these AEs were viral infections, which is consistent with published literature. 41 There was also a greater incidence of SAEs in the adalimumab group than the placebo group (21.7% vs. 6.7%, respectively). The most frequently reported SAEs were infections and infestations (11 events in 9 participants in the adalimumab group). The majority of the SAEs were mild or moderate in severity. There were no cases of malignancies, demyelinating events or deaths during the course of this study.
The economic analysis has strength in that it estimates the cost-effectiveness of adalimumab over a time period beyond that of SYCAMORE, up until the participants reach the age of 18 years. However, key limitations included incomplete data on health utilities and crossover effects resulting from early trial closure. These were addressed using robust methods (multiple imputation and instrumental variable regression) but, nevertheless, might have introduced bias to the analysis. We also deviated from the protocol by using HUI3 [instead of the Health Utility Index Mark 2 (HUI2)] utilities. This was carried out for two justifiable reasons, which had not become apparent (or which we had not appreciated) during the writing of the protocol. First, the attributes of the two HUIs are different, the HUI2 does not include an attribute specific to vision, which, given the context of SYCAMORE, would be a major disadvantage. Second, HUI3 yields more information than HUI2 when there are missing data. Taking the HUI2 sensation attribute as an example, any single missing response to one of the three subattributes (e.g. speech) would result in the whole sensation score being classed as missing in HUI2, whereas in HUI3 it would only affect the speech attribute but vision and hearing could still be counted as they are single attributes. It is acknowledged that one consequence of this change is that although there is UK health state valuation for the HUI2 there is none for HUI3, and so this is based on Canadian values. 73
A further limitation, related to the outcome measure for the analysis being VI, that differs from the primary outcome measure in SYCAMORE was time to treatment failure. This was necessary in order to model the impact of adalimumab over a time frame valid for JIA-associated uveitis and given the need to link to the Bristol cohort data. Regarding the generalisability of the Bristol cohort, both age and sex were representative of those in SYCAMORE; however, the Bristol cohort included idiopathic uveitis and patients experienced more VI than SYCAMORE participants. Moreover, there were probably differences in treatment plans, such as the choice of DMARDs and thresholds for initiating biologics. Bristol also pioneered combined (ophthalmology and rheumatology) clinics, which needed to be developed during SYCAMORE in many centres or optimised in many others to aid successful delivery of the trial to time and target.
The extrapolation model has several limitations, mainly because of a paucity of data. The structure of the model was limited by the number of participants and the incompleteness of the Bristol data set. 74 The model structure reflects visual acuity in a patient’s worst eye, which was deemed to be most clinically relevant. A further stratification of logMAR ≥ 1 as severe VI may have added to the interpretation of the progression of uveitis; however, within SYCAMORE, a logMAR score of 1 was recorded only once in over 900 recorded visits, and transitioning to severe VI from either no VI or VI in the Bristol data set74 was recorded in < 3% of transitions.
It was assumed that the transition probabilities in the model were independent of trial arm. Although the longitudinal (Bristol) data set74 included information on adalimumab prescription, it appeared that adalimumab was only prescribed to those patients in a worse health state (confounding by indication), which would bias the results, hence a single set of transition probabilities were derived based on data from all patients.
A further limitation is acknowledged: 15 participants from the SYCAMORE cohort are included in the longitudinal data set, contributing to 26 out of 268 total recorded transitions. To retain as much power as possible, these participants were not removed; however, this could be considered double counting.
This approach to modelling the long-term costs and consequences of adalimumab might have been structured differently. The model considered the progression of uveitis and associated complications requiring surgery, but does not explicitly reflect the impact of treatment on the progression of JIA. However, health state costs and QALYs determined from the trial implicitly included the benefits of treatment on mobility and pain, which are captured within the HUI3 utilities. The model also did not include the state of severe VI or blindness. A model based on the association between AC cell count and blindness might have offered an alternative approach, although this would not have reduced the need for considerable assumptions relating to the magnitude of long-term treatment benefits. Expected rates of blindness may be low in SYCAMORE participants as they had mild or moderate uveitis, with 91% of participants having AC cell counts of 1+ or 2+ at baseline. It may, alternatively, have been possible to calculate utilities and costs for the states of treatment failure and no treatment failure, and project the survival curves relating to the primary clinical end point of time to treatment failure, with the differences in costs and QALYs between treatment groups being driven by the survival function. This would have required an assumption that all the health-related quality-of-life benefits and costs (other than those of adalimumab) are driven by treatment success/failure, which might not necessarily be the case.
Despite these limitations, the analysis is robust to several assumptions. In an extreme scenario of every participant randomised to the placebo group moving to the state of VI in the modelled extrapolation, the ICER reduces only to £78,524 per QALY gained, which still exceeds the cost-effectiveness threshold. In order to demonstrate cost-effectiveness, participants receiving adalimumab would need to experience 1.00 additional QALY gain over the 10-year extrapolation, which seems unlikely, given that the QALY gain over the course of the trial was only 0.11.
In summary, this is the first large, randomised, double-blind, placebo-controlled trial of children with MTX-refractory JIA-associated uveitis, which shows that treatment with adalimumab combined with MTX is both effective and safe but, at £129,025 per QALY gained, is unlikely to be cost-effective in the UK NHS setting.
Implications for practice
This trial provides robust evidence regarding the use of adalimumab in the management of children and adolescents with JIA-associated uveitis refractory to MTX. The results show that adalimumab therapy in combination with MTX controlled inflammation and had a lower rate of treatment failure than placebo in this patient group. This finding, taken alongside the expected increased incidence of AEs in the adalimumab group and the economic analysis, can potentially inform current clinical practice with the aim of reducing uveitis-related ocular morbidity, including VI.
Recommendations for research
This trial answered some key questions for the management of JIA-associated uveitis refractory to MTX. However, it also identified a number of crucial next-step research challenges. These include the following:
-
defining the optimum time after control of uveitis to withdraw/stop adalimumab
-
defining the development and clinical utility of testing for antidrug antibodies in patients with waning of response to adalimumab
-
consideration of the option of increasing the frequency of administration of adalimumab for those with a suboptimal response
-
developing biomarkers for predicting early and complete response to adalimumab so that patients can be stratified more quickly to the appropriate treatment.
Overall conclusion
Adalimumab in combination with MTX is safe and effective in the management of JIA-associated uveitis. However, the likelihood of cost-effectiveness is < 1% at the £30,000-per-QALY threshold. Several limitations of the trial are noted. However, overall, this trial is, to our knowledge, the first randomised, double-blind, placebo-controlled, multicentre trial with an integrated economic evaluation comparing the efficacy, safety and cost-effectiveness of adalimumab in combination with MTX versus placebo with MTX alone, with regard to controlling disease activity in refractory uveitis associated with JIA. It also notes important future research priorities, including a future clinical trial to define the most effective time to stop therapy and to identify prognostic biomarkers of early and complete response.
Acknowledgements
We would like to thank all of the children and their families who gave their time and participated in SYCAMORE. We thank all the members of the oversight committees (see Appendix 1), all those from the CTRC who contributed to trial activities at various times of the trial (see Appendix 4) and all those who contributed to recruitment and follow-up from the participating sites.
We would like to thank the staff of the National Institute for Health Research Clinical Research Network Children for their assistance in the set-up and delivery of this trial.
We would also like to thank Dr Colin Ridyard and Dr Lorna Tuersley for their contributions to the health economic analyses, and Dr Megan Cann for help in collating data from the Regional Tertiary Paediatric Uveitis clinic that contributed to the health economic evaluation.
Contributions of authors
Athimalaipet V Ramanan and Michael W Beresford were chief investigators of the trial and provided rheumatology expertise for the trial design and throughout the trial period, with input from Sandrine Compeyrot-Lacassagne and Patricia Woo.
Athimalaipet V Ramanan, Ashley P Jones, Dyfrig A Hughes and Michael W Beresford wrote early versions of the manuscript with significant input from all co-authors. The manuscript was finalised and revisions addressed by Athimalaipet V Ramanan and Michael W Beresford with all co-authors’ consent.
Andrew D Dick and Clive Edelsten provided ophthalmology support and expertise during the design of the trial and throughout the trial period.
The study was conceived by Athimalaipet V Ramanan and Michael W Beresford, and the protocol and design were by Athimalaipet V Ramanan, Clive Edelsten, Dyfrig A Hughes, Ashley P Jones, Ben Hardwick, Helen Hickey and Michael W Beresford with support from all co-authors.
The day-to-day management of the trial was undertaken by Ashley P Jones, Andrew McKay, Paula R Williamson, Ben Hardwick and Helen Hickey, with ongoing support from Athimalaipet V Ramanan, Michael W Beresford and all co-authors.
Data were gathered and analysed by Athimalaipet V Ramanan, Ashley P Jones, Dyfrig A Hughes, Andrew McKay, Anna Rosala-Hallas, Paula R Williamson, Naomi Rainford, Graeme Hickey, Ruwanthi Kolamunnage-Dona, Giovanna Culeddu, Catrin Plumpton, Eifiona Wood and Michael W Beresford, and who vouch for the data and the analyses.
The authors assume responsibility for the accuracy and completeness of the data and vouch for the fidelity of the trial to the protocol.
Publications
Ramanan AV, Dick AD, Jones AP, McKay A, Williamson PR, Compeyrot-Lacassagne S, et al. Adalimumab plus methotrexate for uveitis in juvenile idiopathic arthritis. N Eng J Med 2017;376:1637–46.
Hughes DA, Culeddu G, Plumpton CO, Wood E, Dick AD, Jones AP, et al. Cost-effectiveness analysis of adalimumab for the treatment of uveitis associated with juvenile idiopathic arthritis [published online ahead of print 15 October 2018]. Ophthalmology 2018.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
University Hospitals Bristol NHS Foundation Trust, as the sponsor of this trial, has a data-sharing agreement with AbbVie Inc. in support of regulatory purposes.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Edelsten C, Reddy MA, Stanford MR, Graham EM. Visual loss associated with pediatric uveitis in English primary and referral centers. Am J Ophthalmol 2003;135:676-80. https://doi.org/10.1016/S0002-9394(02)02148-7.
- Smith JA, Mackensen F, Sen HN, Leigh JF, Watkins AS, Pyatetsky D, et al. Epidemiology and course of disease in childhood uveitis. Ophthalmology 2009;116:1544-51e1. https://doi.org/10.1016/j.ophtha.2009.05.002.
- Kanski JJ. Uveitis in juvenile chronic arthritis: incidence, clinical features and prognosis. Eye 1988;2:641-5. https://doi.org/10.1038/eye.1988.118.
- Kotaniemi K, Kautiainen H, Karma A, Aho K. Occurrence of uveitis in recently diagnosed juvenile chronic arthritis: a prospective study. Ophthalmology 2001;108:2071-5. https://doi.org/10.1016/S0161-6420(01)00773-4.
- Saurenmann RK, Levin AV, Feldman BM, Rose JB, Laxer RM, Schneider R, et al. Prevalence, risk factors, and outcome of uveitis in juvenile idiopathic arthritis: a long-term follow-up study. Arthritis Rheum 2007;56:647-57. https://doi.org/10.1002/art.22381.
- Chia A, Lee V, Graham EM, Edelsten C. Factors related to severe uveitis at diagnosis in children with juvenile idiopathic arthritis in a screening program. Am J Ophthalmol 2003;135:757-62. https://doi.org/10.1016/S0002-9394(03)00225-3.
- Holland GN, Denove CS, Yu F. Chronic anterior uveitis in children: clinical characteristics and complications. Am J Ophthalmol 2009;147:667-78.e5. https://doi.org/10.1016/j.ajo.2008.11.009.
- Woreta F, Thorne JE, Jabs DA, Kedhar SR, Dunn JP. Risk factors for ocular complications and poor visual acuity at presentation among patients with uveitis associated with juvenile idiopathic arthritis. Am J Ophthalmol 2007;143:647-55. https://doi.org/10.1016/j.ajo.2006.11.025.
- Edelsten C, Lee V, Bentley CR, Kanski JJ, Graham EM. An evaluation of baseline risk factors predicting severity in juvenile idiopathic arthritis-associated uveitis and other chronic anterior uveitis in early childhood. Br J Ophthalmol 2002;86:51-6. https://doi.org/10.1136/bjo.86.1.51.
- Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of Uveitis Nomenclature (SUN) Working Group . Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005;140:509-16. https://doi.org/10.1016/j.ajo.2005.03.057.
- Thorne JE, Woreta F, Kedhar SR, Dunn JP, Jabs DA. Juvenile idiopathic arthritis-associated uveitis: incidence of ocular complications and visual acuity loss. Am J Ophthalmol 2007;143:840-6. https://doi.org/10.1016/j.ajo.2007.01.033.
- Wolf MD, Lichter PR, Ragsdale CG. Prognostic factors in the uveitis of juvenile rheumatoid arthritis. Ophthalmology 1987;94:1242-8. https://doi.org/10.1016/S0161-6420(87)80007-6.
- de Boer J, Wulffraat N, Rothova A. Visual loss in uveitis of childhood. Br J Ophthalmol 2003;87:879-84. https://doi.org/10.1136/bjo.87.7.879.
- Takken T, Van der Net J, Helders PJ. Methotrexate for treating juvenile idiopathic arthritis. Cochrane Database Syst Rev 2001;3. https://doi.org/10.1002/14651858.CD003129.
- Beresford MW, Baildam EM. New advances in the management of juvenile idiopathic arthritis – 1: non-biological therapy. Arch Dis Child Educ Pract Ed 2009;94:144-50. https://doi.org/10.1136/adc.2008.144576.
- Foeldvari I, Wierk A. Methotrexate is an effective treatment for chronic uveitis associated with juvenile idiopathic arthritis. J Rheumatol 2005;32:362-5.
- Weiss AH, Wallace CA, Sherry DD. Methotrexate for resistant chronic uveitis in children with juvenile rheumatoid arthritis. J Pediatr 1998;133:266-8. https://doi.org/10.1016/S0022-3476(98)70232-X.
- Yu EN, Meniconi ME, Tufail F, Baltatzis S, Foster CS. Outcomes of treatment with immunomodulatory therapy in patients with corticosteroid-resistant juvenile idiopathic arthritis-associated chronic iridocyclitis. Ocul Immunol Inflamm 2005;13:353-60. https://doi.org/10.1080/09273940590951061.
- Sharma SM, Dick AD, Ramanan AV. Non-infectious pediatric uveitis: an update on immunomodulatory management. Paediatr Drugs 2009;11:229-41. https://doi.org/10.2165/00148581-200911040-00002.
- Kalinina Ayuso V, van de Winkel EL, Rothova A, de Boer JH. Relapse rate of uveitis post-methotrexate treatment in juvenile idiopathic arthritis. Am J Ophthalmol 2011;151:217-22. https://doi.org/10.1016/j.ajo.2010.08.021.
- Doycheva D, Deuter C, Stuebiger N, Biester S, Zierhut M. Mycophenolate mofetil in the treatment of uveitis in children. Br J Ophthalmol 2007;91:180-4. https://doi.org/10.1136/bjo.2006.094698.
- Kilmartin DJ, Forrester JV, Dick AD. Cyclosporin A therapy in refractory non-infectious childhood uveitis. Br J Ophthalmol 1998;82:737-42. https://doi.org/10.1136/bjo.82.7.737.
- Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU). Prog Retin Eye Res 2004;23:617-37. https://doi.org/10.1016/j.preteyeres.2004.06.005.
- Imrie FR, Dick AD. Biologics in the treatment of uveitis. Curr Opin Ophthalmol 2007;18:481-6. https://doi.org/10.1097/ICU.0b013e3282f03d42.
- Smith JR, Hart PH, Coster DJ, Williams KA. Mice deficient in tumor necrosis factor receptors p55 and p75, interleukin-4, or inducible nitric oxide synthase are susceptible to endotoxin-induced uveitis. Invest Ophthalmol Vis Sci 1998;39:658-61.
- Koizumi K, Poulaki V, Doehmen S, Welsandt G, Radetzky S, Lappas A, et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Invest Ophthalmol Vis Sci 2003;44:2184-91. https://doi.org/10.1167/iovs.02-0589.
- Diaz-Llopis M, García-Delpech S, Salom D, Udaondo P, Bosch-Morell F, Quijada A, et al. High-dose infliximab prophylaxis in endotoxin-induced uveitis. J Ocul Pharmacol Ther 2007;23:343-50. https://doi.org/10.1089/jop.2006.0148.
- Biester S, Deuter C, Michels H, Haefner R, Kuemmerle-Deschner J, Doycheva D, et al. Adalimumab in the therapy of uveitis in childhood. Br J Ophthalmol 2007;91:319-24. https://doi.org/10.1136/bjo.2006.103721.
- Foeldvari I, Nielsen S, Kümmerle-Deschner J, Espada G, Horneff G, Bica B, et al. Tumour necrosis factor-alpha blocker in treatment of juvenile idiopathic arthritis-associated uveitis refractory to second-line agents: results of a multinational survey. J Rheumatol 2007;34:1146-50.
- Gallagher M, Quinones K, Cervantes-Castañeda RA, Yilmaz T, Foster CS. Biological response modifier therapy for refractory childhood uveitis. Br J Ophthalmol 2007;91:1341-4. https://doi.org/10.1136/bjo.2007.124081.
- Saurenmann RK, Levin AV, Rose JB, Parker S, Rabinovitch T, Tyrrell PN, et al. Tumour necrosis factor-alpha inhibitors in the treatment of childhood uveitis. Rheumatology 2006;45:982-9. https://doi.org/10.1093/rheumatology/kel030.
- Sharma SM, Ramanan AV, Riley P, Dick AD. Use of infliximab in juvenile onset rheumatological disease-associated refractory uveitis: efficacy in joint and ocular disease. Ann Rheum Dis 2007;66:840-1. https://doi.org/10.1136/ard.2006.065441.
- Tynjälä P, Kotaniemi K, Lindahl P, Latva K, Aalto K, Honkanen V, et al. Adalimumab in juvenile idiopathic arthritis-associated chronic anterior uveitis. Rheumatology 2008;47:339-44. https://doi.org/10.1093/rheumatology/kem356.
- Schmeling H, Minden K, Foeldvari I, Ganser G, Hospach T, Horneff G. Efficacy and safety of adalimumab as the first and second biologic agent in juvenile idiopathic arthritis: the German Biologics JIA Registry. Arthritis Rheum 2014;66:2580-9. https://doi.org/10.1002/art.38741.
- Smith JA, Thompson DJ, Whitcup SM, Suhler E, Clarke G, Smith S, et al. A randomized, placebo-controlled, double-masked clinical trial of etanercept for the treatment of uveitis associated with juvenile idiopathic arthritis. Arthritis Rheum 2005;53:18-23. https://doi.org/10.1002/art.20904.
- Hale S, Lightman S. Anti-TNF therapies in the management of acute and chronic uveitis. Cytokine 2006;33:231-7. https://doi.org/10.1016/j.cyto.2005.12.012.
- Saurenmann R, Levin A, Feldman B, Laxer R, Schneider R, Silverman E. Risk of new-onset uveitis in patients with juvenile idiopathic arthritis treated with anti-TNF-alpha agents. J Pediatr 2006;149:833-6. https://doi.org/10.1016/j.jpeds.2006.08.044.
- Taban M, Dupps WJ, Mandell B, Perez VL. Etanercept (enbrel)-associated inflammatory eye disease: case report and review of the literature. Ocul Immunol Inflamm 2006;14:145-50. https://doi.org/10.1080/09273940600659393.
- Lim LL, Fraunfelder FW, Rosenbaum JT. Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum 2007;56:3248-52. https://doi.org/10.1002/art.22918.
- Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal anti-body) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum 2004;50:1400-11. https://doi.org/10.1002/art.20217.
- Lovell DJ, Ruperto N, Goodman S, Reiff A, Jung L, Jarosova K, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med 2008;359:810-20. https://doi.org/10.1056/NEJMoa0706290.
- Vazquez-Cobian LB, Flynn T, Lehman TJ. Adalimumab therapy for childhood uveitis. J Pediatr 2006;149:572-5. https://doi.org/10.1016/j.jpeds.2006.04.058.
- Shepherd J, Cooper K, Harris P, Picot J, Rose M. The clinical effectiveness and cost-effectiveness of abatacept, adalimumab, etanercept and tocilizumab for treating juvenile idiopathic arthritis: a systematic review and economic evaluation. Health Technol Assess 2016;20. https://doi.org/10.3310/hta20340.
- Cummins C, Connock M, Fry-Smith A, Burls A. A systematic review of effectiveness and economic evaluation of new drug treatments for juvenile idiopathic arthritis: etanercept. Health Technol Assess 2002;6. https://doi.org/10.3310/hta6170.
- Ungar WJ, Costa V, Hancock-Howard R, Feldman BM, Laxer RM. Cost-effectiveness of biologics in polyarticular-course juvenile idiopathic arthritis patients unresponsive to disease-modifying antirheumatic drugs. Arthritis Care Res 2011;63:111-19. https://doi.org/10.1002/acr.20337.
- Simpson K, Marlow N, Shaw J, Rudakova A. Pharmacoeconomic issues of adalimumab therapy in juvenile idiopathic arthritis. Pediatr Pharmacol 2012;9:48-52. https://doi.org/10.15690/pf.v9i4.390.
- Prince FH, de Bekker-Grob EW, Twilt M, van Rossum MA, Hoppenreijs EP, ten Cate R, et al. An analysis of the costs and treatment success of etanercept in juvenile idiopathic arthritis: results from the Dutch Arthritis and Biologicals in Children register. Rheumatology 2011;50:1131-6. https://doi.org/10.1093/rheumatology/keq432.
- Squires H, Poku E, Bermejo I, Cooper K, Stevens J, Hamilton J, et al. A systematic review and economic evaluation of adalimumab and dexamethasone for treating non-infectious intermediate uveitis, posterior uveitis or panuveitis in adults. Health Technol Assess 2017;21. https://doi.org/10.3310/hta21680.
- Ramanan AV, Dick AD, Benton D, Compeyrot-Lacassagne S, Dawoud D, Hardwick B, et al. A randomised controlled trial of the clinical effectiveness, safety and cost-effectiveness of adalimumab in combination with methotrexate for the treatment of juvenile idiopathic arthritis associated uveitis (SYCAMORE Trial). Trials 2014;15. https://doi.org/10.1186/1745-6215-15-14.
- Medicines and Healthcare products Regulatory Agency . Good Clinical Practice for Clinical Trials. 2014. www.gov.uk/guidance/good-clinical-practice-for-clinical-trials (accessed 4 February 2019).
- The Medicines for Human Use (Clinical Trials) Regulations 2004. London: The Stationery Office; 2004.
- Oliveira S, Ravelli A, Pistorio A, Castell E, Malattia C, Prieur AM, et al. Proxy-reported healthrelated quality of life of patients with juvenile idiopathic arthritis: the paediatric rheumatology international trials organisation multinational quality of life cohort study. Arthritis Care Res 2007;1:35-43. https://doi.org/10.1002/art.22473.
- Nugent J, Ruperto N, Grainger J, Machado C, Sawhney S, Baildam E, et al. The British version of the Childhood Health Assessment Questionnaire (CHAQ) and the Child Health Questionnaire (CHQ). Clin Exp Rheumatol 2001;19:S163-7.
- Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A. Preliminary definition of improvement in juvenile arthritis. Arthritis Rheum 1997;40:1202-9. https://doi.org/10.1002/1529-0131(199707)40:7.lt;1202::AID-ART3.gt;3.0.CO;2-R.
- Brunner HI, Lovell DJ, Finck BK, Giannini EH. Preliminary definition of disease flare in juvenile rheumatoid arthritis. J Rheumatol 2002;29:1058-64.
- Wallace CA, Ruperto N, Giannini E. Childhood Arthritis and Rheumatology Research Alliance . Preliminary criteria for clinical remission for select categories of juvenile idiopathic arthritis. J Rheumatol 2004;31:2290-4.
- Magni-Manzoni S, Ruperto N, Pistorio A, Sala E, Solari N, Palmisani E, et al. Development and validation of a preliminary definition of minimal disease activity in patients with juvenile idiopathic arthritis. Arthritis Rheum 2008;59:1120-7. https://doi.org/10.1002/art.23916.
- Consolaro A, Ruperto N, Bazso A, Pistorio A, Magni-Manzoni S, Filocamo G, et al. Development and validation of a composite disease activity score for juvenile idiopathic arthritis. Arthritis Rheum 2009;61:658-66. https://doi.org/10.1002/art.24516.
- Ilowite NT. Update on biologics in juvenile idiopathic arthritis. Curr Opin Rheumatol 2008;20:613-18. https://doi.org/10.1097/BOR.0b013e3283060778.
- Ruperto N, Lovell DJ, Cuttica R, Wilkinson N, Woo P, Espada G, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum 2007;56:3096-106. https://doi.org/10.1002/art.22838.
- Arthritis UK . Research Policy – Observational Data and Sample Collection n.d. www.versusarthritis.org/media/1384/observational-data-and-sample-collection-policy.pdf (accessed 4 February 2019).
- Pocock SJ. Current controversies in data monitoring for clinical trials. Clin Trials 2006;3:513-21. https://doi.org/10.1177/1740774506073467.
- Ramanan AV, Dick AD, Jones AP, McKay A, Williamson PR, Compeyrot-Lacassagne S, et al. for the SYCAMORE Study Group . Adalimumab plus methotrexate for uveitis in juvenile idiopathic arthritis. N Engl J Med 2017;376:1637-46. https://doi.org/10.1056/NEJMoa1614160.
- Schulz KF, Altman DG, Moher D. CONSORT Group . CONSORT 2010 Statement: updated guidelines for reporting parallel-group randomised trials. BMC Med 2010;8. https://doi.org/10.1186/1741-7015-8-18.
- Henderson R, Diggle P, Dobson A. Joint modelling of longitudinal measurements and event time data. Biostatistics 2000;1:465-80. https://doi.org/10.1093/biostatistics/1.4.465.
- Prescription Cost Analysis Data. Newcastle upon Tyne: NHS BSA; n.d.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2017.
- NHS National Tariff Payment System 2016/17. Leeds: NHS England; 2016.
- A Simple Guide to Payment by Results. Leeds: NHS England; 2013.
- NHS Reference Costs 2015 to 2016. Leeds: NHS England; 2016.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2015. Canterbury: Personal Social Services Research Unit, University of Kent; 2015.
- Geue C, Lewsey J, Lorgelly P, Govan L, Hart C, Briggs A. Spoilt for choice: implications of using alternative methods of costing hospital episode statistics. Health Econ 2012;21:1201-16. https://doi.org/10.1002/hec.1785.
- Furlong W, Feeny D, Torrance G, Goldsmith C, DePauw S, Zhu Z, et al. Multiplicative Multi-Attribute Utility Function for the HUI Mark 3 (HUI3) System: a Technical Report CHEPA Working Paper. Hamilton, ON: Centre for Health Economics and Policy Analysis (CHEPA); 1998.
- Cann M, Ramanan AV, Crawford A, Dick AD, Clarke SLN, Rashed F, et al. Outcomes of non-infectious paediatric uveitis in the era of biologic therapy. Pediatr Rheumatol Online J 2018;16. https://doi.org/10.1186/s12969-018-0266-5.
- Shkolnikov V, Barbieri MWJ. The Human Mortality Database 2017. www.mortality.org (accessed 3 October 2017).
- Davies R, Southwood T, Kearsley-Fleet L, Lunt M, Hyrich KL. FRI0557 standardized mortality rates are increased in patients with severe juvenile idiopathic arthritis. Ann Rheum Dis 2014;73. https://doi.org/10.1093/rheumatology/keu496.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. https://doi.org/10.1002/sim.4067.
- Sussman JB, Hayward RA. An IV for the RCT: using instrumental variables to adjust for treatment contamination in randomised controlled trials. BMJ 2010;340. https://doi.org/10.1136/bmj.c2073.
- Fenwick E, Claxton K, Sculpher M. Representing uncertainty: the role of cost-effectiveness acceptability curves. Health Econ 2001;10:779-87. https://doi.org/10.1002/hec.635.
- Guide to the Methods Of Technology Appraisal 2013. Process and Methods [PMG9]. London: NICE; 2013.
- Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) – explanation and elaboration: a report of the ISPOR Health Economic Evaluation Publication Guidelines Good Reporting Practices Task Force. Value Health 2013;16:231-50. https://doi.org/10.1016/j.jval.2013.02.002.
- Sen ES, Dick AD, Ramanan AV. Uveitis associated with juvenile idiopathic arthritis. Nat Rev Rheumatol 2015;11:338-48.
Appendix 1 Trial oversight committees
Trial Steering Committee
Independent members
Professor Ian Bruce (chairperson), Professor of Rheumatology, Arthritis Research UK Centre for Epidemiology, University of Manchester, UK.
Dr Carlos Pavesio, Consultant Ophthalmologist, Moorfields Eye Hospital NHS Foundation Trust, London, UK.
Mr Matt Sydes, Senior Scientist and Senior Medical Statistician, MRC Clinical Trials Unit, London, UK.
Dr Janine Gray, Principal Medical Statistician, Leeds Institute of Clinical Trials Research, University of Leeds, UK (Dr Gray left the TSC on 29 January 2015).
Non-independent members
Professor Athimalaipet Vaidyanathan Ramanan* (Co-Chief Investigator), Consultant Paediatric Rheumatologist, Bristol Royal Hospital for Children & Royal National Hospital for Rheumatic Diseases, UK.
Professor Michael W Beresford* (Co-Chief Investigator), Professor of Child Health, University of Liverpool, Liverpool, UK.
Dr Ashley P Jones, Senior Statistician, CTRC, University of Liverpool, Liverpool, UK.
Professor Andrew Dick, Professor of Ophthalmology, Department of Clinical Sciences, University of Bristol, Bristol, UK.
Dr Clive Edelsten, Consultant Ophthalmologist, Great Ormond Street Hospital for Children, London, UK.
Mr Ben Hardwick, Trial Co-ordinator, CTRC, University of Liverpool, Liverpool, UK.
*Trial management group members eligible to vote. Only one non-independent member was eligible to vote.
Independent Data and Safety Monitoring Committee
Professor John Sparrow (Chairperson), Consultant Ophthalmologist, Bristol Eye Hospital, Bristol, UK.
Professor Justine Smith, Consultant Ophthalmologist, School of Medicine, Adelaide, SA, Australia.
Dr Nico Wulffraat, Paediatric Rheumatologist, UMC Utrecht, Department of Paediatrics, Utrecht, Netherlands.
Professor Steff Lewis, Personal Chair in Medical Statistics, Public Health Sciences section, Centre for Population Health Sciences, The University of Edinburgh, Medical School, Edinburgh, UK.
Trial Management Group
Professor Athimalaipet Vaidyanathan Ramanan (Co-Chief Investigator), Consultant Paediatric Rheumatologist, Bristol Royal Hospital for Children & Royal National Hospital for Rheumatic Diseases, UK.
Professor Michael W Beresford (Co-Chief Investigator), Professor of Child Health, University of Liverpool, Liverpool, UK.
Diana Benton, Research & Innovation, University Hospitals Bristol NHS Foundation Trust, Bristol, UK.
Jessica Bisset, Research & Innovation, University Hospitals Bristol NHS Foundation Trust, Bristol, UK.
Professor Andrew Dick, Professor of Ophthalmology, Department of Clinical Sciences, University of Bristol, Bristol, UK.
Dr Clive Edelsten, Consultant Ophthalmologist, Great Ormond Street Hospital for Children, London, UK.
Mr Ben Hardwick, Trial Co-ordinator, CTRC, University of Liverpool, Liverpool, UK.
Ms Helen Hickey, Head of Trial Management, CTRC, University of Liverpool, Liverpool, UK.
Professor Dyfrig Hughes, Professor of Pharmaeconomics, Centre for Health Economics and Medicines Evaluation, Bangor University, Bangor, UK.
Debbie Janson, patient representative.
Dr Ashley P Jones, Senior Statistician, CTRC, University of Liverpool, Liverpool, UK.
Dr Sandrine Lacassagne, Paediatric Rheumatologist, Great Ormond Street Hospital for Children NHS Trust, London, UK.
Mr Andrew McKay, Trial Statistician, CTRC, University of Liverpool, Liverpool, UK.
Jennifer Oliver, Bristol Children’s Vaccine Centre, Bristol, UK.
Emma Stoica, Research & Innovation, University Hospitals Bristol NHS Foundation Trust, Bristol, UK.
Ms Giovanna Culeddu, Research Project Support Officer, Centre for Health Economics and Medicines Evaluation, Bangor University, Bangor, UK.
Professor Paula Williamson, Director, CTRC, University of Liverpool, Liverpool, UK.
Professor Patricia Woo, Emeritus Professor of Paediatric Rheumatology, University College London.
Appendix 2 Differences between The New England Journal of Medicine manuscript63 and Chapter 5 results of this report
| Outcome | NEJM paper63 results | Chapter 5 results | Notes |
|---|---|---|---|
| Time to treatment failure | HR 0.25, 95% CI 0.12 to 0.49; p < 0.0001 | HR 0.25, 95% CI 0.12 to 0.51; p < 0.0001 | Three participants who were classified as treatment failures in the first snapshot were classified incorrectly and, therefore, should have been withdrawals (participants from the adalimumab group, anonymised identification numbers: 0249031, 0249040; placebo: 0243049). Additionally, in the treatment failure line listings, 0133058’s further details changed from ‘No follow up, consent to use collected data’ to ‘Continue follow up’ and 0116008’s first of the consecutive visits changed from ‘Visit 3 – 3 months’ treatment: 2+ on 31 May 2012’ to ‘Unscheduled visit: 2+ on 12 July 2012’ |
| Number of participants failing treatment | RR 0.40, 95% CI 0.22 to 0.73; p = 0.002 | RR 0.40, 95% CI 0.23 to 0.72; p = 0.002 | See above for an explanation of the reclassification of the three treatment failures |
| AEs |
588 AEs in 53 participants in the adalimumab group and 103 AEs in 25 participants in the placebo group Rate (95% CI) in the adalimumab group 10.07, 95% CI 9.26 to 10.89 Rate in the placebo group 6.51, 95% CI 5.26 to 7.77 |
619 AEs in 59 participants in the adalimumab group and 114 AEs in 26 participants in the placebo group Rate (95% CI) in the adalimumab group 10.60, 95% CI 9.77 to 11.44 Rate in the placebo group 7.21, 95% CI 5.89 to 8.53 |
The number of AEs increased owing to responses to data queries that were received in relation to concomitant medications. When concomitant medications were queried, extra rows of concomitant medications that were not expected were submitted, which then indicated that there were further AEs (as they had AE numbers indicated on the form that had not been received) |
| SAEs |
17 SAEs in 13 participants in the adalimumab group and 3 SAEs in two participants in the placebo group Rate (95% CI) in the adalimumab group 0.29, 95% CI 0.15 to 0.43 Rate in the placebo group 0.19, 95% CI 0.00 to 0.40 |
No change | N/A |
| Compliance |
Participant diaries: IMP – Adalimumab, 82.87%; placebo, 74.11% MTX – Adalimumab, 60.46%; placebo, % Accountability logs: IMP – Adalimumab, 94.00%; placebo, 90.24% |
Participant diaries: IMP – Adalimumab, 84.00%; placebo, 74.00% MTX – Adalimumab, 62.50%; placebo, % Accountability logs: IMP – Adalimumab, 94.00%; placebo, 90.00% |
Minor differences between two sets of result |
| Total oral corticosteroid dose | The analysis of this outcome was not reported in the NEJM manuscript or supplementary table | N/A | N/A |
| Reduction in systemic corticosteroid dose from entry dose to 0 mg | Analyses of these data were not possible owing to the statistical algorithm not converting | No change | N/A |
| Reduction in systemic corticosteroid dose from entry dose to < 5 mg | Analyses of these data were not possible owing to the statistical algorithm not converting | No change | N/A |
| Time to reduction to fewer than two drops in topical corticosteroid | HR 3.72, 95% CI 1.09 to 12.71; p = 0.04 | HR 3.99, 95% CI 1.18 to 25.20; p = 0.03 |
00243049: originally failure code 2 and now failure code 1. Changed from 49 days to 147 days owing to incorrect treatment failure. Placebo participant 00249040: originally failure code 2 and now failure code 1. Changed from 202 days to 343 days owing to incorrect treatment failure. Adalimumab participant |
| Time to reduction to zero drops in topical steroid (post hoc analysis) | HR 3.58, 95% CI 1.24 to 10.32; p = 0.02 | HR 4.02, 95% CI 1.40 to 11.50; p = 0.01 |
00243049: originally failure code 2 and now failure code 1. Changed from 49 days to 147 days owing to incorrect treatment failure. Placebo participant 00249040: originally failure code 2 and now failure code 1. Changed from 202 days to 475 days owing to incorrect treatment failure. Adalimumab participant 00249048: originally failure code 0 and now failure code 1. Changed from 504 days to 483 days. One concomitant medication added. Originally, the last entry had a missing end date, so this was imputed as the PO date, but now one concomitant medication has been added, which has an end date and so the participant reached zero drops before their PO date. Adalimumab participant |
| Number of participants requiring pulsed therapy | RR 0.50, 95% CI 0.03 to 7.72; p > 0.99 | RR 1.00, 95% CI 0.09 to 10.59, p > 0.99 | Two participants required pulsed therapy during the blinded phase in the adalimumab group; only one was reported in the NEJM manuscript |
| Number of participants having uveitis disease flares following 3 months of disease control | The analysis of this outcome was not reported in the NEJM manuscript or supplementary table | N/A | |
| Number of participants having disease flares within the first 3 months | RR 0.07, 95% CI 0.00 to 1.36 | No change | N/A |
| Visual acuity measured by age-appropriate logMAR assessment |
Best case: –0.01, 95% CI –0.06 to 0.03; p = 0.59 Worst case: –0.02, 95% CI –0.07 to 0.03; p = 0.53 |
Best case: –0.01, 95% CI –0.07 to 0.02; p = 0.51 Worst case: –0.02, 95% CI –0.07 to 0.02; p = 0.36 |
An incorrect assumption had been made with regard to how premature discontinuations were handled in the analysis. In the NEJM manuscript, these were incorrectly handled as treatment failures |
| Number of participants with resolution of associated optic nerve or macular oedema |
Analysis of the optic nerve data was not possible owing to the low number of participants who had this Macular oedema: RR 5.00, 95% CI 0.34 to 74.52 |
No changes | N/A |
| Number of participants with disease control (in all eligible eyes) |
3 months: RR 11.00, 95% CI 1.56 to 77.74; p < 0.001 6 months: RR 8.50, 95% CI 1.19 to 60.87; p = 0.005 |
3 months: RR 5.75, 95% CI 1.45 to 22.78; p = 0.001 6 months: no change |
One more participant in the adalimumab arm had disease control for 3 months |
| Number of participants entering disease remission (in all eligible eyes) |
3 months: RR 7.50, 95% CI 1.04 to 54.12; p = 0.02 6 months: RR 13.72, 95% CI 0.84 to 223.26; p = 0.004 |
3 months: no change 6 months: no change |
N/A |
| Duration of sustaining inactive disease | MD 164.79, 95% CI 104.41 to 225.16; p < 0.0001 | MD 164.55, 95% CI 104.41 to 224.69; p < 0.0001 | Very minor difference in MD |
| CHQ |
PsS 2.69, 95% CI –0.26 to 5.86; p = 0.06 PhS 1.36, 95% CI –2.28 to 5.05; p = 0.49 |
PsS 2.31, 95% CI –0.44 to 5.40; p = 0.15 PhS 1.16, 95% CI –2.41 to 5.05; p = 0.55 |
An incorrect assumption had been made with regard to how premature discontinuations were handled in the analysis. In the NEJM manuscript, these were incorrectly handled as treatment failures |
| CHAQ | –0.14, 95% CI –0.32 to 0.01; p = 0.08 | –0.14, 95% CI –0.31 to 0.02; p = 0.09 | An incorrect assumption had been made with regard to how premature discontinuations were handled in the analysis. In the NEJM manuscript these were incorrectly handled as treatment failures |
| ACR |
ACR30: –0.70, 95% CI –1.86 to 0.52; p = 0.23 ACR50: –0.65, 95% CI –1.74 to 0.44; p = 0.25 ACR70: –0.61, 95% CI –1.89 to 0.74; p = 0.34 ACR90: –0.46, 95% CI –3.38 to 2.46; p = 0.76 ACR100: –1.23, 95% CI –2.34 to –0.14; p = 0.03 |
ACR30: –0.04, 95% CI –1.37 to 1.59; p = 0.98 ACR50: –0.70, 95% CI –2.15 to 0.77; p = 0.37 ACR70: –1.08, 95% CI –2.70 to 0.46; p = 0.16 ACR90: –0.33, 95% CI –2.22 to 1.39; p = 0.72 ACR100: –0.32, 95% CI –1.85 to 1.17; p = 0.65 |
An incorrect assumption had been made with regard to how premature discontinuations were handled in the analysis. In the NEJM manuscript, these were incorrectly handled as treatment failures The significance of ACR100 did change, but this is not regarded as clinically important because there were such small numbers in the analysis |
| Number of participants undergoing arthritis disease flare, in remission on and/or off medication for their JIA, and with minimum disease activity |
Disease flare: RR 0.07, 95% CI 0.00 to 1.36; p = 0.03 Remission: this outcome was only reportable during the follow-up phase and, therefore, was not reported in the NEJM paper Minimum disease activity: RR 2.33, 95% CI 0.87 to 6.24; p = 0.08 |
Disease flare: no change Remission: N/A Minimum disease activity: no change |
N/A |
| Number of participants requiring change in biologics or DMARD therapy owing to failure to respond from arthritis | RR 1.00, 95% CI 0.20 to 5.09; p = 0.99 | No change | N/A |
| JADAS |
JADAS10: –0.35, 95% CI –0.79 to –0.01; p = 0.07 JADAS27: –0.34, 95% CI –0.78 to 0.03; p = 0.08 JADAS71: –0.36, 95% CI –0.80 to –0.0004; p = 0.07 |
JADAS10: –0.35, 95% CI –0.78 to 0.01; p = 0.07 JADAS27: –0.34, 95% CI –0.76 to 0.03; p = 0.08 JADAS71: –0.36, 95% CI –0.78 to 0.004; p = 0.07 |
An incorrect assumption had been made with regard to how premature discontinuations were handled in the analysis. In the NEJM manuscript these were incorrectly handled as treatment failures |
Appendix 3 Additional clinical effectiveness data
FIGURE 13.
Overall recruitment graph.
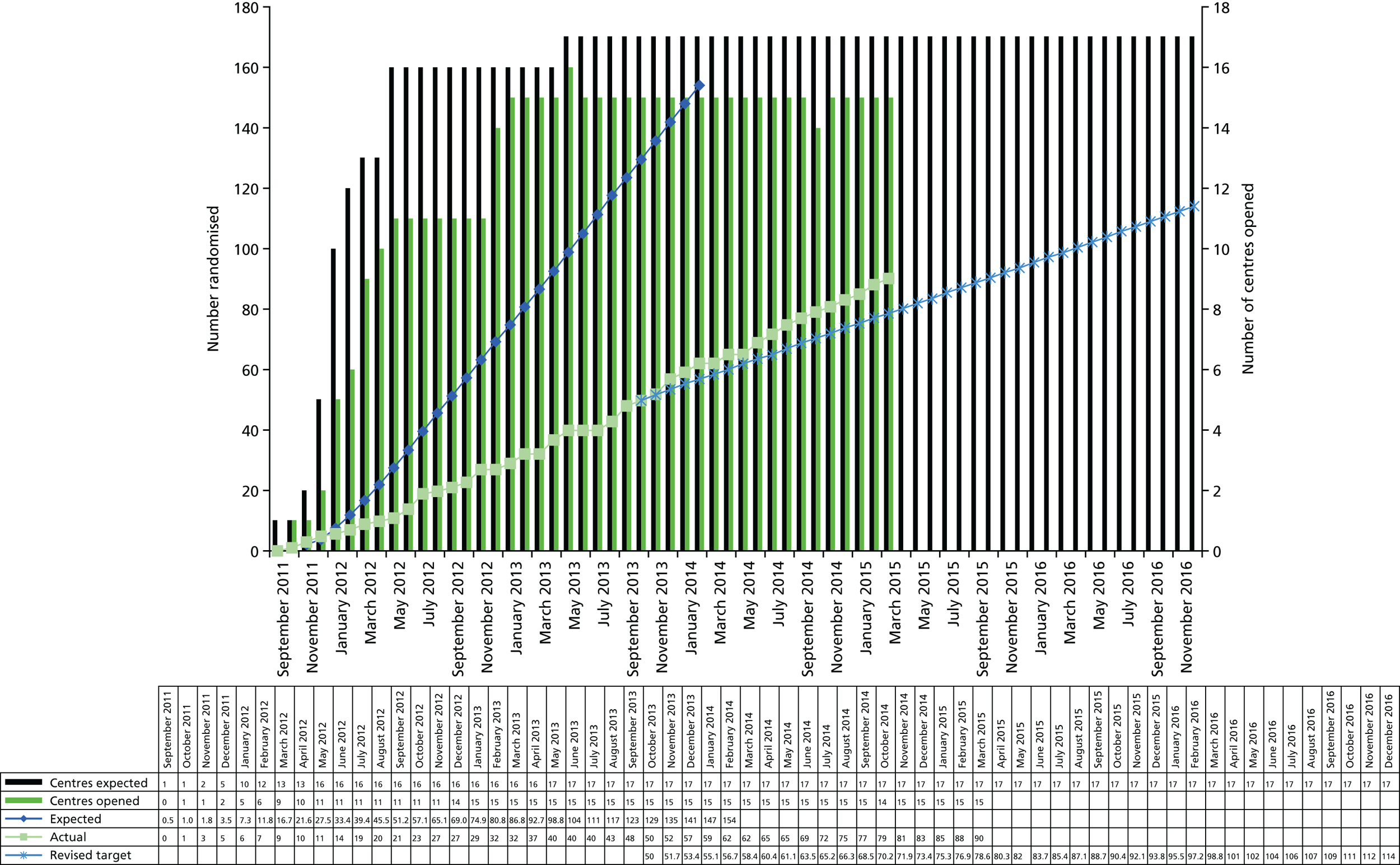
| Site | Patients, n (n visits) | Not consented (n) | Randomised (n) | |||
|---|---|---|---|---|---|---|
| Screened | Eligible | Non-eligible | Eligibility unclear | |||
| University Hospitals Bristol NHS Foundation Trust | 43 (59) | 28 (28) | 26 (30) | 1 (1) | 0 | 28 |
| Great Ormond Street Hospital for Children NHS Trust | 26 (34) | 22 (24) | 10 (10) | 2 | 22 | |
| Alder Hey Children’s NHS Foundation Trust Hospital | 30 (33) | 12 (13) | 20 (20) | 6 | 7 | |
| The Newcastle upon Tyne Hospitals NHS Foundation Trust | 18 (23) | 9 (9) | 13 (14) | 4 | 5 | |
| Norfolk and Norwich University Hospitals NHS Foundation Trust | 25 (49) | 7 (8) | 24 (41) | 3 | 5 | |
| Central Manchester University Hospitals NHS Foundation Trust | 8 (15) | 6 (6) | 6 (9) | 2 | 4 | |
| University Hospital Southampton NHS Foundation Trust | 8 (13) | 6 (6) | 3 (6) | 1 (1) | 2 | 4 |
| Sheffield Children’s NHS Foundation Trust | 33 (52) | 11 (13) | 25 (39) | 9 | 3 | |
| Royal Belfast Hospital for Sick Children | 11 (37) | 4 (4) | 10 (33) | 1 | 3 | |
| University Hospitals of Leicester NHS Trust | 5 (5) | 3 (3) | 2 (2) | 0 | 2 | |
| The Leeds Teaching Hospitals NHS Trust | 17 (29) | 9 (9) | 13 (20) | 7 | 1 | |
| Birmingham Children’s Hospital NHS Foundation Trust | 90 (151) | 8 (11) | 87 (137) | 3 (3) | 10 | 1 |
| Hull and East Yorkshire Hospitals NHS Trust | 3 (4) | 1 (1) | 3 (3) | 0 | 1 | |
| Royal Hospital for Sick Children Edinburgh – NHS Lothian | 6 (6) | 2 (2) | 4 (4) | 1 | 0 | |
| Royal Hospital for Sick Children, Glasgow – NHS Greater Glasgow and Clyde | 9 (9) | 2 (2) | 7 (7) | 2 | 2 | |
| Total | 332 (519) | 130 (139) | 253 (375) | 5 (5) | 49 | 90 |
| Site | Total (N) | Reason | n | Percentage of total |
|---|---|---|---|---|
| The Leeds Teaching Hospitals NHS Trust | 7 | Does not like injections | 1 | 14.3 |
| Wanted alternative treatment | 3 | 42.9 | ||
| Could not comply with trial | 1 | 14.3 | ||
| Clinician did not think appropriate | 1 | 14.3 | ||
| Wanted to start adalimumab outside trial | 1 | 14.3 | ||
| Norfolk and Norwich University Hospitals NHS Foundation Trust | 3 | Other family commitments | 2 | 66.7 |
| Declined; no reason | 1 | 33.3 | ||
| The Newcastle upon Tyne Hospitals NHS Foundation Trust | 4 | Declined; no reason | 4 | 100.0 |
| University Hospital Southampton NHS Foundation Trust | 2 | Declined; no reason | 1 | 50.0 |
| Did not want placebo | 1 | 50.0 | ||
| Birmingham Children’s Hospital | 10 | Declined; no reason | 3 | 30.0 |
| Did not want placebo | 2 | 20.0 | ||
| Does not like injections | 1 | 10.0 | ||
| Wanted to start adalimumab outside trial | 1 | 10.0 | ||
| Other | 3 | 30.0 | ||
| Alder Hey Children’s NHS Foundation Trust Hospital | 6 | Declined; no reason | 3 | 50.0 |
| Did not want placebo | 1 | 16.7 | ||
| Does not like injections | 1 | 16.7 | ||
| Other | 1 | 16.7 | ||
| Central Manchester University Hospitals NHS Foundation Trust | 2 | Did not want placebo | 1 | 50.0 |
| Could not comply with trial | 1 | 50.0 | ||
| Sheffield Children’s NHS Foundation Trust | 9 | Other family commitments | 4 | 44.4 |
| Declined; no reason | 3 | 33.3 | ||
| Did not want placebo | 1 | 11.1 | ||
| Other | 1 | 11.1 | ||
| Great Ormond Street Hospital for Children NHS Trust | 2 | Other | 2 | 100.0 |
| Royal Hospital for Sick Children Edinburgh – NHS Lothian | 1 | Did not want placebo | 1 | 100.0 |
| Royal Hospital for Sick Children, Glasgow – NHS Greater Glasgow and Clyde | 2 | Declined; no reason | 1 | 50.0 |
| Did not want placebo | 1 | 50.0 | ||
| Royal Belfast Hospital for Sick Children | 1 | Does not like injections | 1 | 100.0 |
| Participant ID | Site | Treatment | Date of randomisation | Date of treatment failure | Time to fail (days/months) | Reason for treatment failure | Further details | Participant unblinded at time of treatment failure (date) | Eye treatment failure occurred in |
|---|---|---|---|---|---|---|---|---|---|
| 0116003 | Bristol Children’s Hospital | Placebo | 23 November 2011 | 19 January 2012 | 57/1.87 | Intermittent or continuous suspension of trial treatment (placebo) | Continue follow-up | No | Right |
| 0116008 | Bristol Children’s Hospital | Placebo | 15 March 2012 | 16 August 2012 | 154/5.06 | Sustained scores as recorded at entry grade, measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (1+) Previous visit = SYCAMORE Unscheduled visit – (2+ on 12 July 2012) Failure visit = SYCAMORE Premature withdrawal – (1+ on 16 August 2012) |
No | Left |
| 0249019 | Great Ormond Street Hospital | Placebo | 27 July 2012 | 27 September 2012 | 62/2.04 | Non-permitted used | No follow-up, consent to use collected data | Yes; 1 October 2012 | Left |
| 0114011 | Southampton General Hospital | Adalimumab | 11 April 2012 | 2 January 2013 | 266/8.74 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | Yes; 3 January 2013 | Left |
| 0036018 | Norfolk and Norwich University Hospitals | Placebo | 17 July 2012 | 21 January 2013 | 188/6.18 | Sustained scores as recorded at entry grade, measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (2+) Previous visit = SYCAMORE Treatment visit 3 – 3-months’ treatment (3+ on 16 October 2012) Failure visit = SYCAMORE Premature withdrawal – (2+ on 21 January 2013) |
Yes; 11 March 2013 | Right |
| 0249025 | Great Ormond Street Hospital | Placebo | 2 November 2012 | 13 February 2013 | 103/3.38 | Non-permitted concomitant medications used | No follow-up, consent to use collected data | Yes; 20 November 2013 | Both |
| 0116006 | Bristol Children’s Hospital | Adalimumab | 26 January 2012 | 14 March 2013 | 413/13.57 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | Yes; 15 March 2013 | Left |
| 0114033 | Southampton General Hospital | Placebo | 13 February 2013 | 11 April 2013 | 57/1.87 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | Yes; 17 April 2013 | Left |
| 0246030 | Royal Manchester Children’s Hospital | Placebo | 29 January 2013 | 24 April 2013 | 85/2.79 | Non-permitted concomitant medications used | Continue follow-up | Yes; 26 April 2013 | Both |
| 0116026 | Bristol Children’s Hospital | Adalimumab | 8 November 2012 | 25 April 2013 | 168/5.52 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | Yes; 5 April 2013 | Both |
| 0243023 | Alder Hey Children’s Hospital | Adalimumab | 12 October 2012 | 19 October 2013 | 250/8.21 | Non-permitted concomitant medications used | Continue follow-up | Yes; 19 June 2013 | Left |
| 0249024 | Great Ormond Street Hospital | Adalimumab | 16 October 2012 | 21 June 2013 | 248/8.15 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | No | Right |
| 0116005 | Bristol Children’s Hospital | Placebo | 15 December 2011 | 4 July 2013 | 567/18.63 | Sustained scores as recorded at entry grade, measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (1+) Previous visit = SYCAMORE End of treatment – follow-up visit 2 (1+ on 2 May 2013) Failure visit = SYCAMORE Unscheduled visit – (2+ on 4 July 2013) |
Yes; 16 November 2012 | Right |
| 0243032 | Alder Hey Children’s Hospital | Placebo | 11 February 2013 | 17 July 2013 | 156/5.13 | Non-permitted concomitant medications used | Continue follow-up | No | Right |
| 0540037 | Royal Belfast Hospital for Sick Children | Placebo | 30 April 2013 | 24 July 2013 | 85/2.79 | Non-permitted concomitant medications used | Continue follow-up | Yes; 24 July 2013 | Both |
| 0249043 | Great Ormond Street Hospital | Adalimumab | 16 August 2013 | 28 October 2013 | 73/2.4 | Non-permitted concomitant medications used | Continue follow-up | No | Right |
| 0249029 | Great Ormond Street Hospital | Placebo | 25 January 2013 | 20 December 2013 | 329/10.81 | Sustained scores as recorded at entry grade, measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (1+) Previous visit = SYCAMORE Treatment visit 5 – 9 months’ treatment (2+ on 27 September 2013) Failure visit = SYCAMORE Treatment visit 6 – 12 months’ treatment (1+ on 20 December 2013) |
Yes; 26 June 2014 | Left |
| 0069039 | Great North Children’s Hospital | Adalimumab | 2 May 2013 | 27 January 2014 | 270/8.87 | Intermittent or continuous suspension of study treatment (adalimumab) | Continue follow-up | Yes; 16 January 2014 | Right |
| 0249047 | Great Ormond Street Hospital | Placebo | 12 September 2013 | 28 February 2014 | 169/5.55 | Sustained scores as recorded at entry grade, measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (1+) Previous visit = SYCAMORE Treatment visit 2 – 2 months’ treatment (1+ on 15 November 2013) Failure visit = SYCAMORE End of treatment – follow-up visit 1 (3+ on 28 February 2014) |
Yes; 18 November 2013 | Right |
| 0133058 | Birmingham Children’s Hospital | Adalimumab | 31 December 2013 | 24 February 2014 | 83/2.73 | Non-permitted concomitant medications used | Continue follow-up | Yes; 30 May 2014 | Right |
| 0116051 | Bristol Children’s Hospital | Placebo | 31 October 2013 | 17 April 2014 | 168/5.52 | Sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (1+) Previous visit = SYCAMORE Treatment visit 3 – 3 months of treatment (1+ on 23 January 2014) Failure visit = SYCAMORE Treatment visit 4 – 6 months of treatment (1+ on 17 April 2014) |
No | Left |
| 0246055 | Royal Manchester Children’s Hospital | Adalimumab | 5 December 2013 | 23 April 2014 | 139/4.57 | Sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (2+) Previous visit = SYCAMORE End of treatment – follow-up visit 1 (1+ on 7 March 2014) Failure visit = SYCAMORE Unscheduled visit (1+ on 23 April 2014) |
No | Left |
| 0249059 | Great Ormond Street Hospital | Placebo | 2 January 2014 | 20 June 2014 | 169/5.55 | Sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
No follow-up, consent to use collected data: Baseline visit – (1+) Previous visit = SYCAMORE Unscheduled visit – (2+ on 16 May 2014) Failure visit = SYCAMORE Treatment visit 4 – 6 months of treatment (3+ on 20 June 2014) |
No | Left |
| 0116067 | Bristol Children’s Hospital | Placebo | 5 June 2014 | 24 July 2014 | 49/1.61 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | No | Both |
| 0030035 | Leeds General Infirmary | Adalimumab | 19 April 2013 | 5 September 2014 | 504/16.56 | Permitted concomitant medications used against acceptable criteria and intermittent or continuous suspension of study treatment (adalimumab) | Continue follow-up | Yes; 28 August 2014 | Both |
| 0393075 | Royal Hospital for Sick Children (Edinburgh) | Placebo | 21 August 2014 | 16 October 2014 | 56/1.84 | Non-permitted concomitant medications used | Continue follow-up | No | Right |
| 0030073 | Leeds General Infirmary | Adalimumab | 30 July 2014 | 17 November 2014 | 110/3.61 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | Yes; 3 December 2014 | Both |
| 0069076 | Great North Children’s Hospital | Adalimumab | 28 August 2014 | 17 February 2015 | 173/5.68 | Sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy |
Continue follow-up: Baseline visit – (1+) Previous visit = SYCAMORE Unscheduled visit (1+ on 6 January 2015) Failure visit = SYCAMORE Treatment visit 4 – 6 months of treatment (1+ on 17 February 2015) |
Yes; 25 February 2015 | Right |
| 0249086 | Great Ormond Street Hospital | Placebo | 5 January 2015 | 2 April 2015 | 87/2.86 | Non-permitted concomitant medications used | Continue follow-up | Yes; 8 May 2015 | Left |
| 0116062 | Bristol Children’s Hospital | Adalimumab | 17 February 2014 | 9 April 2015 | 416/13.67 | Intermittent or continuous suspension of study treatment (adalimumab) | Continue follow-up | Yes; 10 April 2015 | Right |
| 0116066 | Bristol Children’s Hospital | Adalimumab | 25 April 2014 | 9 April 2015 | 349/11.47 | Intermittent or continuous suspension of study treatment (adalimumab) | Continue follow-up | Yes; 30 March 2015 | Right |
| Subject number | Age at onset (years)/sex | Onset date | Resolution date | MedDRA PT | Severity | Relationship to adalimumab/placebo (per the investigator) |
|---|---|---|---|---|---|---|
| Adalimumab | ||||||
| 0114011 | 4.89/female | 9 June 2012 | 13 June 2012 | Varicella | Moderate | Possibly/possibly |
| 0114016 | 3.56/female | 23 July 2012 | 15 August 2012 | Streptococcal infection | Moderate | Possibly/possibly |
| 0249021 | 7.63/male | 21 October 2012 | 25 October 2012 | Diarrhoea | Moderate | Possibly/possibly |
| Syncope | Moderate | Possibly/possibly | ||||
| 0116015 | 5.14/female | 14 November 2012 | 15 November 2012 | Viral infection | Moderate | Possibly/possibly |
| 0116004 | 9/male | 24 March 2013 | 3 April 2013 | Scarlet fever | Moderate | Possibly/possibly |
| 0246022 | 9.22/female | 9 August 2013 | 20 August 2013 | Cellulitis | Mild | Possibly/possibly |
| Infected bites | Mild | Possibly/possibly | ||||
| 0116015 | 5.85/female | 1 August 2013 | 1 September 2013 | Lower respiratory tract infection | Moderate | Probably/possibly |
| 0116026 | 9.43/female | 24 April 2013 | 1 May 2013 | Cataract | Moderate | Unrelated/unrelated |
| 0243056 | 5.05/female | 25 January 2014 | 28 January 2014 | Varicella | Moderate | Possibly/possibly |
| 0116044 | 14.37/male | 27 February 2014 | 15 June 2014 | Testes exploration | Moderate | Unrelated/unrelated |
| 0248064 | 6.17/male | 7 July 2014 | 9 July 2014 | Streptococcal infection | Moderate | Probably/possibly |
| 0030035 | 7.47/female | 25 August 2014 | 27 August 2014 | Viral infection | Mild | Possibly/possibly |
| 0116068 | 8.31/female | 18 October 2014 | 18 October 2014 | Antiviral prophylaxis | Mild | Unrelated/unrelated |
| 0116052 | 7.56/male | 9 November 2014 | 17 November 2014 | Food poisoning | Moderate | Unrelated/unrelated |
| 0248064 | 6.76/male | 6 February 2015 | 7 February 2015 | Tonsillar hypertrophy | Moderate | Possibly/possibly |
| 0116061a | 5.34/female | 30 June 2015 | 1 July 2015 | Tonsillitis | Mild | Possibly/possibly |
| 0540060b | 12.82/female | 13 May 2015 | 14 May 2015 | Joint swelling | Mild | Unlikely/unlikely |
| Placebo | ||||||
| 0249025 | 7.07/female | 13 February 2013 | 13 February 2013 | Anterior chamber flare | Mild | Possibly/unrelated |
| Anterior chamber flare | Mild | Possibly/unrelated | ||||
| 0248070 | 6.24/male | 7 July 2014 | 20 August 2014 | Uveitis | Severe | Possibly/unrelated |
| Participant ID | Date of first dose | Date of expected last dose | Reason for expected last dose | Treatment diaries | Accountability logs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expected number of study drug doses | Recorded number of study drug doses | Compliance (%) | Missing entry in diary (n) | Number of vials issued | Number of vials returned used | Number of vials returned unused | Missing vials (n) | Compliance (%) | Additional information on vials returned unused | ||||
| 0030035 | 19 April 2013 | 5 September 2014 | Failed treatment: permitted concomitant medications used against acceptable criteria and intermittent or continuous suspension of study treatment (adalimumab) | 37 | 32 | 86.49 | 5 | 36 | 31 | 4 | 1 | 88.89 | |
| 0030073 | 1 August 2014 | 17 November 2014 | Failed treatment: permitted concomitant medications used against acceptable criteria | 8 | 7 | 87.50 | 1 | 8 | 7 | 0 | 1 | 100.00 | |
| 0031078 | 1 October 2014 | 15 September 2015 | Last treatment visit in blinded phase | 25 | 25 | 100.00 | 0 | 30 | 0 | 0 | 30 | 100.00 | |
| 0031079 | 16 October 2014 | 26 November 2015 | Last treatment visit in blinded phase | 30 | 26 | 86.67 | 4 | 38 | 0 | 0 | 38 | 100.00 | |
| 0036038 | 2 May 2013 | 13 October 14 | Completed 18 months of treatment | 38 | 34 | 89.47 | 4 | 38 | 26 | 2 | 10 | 94.74 | |
| 0036053 | 11 November 2013 | 7 April 2014 | Withdrew | 11 | 11 | 100.00 | 0 | 14 | 11 | 3 | 0 | 78.57 | |
| 0036081 | 21 November 2014 | 11 January 2016 | Last treatment visit in blinded phase | 30 | 24 | 80.00 | 6 | 30 | 20 | 0 | 10 | 100.00 | |
| 0069020 | 30 July 2012 | 10 February 2014 | Completed 18 months of treatment | 41 | 40 | 97.56 | 1 | 42 | 20 | 0 | 22 | 100.00 | |
| 0069039 | 8 May 2013 | 27 January 2014 | Failed treatment: intermittent or continuous suspension of study treatment (adalimumab) | 19 | 13 | 68.42 | 6 | 18 | 0 | 6 | 12 | 66.67 | |
| 0069076 | 2 September 2014 | 17 February 2015 | Failed treatment: sustained scores as recorded at entry grade, measured over two consecutive readings (grades 1 to 2) still present after 6 months of therapy | 13 | 13 | 100.00 | 0 | 18 | 13 | 5 | 0 | 72.22 | Four vials were issued on 10 February 2015. One vial was used prior to the participant failing treatment on 17 February 2015. The remaining three were returned unused |
| 0078071 | 3 July 2014 | 21 August 2014 | Withdrew | 4 | 2 | 50.00 | 2 | 4 | 0 | 0 | 4 | 100.00 | |
| 0114011 | 11 April 2012 | 2 January 2013 | Failed treatment | 20 | 19 | 95.00 | 1 | 22 | 9 | 1 | 12 | 95.45 | |
| 0114016 | 4 July 2012 | 15 August 2012 | Withdrew | 4 | 1 | 25.00 | 3 | 4 | 0 | 0 | 4 | 100.00 | |
| 0114041 | 22 May 2013 | 13 February 2014 | Withdrew | 20 | 19 | 95.00 | 1 | 24 | 20 | 0 | 4 | 100.00 | |
| 0116001 | 27 October 2011 | 14 March 2013 | Completed 18 months of treatment | 37 | 34 | 91.89 | 3 | 36 | 19 | 0 | 17 | 100.00 | |
| 0116002 | 10 November 2011 | 28 March 2013 | Completed 18 months of treatment | 37 | 33 | 89.19 | 4 | 36 | 32 | 0 | 4 | 100.00 | |
| 0116004 | 15 December 2011 | 2 May 2013 | Completed 18 months of treatment | 37 | 35 | 94.59 | 2 | 36 | 0 | 0 | 36 | 100.00 | |
| 0116006 | 26 January 2012 | 14 March 2013 | Failed treatment | 30 | 30 | 100.00 | 0 | 30 | 0 | 0 | 30 | 100.00 | |
| 0116007 | 8 February 2012 | 27 June 2013 | Completed 18 months of treatment | 37 | 36 | 97.30 | 1 | 36 | 33 | 0 | 3 | 100.00 | |
| 0116013 | 21 June 2012 | 7 November 2013 | Completed 18 months of treatment | 37 | 36 | 97.30 | 1 | 38 | 2 | 0 | 36 | 100.00 | |
| 0116014 | 28 June 2012 | 14 November 2013 | Completed 18 months of treatment | 37 | 36 | 97.30 | 1 | 36 | 33 | 0 | 3 | 100.00 | |
| 0116015 | 28 June 2012 | 14 November 2013 | Completed 18 months of treatment | 37 | 34 | 91.89 | 3 | 42 | 17 | 0 | 25 | 100.00 | |
| 0116026 | 8 November 2012 | 25 April 2013 | Failed treatment | 13 | 11 | 84.62 | 2 | 14 | 0 | 0 | 14 | 100.00 | |
| 0116034 | 18 April 2013 | 11 September 2014 | Completed 18 months of treatment | 37 | 36 | 97.30 | 1 | 36 | 1 | 0 | 35 | 100.00 | |
| 0116044 | 23 August 2013 | 15 January 2015 | Completed 18 months of treatment | 37 | 24 | 64.86 | 13 | 36 | 0 | 0 | 36 | 100.00 | |
| 0116052 | 7 November 2013 | 18 November 2014 | Withdrew | 27 | 23 | 85.19 | 4 | 30 | 20 | 3 | 7 | 90.00 | |
| 0116061 | 14 February 2014 | 18 June 2015 | Completed 18 months of treatment | 35 | 34 | 97.14 | 1 | 38 | 30 | 2 | 6 | 94.74 | |
| 0116062 | 18 February 2014 | 9 April 2015 | Failed treatment: intermittent or continuous suspension of study treatment (adalimumab) | 30 | 27 | 90.00 | 3 | 32 | 24 | 4 | 4 | 87.50 | |
| 0116066 | 28 April 2014 | 9 April 2015 | Failed treatment: intermittent or continuous suspension of study treatment (adalimumab) | 25 | 15 | 60.00 | 10 | 28 | 21 | 6 | 1 | 78.57 | |
| 0116068 | 5 June 2014 | 30 July 2015 | Last treatment visit in blinded phase (unscheduled) | 31 | 17 | 54.84 | 14 | 32 | 13 | 3 | 16 | 90.63 | |
| 0116069 | 5 June 2014 | 17 September 2015 | Last treatment visit in blinded phase (unscheduled) | 34 | 29 | 85.29 | 5 | 36 | 0 | 0 | 36 | 100.00 | |
| 0116072 | 10 July 2014 | 26 November 2015 | Completed 18 months of treatment | 37 | 36 | 97.30 | 1 | 36 | 2 | 0 | 34 | 100.00 | |
| 0116088 | 5 February 2015 | 15 October 2015 | Last treatment visit in blinded phase | 19 | 12 | 63.16 | 7 | 24 | 3 | 0 | 21 | 100.00 | |
| 0133058 | 31 December 2013 | 24 March 2014 | Failed treatment: non-permitted concomitant medications used | 6 | 1 | 16.67 | 5 | 12 | 2 | 0 | 10 | 100.00 | |
| 0243009 | 26 March 2012 | 31 July 2013 | Completed 18 months of treatment | 36 | 34 | 94.44 | 2 | 36 | 35 | 1 | 0 | 97.22 | |
| 0243023 | 12 October 2012 | 19 June 2013 | Failed treatment: non-permitted concomitant medications used | 18 | 17 | 94.44 | 1 | 18 | 18 | 0 | 0 | 100.00 | |
| 0243042 | 17 August 2013 | 17 December 2014 | Completed 18 months of treatment | 35 | 28 | 80.00 | 7 | 36 | 36 | 0 | 0 | 100.00 | |
| 0243056 | 16 December 2013 | 6 May 2015 | Completed 18 months of treatment | 37 | 34 | 91.89 | 3 | 36 | 36 | 0 | 0 | 100.00 | |
| 0243089 | 11 February 2015 | 13 January 2016 | Last treatment visit in blinded phase | 25 | 23 | 92.00 | 2 | 24 | 24 | 0 | 0 | 100.00 | |
| 0246022 | 11 September 2012 | 14 February 2014 | Completed 18 months of treatment | 38 | 34 | 89.47 | 4 | 38 | 22 | 0 | 16 | 100.00 | |
| 0246054 | 4 December 2013 | 8 May 2015 | Completed 18 months of treatment | 38 | 36 | 94.74 | 2 | 38 | 34 | 1 | 3 | 97.37 | |
| 0246055 | 10 December 2013 | 19 February 2014 | Withdrew | 6 | 3 | 50.00 | 3 | 4 | 3 | 1 | 0 | 75.00 | |
| 0248050 | 10 October 2013 | 9 March 2015 | Completed 18 months of treatment | 37 | 15 | 40.54 | 22 | 36 | 33 | 1 | 2 | 97.22 | |
| 0248064 | 10 April 2014 | 16 March 2015 | Withdrew | 25 | 22 | 88.00 | 3 | 26 | 23 | 3 | 0 | 88.46 | |
| 0248087 | 5 January 2015 | 7 December 2015 | Last treatment visit in blinded phase | 25 | 13 | 52.00 | 12 | 24 | 18 | 0 | 6 | 100.00 | |
| 0249021 | 24 August 2012 | 10 January 2014 | Completed 18 months treatment | 37 | 35 | 94.59 | 2 | 36 | 36 | 0 | 0 | 100.00 | |
| 0249024 | 18 October 2012 | 21 June 2013 | Failed treatment: permitted concomitant medications used against acceptable criteria | 18 | 16 | 88.89 | 2 | 24 | 16 | 8 | 0 | 66.67 | On 11 June 2013, six vials were issued; all were returned unused as the participant withdrew on 21 June 2013 |
| 0249028 | 23 November 2012 | 28 March 2014 | Completed 18 months of treatment | 36 | 34 | 94.44 | 2 | 36 | 36 | 0 | 0 | 100.00 | |
| 0249031 | 1 February 2013 | 9 May 2014 | Failed treatment: permitted concomitant medications used against acceptable criteria | 34 | 4 | 11.76 | 30 | 36 | 29 | 5 | 2 | 86.11 | On 19 March 2014, six vials were issued. One vial was used prior to the participant failing treatment on 9 May 2016; the rest were returned unused |
| 0249036 | 26 April 2013 | 12 September 2014 | Completed 18 months of treatment | 37 | 34 | 91.89 | 3 | 36 | 36 | 0 | 0 | 100.00 | |
| 0249040 | 17 May 2013 | 5 December 2013 | Failed treatment: permitted concomitant medications used against acceptable criteria | 15 | 12 | 80.00 | 3 | 18 | 12 | 6 | 0 | 66.67 | On 28 October 2013, six vials were issued. All six vials were returned unused |
| 0249043 | 16 August 2013 | 28 October 2013 | Failed treatment: non-permitted concomitant medications used | 6 | 4 | 66.67 | 2 | 12 | 6 | 6 | 0 | 50.00 | On 18 October 2013, six vials were issued. All six vials were returned unused |
| 0249046 | 12 September 2013 | 30 January 2015 | Completed 18 months of treatment | 37 | 36 | 97.30 | 1 | 40 | 35 | 3 | 2 | 92.50 | |
| 0249048 | 13 September 2013 | 30 January 2015 | Completed 18 months of treatment | 37 | 32 | 86.49 | 5 | 36 | 36 | 0 | 0 | 100.00 | |
| 0249065 | 25 April 2014 | 11 September 2015 | Completed 18 months of treatment | 37 | 35 | 94.59 | 2 | 36 | 36 | 0 | 0 | 100.00 | |
| 0249077 | 5 September 2014 | 16 January 2015 | Last treatment visit in blinded phase (unscheduled) | 10 | 5 | 50.00 | 5 | 12 | 9 | 0 | 3 | 100.00 | |
| 0249083 | 5 December 2014 | 6 November 2015 | Last treatment visit in blinded phase | 25 | 18 | 72.00 | 7 | 28 | 9 | 0 | 19 | 100.00 | |
| 0249091 | 27 March 2015 | 4 December 2015 | Last treatment visit in blinded phase | 19 | 18 | 94.74 | 1 | 18 | 12 | 0 | 6 | 100.00 | |
| 0540060 | 31 January 2014 | 7 January 2015 | Withdrew | 25 | 14 | 56.00 | 11 | 24 | 0 | 5 | 19 | 79.17 | On 7 October 2014, six vials were issued. Five of these vials were returned unused. Participant withdrew as a result of missing MTX doses before failing treatment for missing adalimumab doses |
| 0540080 | 21 October 2014 | 9 December 2015 | Last treatment visit in blinded phase | 30 | 29 | 96.67 | 1 | 28 | 0 | 0 | 28 | 100.00 | |
| Mean compliance | 81.01% | 93.91% | |||||||||||
| Participant ID | Date of first dose | Date of expected last dose | Reason for expected last dose | Treatment diaries | Accountability logs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expected number of study drug doses | Recorded number of study drug doses | Compliance (%) | Missing entry in diary (n) | Number of vials issued | Number of vials returned used | Number of vials returned unused | Compliance (%) | Missing vials (n) | Additional information on vials returned unused | ||||
| 0031074 | 14 August 2014 | 5 May 2015 | Last treatment visit in blinded phase (unscheduled) | 19 | 18 | 94.74 | 1 | 20 | 0 | 0 | 100.00 | 20 | |
| 0036018 | 18 July 2012 | 21 January 2013 | Failed treatment: sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2), still present after 6 months of therapy | 14 | 15 | 107.14 | 0 | 14 | 13 | 1 | 92.86 | 0 | |
| 0036057 | 16 December 2013 | 31 March 2014 | Withdrew | 8 | 8 | 100.00 | 0 | 12 | 8 | 4 | 66.67 | 0 | Six vials were issued on 6 March 2014. Two vials were used prior to the participant withdrawing on 31 March 2014. The remaining four vials were returned unused |
| 0069017 | 5 July 2012 | 13 January 2014 | Completed 18 months of treatment | 40 | 39 | 97.50 | 1 | 44 | 6 | 2 | 95.45 | 36 | |
| 0069092 | 31 March 2015 | 5 May 2015 | Last treatment visit in blinded phase (unscheduled) | 3 | 2 | 66.67 | 1 | 4 | 2 | 0 | 100.00 | 2 | |
| 0114033 | 13 February 2013 | 11 April 2013 | Failed treatment | 5 | 3 | 60.00 | 2 | 6 | 4 | 0 | 100.00 | 2 | |
| 0116003 | 24 November 2011 | 19 January 2012 | Failed treatment | 5 | 1 | 20.00 | 4 | 6 | 0 | 0 | 100.00 | 6 | |
| 0116005 | 15 December 2011 | 15 November 2012 | Withdrew | 25 | 22 | 88.00 | 3 | 30 | 16 | 0 | 100.00 | 14 | |
| 0116008 | 15 March 2012 | 16 August 2012 | Failed treatment: sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2), still present after 6 months of therapy | 12 | 11 | 91.67 | 1 | 16 | 1 | 6 | 62.50 | 9 | Six vials were issued on 16 August 2012 prior to the participant failing treatment (on the same day). These vials were all returned unused |
| 0116012 | 17 May 2012 | 10 October 2013 | Completed 18 months of treatment | 37 | 35 | 94.59 | 2 | 36 | 27 | 1 | 97.22 | 8 | |
| 0116027 | 8 November 2012 | 17 January 2013 | Withdrew | 6 | 2 | 33.33 | 4 | 4 | 0 | 0 | 100.00 | 4 | |
| 0116051 | 31 October 2013 | 17 April 2014 | Failed treatment | 13 | 12 | 92.31 | 1 | 14 | 9 | 0 | 100.00 | 5 | |
| 0116063 | 27 February 2014 | 12 February 2015 | Withdrew | 26 | 24 | 92.31 | 2 | 34 | 19 | 9 | 73.53 | 6 |
Two vials were issued on 6 May 2014 and a further six were issued on 21 May 2014. The extra two vials issued in error were returned unused Six vials were issued on 28 January 2015 and all six were returned unused; one of these vials should have been used |
| 0116067 | 5 June 2014 | 24 July 2014 | Failed treatment: permitted concomitant medications used against acceptable criteria | 4 | 4 | 100.00 | 0 | 6 | 1 | 2 | 66.67 | 3 | |
| 0116085 | 18 December 2014 | 29 January 2015 | Withdrew | 4 | 2 | 50.00 | 2 | 4 | 2 | 0 | 100.00 | 2 | |
| 0243032 | 12 February 2013 | 18 December 2013 | Withdrew | 23 | 22 | 95.65 | 1 | 26 | 22 | 4 | 84.62 | 0 | Two extra vials issued on 3 December 2013 were returned unused |
| 0243049 | 24 September 2013 | 6 November 2013 | Failed treatment: permitted concomitant medications used against acceptable criteria | 4 | 3 | 75.00 | 1 | 4 | 3 | 1 | 75.00 | 0 | |
| 0246030 | 30 January 2013 | 24 April 2013 | Failed treatment: non-permitted concomitant medications used | 7 | 6 | 85.71 | 1 | 12 | 0 | 0 | 100.00 | 12 | |
| 0248070 | 10 June 2014 | 7 July 2014 | Withdrew | 2 | 2 | 100.00 | 0 | 4 | 2 | 2 | 50.00 | 0 | |
| 0249019 | 2 August 2012 | 27 September 2012 | Failed treatment: non-permitted concomitant medications used | 5 | 4 | 80.00 | 1 | 6 | 4 | 2 | 66.67 | 0 | |
| 0249025 | 2 November 2012 | 13 February 2013 | Failed treatment: non-permitted concomitant medications used | 8 | 5 | 62.50 | 3 | 12 | 4 | 4 | 66.67 | 4 | On 29 January 2013, six vials were issued; two were used prior to participant failing treatment on 13 February 2013 and the remaining four were returned unused |
| 0249029 | 25 January 2013 | 20 December 2013 | Failed treatment: sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2), still present after 6 months of therapy | 24 | 9 | 37.50 | 15 | 30 | 22 | 6 | 80.00 | 2 | On 16 December 2013, six vials were issued, all were returned unused as participant failed treatment on 20 December 2013 |
| 0249045 | 13 September 2013 | 30 January 2015 | Completed 18 months of treatment | 37 | 14 | 37.84 | 23 | 40 | 35 | 1 | 97.50 | 4 | |
| 0249047 | 12 September 2013 | 15 November 2013 | Withdrew | 5 | 2 | 40.00 | 3 | 6 | 3 | 2 | 66.67 | 1 | |
| 0249059 | 3 January 2014 | 20 June 2014 | Failed treatment: sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2), still present after 6 months of therapy | 13 | 1 | 7.69 | 12 | 18 | 12 | 6 | 66.67 | 0 | On 16 June 2014, six vials were issued. All six vials were returned unused |
| 0249082 | 28 November 14 | 31 July 2015 | Last treatment visit in blinded phase (unscheduled) | 18 | 20 | 111.11 | 0 | 20 | 9 | 0 | 100.00 | 11 | |
| 0249086 | 5 January 2015 | 2 April 2015 | Failed treatment: non-permitted concomitant medications used | 7 | 6 | 85.71 | 1 | 10 | 6 | 4 | 60.00 | 0 | On 5 February 2015, six vials were issued; a further two vials were issued on 6 March 2015. Four vials were used prior to the participant failing treatment on 2 April 2014. The remaining four vials were returned unused |
| 0249090 | 13 February 2015 | 8 May 2015 | Last treatment visit in blinded phase | 7 | 6 | 85.71 | 1 | 6 | 4 | 0 | 100.00 | 2 | |
| 0393075 | 21 August 2014 | 16 October 2014 | Failed treatment: non-permitted concomitant medications used | 5 | 3 | 60.00 | 2 | 6 | 1 | 1 | 83.33 | 4 | |
| 0540037 | 1 May 2013 | 24 July 2013 | Failed treatment: non-permitted concomitant medications used | 7 | 6 | 85.71 | 1 | 12 | 0 | 6 | 50.00 | 6 | On 24 July 2013, six vials were issued prior to the participant failing treatment. All six vials were returned unused |
| Mean compliance | 74.61% | 83.40% | |||||||||||
| Visit | Treatment group | Total | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | |||||||||||||||||
| Best score | Worst score | Best score | Worst score | Best score | Worst score | |||||||||||||
| n | Mean (SD) | Median (range) | n | Mean (SD) | Median (range) | n | Mean (SD) | Median (range) | n | Mean (SD) | Median (range) | n | Mean (SD) | Median (range) | n | Mean (SD) | Median (range) | |
| Baseline | 60 | 0.04 (0.15) | 0.00 (–0.23 to 0.56) | 60 | 0.05 (0.16) | 0.00 (–0.23 to 0.56) | 30 | 0.06 (0.12) | 0.05 (–0.13 to 0.40) | 30 | 0.08 (0.12) | 0.06 (–0.10 to 0.40) | 90 | 0.04 (0.14) | 0.00 (–0.23 to 0.56) | 90 | 0.06 (0.14) | 0.03 (–0.23 to 0.56) |
| 1 month | 60 | 0.03 (0.17) | 0.00 (–0.30 to 0.80) | 60 | 0.04 (0.18) | 0.00 (–0.30 to 0.80) | 30 | 0.02 (0.16) | 0.00 (–0.28 to 0.38) | 30 | 0.06 (0.17) | 0.04 (–0.28 to 0.38) | 90 | 0.02 (0.17) | 0.00 (–0.30 to 0.80) | 90 | 0.05 (0.18) | 0.00 (–0.30 to 0.80) |
| 2 months | 57 | 0.02 (0.17) | 0.00 (–0.20 to 0.56) | 57 | 0.04 (0.19) | 0.00 (–0.20 to 0.75) | 25 | 0.05 (0.18) | 0.00 (–0.15 to 0.76) | 25 | 0.06 (0.18) | 0.02 (–0.15 to 0.76) | 82 | 0.03 (0.17) | 0.00 (–0.20 to 0.76) | 82 | 0.05 (0.19) | 0.00 (–0.20 to 0.76) |
| 3 months | 56 | 0.00 (0.16) | 0.00 (–0.20 to 0.80) | 56 | 0.02 (0.20) | 0.00 (–0.20 to 0.88) | 19 | 0.01 (0.11) | 0.00 (–0.13 to 0.24) | 19 | 0.03 (0.12) | 0.00 (–0.13 to 0.28) | 75 | 0.00 (0.14) | 0.00 (–0.20 to 0.80) | 75 | 0.02 (0.18) | 0.00 (–0.20 to 0.88) |
| 6 months | 47 | 0.02 (0.20) | –0.02 (–0.20 to 0.88) | 47 | 0.03 (0.20) | 0.00 (–0.20 to 0.88) | 12 | 0.05 (0.16) | 0.02 (–0.18 to 0.30) | 12 | 0.07 (0.19) | 0.02 (–0.18 to 0.38) | 59 | 0.03 (0.19) | 0.00 (–0.20 to 0.88) | 59 | 0.03 (0.19) | 0.00 (–0.20 to 0.88) |
| 9 months | 42 | –0.01 (0.14) | 0.00 (–0.25 to 0.40) | 42 | –0.01 (0.14) | 0.00 (–0.25 to 0.40) | 7 | 0.00 (0.17) | –0.08 (–0.10 to 0.36) | 7 | 0.04 (0.20) | –0.08 (–0.10 to 0.36) | 49 | –0.01 (0.14) | 0.00 (–0.25 to 0.40) | 49 | 0.00 (0.15) | 0.00 (–0.25 to 0.40) |
| 12 months | 34 | –0.01 (0.14) | –0.01 (–0.23 to 0.34) | 34 | 0.00 (0.14) | 0.00 (–0.23 to 0.34) | 5 | 0.03 (0.14) | 0.02 (–0.10 to 0.26) | 5 | 0.08 (0.17) | 0.03 (–0.10 to 0.26) | 39 | –0.01 (0.14) | 0.00 (–0.23 to 0.34) | 39 | 0.01 (0.14) | 0.00 (–0.23 to 0.34) |
| 15 months | 27 | 0.00 (0.14) | 0.00 (–0.25 to 0.40) | 27 | 0.00 (0.13) | 0.00 (–0.25 to 0.40) | 3 | 0.00 (0.26) | –0.10 (–0.20 to 0.30) | 3 | 0.00 (0.26) | –0.10 (–0.20 to 0.30) | 30 | 0.00 (0.15) | –0.02 (–0.25 to 0.40) | 30 | 0.00 (0.15) | –0.02 (–0.25 to 0.40) |
| 18 months | 23 | 0.02 (0.13) | 0.00 (–0.20 to 0.28) | 23 | 0.04 (0.11) | 0.02 (–0.20 to 0.28) | 3 | 0.02 (0.21) | –0.10 (–0.10 to 0.26) | 3 | 0.02 (0.21) | –0.10 (–0.10 to 0.26) | 26 | 0.02 (0.13) | 0.00 (–0.20 to 0.28) | 26 | 0.04 (0.12) | 0.01 (–0.20 to 0.28) |
FIGURE 14.
Mean profile plot for logMAR best score.
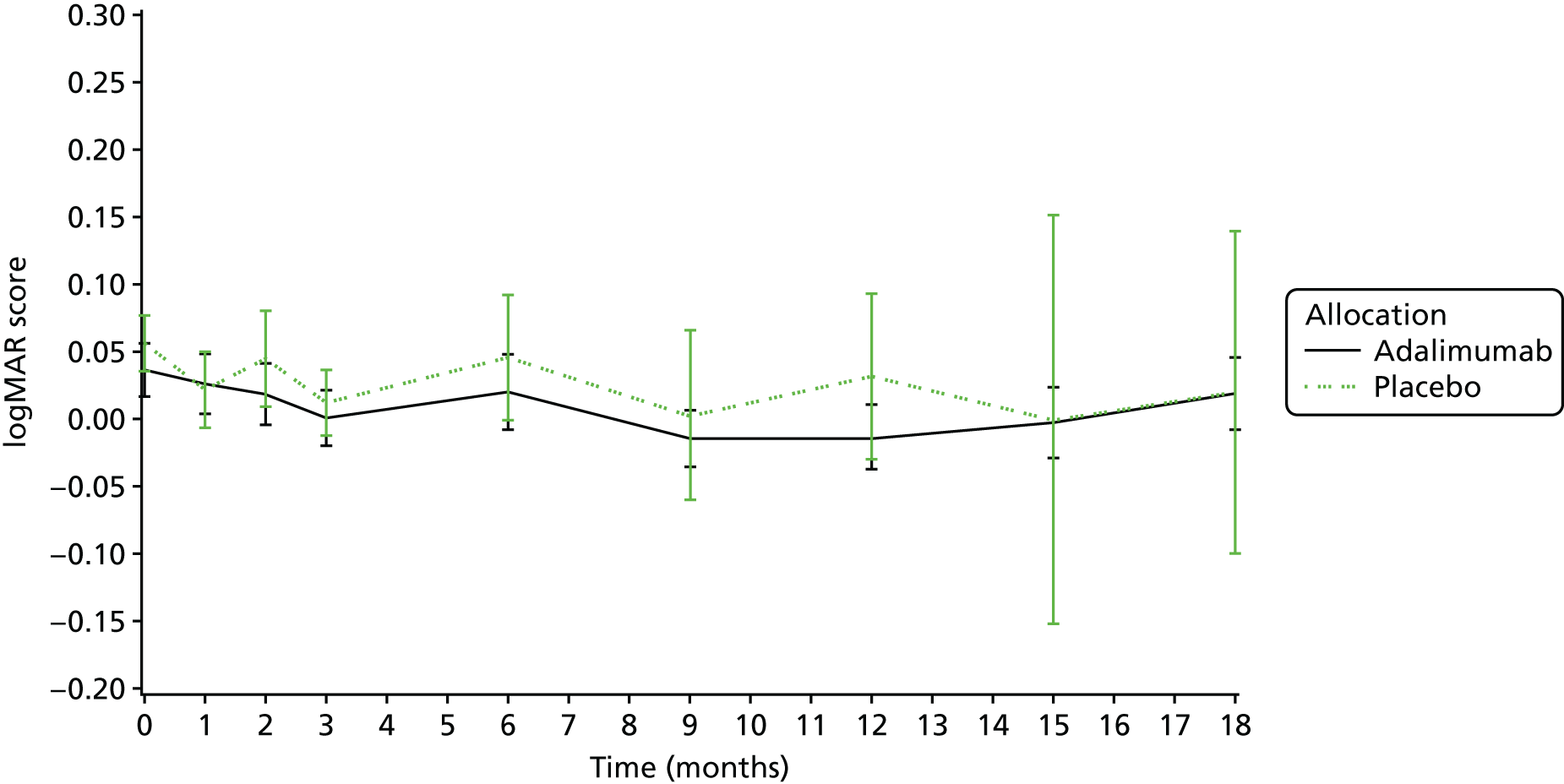
FIGURE 15.
Mean profile plot for logMAR worst score.
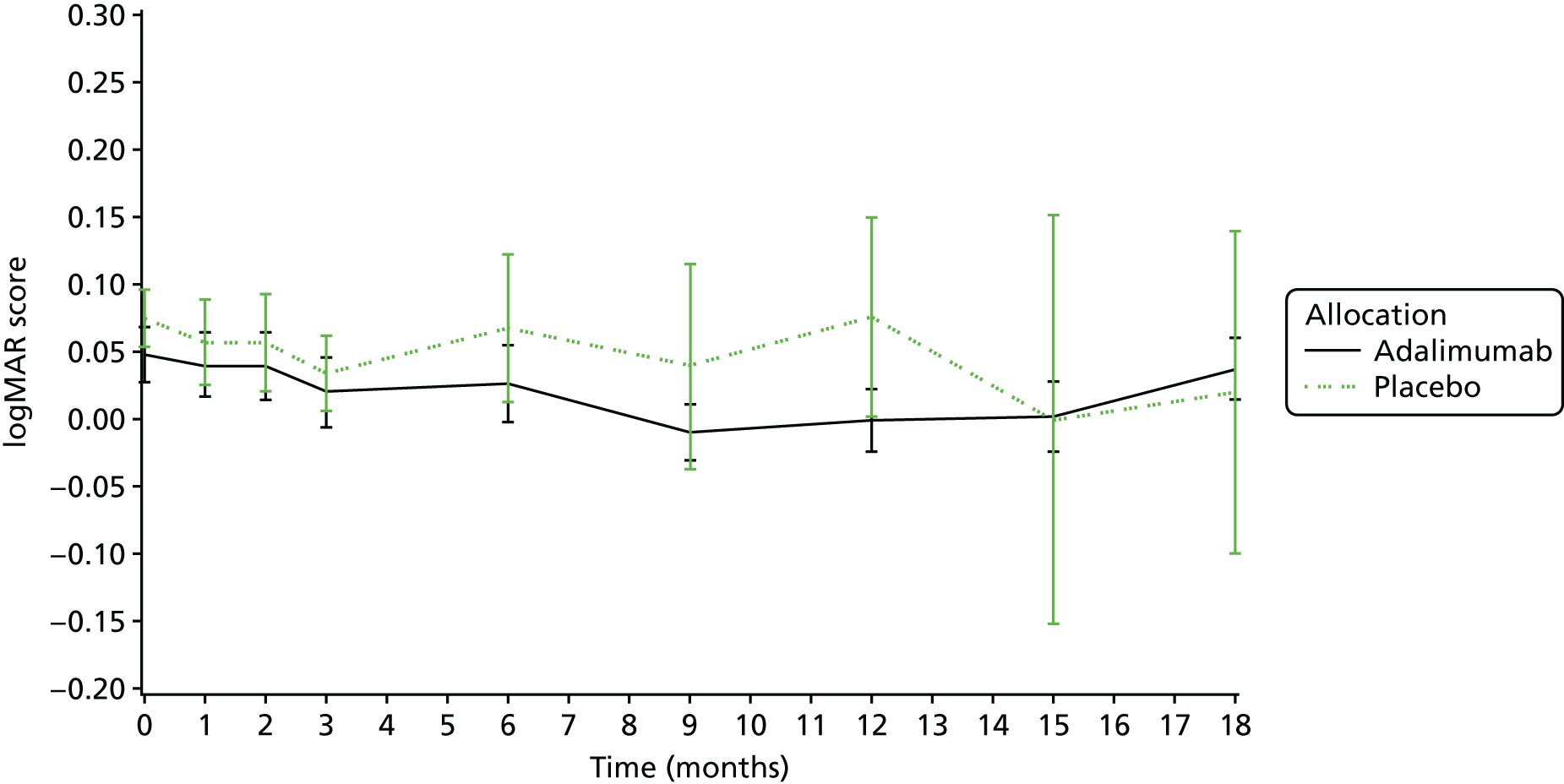
| Visit | Treatment group | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | |||||||||||
| n | Missing | Mean (SD) | Median (range) | n | Missing | Mean (SD) | Median (range) | n | Missing | Mean (SD) | Median (range) | |
| Baseline | 53 | 7 | 51.17 (9.53) | 52.74 (30.69–65.85) | 22 | 8 | 49.48 (7.55) | 49.67 (37.25–60.76) | 75 | 15 | 50.68 (8.98) | 51.99 (30.69–65.85) |
| 1 month | 53 | 7 | 51.06 (10.36) | 53.41 (18.72–65.19) | 27 | 3 | 50.01 (10.27) | 50.58 (20.75–63.60) | 80 | 10 | 50.71 (10.28) | 53.24 (18.72–65.19) |
| 2 months | 49 | 9 | 53.02 (10.00) | 54.62 (24.01–64.52) | 19 | 6 | 50.20 (10.75) | 52.42 (30.57–64.29) | 68 | 15 | 52.23 (10.21) | 54.47 (24.01–64.52) |
| 3 months | 50 | 6 | 54.12 (9.02) | 57.52 (26.86–64.30) | 16 | 3 | 54.21 (8.57) | 55.38 (38.63–64.47) | 66 | 9 | 54.14 (8.85) | 57.32 (26.86–64.47) |
| 6 months | 40 | 8 | 53.94 (9.79) | 57.00 (15.14–65.20) | 11 | 1 | 49.68 (11.56) | 54.18 (23.16–61.32) | 51 | 9 | 53.02 (10.23) | 56.19 (15.14–65.20) |
| 9 months | 35 | 7 | 55.82 (6.84) | 58.04 (39.68–65.82) | 7 | 0 | 50.26 (13.72) | 56.14 (25.06–62.08) | 42 | 7 | 54.89 (8.41) | 56.93 (25.06–65.82) |
| 12 months | 33 | 1 | 54.08 (9.22) | 57.61 (27.32–65.30) | 4 | 1 | 54.18 (8.83) | 55.36 (42.61–63.39) | 37 | 2 | 54.09 (9.06) | 57.61 (27.32–65.30) |
| 15 months | 25 | 2 | 53.56 (7.76) | 53.61 (36.75–65.35) | 3 | 0 | 53.27 (11.83) | 55.45 (40.50–63.87) | 28 | 2 | 53.53 (8.00) | 54.53 (36.75–65.35) |
| 18 months | 20 | 3 | 53.58 (11.71) | 55.14 (10.02–64.60) | 3 | 0 | 47.25 (18.64) | 55.49 (25.91–60.35) | 23 | 3 | 52.76 (12.44) | 55.49 (10.02–64.60) |
FIGURE 16.
The PsS mean profile plot.
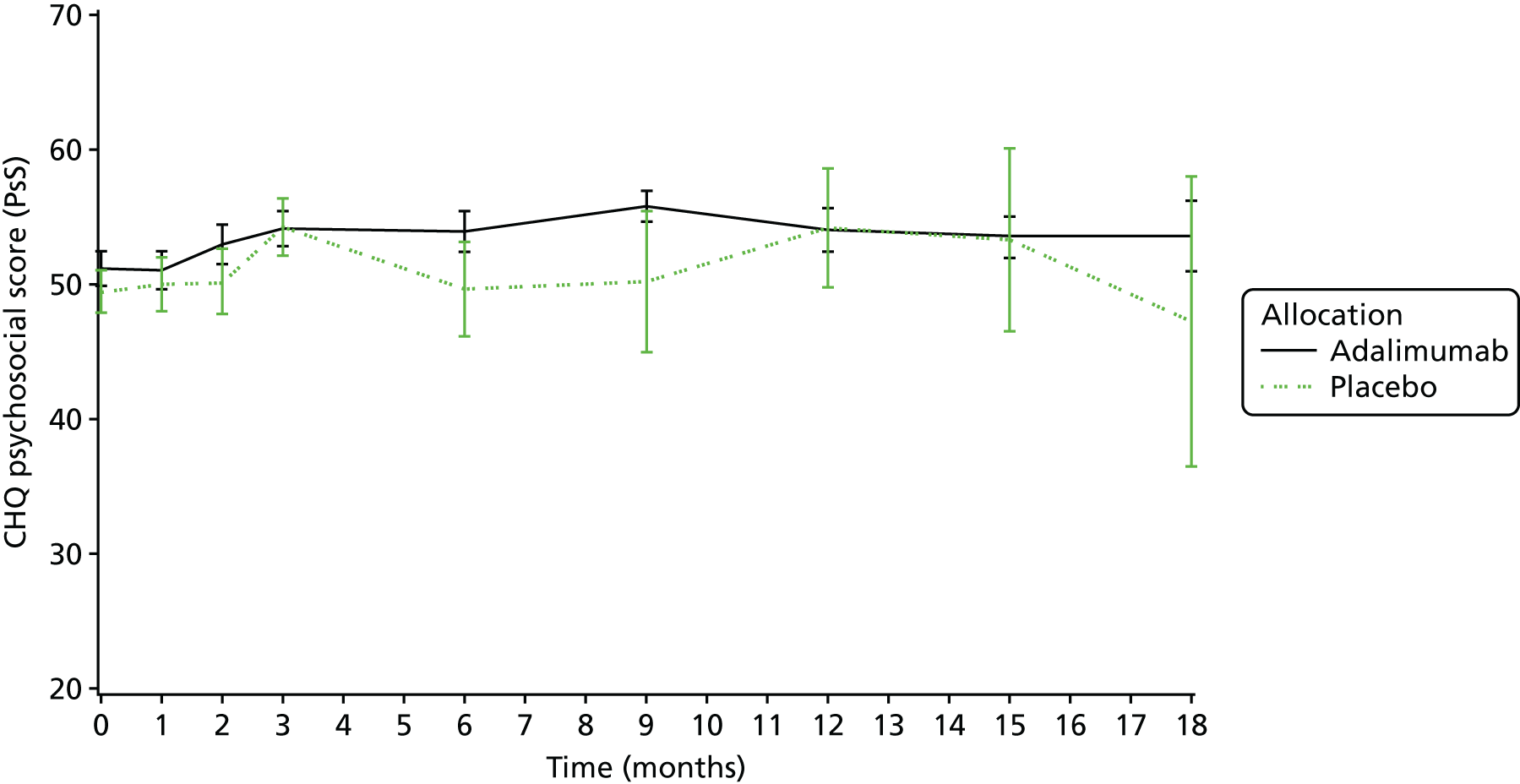
| Visit | Treatment group | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | |||||||||||
| n | Missing | Mean (SD) | Median (range) | n | Missing | Mean (SD) | Median (range) | n | Missing | Mean (SD) | Median (range) | |
| Baseline | 53 | 7 | 43.20 (11.84) | 46.39 (–3.81 to 59.97) | 22 | 8 | 40.48 (16.36) | 43.54 (6.44 to 58.87) | 75 | 15 | 42.40 (13.26) | 46.28 (–3.81 to 59.97) |
| 1 month | 53 | 7 | 45.54 (11.29) | 47.61 (1.97 to 61.07) | 27 | 3 | 44.73 (12.10) | 49.02 (7.47 to 59.60) | 80 | 10 | 45.27 (11.50) | 47.69 (1.97 to 61.07) |
| 2 months | 49 | 9 | 47.54 (10.69) | 52.06 (10.96 to 59.65) | 19 | 6 | 43.65 (15.56) | 46.85 (–2.61 to 59.65) | 68 | 15 | 46.46 (12.25) | 51.81 (–2.61 to 59.65) |
| 3 months | 50 | 6 | 46.50 (13.13) | 50.76 (3.62 to 60.76) | 16 | 3 | 47.35 (7.97) | 48.89 (31.61 to 59.65) | 66 | 9 | 46.70 (12.03) | 49.70 (3.62 to 60.76) |
| 6 months | 40 | 8 | 47.16 (11.84) | 52.06 (9.30 to 58.64) | 11 | 1 | 41.95 (15.79) | 43.69 (1.20 to 59.65) | 51 | 9 | 46.03 (12.80) | 50.40 (1.20 to 59.65) |
| 9 months | 35 | 7 | 47.50 (11.26) | 51.57 (11.12 to 59.00) | 7 | 0 | 45.20 (14.77) | 51.67 (22.56 to 57.01) | 42 | 7 | 47.12 (11.74) | 51.62 (11.12 to 59.00) |
| 12 months | 33 | 1 | 47.29 (13.06) | 52.92 (3.54 to 59.53) | 4 | 1 | 53.09 (4.79) | 54.07 (46.83 to 57.40) | 37 | 2 | 47.92 (12.53) | 52.92 (3.54 to 59.53) |
| 15 months | 25 | 2 | 42.85 (15.88) | 48.98 (6.03 to 60.05) | 3 | 0 | 55.75 (2.48) | 54.86 (53.83 to 58.55) | 28 | 2 | 44.23 (15.53) | 49.97 (6.03 to 60.05) |
| 18 months | 20 | 3 | 45.92 (12.06) | 51.93 (17.26 to 55.79) | 3 | 0 | 53.77 (9.71) | 59.09 (42.56 to 59.65) | 23 | 3 | 46.94 (11.89) | 52.78 (17.26 to 59.65) |
FIGURE 17.
The PhS mean profile plot.
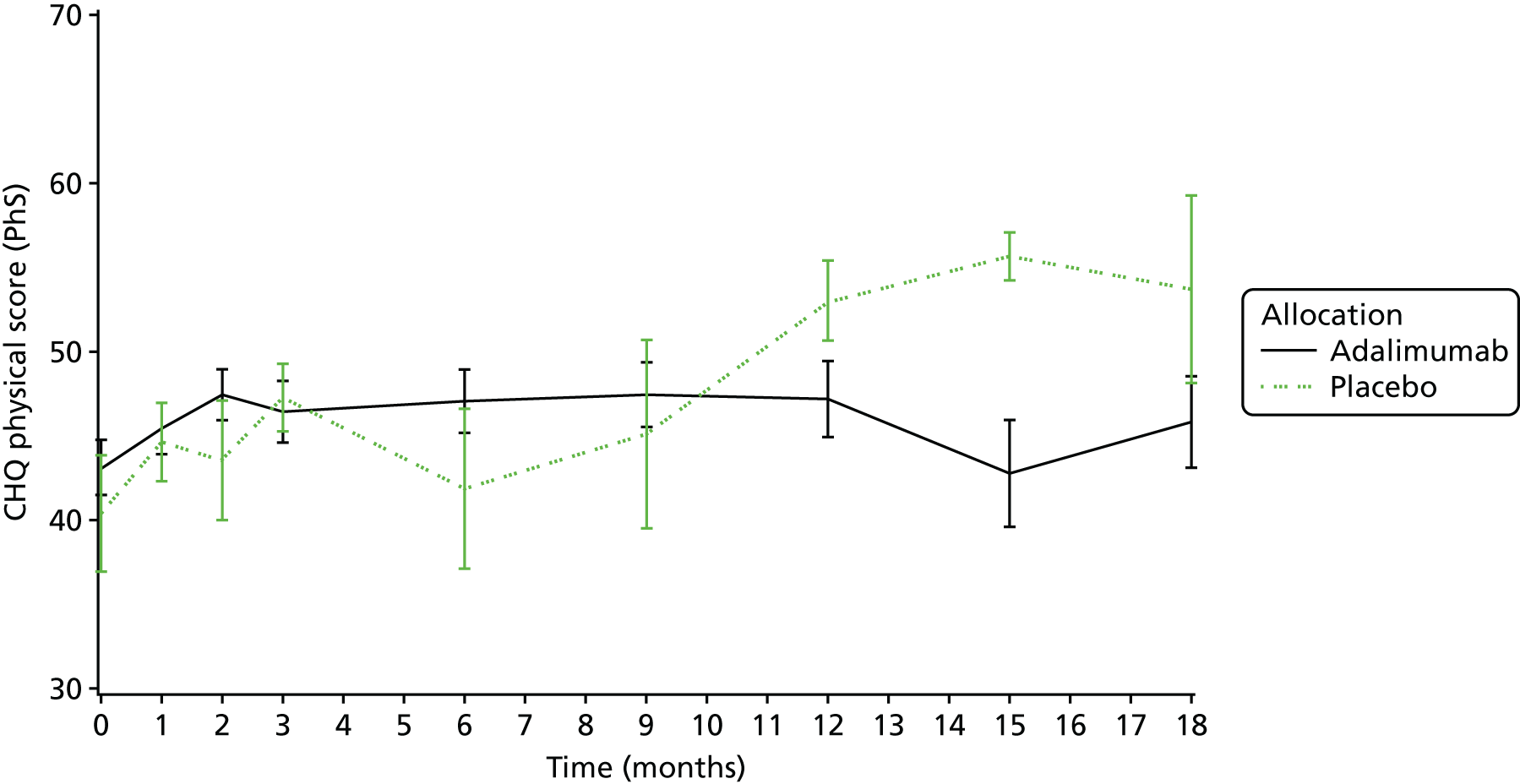
| Visit | Treatment group | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adalimumab | Placebo | |||||||||||
| n | Missing | Mean (SD) | Median (range) | n | Missing | Mean (SD) | Median (range) | n | Missing | Mean (SD) | Median (range) | |
| Baseline | 59 | 1 | 0.52 (0.64) | 0.21 (0.00–2.49) | 28 | 2 | 0.48 (0.49) | 0.45 (0.00–1.57) | 87 | 3 | 0.51 (0.59) | 0.33 (0.00–2.49) |
| 1 month | 59 | 1 | 0.41 (0.56) | 0.13 (0.00–2.29) | 30 | 0 | 0.60 (0.55) | 0.55 (0.00–1.61) | 89 | 1 | 0.47 (0.56) | 0.22 (0.00–2.29) |
| 2 months | 57 | 1 | 0.38 (0.53) | 0.11 (0.00–1.96) | 24 | 1 | 0.54 (0.59) | 0.45 (0.00–2.32) | 81 | 2 | 0.43 (0.55) | 0.20 (0.00–2.32) |
| 3 months | 54 | 2 | 0.36 (0.58) | 0.03 (0.00–2.49) | 18 | 1 | 0.37 (0.47) | 0.19 (0.00–1.50) | 72 | 3 | 0.36 (0.55) | 0.05 (0.00–2.49) |
| 6 months | 45 | 3 | 0.36 (0.61) | 0.02 (0.00–2.49) | 12 | 0 | 0.46 (0.63) | 0.11 (0.00–2.00) | 57 | 3 | 0.38 (0.61) | 0.05 (0.00–2.49) |
| 9 months | 39 | 3 | 0.35 (0.63) | 0.06 (0.00–2.28) | 7 | 0 | 0.36 (0.57) | 0.09 (0.00–1.58) | 46 | 3 | 0.35 (0.62) | 0.06 (0.00–2.28) |
| 12 months | 34 | 0 | 0.33 (0.60) | 0.02 (0.00–2.04) | 4 | 1 | 0.09 (0.15) | 0.02 (0.00–0.31) | 38 | 1 | 0.31 (0.57) | 0.02 (0.00–2.04) |
| 15 months | 26 | 1 | 0.43 (0.58) | 0.11 (0.00–2.00) | 3 | 0 | 0.03 (0.04) | 0.01 (0.00–0.07) | 29 | 1 | 0.39 (0.56) | 0.10 (0.00–2.00) |
| 18 months | 22 | 1 | 0.30 (0.48) | 0.02 (0.00–1.55) | 3 | 0 | 0.03 (0.05) | 0.01 (0.00–0.09) | 25 | 1 | 0.27 (0.46) | 0.02 (0.00–1.55) |
FIGURE 18.
Mean profile plots for CHAQ score by treatment group.
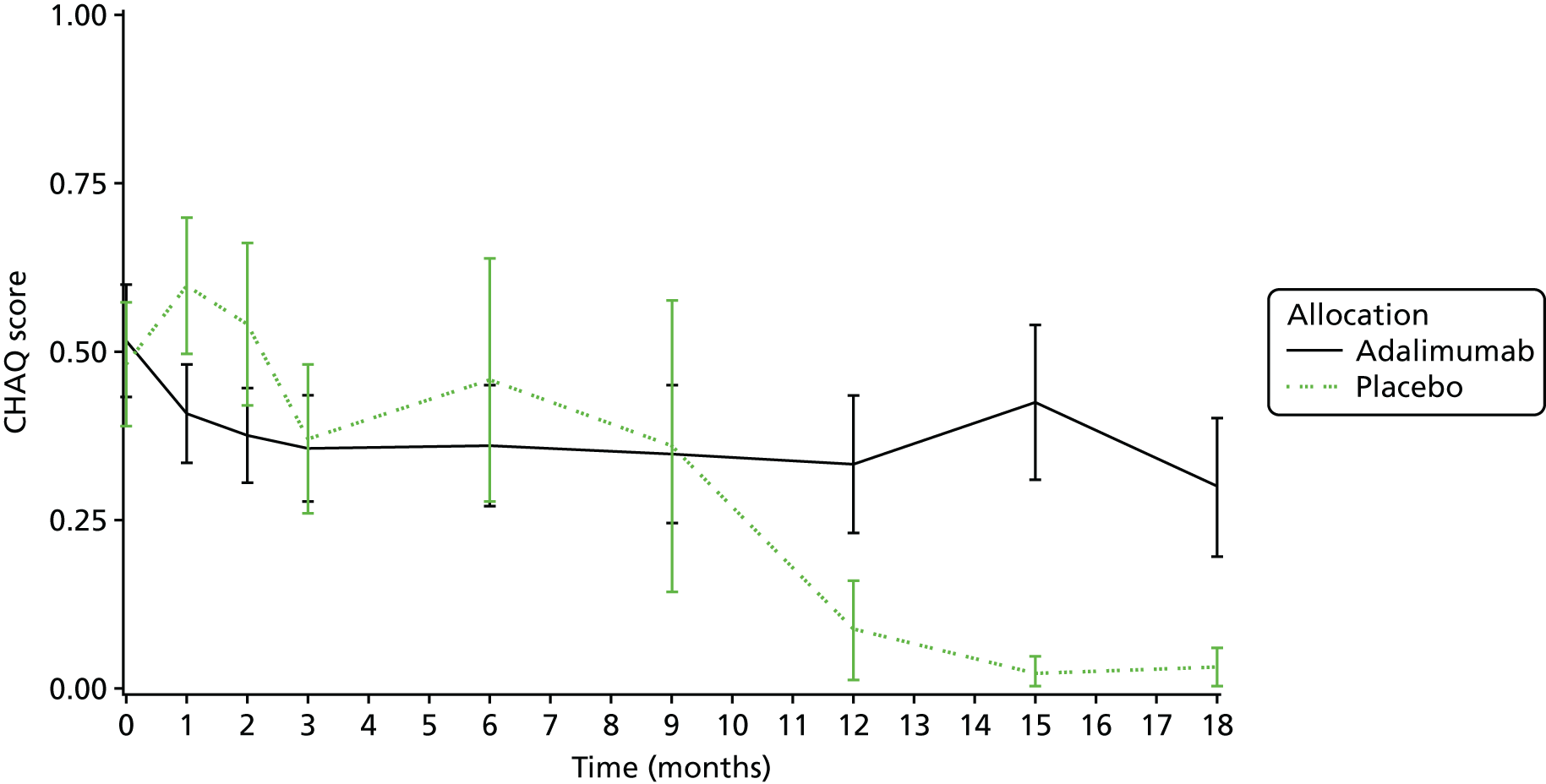
| Participant ID | Site | Date of randomisation | Date of treatment failure | Time to failure (days/months) | Reason for treatment failure | Further details | Patient unblinded at time of treatment failure (date) | Eye failed |
|---|---|---|---|---|---|---|---|---|
| 0116068 | Bristol Children’s Hospital | 5 June 2014 | 18 June 2015 | 378/12.42 | Permitted concomitant medications used against acceptable criteria | Continue follow-up | Already in open-label phase | Left |
| 0116069 | Bristol Children’s Hospital | 5 June 2014 | 20 August 2015 | 441/14.49 | Sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2), still present after 6 months of therapy |
Continue follow-up: Base – (1+) Previous = SYCAMORE Treatment visit 7 – 15 months’ treatment (1+ on 30 July 2015) Fail = SYCAMORE Unscheduled visit – (1+ on 20 August 2015) |
Already in open-label phase | Right |
| 0249091 | Great Ormond Street Hospital | 27 March 2015 | 11 September 2015 | 168/5.52 | Sustained scores as recorded at entry grade measured over two consecutive readings (grades 1 to 2), still present after 6 months of therapy |
Continue follow-up: Base – (2+) Previous = SYCAMORE Treatment visit 3 – 3 months’ treatment (1+ on 19 June 2015) Fail = SYCAMORE Treatment visit 4 – 6 months’ treatment (2+ on 11 September 2015) |
Already in open-label phase | Left |
Appendix 4 Trial management team
All trial management was conducted by the CTRC, University of Liverpool, Liverpool, UK.
Senior statisticians
Professor Paula Williamson.
Dr Ashley P Jones.
Head of trial management
Ms Helen Hickey.
Senior data manager
Mrs Clare Jackson.
Information systems manager
Dr Duncan Appelbe.
Trial co-ordinator
Mr Ben Hardwick.
Statisticians, database developers and administration
Mrs Anna Rosala-Hallas.
Miss Naomi Rainford.
Dr Graeme Hickey.
Dr Ruwanthi Kolamunnage-Dona.
Miss Elizabeth Conroy.
Dr Kerry Dawn.
Mrs Michaela Brown.
Mr Meirion Thomas.
Mrs Janet Harrison.
Miss Catherine Forrest.
List of abbreviations
- AC
- anterior chamber
- ACR
- American College of Rheumatology
- A&E
- accident and emergency
- AE
- adverse event
- AIC
- Akaike information criterion
- CHAQ
- Childhood Health Assessment Questionnaire
- CHQ
- Childhood Health Questionnaire
- CI
- confidence interval
- CR
- credible range
- CRF
- case report form
- CTRC
- Clinical Trials Research Centre
- DIC
- deviance information criterion
- DMARD
- disease-modifying antirheumatic drugs
- GFR
- glomerular filtration rate
- GP
- general practitioner
- HR
- hazard ratio
- HRG
- Healthcare Resource Group
- HUI
- Health Utilities Index
- HUI2
- Health Utility Index Mark 2
- HUI3
- Health Utility Index Mark 3
- ICER
- incremental cost-effectiveness ratio
- IDSMC
- Independent Data and Safety Monitoring Committee
- IMP
- investigational medicinal product
- IOP
- intraocular pressure
- ITT
- intention to treat
- JADAS
- Juvenile Arthritis Disease Activity Score
- JIA
- juvenile idiopathic arthritis
- LMM
- linear missed model
- logMAR
- logarithm of the minimum angle of resolution
- MTX
- methotrexate
- PAS
- patient administration systems
- PhS
- physical subscale
- PLICS
- patient-level information and costing systems
- PsS
- psychosocial subscale
- QALY
- quality-adjusted life-year
- RF
- rheumatoid factor
- RR
- relative risk
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SAS PROC
- Statistical Analysis Systems Procedure
- SD
- standard deviation
- SE
- standard error
- SUN
- Standardisation of Uveitis Nomenclature
- SYCAMORE
- a randomised controlled trial of the clinical effectiveness, SafetY and Cost-effectiveness of Adalimumab in Combination with MethOtRExate for the treatment of juvenile idiopathic arthritis associated uveitis
- TMG
- Trial Management Group
- TNF-α
- tumour necrosis factor alpha
- TSC
- Trial Steering Committee
- VI
- visual impairment