

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 12/29/01. The contractual start date was in July 2014. The draft report began editorial review in May 2018 and was accepted for publication in December 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Fiona C Denison has received funding from Dilafor AB (Solna, Sweden) outside the submitted work. Jane E Norman has received funding from Dilafor AB and GlaxoSmithKline plc (Middlesex, UK) outside the submitted work and declares membership of the National Institute for Health Research (NIHR) Health Technology Assessment (HTA) Maternal Newborn and Child Health Panel. Jane Norman was a member of the HTA and Efficacy and Mechanism Evaluation (EME) Editorial Board. John Norrie declares grants from the University of Aberdeen and the University of Edinburgh during the conduct of the study and membership of the following NIHR boards: Cardiopulmonary Resuscitation Decision-making Committee, HTA Commissioning Board, HTA Commissioning Sub-Board (Expression of Interest), HTA Funding Boards Policy Group, HTA General Board, HTA post-board funding teleconference, NIHR Clinical Trials Unit Standing Advisory Committee, NIHR HTA and EME Editorial Board and Pre-exposure Prophylaxis Impact Review Panel. Julia Lawton declares membership of the HTA General Board.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Denison et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background and rationale
Retained placenta
A retained placenta is diagnosed when the placenta is not delivered within 30 minutes following active management of the third stage of labour after delivery of the baby (comprising routine use of uterotonic drugs, early clamping and cutting of the cord and controlled cord traction) or within 60 minutes following physiological management of the third stage (no routine use of uterotonic drugs, no clamping of the cord until pulsation has ceased and delivery of the placenta by maternal effort) followed by active management as per the National Institute for Health and Care Excellence (NICE) Clinical Guideline 190. 1 The incidence of retained placenta is rising in the UK because of changes in maternal demographics, increased intrapartum interventions and operations involving exploration of the uterine cavity (e.g. surgical termination of pregnancy and dilatation and curettage). 2 It affects 2% of vaginal deliveries,2 which equates to nearly 11,000 women in the UK per annum. Retained placenta is a major cause of postpartum haemorrhage,3,4 with major obstetric haemorrhage affecting nearly 1 in 180 women and being the most common cause of significant maternal morbidity. 5 Following the failure of active or physiological management, NICE recommends that retained placenta should be treated by manual removal of placenta. 1 Compared with spontaneous placental delivery, this surgical procedure (which requires an operative environment and trained personnel) has its own risks, including bleeding6 and infection. 7 In addition, delays incurred while arranging this operative procedure increases risk of significant haemorrhage. 4 Furthermore, the infrastructure required for this operative intervention is not available in all delivery settings and the invasive nature of this procedure has the potential to delay or interrupt mother–baby bonding in the immediate postpartum period. The Cochrane Group8 and NICE1 both recognise that non-surgical management options for retained placenta are limited and have recommended that research is needed into new medical strategies of retained placenta. New (and effective) treatments for retained placenta would dramatically reduce the number of women requiring manual removal of placenta, with the operation being restricted to the small minority of women with particularly adherent placentae (partial placenta accreta). The reduction in operative interventions would have cost benefits for the NHS and also benefits for women in terms of increased satisfaction, less separation of mother and baby immediately after birth and reduced morbidity.
Rationale
Rationale for glyceryl trinitrate (nitric oxide donor) for retained placenta
The first report of a nitric oxide donor being used as a treatment for retained placenta was in 1811 when inhalation amyl nitrate was used to treat a uterine constriction ring, thus facilitating manual removal of placenta. 9 Since then, numerous observational studies have suggested that nitric oxide donors including glyceryl trinitrate (GTN) might be effective for the management of retained placenta. Many of these studies used intravenous boluses of GTN10 ranging from 50 µg to 200 µg, with uterine relaxation occurring about 60 seconds after injection and lasting for 2 minutes. Of the five observational studies that report the use of intravenous GTN for management of retained placenta (50–200 µg, up to two doses; n = 87 women), success rates range from 94% to 100%. 11–14 In these small studies, intravenous GTN appears to be efficacious for retained placenta; however, intravenous administration can cause problematic side effects, including symptomatic hypotension, particularly when given at higher doses, and it is not possible to administer GTN by this route in all settings. 15
More recently, alternative formulations of sublingual GTN have been trialled. All studies have used sublingual GTN at 1 mg. Two small studies16,17 (n = 48 women) suggested that GTN given as a sublingual tablet was effective in treating retained placenta. However, a larger study [International Standard Randomised Controlled Trial Number (ISRCTN) 34755982; n = 105 women] showed no benefit of GTN over placebo for treatment of retained placenta (37.3% vs. 20.4%; p > 0.05). 16 Compared with the tablet preparation, sublingual GTN spray18 has several advantages including stability at room temperature, significant reduction in latency of onset (with onset beginning at 30–45 seconds, peaking at 90–120 seconds and lasting up to 5 minutes)19–21 and fewer problematic subjective and objective side effects. 22 Anecdotal reports allude to GTN spray having a potential utility in the medical treatment of retained placenta. 18 GTN spray is used for other obstetric emergencies when uterine relaxation is required, such as releasing a trapped head at caesarean section or in breech delivery. 18 Clinicians are therefore familiar with the use of sublingual GTN spray in the emergency obstetric setting, which potentially lowers the barrier for trialling its use for retained placenta.
Biological plausibility of glyceryl trinitrate for the management of retained placenta
It is likely that failure of myometrial contractions, placental trapping and adherence to the myometrium contribute variably to the ultimate clinical diagnosis of retained placenta. In placentae that are detached but trapped behind a myometrial contraction ring, GTN could potentially treat retained placenta simply by relaxing local uterine muscle constriction and, thereby, effecting placental release. 8 For adherent placenta, Farley et al. 23 have suggested that nitric oxide-mediated contraction and relaxation of human chorionic villi along their longitudinal axis might serve as a GTN-mediated mechanism for placental separation. For placentae that are retained because of partial placenta accreta, currently available nitric oxide drugs (including GTN) are unlikely to effect release and surgical management is likely to remain the preferred method of treatment.
Conclusion
Although there is an increasing amount of evidence that supports the use of GTN for treatment of retained placenta, much of this evidence is based on anecdotal case reports or clinical ‘trials’ that are non-randomised, do not contain a placebo group and are underpowered. 16,17 In addition, in the context of constrained maternity resources in a publicly funded health system, it is important to quantify the costs associated with GTN (including any subsequent monitoring costs and costs associated with complications) in relation to its effectiveness and any subsequent cost savings it may deliver over standard practice. There is therefore an urgent need for a pragmatic clinical trial of GTN for those with a retained placenta to determine whether or not GTN is efficacious, safe, acceptable and cost-effective as a treatment for retained placenta before a treatment, which may (or may not) work, is embedded within routine clinical practice. In response to a Health Technology Assessment (HTA) commissioned call, we aimed to determine whether or not sublingual GTN was clinically effective and cost-effective for the medical management of retained placenta in a randomised placebo-controlled double-blind pragmatic UK-wide Glyceryl trinitrate fOr reTaIned placenTa (GOT-IT) trial.
Objectives
The overall aim of the randomised placebo-controlled double-blind pragmatic UK-wide GOT-IT trial (with internal pilot study) was to determine the clinical effectiveness and cost-effectiveness of sublingual GTN spray compared with placebo in reducing the need for manual removal of placenta in women with retained placenta after vaginal delivery following the failure of current management. Outcomes were measured over four inter-related domains: clinical, safety, patient sided and economic.
The primary research objectives of the internal pilot randomised controlled trial (RCT) were as follows:
-
to demonstrate trial processes for approaching women, gaining consent, randomising, treating and assessing outcomes were optimal, and to implement improvements as required
-
to determine achievable recruitment rates
-
to determine the probable effect size, to inform calculation on whether or not the planned sample size could be reduced while maintaining study power
-
to pilot and modify if required the postpartum questionnaires (assessment of patient satisfaction and collection of health service use outcomes).
The primary research objectives of the substantive GOT-IT RCT were as follows:
-
to determine the clinical effectiveness of sublingual GTN in treating retained placenta and avoiding manual removal of placenta in women with vaginal delivery following failure of current management (defined as the third stage of labour lasting more than 30 minutes after active management or 60 minutes after physiological management followed by active management respectively) (clinical domain)
-
to determine the side-effect profile for GTN given to treat retained placenta (safety domain)
-
to assess patient satisfaction with GTN given for retained placenta (patient-sided domain)
-
to assess the net costs (or cost savings) to the NHS of using GTN for the treatment of retained placenta compared with standard practice (economic domain).
The secondary objectives were to assess NHS costs in relation to the primary outcome and a range of secondary outcomes expected to differ between the groups in the trial, using a cost–consequences balance sheet approach.
Chapter 2 Methods
Trial design
The GOT-IT trial was a multicentre, pragmatic group-sequential, placebo-controlled, randomised trial with cost-effectiveness analysis. The study protocol can be accessed from www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/ (accessed 31 May 2019).
Setting
The trial was conducted in the delivery suites of 29 hospitals with obstetric units in 27 NHS trusts and boards in England and Scotland. The delivery wards were of varying size and location to ensure that the results of the trial were generalisable to the UK. Women did not have to have delivered in obstetric units to be eligible for trial entry: if a woman developed a retained placenta following failure of current management after giving birth at home or at a stand-alone or along-side midwifery delivery unit, she was still eligible for trial entry once admitted to one of the recruiting centres.
Participants
The trial was designed to include an internal pilot trial, which aimed to recruit 75 women from eight pilot sites. Once recruited to the pilot trial, those women also formed part of the main substantive RCT. The number of sites planned for the main substantive trial was a minimum of 20, and the final number of sites included was 29. The number of participants required to be recruited into the main trial was informed by the group-sequential design. If the trial recruited to maximum size, 1100 participants were required to be recruited.
Eligibility criteria
Inclusion criteria
-
Women with retained placenta.
-
Women aged ≥ 16 years.
-
Women with vaginal delivery in the incident pregnancy (including women with past obstetric history of caesarean section in a previous pregnancy).
-
Women who were haemodynamically stable, defined as having a systolic blood pressure level of > 100 mmHg and a pulse of < 110 beats per minute (b.p.m.).
-
Women who delivered at > 14 weeks’ gestation in the incident pregnancy.
Exclusion criteria
-
Women who were unable to give informed consent.
-
Women with suspected placenta accreta, increta or percreta in the incident pregnancy.
-
Multiple pregnancy in the incident pregnancy.
-
Women who had an instrumental vaginal delivery in the incident pregnancy.
-
Women with an allergy or hypersensitivity to nitrates or to any other constituent of the formulation of the study medication.
-
Women who had taken alcohol in the 24 hours prior to delivery.
-
Women with a history of phosphodiesterase inhibitor use during pregnancy.
-
Women with a contraindication to GTN administration because of one or more of the following: incipient glaucoma, severe anaemia, profound bradycardia, glucose-6-phosphatedehyrogenase deficiency, brain trauma and cerebral haemorrhage, angina, mitral and/or aortic stenosis caused by hypertrophic obstructive cardiomyopathy, constrictive pericarditis, circulatory collapse due to cardiogenic shock and toxic pulmonary oedema.
The trial was designed to be pragmatic, and the inclusion and exclusion criteria were therefore as broad and as inclusive as possible. However, we decided to exclude women with multiple pregnancies and women who were in theatre having an instrumental vaginal delivery for the following reasons:
-
Measured blood loss between randomisation and transfer to the postnatal ward was our prespecified primary safety outcome. Multiple pregnancy is an independent risk factor for haemorrhage and these women were likely to have significantly higher blood loss than women with singleton pregnancies.
-
If women were already in theatre having an instrumental delivery with adequate analgesia, we thought that it would be highly unlikely that the obstetrician delivering the baby would wait for 30 minutes after active management or 60 minutes after physiological management plus a further 30 minutes of active management before diagnosing a retained placenta. In such an operative environment in which skilled personnel and appropriate analgesia were already in place, the threshold to proceed to manual removal of placenta would be much lower. Furthermore, our midwifery and lay representatives felt strongly that in this situation it would be unethical and undignified for a woman to remain in theatre for longer than required when the only reason to do so would be to fulfil eligibility criteria.
-
If we had included these two groups we would have required to stratify the data and significantly increase the sample size, which may have made the trial unfeasible.
Co-enrolment
Participation in another clinical trial of an investigational medicinal product (CTIMP) was initially an exclusion criterion. However, as enrolment to the GOT-IT trial could occur only once the diagnosis of retained placenta had been made, many women who would otherwise have been eligible were being ruled out as they were already participating in another CTIMP. We therefore submitted a substantial amendment that permitted co-enrolment between CTIMP studies providing there was a CTIMP-to-CTIMP agreement. This involved proposals for co-enrolment between CTIMP studies to be captured in a written, authorised agreement between the sponsors and chief investigators of each study.
Recruitment procedure
Recruitment was initiated on 13 October 2014 and completed by 26 July 2017 in the delivery wards of 29 UK maternity hospitals.
Screening and consent
Clinicians undertook the initial eligibility screening. Women were identified as being potentially eligible for the trial if, following vaginal birth, they had a retained placenta and were at risk of needing a manual removal of placenta after failure of current management of the third stage of labour, defined as the placenta remaining undelivered after (1) 30 minutes of active management or (2) 60 minutes of physiological management followed by 30 minutes of active management.
Following the diagnosis of a retained placenta and successful screening, women were approached by either a clinician or a midwife who had received the approved GOT-IT training package that had sponsor-approved good clinical practice training embedded within it. Potential participants were given verbal and written information about the trial, with the latter consisting of a summary and a full patient information sheet. Women were given an opportunity to ask questions relating to any aspect of the trial in order to gain a full understanding of what was required of them. The patient information leaflets contained information that explained that sublingual GTN was being investigated as a possible treatment for retained placenta. If women wished to participate then verbal or written consent was obtained. If consent was gained verbally, written consent had to be taken as soon as possible after the trial. This approach has been endorsed by the Royal College of Obstetricians and Gynaecologists in recent guidance about obtaining consent in perinatal research where consent time is critical. 24
Written consent involved obtaining a dated signature from the participant and a dated signature from the consenter. The consent form included a clear explanation that the woman could withdraw from the trial at any time without providing a reason if she wished, and that if she chose to do so it would not influence any future medical care. A copy of the signed informed consent form was returned to the woman, another copy filed in her medical notes and the original was obtained in the investigator site file.
Consent and recruitment of subjects undertaken in the qualitative pilot trial25–27 led us to improve the consent and recruitment procedure for those subjects recruited to the substantive trial (see Chapter 3 for further details and www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/). In response to feedback from women, information, with researcher contact details, was provided to women antenatally, if they were thought to be at a high risk of having a retained placenta, for example a history of previous retained placenta. 28 This is an example of how the qualitative trial strengthened the substantive trial.
Randomisation, concealment and blinding
Study medication was provided to site pharmacies in pre-packed randomised permuted blocks. Drug packs were ordered from the pharmacy and stored in the labour ward setting. Eligible participants were randomly assigned to active treatment with GTN or an identical-looking placebo.
The method of randomisation was via the next available treatment pack from the shelf. The drug pack number on the pack and the vial was recorded in the women’s medical notes, in study documentation and entered into the electronic database. Subjects were randomised in a 1 : 1 ratio of GTN to placebo. Study staff advised the women on how to deliver the study medication and provided, prior to drug administration, a leaflet demonstrating its use.
As the design of the study was double blind, neither the women nor the onsite study staff knew which treatment had been allocated. An unblinding mechanism was available if required for emergency procedures. This was via an interactive voice response system to the Centre for Healthcare Randomised Trials that held the randomisation list for the trial, which contained study pack numbers and treatment allocation. Emergency unblinding was required to be performed by a senior clinician.
Treatment group allocation
The study was designed to achieve concealment of allocation. Active GTN sprays were identical to placebo sprays. Study outcomes were recorded by clinicians and midwives blinded to treatment allocation. Unblinding was not performed until after data entry was complete, the database was fully checked and validated and all queries were resolved.
Intervention
Women were required to take two puffs sublingually of either GTN or placebo. Each drug canister contained a pump mechanism that delivered a metered dose of 400 µg of either GTN or placebo. We chose a sublingual route because administering GTN via a sublingual spray had previously not been tested and was required by the HTA during the commissioning of our trial. The intervention was self-administered as a single intervention (two puffs of 800 µg active drug or two puffs of placebo spray) as soon as possible after diagnosis of retained placenta. Both the study drug and the matching placebo were manufactured by Pharmasol Ltd (Liverpool, UK) and labelled by Sharp Clinical Services (UK) Ltd (Ashby-de-la-Zouch, UK).
Data collection and management
The GOT-IT trial met the requirements of the Data Protection Act 1998. 29 Data were collected at each site by a GOT-IT trained researcher using a standardised case report form. Initially, data were captured in a paper case report form as well as being entered into the electronic case report form. However, to minimise duplication, the sponsor requested that use of a full paper case report form should be discontinued and replaced by an abbreviated paper case report form. Following this change, previously required information no longer collected on the abbreviated case report form was captured straight from source documentation and entered directly into the database.
The study collected pre-baseline data (i.e. subject log, eligibility and consent), baseline data (i.e. clinical observations at baseline, demographics, obstetric history, current pregnancy information, medical history and medications), clinical observations (at 5 and 15 minutes) and details of placenta delivery, first post-natal day and discharge information. All data, including discharge questionnaires (see www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/), safety data and a 6-week postnatal check data, were entered by research midwives or clinical data administrators based at each of the sites. The exception to this was postnatal questionnaire data that were entered by administrators in the trial office (see www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/). Participants were identified by a unique five-digit code.
Data validation checks developed within the database flagged missing or erroneous data. In addition, the trial office undertook regular manual checks of the database and any discrepancies noted were queried with individual sites. The study had a specific monitoring plan developed by the lead study monitor for Academic and Clinical Central Office for Research & Development (ACCORD) (joint office for the University of Edinburgh and NHS Lothian). All sites received an onsite monitoring visit following the recruitment of four subjects to the trial. The monitoring visit involved reviewing consent forms, confirming participant eligibility, checking that staff allocated to undertake delegated tasks were appropriately qualified to do so, checking the quality of data abstraction and visiting the local pharmacy. Outstanding monitoring actions were logged and a written report was forwarded to the principal investigator following the visit. The onsite study staff then completed any actions highlighted.
Study assessments
All women received the following assessments prior to randomisation: screening, confirmation of eligibility (including a brief medical history and concomitant medication check), informed consent, baseline observations (blood pressure, heart rate, temperature) and a full blood count had to have been taken within the past 24 hours to obtain a baseline haemoglobin level.
Following randomisation (administration of the study intervention), blood pressure, heart rate and temperature were measured at 5 minutes and 15 minutes. Blood loss was measured from the point of study drug administration until the woman was transferred to the postnatal area. Prior to hospital discharge, a full blood count was again collected to measure haemoglobin level and a questionnaire was given to each woman to complete. The questionnaire was designed to measure patient-rated side effects and patient-rated satisfaction. Any adverse events (AEs) noted were also recorded.
Women received a further questionnaire 6 weeks later from the trial office. This postnatal questionnaire was also designed to measure patient satisfaction, side effects and health resource use. Women were asked to complete and return it to the trial office in a provided prepaid envelope.
Outcomes
Primary outcome
The primary outcomes were measured over four inter-related domains of clinical, safety, patient sided and economic.
-
Clinical: the need for manual removal of placenta, defined as the placenta remaining undelivered 15 minutes post study treatment and/or delivery being required within 15 minutes of treatment because of safety concerns.
-
Safety: measured blood loss between administration of treatment and transfer to the postnatal ward or another clinical area (e.g. labour ward high dependency).
-
Patient sided: satisfaction with treatment and side-effect profile assessed by questionnaire.
-
Economic: evaluation of possible net incremental costs (or cost savings) to the NHS of using GTN versus standard practice. Costs included GTN (dose and time to administer the study drug, monitor a woman and deliver the placenta if effective), manual removal of placenta and further health service resource use up to 6 weeks post childbirth (as measured by the health service resource-use questionnaire).
Secondary outcomes
-
Clinical outcomes:
-
fall in haemoglobin level of > 15% between recruitment and the first postnatal day
-
time from randomisation to delivery of placenta
-
manual removal of placenta in theatre
-
need for earlier than planned manual removal of placenta because of clinical concerns
-
fall in diastolic or systolic blood pressure of > 15 mmHg and/or increase in pulse of > 20 b.p.m. between baseline observations and 5 and 15 minutes post administration of study treatment
-
requirement for general anaesthesia
-
requirement for blood transfusion between the time of delivery and postnatal discharge from hospital
-
maternal pyrexia, defined as at least a temperature reading of > 38 °C within 72 hours of delivery or discharge from hospital, if the hospital discharge occurs prior to 72 hours following delivery
-
sustained uterine relaxation after the placenta has been removed requiring treatment with uterotonics.
-
-
Costs: the mean costs were summarised by treatment allocation group, and the incremental cost (cost-saving) associated with the use of GTN was estimated using an appropriately specified general linear model. The cost data was presented alongside the primary and secondary outcome data in a cost–consequence balance sheet, which indicated which strategy each outcome favoured.
Safety considerations
Adverse events were defined as any untoward medical occurrence in a clinical trial, which does not necessarily have a causal relationship with an investigational medicinal product.
Adverse reactions were defined as any untoward and unintended response to an investigational medicinal product that is related to any dose administered to a participant.
A serious adverse event (SAE), or serious adverse reaction, was defined as any AE or adverse reaction that at any dose:
-
results in the death of the clinical trial participant
-
is life-threatening (life-threatening in the definition of a SAE or serious adverse reaction refers to an event during which the participant was at risk of death at the time of the event; it does not refer to an event that hypothetically might have caused death if it were more severe)
-
requires inpatient hospitalisation, or prolongation of existing hospitalisation
-
results in persistent or significant disability or incapacity
-
consists of a congenital anomaly or birth defect
-
results in any other significant medical event not meeting the criteria above.
Hospitalisations for treatment planned prior to randomisation and hospitalisation for elective treatment of a pre-existing condition were not considered serious adverse reactions.
A suspected unexpected serious adverse reaction was defined as any adverse reaction that was classified as serious and was suspected to be caused by the investigational medicinal product. In addition, the reaction was not consistent with the information about the investigational medicinal product in the summary of product characteristics. 30
Adverse events and SAEs were documented only if they occurred between when the participant signed the consent form to take part in the study and the 6-week postnatal outcome assessment point. Only AEs and SAEs that related to the mother were reported. Participants were asked about the occurrence of AEs and SAEs prior to discharge from the hospital and in the 6-week postnatal questionnaire. The 6-week postnatal questionnaire also asked participants if they had seen their general practitioner (GP), been admitted to a hospital or been prescribed any medication. The local principal investigator responsible for the care of the participant (or delegated clinician) was responsible for assessing the severity, causality and expectedness of an AE and whether or not the event fulfilled the criteria for reporting it as serious.
Adverse events were documented in the participant’s medical case notes and on the electronic case report form. All reported AEs were collated and coded by the trial office and the chief investigator had regular oversight of them.
All SAEs that were observed during the trial were reported within 24 hours of the site becoming aware of the event. They were reported by the principal site investigator to the sponsor and the trial office and also entered into the electronic database. All SAEs were followed up until resolution. The chief investigator was notified of all SAE reports.
For the purposes of this study the following events were not considered SAEs:
-
pregnancy
-
hospitalisations for treatment planned prior to hospitalisation
-
hospitalisations for elective treatment of a pre-existing condition
-
decrease in haemoglobin level of > 15% between recruitment and the first postnatal day
-
manual removal of placenta in theatre
-
the need for earlier than planned manual removal of placenta
-
decrease in either systolic or diastolic blood pressure of > 15 mmHg and/or increase in heart rate of > 20 b.p.m. between baseline and 5 and 15 minutes post administration of active/placebo treatment
-
the need for blood transfusion between time of delivery and discharge from hospital
-
the need for general anaesthesia
-
maternal pyrexia (one or more temperature readings of > 38 °C within 72 hours of delivery or discharge from hospital if discharge occurs sooner)
-
sustained uterine relaxation after removal of placenta requiring uterotonics.
The SAEs were reported to the Data Monitoring Committee (DMC) and the Trial Steering Committee (TSC) at regular 6-monthly meetings. The DMC reviewed the data with an unblinded status. If any serious concerns had arisen about the safety of the data, the chairperson of the DMC would have recommended to the chairperson of the TSC that the study should be discontinued.
Governance and oversight
The GOT-IT trial was registered on the ISRCTN registry as ISRCTN88609453. Ethics approval was obtained from the North East – Newcastle and North Tyneside 2 Research Ethics Committee (13/NE/0339). Approval was also obtained from the Medicines and Health products Regulatory Agency (2013-003819-42) and the Health Research Authority as well as from the local trust Research and Development Offices. The study was a commissioned trial funded by the National Institute for Health Research (NIHR) as part of the HTA programme commissioned call funding stream (reference number 12/29/01).
Statistical methods and trial analysis
Too much uncertainty existed in two crucial parameters to commit to a fixed sample size design. We believed that the most appropriate primary outcome was the proportion of women needing surgical intervention for removal of the placenta. However, there was considerable uncertainty as to how many women who may have been eligible for trial entry would actually go on to require surgery owing to (1) a lack of knowledge of frequency of spontaneous delivery of the placenta beyond the time frame and (2) variations in local clinical practice (e.g. logistics and time taken to organise theatre space and skilled staff to perform manual removal of placenta). The routinely recorded statistics were not sufficiently detailed for these variables to be accurately determined. In addition, we were very unsure what the magnitude of the benefit would be from GTN spray. To reflect these uncertainties and give us a design that was flexible enough to maximise the chance that we would efficiently detect and estimate the true benefit of treatment in the quickest time with the right number of participants, but equally give ourselves the controlled opportunity to abandon the trial if it turned out that no worthwhile treatment effect actually existed (via futility analyses), we proposed a group-sequential design. We believed that the GOT-IT trial had the ideal design because it enabled us to present the maximum size of the trial that was needed, alongside a flexible group-sequential approach that would allow the trial to terminate early for one of two scenarios. The first scenario would have been overwhelming evidence of benefit (owing to a large treatment effect and/or less variability in the outcome measure). The second scenario would have been due to a suitably defined futility (i.e. having partially obtained the maximum trial size); we were confident that a large treatment effect was implausible and that the current estimate of the treatment effect was sufficiently precise to be convincing, thus allowing the trial to be terminated early.
Ground rules for the statistical analysis
The statistical analysis followed a statistical analysis plan (see www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/) that was agreed by the TSC. The interim analyses for the DMC were specified within the DMC charter, and results of these interim analyses were in strict confidence (no member of the research group apart from the study statistician was aware of the contents of these analyses). The analysis was based on the intention-to-treat (i.e. analysed as randomised) principle. Statistical significance was at the two-sided 5% level with corresponding confidence intervals (CIs) derived.
Sample size
From discussing with clinical colleagues and listening to mothers and pregnant women, a 10% decrease in women needing manual removal of placenta would be a sufficient advantage to make it worthwhile implementing the GTN spray in clinical practice. From a statistical perspective, we knew that the maximum variability in a binary outcome (i.e. need for surgical intervention: yes or no) would occur at a 50% rate in the placebo group. On a fixed sample approach with a 90% power and 5% significance level, we would need 1038 women to demonstrate a 10% change from 50% on placebo to 40% on GTN spray. Allowing for the multiple sequential looks at the data, a possible maximum sample size of 1078 participants was needed. Because the primary clinical outcome was recorded within minutes of the intervention being administered, we anticipated minimal (if any) loss to follow-up and, therefore, no adjustment for missing data was made.
Group-sequential design
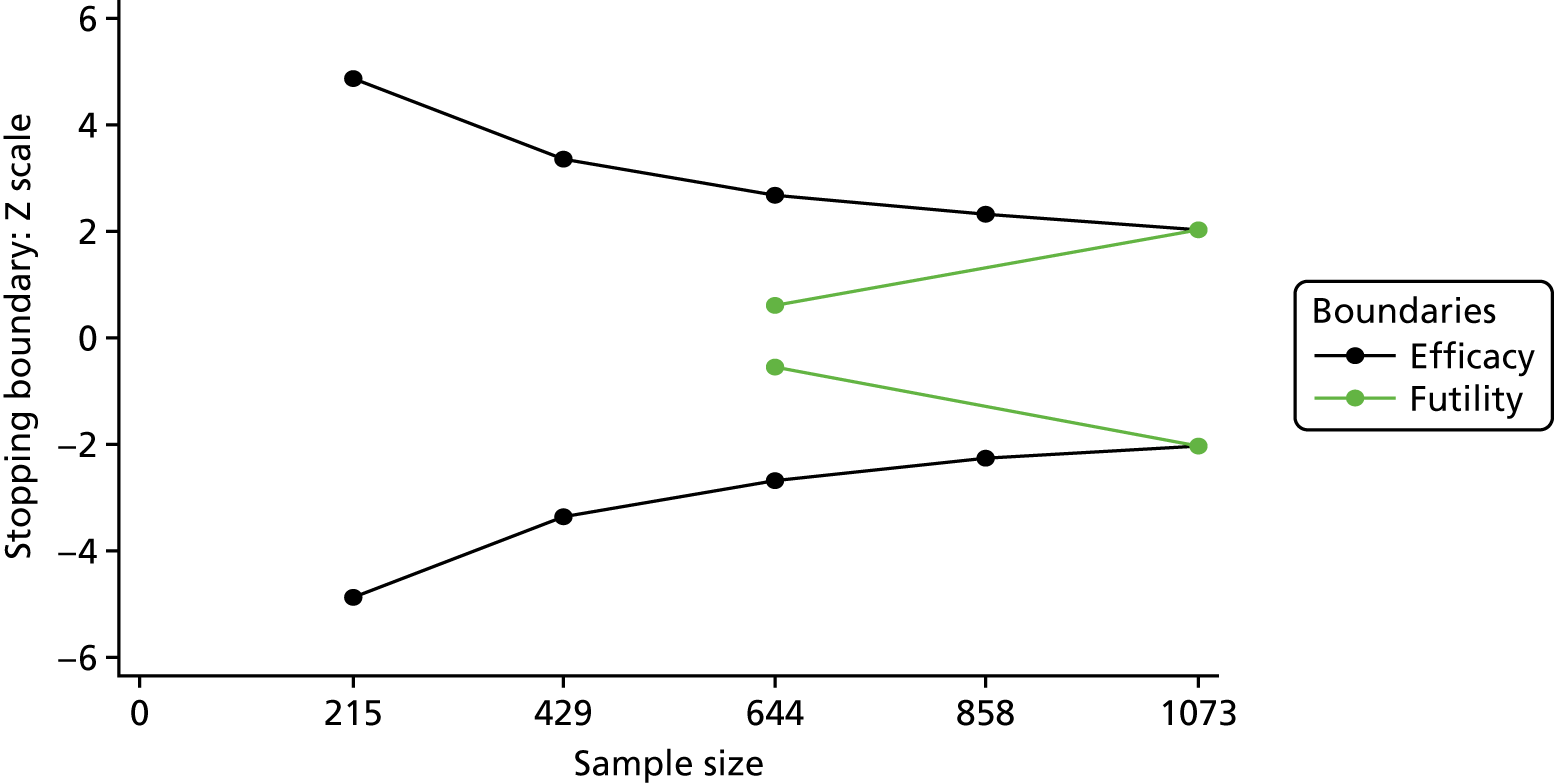
There were many options for deciding on a group-sequential design and the DMC was central to its implementation and interpretation. Detailed discussions were held with the independent statistician over the group-sequential design and it was decided to use a Lan–DeMets alpha spending approach31 with O’Brien–Fleming boundaries. 32 We specified a two-sided test, with efficacy and futility boundaries with five interim looks (the last being the final analysis), equally spaced at 215, 429, 644, 858 and 1073 patients. Figure 1 shows the stopping boundaries.
FIGURE 1.
Stopping boundaries. Black, efficacy boundaries; green, futility boundaries.
Primary/secondary outcome analysis
The group-sequential analysis for the primary clinical outcome was analysed using the statistical programme East® 6.4.1 (2016; Cytel Inc., Cambridge, MA, USA). For the other primary outcomes, safety was analysed using ordered logistic regression and patient-sided outcomes were analysed using logistic regression. Both accounted centre by using cluster robust standard errors. Secondary outcomes were analysed in a similar way using either a logistic or a linear regression when appropriate. The remainder of the analysis was undertaken using Stata 14® (StataCorp LP, College Station, TX, USA).
Planned subgroup analyses
The planned subgroup analysis was to explore the possible treatment effect modification through the use of treatment-by-subgroup interactions all using a stricter two-sided 1% level of statistical significance. The subgroups were:
-
previous caesarean section
-
gestation at delivery (< 36 and ≥ 36 weeks’ gestation).
Timing and frequency of analysis
Apart from the primary clinical outcome, a single principal analysis was carried out when the final participant reached the 6-week time point.
Summary of changes to the project protocol
Changes were made to the project protocol as follows.
Eligibility criteria
-
Protocol V3.0 – substantial amendment 2
Change of exclusion criteria from women having an instrumental delivery to women having an instrumental delivery in theatre.
-
Protocol V8.0 – substantial amendment 11
The original definition of haemodynamically stable was a systolic blood pressure level of > 100 mmHg and a pulse of < 110 b.p.m. This definition was changed to satisfy all three definitions:
-
haemodynamically stable
-
a heart rate of ≤ 119 b.p.m.
-
a systolic blood pressure of > 100 mmHg.
-
-
Protocol V8.0 – substantial amendment 11
Change in exclusion criteria to allow co-enrolment to exist for CTIMP trials, providing there was a CTIMP–CTIMP agreement between the sponsors and investigators of each trial.
Sample size
-
Minor amendment 9
Minor increase in sample size from 1086 to 1100.
No changes were made to the study objectives, outcomes or statistical parameters throughout the duration of the trial. In total, 16 substantial amendments and 10 minor amendments were sought, which in addition to the changes described above, covered changes to the recruitment materials, clarifications and administrative changes to the protocol.
Chapter 3 Nested qualitative study within the internal pilot
A nested qualitative study was undertaken during the internal pilot RCT, the purpose of which, as outlined in the study protocol, was to adjust strategies to:
-
maximise recruitment into the main trial
-
optimise opportunities for gaining informed consent
-
ensure that staff were given appropriate training and support to help promote the successful delivery of the main trial.
The specific aims and objectives of the qualitative work were as follows.
Aims
-
To explore women’s and staff members’ experiences of, and views about, the information and consent pathway used in the pilot RCT.
-
To establish women’s likes and dislikes of the interventions and procedures received in the pilot RCT.
Objectives
-
To refine/improve the information and consent pathway used in the substantive trial to maximise recruitment and informed consent.
-
To identify better ways of supporting staff involved in recruitment.
An additional objective outlined in the original protocol was ‘to refine the questionnaire used to assess patient satisfaction with GTN given for retained placenta’ (reproduced with permission from Denison et al. 33; contains information licensed under the Non-Commercial Government Licence v2.0). However, as this questionnaire was developed in advance of the pilot trial (and, hence, the qualitative work being undertaken), the work done to develop and refine the questionnaire is reported separately (see Chapter 6).
Overview comments
The qualitative work was started and completed on schedule, which enabled a full evaluation of the pilot RCT to be undertaken from women’s and staff members’ perspectives. As outlined further below, feedback was given to the trial team and participating sites in a timely fashion allowing the incorporation of the findings and recommendations into the main trial.
Given that recruitment into the pilot study was so successful, the qualitative work gave primacy to:
-
exploring women’s and staff views about the recruitment and consent pathway used in the pilot and how it could be refined for use in the main trial to maximise informed consent
-
using findings from interviews with staff to inform the training and support offered to those working on the main trial to promote successful trial delivery.
The qualitative research also considered why recruitment had been successful in the pilot RCT to help ensure ongoing success in the main trial.
In line with the Royal College of Obstetrics and Gynaecology guidelines for undertaking recruitment into perinatal trials,34 and as per the original trial research proposal, approvals were sought and mechanisms put in place by the trial team to enable women to access information about the trial during the antenatal period, using a pathway similar to that developed by Vernon et al. 35 However, owing to the timing of the pilot RCT, it was not possible to interview women who had been exposed to an information and consent pathway in which they had received trial information during the pregnancy. Instead, the pathway evaluated in the pilot was one in which women’s first exposure to information was when they were recruited in the labour wards, following diagnosis of a retained placenta. At the time they were recruited, these women were given written information about the trial in the form of a one-page summary leaflet accompanied by a detailed participant information sheet. Women were also provided with a verbal explanation of the trial by a designated and trained member of recruitment staff that covered all elements in the participant information sheet and consent form. In recognition of the clinical situation that the women were in, the trial permitted verbal consent to be given, provided it was followed by written consent at a later stage.
Qualitative study design
In-depth interviews were used in the qualitative evaluation of the trial’s pilot phase as these afforded the flexibility needed for participants (staff and women) to raise and discuss issues that they perceived as being salient, including those unforeseen at the study’s outset. 36,37 The use of one-to-one interviews also afforded privacy, allowing participants to share negative views about the trial’s processes and procedures, should they have chosen to do so. Data collection and analysis took place concurrently as this enabled the areas explored in the interviews, and also sampling, to be revised in the light of emerging findings. 38,39 Staff and women’s interviews took place in parallel allowing findings from one respondent group to inform issues explored with the other.
Sample and recruitment
Recruitment of staff and women started right at the pilot’s outset (i.e. October 2014) and continued to the end of the pilot (i.e. April 2015). This was done to accommodate the staggered entry of sites into the pilot, to allow for the full spectrum of experiences of delivering and receiving the pilot to be captured and to ensure that there was representation of participants (staff and women) from all eight sites that took part in the pilot. To optimise the recruitment strategies used in the main trial, it had originally been intended to interview women who had declined participation in the pilot RCT as well as those who chose to take part. However, there were very few decliners in the pilot (n = 6): two women could not be approached because the necessary research and development approvals had not been finalised, two were determined by staff to be unsuitable to approach for clinical reasons and the remaining two chose not to opt-in to the qualitative research. Therefore, no decliners were interviewed.
In line with our original study plan, women who had taken part in the pilot trial were given recruitment packs when the research midwives visited them on the wards, or packs were posted out to them if their discharge from hospital had already taken place. These packs contained a written invitation from the local principal investigator, information sheet, opt-in form and consent form accompanied by a prepaid envelope. Of the 49 women who were approached during the pilot, 25 opted in to the study, of whom 22 (45%) were interviewed (the remaining three could not be contacted to arrange an interview time, despite repeated attempts). Staff who had been involved in recruiting and/or consenting women were sent recruitment packs containing opt-in forms. Across the centres, these staff comprised obstetricians, research midwives and midwives. In total, 37 individuals returned their expression of interest and, to the best of our knowledge, only one staff member actively chose not to opt in to the qualitative study. Of these 37 individuals who opted in, 27 (73%) were selected for interviews, with purposive sampling being used to ensure that there was representation of staff from all pilot sites, from all disciplinary backgrounds (i.e. obstetricians and clinical and research midwives) and who had worked day/night/weekend shifts when recruitment had taken place. Staff were also approached and selected for interviews if they had experience of attempting to recruit women into the pilot trial who chose to decline. Although some staff were interviewed early on in the pilot to enable early recruitment experiences to be reflected on, others were interviewed nearer the pilot’s completion to allow them to draw on their experiences of trial delivery across the pilot as a whole.
In both the staff and women samples, recruitment continued until data saturation was achieved (i.e. until no new findings or themes were identified in new data collected).
Data collection
Interviews were conducted between November 2014 and May 2015. To reduce potential problems with recall bias, women were interviewed within 4 weeks of having taken part in the pilot trial. Although women were given the choice of a telephone or face-to-face interview, virtually all (n = 21, 95%) chose to be interviewed by telephone. As these women later explained, they preferred to be interviewed by telephone than receive a home visit as this arrangement made it easier for them to cancel and reschedule at short notice if they had not slept well or needed to attend to their baby. All staff opted to be interviewed by telephone.
Interviews with both staff and women were informed by topic guides, developed in the light of literature reviews, inputs from members of the trial team, implementation group and lay advisors, and revised in the light of emerging findings (see Qualitative study design). 38,39 Interviews with women averaged 25 minutes and those with staff lasted around 45 minutes. In all but two cases (in which the women had to end the interview early to attend to their baby) all key areas in the topic guides were covered and explored in depth. All interviews were digitally recorded and transcribed in full for in-depth analysis.
Data analysis
As indicated above, data analysis commenced as soon as data collection began. Data were analysed thematically (using deductive approaches to capture the material needed to answer our original research questions and inductive approaches to capture findings and themes that emerged from the data) by Julia Lawton and Nina Hallowell using the method of constant comparison. 39 This entailed individual interviews being read through repeatedly before being cross-compared to identify issues and experiences, which cut across different accounts. Comparative analyses of women’s and staff accounts were also undertaken to identify differences and similarities in their views about the recruitment and consent procedures used in the trial and the reasons for these. Julia Lawton and Nina Hallowell undertook independent analyses and wrote separate data analysis reports before meeting to discuss and reach agreement on key findings and themes to develop a coding frame. The qualitative analysis software package NVivo9 (QSR International, Warrington, UK) was used to facilitate data coding and retrieval. Coded data sets were subjected to further, in-depth analysis to identify subthemes and illustrative quotations. Illustrative quotations were given pseudonyms to anonymise them, as follows: women were allocated a name (e.g. Ellie), doctors were referred to as Dr X (e.g. Dr H), midwives were referred to using a letter (e.g. O) and research midwives as researcher (e.g. Research A).
Implementation group
An implementation group was set up to enable fast and effective application of the qualitative findings in the main trial. This group comprised the chief investigator, other trial team co-investigators, the trial manager, a patient representative and representatives (research midwives) from all eight participating centres in the pilot. The implementation group was convened prior to the qualitative data phase to help refine the interview topic guides and to discuss sampling strategies, with input from some members sought by e-mail. A face-to-face meeting took place at the end of the pilot phase to enable the qualitative researchers (Nina Hallowell and Julia Lawton) to feedback their findings to the group so that a series of recommendations would be made for implementation into the main trial.
Findings
Full details of the final samples of women and staff who took part in the interviews are provided in Tables 1 and 2.
| Demographic | Number of participants falling into this demographic (N = 22) | % of total |
|---|---|---|
| Age (years) | ||
| 18–24 | 3 | 14 |
| 25–29 | 5 | 23 |
| 30–34 | 8 | 36 |
| 35–40 | 6 | 27 |
| Mean: range | 30.7 | 18–40 |
| Ethnic group | ||
| White British | 17 | 77 |
| South-east Asian | 2 | 9 |
| Other | 3 | 14 |
| Marital status | ||
| Husband/de facto partner | 19 | 86 |
| No current partner | 3 | 14 |
| Highest education level | ||
| School | 7 | 32 |
| Further education | 2 | 9 |
| Degree | 8 | 36 |
| Higher degree | 5 | 23 |
| Occupation | ||
| No paid employment | 4 | 18 |
| Self-employed | 1 | 5 |
| Office/administrative | 5 | 23 |
| Professional | 4 | 18 |
| Semiprofessional | 8 | 36 |
| Previous birthing experiences | ||
| Primigravida | 9 | 41 |
| Previous retained placenta | 5 | 23 |
| Staff characteristic | Number of participants falling into this characteristic (N = 27) | % of total |
|---|---|---|
| Staff job | ||
| Obstetricians | 10 | 37 |
| Clinical midwives | 6 | 22 |
| Research midwives | 11 | 41 |
| Education | ||
| Professional qualifications | 1 | 4 |
| Degree | 26 | 96 |
| Higher degree | 5 | 19 |
| Time in current post (years) | ||
| 0–2 | 9 | 41 |
| 2.5–5 | 13 | 48 |
| 5.5–10 | 2 | 7 |
| > 10 | 3 | 11 |
| No previous research experience | ||
| Obstetricians | 4 | 15 |
| Clinical midwives | 2 | 7 |
| Research midwives | 0 | 0 |
One of the sites experienced a delay in recruitment with only two women randomised during the period November 2014–April 2015. Both women refused our invitation to interview, so the qualitative study included women (n = 22) from only seven out of the eight pilot sites. Another site did not allow us to recruit until April 2015 so our final sample included between one and seven (mode three) women recruited from each of the seven sites. As can be seen from Table 1, we achieved a diverse sample of women in terms of age (18–40 years, mean 31 years), occupation, education, ethnicity, first/other pregnancy and previous experience of retained placenta.
Similarly, we achieved good representation of different types of staff members: midwives, research midwives and obstetricians (including consultants, registrars and specialist trainees) (see Table 2). A total of 27 staff members were interviewed: between one and six (mode three) at each site. Research midwives from all eight pilot sites were interviewed, recruiting obstetricians (including three consultants) from seven sites and midwives (including two labour ward co-ordinators) from a further four sites. At least one research midwife and one obstetrician were interviewed from each of the pilot sites, with the exception of one site. Although 96% of the sample was educated to degree level or above, the degree of clinical and research experience was variable. Three staff members had dealt with women declining randomisation to the GOT-IT trial.
Research questions
-
What are women’s views about the timing of delivery and content of the information provided during the pilot RCT? In what ways do they think the information and consent pathway could be improved, and why?
-
Why did women agree or decline to take part in the pilot RCT?
-
Does the consent process give women a good understanding of the trial – if not how could this understanding be improved?
-
What are women’s likes and dislikes of the trial interventions and procedures?
-
What are staff member’s experiences and views of recruiting women with retained placenta into the pilot RCT; how do they think the recruitment/consenting procedures might be improved; how, if at all, could they be better supported to undertake future recruitment?
-
Do any unforeseen difficulties/issues arise during the pilot RCT; how might these be overcome in the substantive RCT?
The findings presented below are structured under our original research questions. To safeguard participants’ confidentiality, pseudonyms are used throughout this report and all identifying information has been removed or altered to preserve anonymity.
Since qualitative data analysis was completed, three papers have been published that report key findings from the analyses. 25–27 Given the overlaps between the contents of these papers and the material presented in this chapter, the three papers are cross-referenced in various places. Readers wishing to access more details about particular findings and additional quotes should refer to these papers.
Research questions 1 and 3
The answers to research questions 1 and 3 have been combined in this final report owing to the strong overlaps in the material.
Women’s views about the information and consent pathway used in the pilot phase
Most women suggested that the information and consent pathway used in the pilot could be improved in the main trial to optimise informed consent, the exception being a small minority (n = 3) who had relatively fast and straightforward births with minimal requirement for pain management:
The actual labour was 2 and a half hours, very, very quick in the end. Once she decided she was coming, she came. There was no pain relief, just some gas and air and out she came.
Alison
It all happened within 5 hours . . . I just took paracetamol in the morning before I left. I had a couple of, you know, breaths from the gas and air, but maybe only three or four times and then I pushed and that was that. Sounds easy doesn’t it?
Ellie
These women described how, owing to their straightforward birthing experiences, they had been reasonably rested and mentally alert when they had been approached to take part in the trial. This, as these women went on to suggest, had been reflected in their ability to digest the written and verbal information given by recruiting staff and ask questions before making their decision:
I asked for a little bit more information and read the form . . . I was quite happy to read it through myself and felt I was able to logically make the decision about whether to give it a go.
Heather
So they said that there’s this project with the university . . . and I said, ‘yeah, let’s see the leaflet’. And I read the leaflet. That it would be a placebo or, you know, the medication that they give to me. And then there was a doctor, after I read the leaflet, who explained it again: what was basically in the leaflet . . . and, well, before I made my decision, I asked if the medication has any impact on breastfeeding; if there’s anything that can go into the milk, for example.
Ellie
Although these women conveyed satisfaction with the recruitment/consent pathway and felt that it had worked well in their particular case, they also suggested that other women who had just given birth might not be as well placed as they had been to consider trial information carefully and give fully informed consent. To make this point, Heather, for instance, juxtaposed her most recent birthing experience with that of her previous delivery, during which she had been in labour for > 36 hours:
I was fine signing the form but, to be fair, if I’d been in the situation like with my last delivery I don’t think I’d have been able to understand it all and to really understand what it was about, because I wasn’t quite as alert as this time round . . . Last time I fainted, and this time I was really, you know, I was really feeling well because everything went so quickly. I was absolutely fine, you could have taken me to the pub if you’d wanted to [laughter].
Heather
Indeed, in contrast to the minority described above, most of the women who were interviewed described a birthing experience that had left them physically and emotionally exhausted and for which, in some cases, analgesics had been required. 27 These women also discussed how, owing to all of the activity taking place around them, they had often been very distracted when they had been approached to take part in their trial:
I think they were just in a hurry to get the placenta out . . . they had quite a lot of doctors in and they kept telling me about the risk of infection and that they were having to – if nothing else worked – take me to theatre straight away.
Arlene
As a consequence of these kind of birthing experiences, women described liking and appreciating having been given summary information about the trial at the time of recruitment rather than having to read a full participant information sheet. Many also highlighted the benefits of having had staff present to provide verbal descriptions and explanations of the trial because, as they observed, they had simply been too tired to read written information themselves. 26,27
Women’s views about giving and gaining informed consent
Although women praised the content and mode of delivery of the information given at the time of recruitment, the majority (with the exception of those who had straightforward and relatively pain-free births) also noted that, despite the efforts of staff to convey information about the trial in a clear and accessible way, they had simply been too tired, distracted and/or emotionally overwhelmed to take all this information in:
There wasn’t anything that anybody could have done at that time . . . because giving me anything to sort of read, or consent to was pretty pointless to be honest, because I had no idea what was going on, or what I was agreeing to.
Trina
Just cause after I had him, because of the amount of gas and air that I had had, I was still, I wasn’t really understanding what people were saying to me.
Arlene
Indeed, although most women were able to recall a basic understanding of the trial, including, in many cases, use of a placebo and a randomisation process, most also described how they had not absorbed detailed information about the trial, including information relating to possible risks or side effects of taking GTN:
And did he say anything about what was in the spray and what the risks were, did he explain that?
Probably but I was out of it, I was tired, I was fed up . . . I was trying to feed [baby’s name] and I just had to get it [the placenta] out of me. I remember saying, ‘I don’t care how it works, just get it out’.
As a consequence of poor and limited ability to digest and consider all of the information provided, some women also questioned if, in retrospect, their consent had been fully informed. 27 These women reflected on how, at the time that they were recruited, they had been in what they recognised, with hindsight, to have been a vulnerable situation. Specifically, women discussed how they had been desperate to avoid going to theatre as this would have meant leaving their babies and exposing themselves to what they saw as invasive and sometimes frightening medical procedures:
And she said that I could try this new drug and it’s the last resort before an epidural to take the placenta out. So I just thought, ‘oh my god I don’t want an epidural’ so I tried it . . . The word epidural, you’re going to try anything . . . I was frightened to death of an epidural, and I’d managed to keep everything so natural so far . . . You know, just a few puffs of gas and air.
Hannah
Consequently and, as detailed elsewhere,27 most of these women said that, owing to their being in a vulnerable and/or desperate state, they had tended to make their decision to take part in the trial very quickly, in most cases without asking questions or consulting and seeking the opinion and views of other people in the room, such as their partners:
The thought of going to theatre terrified me, and all I wanted to do was to be with my baby, so I just said yes, I didn’t even hesitate. I really didn’t think about it at all.
Hannah
Because for me it was just a really quick decision that I didn’t really think about, it was just a quick ‘yes’ and, in hindsight, I just think it would have been good to have taken a bit more time to process the information and discuss it with my partner . . . Because he said to me later, you know, ‘I don’t know why you agreed with it’ . . . because for him it was a case of ‘you don’t want to try something that’s unknown’.
Lynne
Thus, again, with hindsight, these kinds of experiences led women to question if their decision to take part had been fully informed, although, as detailed elsewhere, these women were also keen to emphasise that they did feel that they had given their consent willingly and freely to take part in the trial. 26
Improving the information and consent pathway: extending information-giving into the antenatal period
In the light of their recruitment experiences, most women suggested that their decision-making and, hence, ability to give fully informed consent could have potentially been enhanced had they been exposed to trial information during the antenatal period. 25 Specifically, women discussed how this earlier exposure to trial information would have allowed them to consider it at a time when they were better placed mentally and emotionally to digest and reflect on it. Some also suggested that this earlier exposure might have enabled them to discuss their potential trial participation more fully with others and to draw on thinking and decision-making made when they were not in such a panicked and desperate state:
I think it’s probably best off discussed in your maternity bit before . . . you know when you’re in labour and the word of an epidural, you’re going to try it, you’re going to try anything, because you’re panicking and you’re desperate. And I think that really, you know, you’re not in your best frame of mind to listen either, because I was still contracting at that time to get that placenta, I was still contracting but it wasn’t coming out. So, I think if you say, if you just mentioned it in like your maternity, like ‘if this happened, if your placenta got stuck or if . . .’ I think it would be probably best off mentioned then than actually when you’re in your full labour.
Hannah
I think it was a bit late after giving birth cause I wouldn’t, I didn’t concentrate. Like I said, I was tired, just had a baby, didn’t really know what was going to happen next. So, em, that’s what I would have liked, I’d have liked to have the information before, so I could kind of go through it in my mind, know what I’ve been preparing myself for. And obviously if I was willing to take part.
Liz
Some women also discussed the potential benefits of making women more aware of retained placentas during pregnancy so that, as Tricia explained, ‘it’s not as much of a shock, because I had no idea’. As these women suggested, ameliorating shock reactions in this way might help to promote more informed decision-making to take part in the trial because, as Helen observed:
You wouldn’t be panicking so much at the time. I didn’t know what would happen if it got stuck or anything like that. So I think I’d maybe have been better if I knew what could happen as I probably would have been more prepared, obviously not been as worried about it, so I would have been better able to concentrate.
Helen
When asked when the best time would be to give women information before labour, most suggested the later stages of the antenatal period. This is because, as several women noted, earlier on during pregnancy, such as at a booking appointment, would be too premature because, at that point in time, not all women are confident a pregnancy will go to term. 27 It was also suggested that the later stages of pregnancy would be a good time to be exposed to information about the trial, as this was when birth plans were drawn up and, hence, potential birthing complications discussed, including, in some cases, retained placentas:
Em, well the midwife speaks to you about – because obviously she tells you that your placenta can get stuck and I think if she had some information then she would maybe just give you a leaflet and talk to you about it, when she’s talking about the placenta . . . I’m sure she spoke to me about it when I was 30 weeks.
Arlene
Some women who had had a retained placenta previously also suggested that, owing to their increased risk of having one again, women, such as themselves, could be targeted for information-giving about the trial:
And especially since I’d had a retained placenta before, it would have been good to have had someone like just to flag up the kind of high-risk people who might be eligible at that time might be . . . would probably be better.
Tricia
Although most women highlighted the potential benefits of being given information during the antenatal period, there were some who questioned the potential merits and/or efficacy of this. Such women observed that, because a retained placenta is a relatively rare condition, being given information might be unduly burdensome and/or this information might not be engaged with at that time:
I’m not sure what the percentage is, how many women have a retained placenta? It would make sense if it happens a lot to kind of raise the awareness. And, you know, there are so many things that can occur during labour. Em, I’m not sure how many leaflets you can read in advance, you know what I mean, you get, you already get so many things about what can happen. So I don’t know if I would have looked into it in much detail.
Heather
However, as described previously,26 even when women were ambivalent about the potential benefits about being given information during the antenatal period in their particular case, they also emphasised the importance of exposing women to trial information so they could decide for themselves whether or not they wished to engage with it. 27 The majority, in contrast, indicated that, for them, the antenatal period had been a time when they had been ‘information hungry’; that is, they had been very receptive to receiving and accessing information to make informed choices regarding their pregnancy and birth. This included Susie, who described how she had undertaken her own research and found out about retained placentas during her pregnancy, and Trina, who considered discussions about birthing complications to be an important and necessary part of responsible birth planning:
Yeah, I think so because obviously, like I say, I’d done a lot of research into it but I still didn’t know about the GOT-IT and I didn’t know that that was an option. So yeah, I think so because then if I would have been approached with it, you know, me and my husband could have maybe talked about it beforehand and said, you know ‘if I did need the spray, would I have it?’.
Susie
So yeah, I do think with the antenatal stuff and midwives possibly making that a little bit – or even just a leaflet in with your pregnancy notes. Because we get leaflets about looking out for pre-eclampsia. You know we get leaflets about looking out for gestational diabetes and things like that, you know throughout your pregnancy, so we can make informed choices.
Trina
Improving the information and consent pathway: extending information-giving into the early postnatal period
Owing to their limited ability to absorb or retain information about the trial at the time of recruitment, some women also highlighted the benefits of revisiting and rereading written materials after the trial. For this reason, women who had lost or misplaced their packs following recruitment described appreciating having been given new ones prior to their discharge from hospital. 26 Those who had experienced a postpartum haemorrhage, or another complication, attached particular importance to revisiting trial information. Not only did these women describe ensuing upset and distress but they also discussed how their experiences had led them to question and regret their decision to take part in the trial and, more specifically, their failure to engage with possible risks and side effects of taking GTN. 26 As well as valuing opportunities to revisit trial materials, these women described needing and benefiting from debriefing sessions with staff to discuss what had happened to them. They described these sessions as enabling them to understand whether the complications they had experienced had been due to having had a retained placenta or if they could be explained by having taken GTN. 26 As these women indicated, they were principally looking for reassurance that, were they to give birth again, they would be unlikely to encounter similar problems:
I don’t know whether it was due to the spray or because of the condition itself, I just bled, I’ve no idea . . . because what worries me if there is a chance I could have another one [child] and it worries me that if I did have another one, could it happen again? Because, you know, if I thought, well it’s the drug that caused the reaction, I’d feel better because I’d think, ‘well, you know it’s not going to happen again’, hence, a 10-minute conversation really would have been nice, would have made me feel a bit more at ease about it all.
Lynne
In general, women benefited from receiving input from staff following trial participation. In some sites, this occurred because treating clinical staff are required to visit and debrief patients who have had a postpartum complication, such as retained placenta, prior to their discharge. In addition, as part of the trial, research midwives visited trial participants prior to their discharge whenever this was possible to deliver questionnaires, approach women about the qualitative study and check they were doing well. However, if women were admitted on a Friday, they could be discharged over the weekend prior to the research midwife doing her rounds. Thus, in a few cases, such as Lynne’s above, there was the chance that women who experienced a complication could be discharged without having the opportunity to discuss their experiences with a staff member (however, see Research question 6).
Research question 2
Reasons for agreeing
As already indicated above and detailed elsewhere,26 women provided very similar reasons for taking part in the trial. Principally, women described having been very anxious about, and wishing to avoid, going to theatre if at all possible. Indeed, it was for this reason that some women also conveyed the belief that by offering them the chance to take part in the trial, trial staff had been working in their best personal and clinical interests. 26
Related to their desire to avoid going to theatre, women discussed how they had wanted to do everything possible to be there for, and avoid separation from, their newborn baby:
I just wanted to get this placenta out, I was fed up and I just wanted to hold my baby.
Lynne
And to be honest, the reason I said yes, was because obviously the thought of theatre wasn’t nice and I really didn’t want to leave the baby.
Susie
Some women also described their efforts to keep their birthing experience as natural as possible, and conveyed their sense of achievement when they had avoided all but minimal pain management (gas and air) during the birth. Although such women recognised that GTN comprised a medical procedure (see below) they described having been motivated to take part in the trial, as they considered taking a spray to be a much less invasive and medicalised option than a theatre visit and accompanying epidural:
I didn’t really want to go to theatre having managed to go through labour normally and naturally . . . I didn’t really want to have an epidural, I just wanted to get on with it and keep it as natural as possible. So I thought, ‘right, OK’, so that basically was my reasons for consenting to it.
Lynne
Although women were motivated primarily by their own needs and those of their newborn, many were also keen to emphasise that they had welcomed their participation in the trial as they saw the study as a useful and well-intended piece of research that might help prevent other women from having to go to theatre in the future:26
And it being a research programme, it’s more like a research, at least it was good to be part of the research so that was why I went for it.
Why was it good to be part of the research?
Well, it’s good to be part so that at least it will help in, well, I’d say it will help to develop so many things, you know? . . . So I think it’s good for somebody to at least try something new and see whether that something new can be good to help out in so many things. So that’s just like when maybe researches are made with regards to like this Ebola crisis now, there’s so many researches that have been undergone and so many people are contributing to it just to eventually get a drug that would combat the problem. So I think the research is just more helping out to see what can be done to prevent a woman who has a retained placenta, to prevent her from going into the theatre.
So why did you decide to take part in GOT-IT?
‘Cause it’d be great to know if I have another baby and I have – if I have another baby I’m likely to have a retained placenta having had one, so if they answer the question by then . . . and I guess we all benefit from making, finding answers that make babies and mothers safer.
Women also noted that trial participation required very little from them because they had to wait an additional 15 minutes only before a referral to theatre in the event of the spray not working, and taking the spray was considered a very simple and easy thing to do.
Reasons for declining
Six women declined participation in three sites during the pilot phase; however, as described earlier, it was not possible for us to interview any of these women. Unfortunately, we were also unable to interview members of staff who attempted to recruit women at the first site, but did interview staff at the others, including those who had been directly involved in explaining the trial to these women, and others who had either observed these failed attempts at recruitment or heard about them.
Staff reported three main reasons for women declining trial participation. The first was primarily to do with women’s anxieties about their infant’s health. Staff described one instance when a woman refused to take part because her baby had been taken to the neonatal intensive care unit. This woman was described as anxious, overwhelmed and unable or unwilling to make a further decision at that time:
The one that I was around for, her baby was rushed off to neonatal unit straight after delivery in an emergency. So the baby was born in poor condition and was whisked away to the neonatal unit. So then she went on to have a retained placenta. So the registrar that actually tried to consent her, she’s very, very positive, very keen for the study. So she said – and I spoke to her – but she said that the woman just said that it was too much for her to think about it at the time. Obviously she was upset. The baby had just been whisked away. And even though the potential was that it might stop her going to theatre, she was more that she didn’t have – she wasn’t able to think about it and make a decision at that time. So she just said she would rather not.
O
In a couple of cases, women had already tried a number of different methods to deliver the placenta, and staff described the women as too tired and exhausted and/or not wanting to waste more time waiting to deliver their placenta. Staff believed that these women had declined participation in the GOT-IT trial because they felt that this would delay them from being able to hold and bond with their baby. The midwife who had been involved with one of these cases (see below) speculated that if had she mentioned the trial sooner, namely, immediately following the diagnosis of retained placenta, then the woman concerned might have been more willing to consider it after she had exhausted other options:
We tried other options. We tried putting baby to breast, jasmine compress, we’ve got her sat up on the birthing stool. We put her legs into McRoberts. We got her standing, and it just wasn’t happening basically, and eventually it was decided that obviously she would need to be going to theatre to get the placenta out, but when it was offered for the GOT-IT – for the trial, I don’t know if it had just been too long where we’d been trying, but she was just, she’d just had enough basically. She wanted to just be able to be with her baby properly and I think she just probably thought the manual removal had been offered. So I think she probably just thought, ‘I just want it over and done wi’ . . . I just think if I’d have offered it sooner it might have been a success, the lady might have taken it. So in hindsight it’s probably made me realise that I should have probably opened me mouth a bit sooner [both laugh].
B
The final reason given for women refusing trial participation was that some women were more ‘difficult’ to engage than others:
The one who declined I don’t recall that she particularly asked many questions of me because she was so kind of negative about anything it was more of a straight no rather than mulling it over.
Dr S
Staff described a woman at another site who had rebuffed all attempts to describe the trial to her. Arguably, this woman can be seen as an indirect decliner because she received minimal information about the trial and its procedures:
And she [doctor] did say to me, you know, this lady’s been – it’s probably not the right wording what – this lady’s being quite difficult, hasn’t really wanted anything that we’re doing, and I think it’s probably better that you don’t come in the room . . . And then she just came back and said, to be honest, she wasn’t letting me even try and have the conversation. She just kept shutting me up and saying, ‘no I don’t want to be part of it. You’re confusing me. Stop adding more’.
P
Unfortunately, as there were so few women who declined trial participation during the pilot phase of the trial, and none agreed to participate in the qualitative study, we can base our analysis only on staff observations and reports.
Research question 4
Trial procedures
Women raised very few concerns about trial procedures; indeed, as described above, the quality and content of the materials given at the time of recruitment were widely and almost uniformly praised, as was the method of delivery of trial information (i.e. verbal as well as written). However, as also documented above, most women did highlight the potential benefits of changing the timing of the delivery of trial information so that this information could be accessed in the antenatal and postpartum periods.
Most women said that they had been happy to sign a consent form at the time of recruitment and presented this aspect of their trial participation as having been straightforward and unproblematic. However, a small minority conveyed a strong preference for a verbal consent procedure to have been used in their particular case, because of exhaustion or complications such as bleeding.
Trial interventions
In general, women perceived the trial intervention (i.e. ‘the spray’/GTN) to have been easy and simple to administer, and as having been preferable to taking a tablet or being injected:
It was very straightforward, literally just a spray under the tongue, so it wasn’t invasive, it wasn’t painful or anything like that . . . I actually quite liked the fact that it was just you know, quite easy for me to just literally open my mouth and lift up my tongue, kind of thing. You know there was no having to swallow tablets, there was no injection or anything like that.
Kirsty
In some cases, the ease and simplicity of administering the spray was also described as having facilitated a decision to take part in the trial, with some women noting that they might not have agreed to participate (or not agreed so readily) had a significantly more invasive procedure been involved:
Em, well it was – because it was a spray under my tongue it was easy, so it was fine. If they’d said, ‘it’s an injection’ yes I probably would have still had it. If they’d have said, it’s something that’s inserted manually, I would have probably said, ‘maybe not actually’. Do you know what I mean? If it was something that was more, em it was more invasive then probably not. But a spray under my tongue, in the grand scheme of what had just happened was fine.
Alison
Some women also described having prior knowledge of GTN, either by virtue of being a health professional or because they had a friend or relative with angina who used the drug. As these women noted, this prior knowledge had offered confidence and reassurance regarding the drug’s potential safety, with some, including Anna (who was medically trained), also noting how this prior knowledge had facilitated a quick decision to take part:
I think I made it instantly, cause – because it was just, well I say this, but it was just GTN but I mean if it was something a bit weirder, if it was a medication that I maybe wasn’t so familiar with I’d never prescribed myself, that I didn’t really know the side effects, if it was something quite new, I think I would have asked more, well ‘is it going to get into my milk? Will it get into the baby?’ and a bit more about the side effects. And how new it is. If it was something really, really new I might have chickened out because there’s just more unknowns.
Anna
Although women’s views about the intervention drug and its method of administration were mostly very positive, some, as indicated earlier, did question if, in retrospect, they had not paid adequate consideration to the possible risks and side effects of taking GTN. This included those who went on to experience complications during the trial (see above). There were also some women, such as Tricia, who shared a (mis)perception that because the drug was a spray, which was simply administered under the tongue, it had to be relatively benign in terms of its potential impact on the body:
I think because it was just a spray. I know that shouldn’t make a difference but if it had been a jag or a tablet it might have been different I think. I think I was thinking of it as quite . . . not invasive, just the fact you’re spraying my tongue . . . it didn’t seem as scary almost . . . obviously it should be going down to affect where it needs to affect but with a tablet it’s going right into your body. I know it’s a wee bit daft thinking that really.
Tricia
Research question 5
Experiences and views of recruitment
As discussed in research question 6, because randomisation to the GOT-IT trial takes place in the immediate postpartum period, trial recruitment and the delivery of the trial intervention requires the involvement of clinical staff. Clinical staff were very positive about recruiting to the GOT-IT trial. It was seen as an easy study to recruit to because the intervention is straightforward, relatively non-invasive, familiar – and, therefore, perceived as safe – and everyone, both the women and clinical staff, wants it to work:25,26
I feel its quite an easy thing to approach women about, I don’t think it’s a difficult, a difficult sell, if you like I don’t think it’s asking a lot of people and I think most people are very keen to be involved in research and also if they think it’s going to stop them having to go to theatre they’re very keen!
Dr A
Staff described the trial as easy to explain to potential participants owing to the simple nature of the intervention and the trial procedures. Hence, most were very confident that the women they recruited had received enough information so that they had an adequate understanding on which to base their decision about trial participation, despite having just given birth:26
As long as you explain it slowly and clearly and don’t overdo it then I think it’s fine, that’s probably the level you should be aiming for when somebody’s just had a baby. I don’t mean that in a condescending way, I just mean they are exhausted and they’ve often been up for 1 or 2 days so giving them too much information is unfair isn’t it . . . All they need to know is that it’s safe but you may or may not get it, it is a bit of spray and it won’t alter the outcome other than it might reduce [you] going to theatre.
Dr F
Staff were asked about the types and amount of information about the trial that they perceived as necessary and sufficient for women to give informed consent in the postpartum period. They said this usually involved providing women with a brief account of uncertainty regarding the effectiveness of the trial drug, the possibility of visiting theatre for a manual removal, a short account of randomisation and use of placebo and any side effects they might experience from GTN:26
I think they just need to know that it may or may not work, that you may still end up with the same treatment that you’ve had anyway, that it’s a relatively safe drug but you may get some side effects.
Dr F
I think I just had said that, it’s a trial, so you might get the real thing, or you might get a fake. And I don’t know which one you’ll get. We’ll never know which one you had.
Dr B
When asked about which trial materials that they had found useful when recruiting, many staff said that they had used the summary information sheets, rather than the full patient information leaflets, as they felt the former contained the right amount of information about the trial for women to take in at this particular time. They said they had also given women/their birthing partner the full information leaflet either at the time of recruitment (consenting doctors) or before discharge from the hospital (research midwives):
I just keep it as simple as I can because there’s that leaflet that’s really detailed and I think that’s a little bit beyond the women maybe when they’ve just had a baby just because they’re tired and, you know if they’ve had opiates and stuff as well they won’t really be in the mood for reading that will they? There’s a simplified version isn’t there which is much more straight forward and I think that’s probably the level that you should be aiming for isn’t it when somebody has just had a baby.
Dr F
However, although most clinical staff felt that they had obtained informed consent from women they recruited, a few acknowledged the difficulties of establishing valid informed consent when women had just undergone a long and painful labour and were still under the influence of analgesia:
Someone who’s maybe a bit distressed or a bit more tired, the fact that she has to go after they’ve delivered the baby, after going through the delivery, then they might not want to – they might not want to listen. And they might not want to read things. That’s – I think that’s the disadvantage of it that it’s something it’s going to be done after delivery where the woman is exhausted most of the time.
Dr C
One staff member, who had consented a number of women, differentiated between women’s understanding of trial design and the risks of the trial intervention and other non-clinical aspects of the trial. Dr J was particularly worried about women’s ability to understand some of the less immediate risks of participation, such as data extraction, data use and the fact that they would not benefit financially from participation:
I found it really difficult to talk to patients especially when they’re so much in pain. And when you tell them to sign a consent form with so much information in there it’s difficult for them to fully understand what they’re supposed to do, and what they’re signing for . . . when patients have just delivered their baby they mostly go into a bit of like adrenaline rush and then they – shiver and it’s difficult for me to relay and to tell them everything in the consent form and also for them to read the leaflet. So basically I’m just trying to push this trial without them fully understanding what the trial’s all about.
Dr J
As noted above, given the clinical situation, the GOT-IT trial protocol allows for women to give verbal consent to study participation, provided that this is supplemented by written consent at a later time. However, even though the staff were confident that providing women with a verbal description of trial procedures was sufficient for gaining their informed consent at this time,27 some, particularly the more junior doctors, were not prepared to rely solely on women’s verbal consent in this context, although some women said they would have preferred this (see Research question 4). 27 As detailed elsewhere, staff reservations stemmed from concerns about potential litigation and the need to have documented consent to present as evidence. 27
Perhaps unsurprisingly, Dr J, who had expressed concern about women’s understanding of the non-clinical aspects of the trial, was pleased to learn that the GOT-IT consent pathway permitted a staggered consenting process (i.e. verbal followed by written consent), if necessary.
Potential improvements to consent procedures
Although most of the staff interviewed were happy with the consent pathway employed in the pilot phase,26,27 many of the women interviewed said that they would have liked to have been informed about the possibility of having a retained placenta and the trial earlier in their pregnancy. We asked staff members what they thought of this suggestion and also explored their preferences regarding the timing of information giving.
As we have noted elsewhere, in contrast to the women, staff were either strongly opposed to or ambivalent about providing individualised trial information antenatally:26
Well if we’re talking to people beforehand and saying, oh by the way, after you’ve had a vaginal delivery you might have your placenta being stuck and then we’re running this trial whereby you can choose to have a drug or not. And if it doesn’t work then we take you to theatre. I think my feeling would be actually that, given that the vast majority of those women that that won’t happen to, in some ways the sort of increase in maternal anxiety that you would cause by even mentioning these things in advance. My feeling was that it’s appropriate to counsel at the point that we’re doing at the moment.
Dr G
It was suggested that even if information were to be made available to women earlier in pregnancy (e.g. trial leaflets placed in booking packs or discussions about the trial) most would not engage with it because they are given so much information in their antenatal appointments:
From experience in the antenatal period, women are bombarded with so much information they struggle to take it in and there’s certainly good evidence that written information very few people actually read, they go on to the websites which show them nice photos of babies and things and they don’t necessarily read the information.
Dr H
Many staff members pointed out that receiving information about retained placenta and the GOT-IT trial may cause women undue worries about the impending birth:
That’s a difficult one I think. Because they’re given so much information, the women, that it’s hard for them to take it all on board. And I think if you go round – you certainly couldn’t go round telling every single woman, that just in case you have a retained placenta we’re taking part in this trial. ‘Cause then you wouldn’t want to frighten them. And you know, it’s a small number of them that will actually end up having a retained placenta.
G
As G suggests, retained placenta is an infrequent postpartum complication and many of the staff we interviewed talked about the resource implications of ensuring that all women receive information about the trial during the later stages of pregnancy, when only a small proportion might benefit:
It’s just resources as well, isn’t it? I mean if you give everybody information leaflets coming through antenatal clinic about retained placenta for, in comparison, a minimal number that will have a retained placenta, it seems a lot of work and a lot of paperwork. With regards to making them worried, no I think education’s a good thing. I don’t think it’d particularly make women worried. I think women do generally want to be informed of what trials we’re running and what’s on offer to them, so they can make their own decision really. So I don’t think that’s a problem. It’s just really giving everybody some sort of information for a small number of people. I think that’s the only problem.
I
Although staff were resistant to the idea of providing individualised trial information antenatally, many agreed that it would help the consenting process if women were aware that the GOT-IT trial was taking place in the site. Thus, staff suggested raising general awareness about the trial by advertising the presence of the trial in the site by using posters in antenatal clinics, triaging areas and birth centres as well as advertising on the trust’s social media sites. This is a practice that many sites adopted during the pilot phase. This was seen as a compromise that would allow interested women to access more detailed trial information for themselves and other women to avoid what they might perceive as potentially anxiety-provoking or irrelevant information:
It’s a minority of women isn’t it? So I don’t want to unduly worry. You know that’s the problem with giving information about research studies that only are applicable to a small number. You’re worried: are you going to worry people unnecessarily? No I don’t think you will. I think because we had posters up saying ‘we’re doing a study on retained placenta, when the placenta doesn’t come out after delivery’. And, you know, ‘if you go in the study the benefits are: you may not need’ – yeah I think there’s no harm with posters or leaflets around. I think posters might be better in some ways.
Dr I
A few members of staff, in contrast, such as Dr C, felt that the trial should be explicitly brought to women’s attention when they arrived in the labour ward/birth centre:
Do you think there is a different time at which women could be given this information?
Yeah. that’s what I was going to come to. I think they should be informed, you probably tell me oh you shouldn’t give it to all women. But I think they should know that a trial is going on in the delivery unit that they’re in. And if the situation has to happen, then you will be informed about so and so. And I think it’s better to give the information before rather than just after they’ve delivered the baby.
At what point do you think it should happen before?
I think they should be informed on arrival to a labour ward. I don’t think it’s something that you can give antenatally, in the antenatal clinics, because I don’t think it would make any sense to women.
Others felt that information could be given to women even earlier in the care pathway and they could be made aware of the trial during their antenatal appointments. One site had acted on this and said that posters and information about the GOT-IT trial were not only available in the labour wards and/or triage areas in the hospital but also in the community bases:
We’ve got them in every room in the birth centre. We’ve sent posters out antenatally to the community offices. We’ve got bases, where we are, where midwives work from bases in the community. So we’ve sent posters out to the community offices, and information leaflets. So there’s education for the women antenatally, that there is a trial running at [site X] and that the information’s there should they want to read it.
I
Finally, when prompted, most staff members agreed that although explicitly informing all pregnant women about the GOT-IT trial may be neither feasible nor acceptable, triaging antenatal information giving, so that information about the study is targeted at women who have previously had a retained placenta, might be a good idea and is a more acceptable use of resources:
What do you think about sort of targeting those women [with previous retained placenta] in particular?
Oh yeah, absolutely. I think that’d be something that would definitely work well for women that are aware of it, they know what it worked out to be, because they have got that fear . . . So, I think it would definitely be great to target those kind of women that have had that previous experience because they know about it and they know that there’s an increased risk. Because there is, I mean, I’ve had a couple of ladies myself that had had previous retained placentas and this was them again holding on to their placenta, so for whatever reason it just doesn’t come away from the wall.
In one site the staff reported that women who had previously had a retained placenta had been triaged to receive trial information on an ad hoc basis, and this had been well received by the women concerned:
If women have had a retained placenta before we do warn them that it might happen again. We haven’t gone down the lines of talking to them about GOT-IT formally but it certainly has been mentioned informally. I would include myself in that because I have, when we first started I came across somebody and she was very happy with the study but of course she didn’t have a retained placenta but that’s just a one-off story, but informally that goes on but we haven’t formalised that.
Dr H
Support for future recruitment
None of the staff involved in recruitment in the pilot phase identified themselves as requiring further support for recruitment/consenting. Indeed, all of those whom we interviewed were very positive about the support they had received from their local research teams. However, the research midwives who were interviewed identified a number of practical strategies (tips and pointers) that they had devised to improve trial delivery and, thus, indirectly support trial recruitment. These were collated and a training resource to aid implementation of the trial in new sites was developed [see Microsoft PowerPoint® (Microsoft Corporation, Redmond, WA, USA) slides accessible at www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/)].
Research question 6
Getting the trial up and running and keeping it going
Although the rollout of the GOT-IT trial across the pilot sites did not generate any trial-specific issues for staff, implementing the trial took local research teams by surprise in a few of the pilot sites. The GOT-IT trial was the first postpartum trial that some sites had been involved with, and relying on clinical staff to recruit and consent women was seen as a major issue for the research midwives and local principal investigators:
The hardest bit’s probably retained placenta can happen any time 24–7 and we’re only here you know during office hours. So we are having to rely on the clinical staff that have already got a lot of things to remember to think about GOT-IT.
Research A
I think difficult because it’s not the midwives recruiting. And all the studies I’ve worked it’s always been me doing the recruitment. Whereas this has been different because there’s a lot of training the doctors up. And, you know, letting them know about the study. And obviously because it’s quite a big unit there’s a lot of doctors to get trained up. So that’s been different. We’re not the ones actually doing the recruitment. And then so a lot of it’s been sort of chasing. I know a lot of it’s been chasing missing data and things that the doctors have missed out really. And sort of following up which women they were, if we’ve sort of lost track of some of them. So it’s been different to the other studies I’ve worked on.
Research F
Requiring busy labour ward staff to deliver the trial was described as needing a great deal of forward planning and training, particularly in sites with bigger delivery units in which a large number of staff needed to be trained. To facilitate this training, the trial office developed a trial-specific good clinical practice light training package. Compared with full good clinical practice training, which normally takes a minimum of 6 hours, the good clinical practice light package was designed to be completed in < 30 minutes. The training package comprised slides, a protocol synopsis and a summary of the core principles of good clinical practice. The good clinical practice light package was designed so that labour ward staff could complete it online in their own time or it could be delivered face to face by the research midwives. Staff completing the training online were required to complete and score 100% in a multiple-choice test to provide evidence that they had completed the package and learned from its content. The package was formally risk assessed by the trial’s sponsor and was deemed to fulfil the training requirements required by the Medicines and Healthcare products Regulatory Agency for clinical staff to recruit to a drug trial. It was hoped that sites would be able to use this more focused, shorter good clinical practice package to facilitate rapid, comprehensive training of clinical staff.
The level of good clinical practice that staff in individual trusts need to undertake is determined by the local research and development departments. In two out of the eight pilot sites, the local research and development departments required their staff to undergo full as opposed to good clinical practice light training. In a further site, the initial approval to use good clinical practice light was withdrawn. This decision was subsequently overturned after discussions with the local research and development director. In practice, in the pilot trial, the requirement for staff to undertake full as opposed to good clinical practice light training delayed the recruitment in only one out of the eight sites. The requirement to ensure that staff had the locally approved level of good clinical practice training, were familiar with the specific trial processes for the GOT-IT trial and had signed the delegation log delegating them to recruit to the trial presented a significant logistical challenge for the research midwives:
So that was a whole new area that I’d never dealt with before, we’ve never had the sheer amount of doctors that I’ve had to do the training with for this study that I’ve had to do with other studies . . . it’s been a bit of a hard slog, I’ll be honest . . . I mean, it was tough going round and finding them all but now I’ve kind of done that bit it’s OK . . . it was easier than what I thought it was going to be.
Research J
A few things made staff training at larger sites more problematic. First, because of the larger numbers, the research midwives were generally less familiar with clinical staff. Second, these sites also used more locums creating a continuing need for good clinical practice training. Finally, larger sites have a greater volume of ongoing and/or competing research studies at any one time. Thus, although smaller sites may have fewer potential recruits, the trial was reportedly easier to set up in these locations as there were fewer doctors/midwives to train and the research midwives, particularly those who also worked clinical shifts, already knew most of them:
So how has it been training the staff?
Well, like any training it’s hard to find time to really engage with staff without being interrupted. So you’re kind of – thankfully research midwives and myself are both clinical midwives as well. So I think that has benefited this trial. Because if it is quiet on the unit you can badger them. But obviously, they’re like kids, they’ve only got a short time span that you try to get the most important things across to them because, you know, they’ve got other things.
The research midwives at all sites, but particularly the larger sites, talked about the strategies that they had employed to ensure that eligible staff received the good clinical practice that their local trust required them to have prior to recruiting to the trial. For the research midwives who delivered face-to-face training, these strategies included catching medical staff in quiet moments, using a GOT-IT champion (see below) to recruit and train staff, giving talks in educational sessions, e-mailing newly recruited doctors in advance of them starting their rotation and getting the local principal investigator to encourage individuals to complete training online or attend for face-to-face training.
Delivering the GOT-IT trial involves more than just ensuring that medical staff are good clinical practice trained, familiar with the trial processes and on the delegation log. Midwives who look after women during the birth are often the first health professionals to become aware that there is a retained placenta. Although midwives cannot assess eligibility for the trial and prescribe the study drug, they can refer women to medical staff so that they can formally consider their eligibility for trial entry. If this referral does not occur, potentially eligible women may not be given the opportunity to consent to the trial. The research midwives therefore talked about how they needed to engage all labour ward staff members and make them aware of the trial and its eligibility criteria. Thus, in addition to talking about the trial with all ward staff and making sure that the GOT-IT trial was raised in daily safety briefs, the research midwives described how they tried to increase the visibility of the trial on the ward by fly posting on delivery suite walls, triage area walls, in tea-making areas, near the drug cupboard on the labour ward and, most effectively, on toilet doors. In addition, in many sites GOT-IT flow charts and summary sheets were pinned to labour ward walls and placed in or near the drugs cupboard:
I put laminated summary sheets up for GOT-IT as well so it’s just a matter of them peeling it off the wall and giving the summary sheet to the woman to read.
Research J
In some sites all trial materials (i.e. paperwork, checklists for filling in paperwork and trial drugs) were packaged up together and put in easily visible and accessible places (i.e. the drug cupboard) so that staff awareness was maintained and they could quickly and easily find the trial materials and complete them correctly.
As retained placenta is a relatively rare occurrence, sites may not randomise for weeks at a time and this gives research midwives the unenviable task of keeping the GOT-IT trial fresh in people’s minds. A number of strategies were reported, such as broadcasting new randomisations at the site, issuing rewards to recruiting staff, issuing GOT-IT pens, issuing sticky notes and issuing certificates to add to training portfolios:
I think getting a certificate is actually quite useful for your portfolio and things, to say that you’ve been involved in clinical trials or whatever.
Dr B
And each member of staff who’s been involved so far they’ve all had their certificate printed and a bit of a fuss made really. And they seem to be really quite excited. And they’ll ring me up, oh got another case for you. And they seem to be quite excited about process and everybody wants one. Everybody wants to get a case so they can have the certificate.
M
In some sites, the first person to be randomised received a bunch of flowers or perfume from the staff and in one site the principal investigator had provided money for John Lewis vouchers prizes (John Lewis Partnership plc, London, UK) and those who had randomised women were entered into a monthly draw. Finally, research midwives used incentives in the form of edible treats for staff – ‘GOT-IT’ sweets, biscuits and tea.
In addition to publicising recent randomisations in individual sites, the research midwives and trial office also kept staff up to date with general trial progress at departmental meetings and through trial newsletters and e-mails. The research midwives also publicised overall recruitment rates across pilot sites to encourage inter-site competition, a strategy that some said might backfire if your site does not recruit too well. Although all of these strategies were seen as effective, the research midwives said that having a continuing physical presence on the ward was essential for maintaining clinical staff awareness of the need to recruit to the GOT-IT trial. All the research midwives said that they made a great effort to go onto the labour ward/delivery suite and talk about the GOT-IT trial whenever they could:
It’s keeping visible on the delivery suite and going up there frequently, making staff feel appreciated for doing it as well. And not just leaving it to them and saying, right here you are, you do this. You get on wi’ it. Ring me when you’ve got one. It’s like involving them all the time, giving them the certificates, making them feel they’ve done something deserving credit really.
Research M
I make sure that I am visible in in the areas every day that I’m in work, and I think just, you know, walking through labour ward and saying hi to people helps keep the profile going.
Research A
It was suggested that the profile of the trial could be maintained most effectively by having a GOT-IT champion at every site. Dr B reflected on the important role played by the principal investigator in their site in keeping the GOT-IT trial to the forefront of staff awareness:
We’ve obviously got the advantage of having PI [principal investigator] in the department. So, you know, she’s a constant reminder about GOT-IT. There are GOT-IT pencils I think . . . I guess you just need somebody like principal investigator at every place, don’t you? But you must have to have – will you not have a – you know a co-ordinator at each site or will that not happen? If you have somebody that’s the GOT-IT champion in each hospital and they’ll chat to people about it and keep it in everybody’s mind. Cause it is having somebody physically around that every so often brings GOT-IT up in conversation that keeps reminding you.
Dr B
In another site, a junior doctor had been assigned the responsibility of championing the trial; alongside the research midwives they were responsible for ensuring that the staff were good clinical practice trained and keeping the GOT-IT trial at the forefront of people’s minds. The idea of appointing a GOT-IT champion for each site from the local medical team or senior midwifery team was endorsed by the implementation group at their meeting in May 2015.
Debriefing following trial participation
As noted above, women who had a postpartum complication (including postpartum haemorrhage) spoke of their distress and worry that this may have been caused by study participation. 26 These women said that they would value, or had valued, the opportunity to discuss their experience with clinical/research staff. Staff agreed that offering all women an opportunity for debriefing after the trial was a good idea as it helps them to understand what had happened and gives them an opportunity to ask further questions, particularly when they have experienced postpartum complications. In most cases, research midwives said that they had debriefed women as a matter of course before they left the wards. Clinical staff in other sites noted that the patient pathway within their trust already included a debriefing session in the event of birthing interventions/postpartum complications. 26 In one site, the local principal investigator said that in addition to the clinical team who would routinely debrief women following a postpartum complication, she would try to visit any woman who had experienced complications following randomisation to the GOT-IT study:
If anybody has a major postpartum haemorrhage obviously, and they’re in this study I will go and speak – you know show my face as well if I’m around. Obviously, if it happens on a Friday and I’m not on that weekend . . . it might be that they go home. I can’t promise that I could do it with everyone, because I might not be there. I might be on leave, but if I’m aware and I’m there I’ll definitely go speak to them and just say: ‘have you got any’ – is that alright, by the way?
Dr I
Implementation: feedback of findings
As indicated earlier, the study’s implementation group was brought together for a face-to-face meeting soon after completion of the trial’s pilot phase. This meeting was held in Edinburgh on 14 May 2015 and was chaired by Dr Claire Snowdon, a qualitative perinatal trials expert who is also a member of the trial’s steering group. Those attending included representatives from the eight pilot sites, Dr Fiona Denison (chief investigator for the GOT-IT trial) and a patient representative.
The meeting comprised a 45-minute PowerPoint presentation delivered by Julia Lawton and Nina Hallowell that covered all of the key issues and findings presented above. This was followed by a 30-minute facilitated discussion to develop a series of recommendations that could be implemented during the main phase of the trial to maintain successful recruitment, optimise and sustain effective trial delivery and maximise opportunities for gaining full informed consent.
The final document, containing a summary of the recommendations of the implementation group, is contained in Box 1, along with how the recommendations were implemented in practice. The qualitative research team also compiled a set of training slides (see www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/) for use as a training resource when new sites came on board in the main trial. This training pack included implementation group recommendations and a number of tips and practical strategies developed by the pilot sites for optimising successful trial delivery. The trial manager circulated this training resource to all the sites in the main trial.
The implementation group recognised that the qualitative team had not been able to interview women who had received antenatal information about the trial because the women recruited during the pilot would not have had opportunities to be exposed to this information.
Recommendation 2The implementation group discussed the findings from the interviews with women in which they described the potential benefits of trial information being given during the antenatal period. These were weighed against the views of staff interviewed in the study, who were more ambivalent about providing information to women during the antenatal period. The implementation group also noted the cost and logistical implications of following Vernon et al. ’s35 recommendation for giving all pregnant women in the trial sites information leaflets.
Implementation: the local challenges of informing potential trial participants and potential solutions were discussed at site initiation visits and at the regular monthly research midwives’ teleconferences. The latter were organised and led by the research midwives based in Edinburgh.
Recommendation 3The implementation group recommended that the pathway to be used in the main trial should draw on the principles of the pathway developed by Vernon et al. ,35 but be ‘scaled down’. It was proposed that information about the trial should be displayed in settings in which women receive their antenatal care in the form of posters and leaflets.
It was also agreed that these documents should contain clear information about how women could access further information should they wish to do so; for instance, by providing contact details of trial staff and links to websites (GOT-IT or local trust websites) containing more information about the trial and about retained placentas). The potential to use social media such as Twitter (Twitter, Inc., San Francisco, CA, USA) and Facebook (Facebook, Inc., Menlo Park, CA, USA) was also highlighted. It was suggested that the proposed method of information delivery would meet the needs of women who are ‘information hungry’ while not overburdening those who are not. The committee noted that any changes or additions to existing documentation would require ethics approval.
Implementation: sites were encouraged to develop local solutions for information dissemination to women. Examples of good practice were discussed and shared at the midwives’ teleconferences.
Recommendation 4The implementation group recommended that those women who are identified as being at increased risk of having a retained placenta (e.g. because of having had one previously) should, when possible, be targeted during the antenatal period and these individuals should be given a trial information sheet.
Implementation: this particular issue was highlighted and regularly fed back to local sites via the monthly midwives’ teleconferences.
Recommendation 5Some women experienced a postpartum haemorrhage and questioned if this was related to their participation in the trial or if it was because they had a retained placenta. The implementation group recognised that it was good clinical practice for all women with a retained placenta who had a postpartum haemorrhage to be debriefed about their experience. In addition to women with a retained placenta and postpartum haemorrhage, the implementation group recognised the importance of offering a specific debriefing session on an as-needed basis to trial participants who want to know more about retained placentas, the trial and its procedures so they can make sense of their birth experience. Ideally, this should take place before they leave hospital. If this is not possible, follow-up should be provided, although some staff members of the implementation group noted that it is ethically and logistically more difficult to contact women after they are discharged. Therefore, it was advised that local solutions to this problem should be developed.
Implementation: local units were encouraged to develop solutions for this. When practical, the local research midwives tried to debrief all participants before they left hospital.
Recommendation 6The implementation group agreed that women should be thanked for their participation and that, ideally, this should be done when the research midwives visit them on the wards. If this is not possible, women should be sent a thank-you letter/card (see www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/).
Implementation: in addition to face-to-face debriefs, sites were provided with thank-you cards.
Recommendation 7It was observed that in certain sites the appointment of a GOT-IT champion from the clinical staff had been very useful in promoting the trial amongst clinical staff. It was suggested that all sites might benefit by signing up a GOT-IT champion (e.g. junior or senior doctor) to support the research midwives. This person should be awarded a certificate to acknowledge their work (designed and issued by the trial office) and their role publicised locally (see www.journalslibrary.nihr.ac.uk/programmes/hta/122901/#/ for additional information).
Implementation: sites were provided with certificates to give to local site champions. Local sites were also encouraged to thank all those who recruited to the trial. Some sites set up local walls of fame or competitions to encourage trial recruitment.
Recommendation 8The implementation group supported the qualitative research team’s proposal to compile a list of slides for use in the induction of future trial sites. These would list practical suggestions and solutions developed in the pilot sites for facilitating trial delivery.
Implementation: the slides were circulated to new sites and key messages were embedded within the slides used for site initiation.
Concluding comments
All objectives of the nested qualitative study were achieved and all of the original research questions answered. As well as the published papers, the qualitative study led to the generation of two significant outputs for use in the main trial: the recommendations from the implementation group and the staff training resource. The implementation group’s recommendations were used to support delivery of the GOT-IT trial to the full site list.
Chapter 4 Participant baseline characteristics
Trial recruitment
Recruitment into the trial was between 13 October 2014 and 26 July 2017 and was followed up to September 2017. Figure 2 shows the trajectory of the recruitment from all centres. In total, 1107 participants were recruited from 29 UK NHS hospitals (Table 3) for treatment of retained placenta. All centres recruited to both interventions with 543 randomised to GTN and 564 randomised to placebo.
FIGURE 2.
Recruitment over time.
| Centre | Randomised (N = 1107), n (%) | Treatment group, n (%) | |
|---|---|---|---|
| GTN (N = 543) | Placebo (N = 564) | ||
| Royal Infirmary of Edinburgh | 92 (8.3) | 45 (8.3) | 47 (8.3) |
| Queen Elizabeth University Hospital | 24 (2.2) | 11 (2.0) | 13 (2.3) |
| St Mary’s Hospital | 122 (11.0) | 64 (11.8) | 58 (10.3) |
| Royal Victoria Infirmary | 42 (3.8) | 20 (3.7) | 22 (3.9) |
| Royal Preston Hospital | 80 (7.2) | 39 (7.2) | 41 (7.3) |
| Warrington Hospital | 45 (4.1) | 21 (3.9) | 24 (4.3) |
| Chesterfield Royal Hospital | 47 (4.2) | 21 (3.9) | 26 (4.6) |
| Leighton Hospital | 46 (4.2) | 20 (3.7) | 26 (4.6) |
| University Hospital of North Durham | 33 (3.0) | 16 (2.9) | 17 (3.0) |
| West Middlesex University Hospital | 28 (2.5) | 14 (2.6) | 14 (2.5) |
| Stoke Mandeville Hospital | 55 (5.0) | 27 (5.0) | 28 (5.0) |
| Furness General Hospital | 10 (0.9) | 6 (1.1) | 4 (0.7) |
| University Hospital Southampton NHS Foundation Trust | 47 (4.2) | 24 (4.4) | 23 (4.1) |
| Bolton NHS Foundation Trust | 48 (4.3) | 24 (4.4) | 24 (4.3) |
| Sunderland Royal Hospital | 86 (7.8) | 42 (7.7) | 44 (7.8) |
| Oxford University Hospitals | 45 (4.1) | 23 (4.2) | 22 (3.9) |
| Nottingham University Hospitals NHS Trust Queen’s Medical Centre | 28 (2.5) | 13 (2.4) | 15 (2.7) |
| Nottingham University Hospitals NHS Trust City Campus | 37 (3.3) | 18 (3.3) | 19 (3.4) |
| East Lancashire Hospital NHS Trust Burnley General Hospital | 28 (2.5) | 13 (2.4) | 15 (2.7) |
| Ashford and St Peter’s Hospitals NHS Trust | 17 (1.5) | 9 (1.7) | 8 (1.4) |
| North Tees and Hartlepool Hospitals NHS Foundation Trust | 19 (1.7) | 10 (1.8) | 9 (1.6) |
| South Tees Hospital NHS Foundation Trust James Cook Hospital | 17 (1.5) | 9 (1.7) | 8 (1.4) |
| Countess of Chester Hospital NHS Foundation Trust | 18 (1.6) | 8 (1.5) | 10 (1.8) |
| Darlington Memorial Hospital | 8 (0.7) | 4 (0.7) | 4 (0.7) |
| York Teaching Hospital | 21 (1.9) | 9 (1.7) | 12 (2.1) |
| The Royal Berkshire Hospital | 16 (1.4) | 8 (1.5) | 8 (1.4) |
| Milton Keynes University Hospital | 20 (1.8) | 12 (2.2) | 8 (1.4) |
| Shrewsbury and Telford Hospital NHS Trust | 18 (1.6) | 8 (1.5) | 10 (1.8) |
| Frimley Park Hospital | 10 (0.9) | 5 (0.9) | 5 (0.9) |
Participant flow
The Consolidated Standards of Reporting Trials (CONSORT) flow diagram for the trial is shown in Figure 3. Of the 1671 patients screened, 1188 were eligible. For the 483 patients who were excluded, 353 were ineligible, 63 declined, 60 were missed and it was not appropriate to approach 7 patients. Of the 63 patients who declined, 22 (34.9%) did not provide a reason and 15 (24.8%) preferred to go straight to theatre. The main reason why patients were missed was because there were no medical staff on duty that had signed the delegation log. Further details of the reasons for patients being excluded are provided in Table 4. Three participants were excluded post randomisation (two in the GTN group and one in the placebo group) because the baseline observations prior to investigational medicinal product administration made them ineligible for participation. Six participants in the GTN group and seven in the placebo group did not receive the study drug. The reasons are detailed in Table 5.
FIGURE 3.
The CONSORT flow diagram. a, Declined refers to women with retained placenta who declined trial participation before their eligibility criteria were able to be checked.
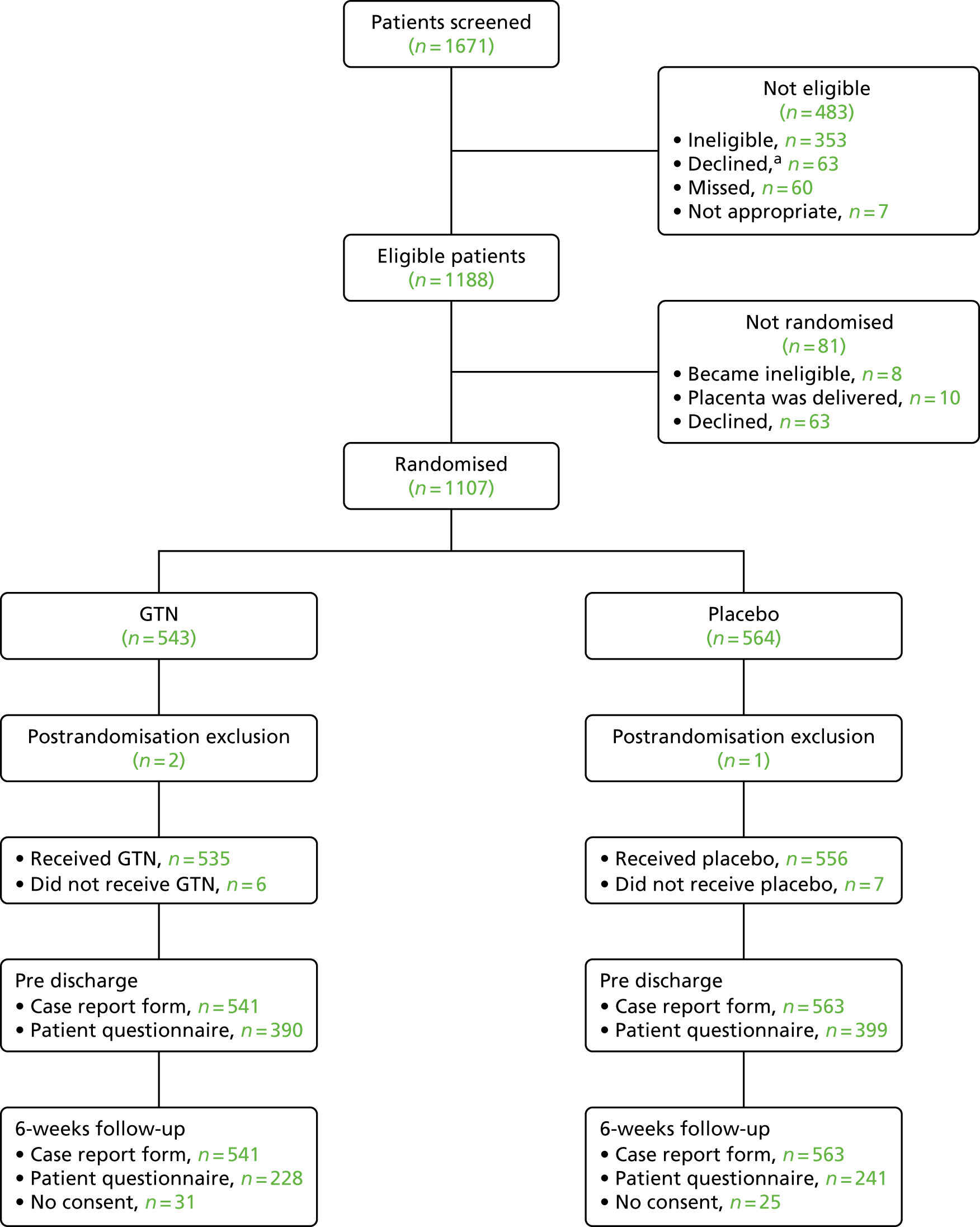
| Reason for being non-eligible | Number of participants | % of total |
|---|---|---|
| Reasons for ineligibility | 353 | |
| Did not meet eligibility criteria | 335 | 94.9 |
| Clinical reasons | 12 | 3.4 |
| Placenta delivered before eligible criteria could be checked | 6 | 1.1 |
| Reasons for declining to take part | 63 | |
| No reason given | 22 | 34.9 |
| Preferred to go straight to theatre | 15 | 23.8 |
| Did not wish to take part | 11 | 17.5 |
| Did not want to participate in research | 5 | 7.9 |
| Randomisation process | 2 | 3.2 |
| Other | 8 | 12.7 |
| Reasons for missing participants | 60 | |
| No medical staff on duty that were on the delegation log | 1 | 1.6 |
| No reason given | 40 | 66.7 |
| Recruitment on halt | 15 | 25.0 |
| Drugs not available | 4 | 6.7 |
| Reasons why study drug was not given | Treatment group | |
|---|---|---|
| GTN (N = 541) | Placebo (n = 563) | |
| Study drug not given, n (%) | 6 (1.1) | 7 (1.2) |
| Reasons | ||
| Placenta was delivered before drug was administered | 0 | 4 |
| Blood pressure was ≤ 100 mmHg | 2 | 1 |
| Blood pressure was ≤ 100 mmHg and heart rate was > 119 b.p.m. when rechecked | 2 | 0 |
| Bleeding | 1 | 1 |
| Heart rate increased | 0 | 1 |
| Medical team misinterpreted 30-to 60-minutes criteria and wrongly assumed spray was given | 1 | 0 |
The pre-discharge questionnaire was completed by 390 participants in the GTN group and 399 in the placebo group. At the 6-week follow-up, 228 completed and returned the questionnaire in the GTN group (31 did not consent to receiving the questionnaire) and 241 completed and returned the questionnaire in the placebo group (25 did not consent to receiving the questionnaire).
Baseline characteristics
The participants’ baseline characteristics are shown in Table 6 and are balanced between the two treatment groups. The mean age was 31 years in both groups and the majority of participants were white (86.5% in the GTN group and 86.5% in the placebo group). The mean systolic and diastolic blood pressure in the GTN group was 123.8 mmHg and 73.3 mmHg, respectively, and in the placebo group was 124.6 mmHg and 75.1 mmHg, respectively. The mean heart rate was 84.6 b.p.m. for participants in the GTN group and 84.7 b.p.m. for participants in the placebo group. Over half the participants had had a previous pregnancy (GTN group, 57.5%; placebo group, 57.4%).
| Baseline characteristics | Treatment group | |
|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | |
| Age (years), mean (SD); n | 30.6 (5.5); 541 | 30.8 (5.1); 563 |
| Body mass index (kg/m2), mean (SD); n | 25.8 (5.4); 526 | 25.4 (5.2); 548 |
| Smoker, n (%) | ||
| Current | 75 (13.9) | 77 (13.7) |
| Ex-smoker | 101 (18.7) | 98 (17.4) |
| Never | 350 (64.7) | 376 (66.8) |
| Missing | 15 (2.8) | 12 (2.1) |
| Alcohol use in pregnancy, n (%) | ||
| Yes | 19 (3.5) | 18 (3.2) |
| No | 505 (93.3) | 521 (92.5) |
| Missing | 17 (3.1) | 24 (4.3) |
| Ethnicity, n (%) | ||
| White | 468 (86.5) | 487 (86.5) |
| Asian | 38 (7.0) | 41 (7.3) |
| Black | 7 (1.3) | 8 (1.4) |
| Mixed | 5 (0.9) | 6 (1.1) |
| Chinese | 5 (0.9) | 6 (1.1) |
| Other | 5 (0.9) | 6 (1.1) |
| Missing | 13 (2.4) | 9 (1.6) |
| Blood pressure (mmHg), mean (SD); n | ||
| Systolic | 123.8 (12.8); 538 | 124.6 (12.6); 559 |
| Diastolic | 73.3 (10.2); 535 | 75.1 (10.1); 559 |
| Heart rate (b.p.m.), mean (SD); n | 84.6 (13.0); 539 | 84.7 (12.9); 559 |
| Temperature (°C), mean (SD); n | 36.8 (0.5); 513 | 36.9 (0.4); 534 |
| Haemoglobin level (g/dl), mean (SD); n | 12.2 (1.3); 468 | 12.3 (1.4); 478 |
| Previous pregnancy, n (%) | 311 (57.5) | 323 (57.4) |
| Previous retained placenta, n (%) | 48 (15.4) | 57 (17.6) |
| Previous placenta praevia/accreta, n (%) | 4 (1.3) | 1 (0.3) |
Chapter 5 Outcomes and results
Primary outcome
The trial did not stop early at any of the interim analysis stages; therefore, the analysis was performed on 1104 participants. The proportion of participants in whom the placenta remained undelivered or required manual removal of placenta within 15 minutes was similar in both groups [n = 505 (93.3%) in the GTN group and n = 518 (92.0%) in the placebo group] (Table 7). Adjusting for multiple looks at the data, there was no difference in the primary clinical outcome between the two groups [odds ratio (OR) 1.01, 95% CI 0.98 to 1.04; p = 0.393].
| Time | Treatment group, n (%) | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | ||||
| Delivered within 15 minutes | 36 (6.7) | 45 (8.0) | |||
| Undelivered within 15 minutes | 505 (93.3) | 518 (92.0) | 1.01 | 0.98 to 1.04 | 0.393 |
Table 8 provides details of the method of removing the placenta. For participants that delivered the placenta within 15 minutes, 86.1% of placentae were delivered by controlled cord traction in the GTN group and 86.7% in the placebo group. For the participants for whom the placenta was delivered after 15 minutes, the majority of participants underwent manual removal of placenta (GTN group, 80.6%; placebo group, 80.5%).
| Method of placenta removal | Treatment group, n/N (%) | |
|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | |
| Placenta delivered within 15 minutes | ||
| Spontaneous | 5/36 (13.9) | 6/45 (13.3) |
| Controlled cord traction | 31/36 (86.1) | 39/45 (86.7) |
| Placenta delivered after 15 minutes | ||
| Spontaneous | 13/505 (2.6) | 16/518 (3.1) |
| Controlled cord traction | 83/505 (16.4) | 84/518 (16.2) |
| Manual removal of placenta | 407/505 (80.6) | 417/518 (80.5) |
| Othera | 1/505 (0.2) | 1/518 (0.2) |
| Unknownb | 1/505 (0.2) | |
Figure 4 shows the stopping boundaries and the pathway of the group-sequential analysis. At each interim look a test statistic based on the accumulated data determined whether or not the trial should be stopped based on whether or not it crossed any of the boundaries (stopping for futility only at the third interim look and at the final analysis). Based on the trial results, we did not cross any of the boundaries at the interim analyses and proceeded to the final analysis.
FIGURE 4.
Stopping boundaries and pathway of group-sequential analysis. Black, efficacy boundaries; green, futility boundaries; blue, test statistic.
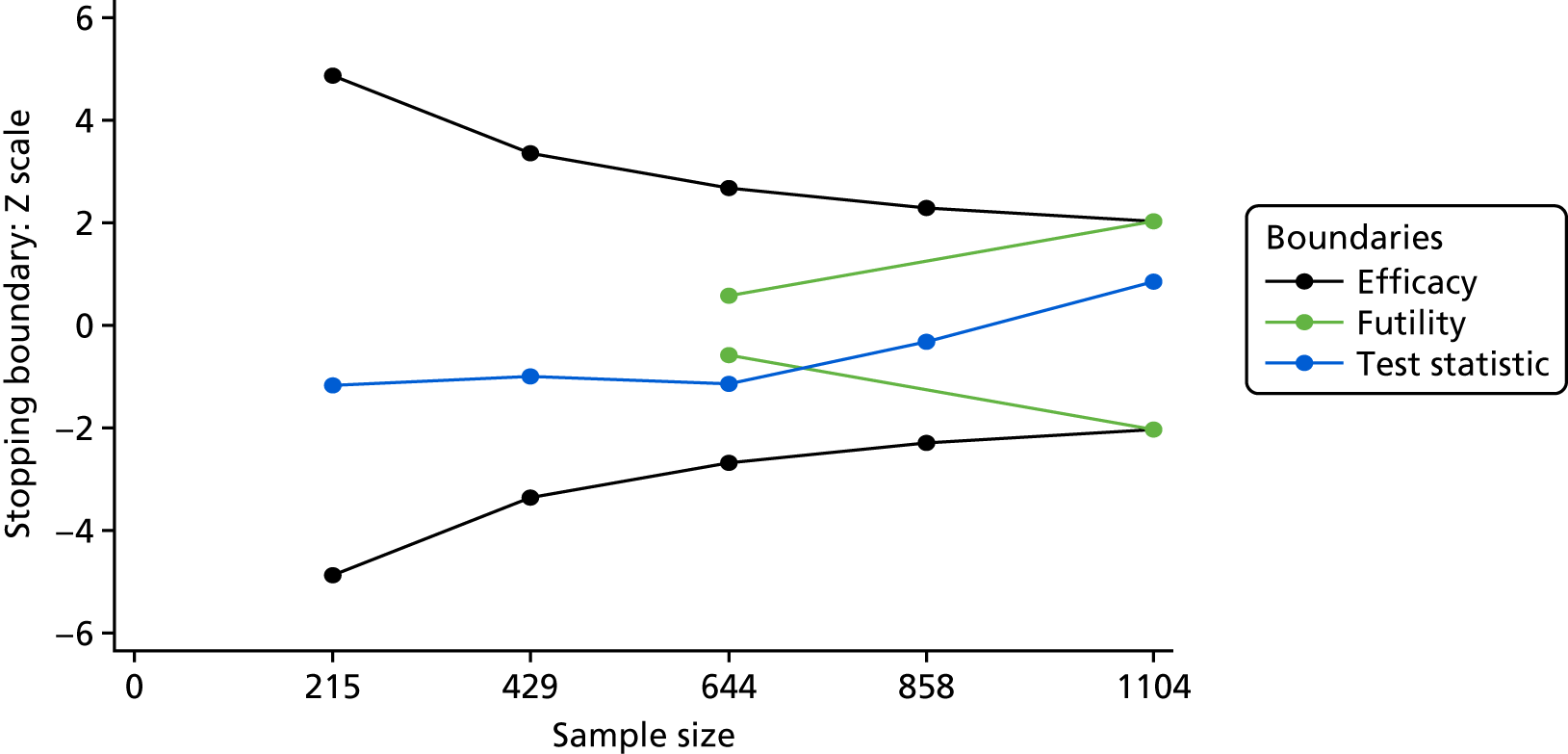
Blood loss between the administration of the study drug and the transfer to a postnatal ward (or other clinical area) is shown in Table 9. Blood loss was < 500 ml in 44.3% of the GTN group and in 44.5% of the placebo group. There was a blood loss of > 1000 ml in 22.2% of participants in the GTN group and in 15.5% of the placebo group. There was no difference in the primary safety outcome of blood loss between the groups (proportional OR 1.14, 95% CI 0.88 to 1.48; p = 0.314).
| Blood loss (ml) | Treatment group, n/N (%) | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| GTN (N = 41) | Placebo (N = 563) | ||||
| < 500 | 238/537 (44.3) | 249/560 (44.5) | |||
| 500–1000 | 180/537 (33.5) | 224/560 (40.0) | |||
| > 1000 | 119/537 (22.2) | 87/560 (15.5) | 1.14 | 0.88 to 1.48 | 0.314 |
Patient satisfaction and side-effect profile before and at 6 weeks are shown in Table 10. There was no evidence of a difference in the primary patient-sided satisfaction outcome of recommending the study drug to a friend/relative pre discharge (OR 0.87, 95% CI 0.62 to 1.22; p = 0.411) and at 6 weeks (OR 1.02, 95% CI 0.66 to 1.56; p = 0.941). For the primary patient-sided safety outcome, feeling sick pre discharge and at 6 weeks both showed no evidence of a difference between the two groups. Palpitations/heart racing showed evidence of a difference at pre discharge in favour of the placebo group (OR 2.60, 95% CI 1.40 to 4.84; p = 0.003); at 6 weeks there was no evidence of a difference.
| Patient satisfaction and side-effect profile | Treatment group, n/N (%) | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | ||||
| Recommend study drug to a friend/relative? | |||||
| Pre discharge | |||||
| No | 94/382 (24.6) | 85/388 (21.9) | |||
| Yes | 288/382 (75.4) | 303/388 (78.1) | 0.87 | 0.62 to 1.22 | 0.411 |
| 6 weeks | |||||
| No | 55/221 (24.9) | 60/238 (25.2) | |||
| Yes | 166/221 (75.1) | 178/238 (74.8) | 1.02 | 0.66 to 1.56 | 0.941 |
| Feeling sick | |||||
| Pre discharge | |||||
| No | 299/377 (79.3) | 323/384 (84.1) | |||
| Yes | 78/377 (20.7) | 61/384 (15.9) | 1.37 | 0.94 to 1.99 | 0.101 |
| 6 weeks | |||||
| No | 180/211 (85.3) | 206/232 (88.8) | |||
| Yes | 31/211 (14.7) | 26/232 (11.2) | 1.40 | 0.80 to 2.47 | 0.239 |
| Palpitations/heart racing | |||||
| Pre discharge | |||||
| No | 332/368 (90.2) | 360/375 (96.0) | |||
| Yes | 36/368 (9.8) | 15/375 (4.0) | 2.60 | 1.40 to 4.84 | 0.003 |
| 6 weeks | |||||
| No | 186/200 (93.0) | 215/225 (95.6) | |||
| Yes | 14/200 (7.0) | 10/225 (4.4) | 1.62 | 0.70 to 3.73 | 0.258 |
Secondary outcome
The secondary clinical outcomes are shown in Table 11. There was no evidence of a difference of a > 15% fall in haemoglobin level between recruitment and the first postnatal day between the two groups. Participants randomised to the GTN group were more likely than those in the placebo group to have a fall in systolic blood pressure, diastolic blood pressure and/or an increase in heart rate (OR 4.90, 95% CI 3.73 to 6.42; p < 0.001). Participants were also more likely to require a blood transfusion between time of delivery and discharge from hospital if they had received GTN (OR 1.53, 95% CI 1.04 to 2.25; p = 0.033). There was no evidence of a difference between the groups for maternal pyrexia time from randomisation to delivery of placenta, manual removal of placenta in theatre, general anaesthesia and sustained uterine relaxation.
| Clinical outcome | Treatment group | Effect sizea | 95% CI | p-value | |
|---|---|---|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | ||||
| > 15% fall in haemoglobin, n/N (%) | |||||
| No | 160/414 (38.6) | 180/421 (42.8) | |||
| Yes | 254/414 (61.4) | 241/421 (57.2) | 1.19 | 0.93 to 1.52 | 0.175 |
| Time from randomisation to delivery of placenta (minutes), mean SD; n | 12.1 (7.3); 539 | 12.2 (7.0); 561 | 0.19 | –0.94 to 0.55 | 0.601 |
| Manual removal of placenta in theatre, n/N (%) | |||||
| No | 141/540 (26.1) | 152/563 (27.0) | |||
| Yes | 399/540 (73.9) | 411/563 (73.0) | 1.05 | 0.80 to 1.36 | 0.736 |
| Need for earlier than planned manual removal of placenta, n/N (%) | |||||
| No | 407/416 (97.8) | 420/431 (97.4) | |||
| Yes | 9/416 (2.2) | 11/431 (2.6) | 0.84 | 0.30 to 2.35 | 0.746 |
| Fall in systolic or diastolic blood pressure of > 15 mmHg and/or increase in heart rate of 20 b.p.m., n/N (%) | |||||
| No | 208/531 (39.2) | 413/544 (75.9) | |||
| Yes | 323/531 (60.8) | 131/544 (24.1) | 4.90 | 3.73 to 6.42 | < 0.001 |
| Blood transfusion, n/N (%) | |||||
| No | 472/533 (88.6) | 508/551 (92.2) | |||
| Yes | 61/533 (11.4) | 43/551 (7.8) | 1.53 | 1.04 to 2.25 | 0.033 |
| General anaesthesia, n/N (%) | |||||
| No | 390/438 (89.0) | 398/443 (89.8) | |||
| Yes | 48/438 (11.0) | 45/443 (10.2) | 1.09 | 0.66 to 1.80 | 0.741 |
| Maternal pyrexia, n/N (%) | |||||
| No | 516/527 (97.9) | 530/551 (96.2) | |||
| Yes | 11/527 (2.1) | 21/551 (3.8) | 0.54 | 0.26 to 1.11 | 0.092 |
| Sustained uterine relaxation, n/N (%) | |||||
| No | 460/528 (87.1) | 482/550 (87.6) | |||
| Yes | 68/528 (12.9) | 68/550 (12.4) | 1.05 | 0.76 to 1.44 | 0.771 |
Subgroup analysis
As there was a low event rate in the clinical primary outcome, the event rate in the subgroups was too low to perform the planned subgroup analysis.
Adverse events
In total, there were 52 SAEs during the trial, with all participants who experienced a SAE only experiencing one event [GTN group, n = 27 (5.0%); placebo group, n = 25 (4.4%)]. Most of the events required hospitalisation (GTN group, n = 24; placebo group, n = 24). The severity of the events was mainly moderate (GTN group, n = 16; placebo group, n = 16) with three events in the placebo group being life-threatening (two were postpartum haemorrhage and one was anaphylaxis due to suxamethonium chloride). Most of the SAEs were due to postpartum haemorrhage (GTN group, n = 23; placebo group, n = 16). The only other SAEs of note were retained products of conception (GTN group, n = 1; placebo group, n = 3). Further details are provided in Table 12.
| SAEs | Treatment group, n (%) | |
|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | |
| Number of SAEsa | 27 (5.0) | 25 (4.4) |
| Hospitalisation | 24 | 24 |
| Life-threatening | 0 | 3 |
| Other significant medical event | 6 | 2 |
| Severity of event | ||
| Mild | 7 | 4 |
| Moderate | 19 | 16 |
| Severe | 1 | 5 |
| Event due to progression of underlying disease | ||
| Yes | 5 | 2 |
| No | 21 | 23 |
| Not applicable | 1 | 0 |
| Event due to a lack of efficacy of investigational medicinal product | ||
| Yes | 1 | 0 |
| No | 25 | 25 |
| Not applicable | 1 | 0 |
| Details of SAE | ||
| Postpartum haemorrhage | 23 | 16 |
| Retained products of conception | 1 | 3 |
| Endometritis | 1 | 0 |
| Sepsis | 0 | 1 |
| Chest infection | 1 | 0 |
| Gall stone pancreatitis | 1 | 0 |
| Anaphylaxis due to suxamethonium | 0 | 1 |
| Escherichia coli and Clostridium difficile infection | 0 | 1 |
| Elevated blood pressure | 0 | 1 |
| Emergency hysterectomy | 0 | 1 |
| Postdural headache | 0 | 1 |
Adverse events are shown in Table 13. There were 430 events of which 352 participants (GTN group, n = 167; placebo group, n = 185) had an event. The majority were unrelated, of mild severity and were expected. There was a variety of different AEs, most were postpartum haemorrhage (GTN group, n = 173; placebo group, n = 175), hypotensive (GTN group, n = 6; placebo group, n = 21) and sepsis (GTN group, n = 7; placebo group, n = 10).
| AEs | Treatment group, n | |
|---|---|---|
| GTN (N = 541) | Placebo (N = 563) | |
| Number of participants with an AE | 166 | 185 |
| Number of AEs | 204 | 226 |
| Causality | ||
| Unrelated | 53 | 54 |
| Possibly related | 151 | 172 |
| Severity | ||
| Mild | 144 | 147 |
| Moderate | 57 | 72 |
| Severe | 3 | 7 |
| Expectedness | ||
| Expected | 166 | 184 |
| Unexpected | 38 | 41 |
| Not applicable | 0 | 1 |
| Details of AEs | ||
| Postpartum haemorrhage | 173 | 175 |
| Hypotensive | 6 | 21 |
| Sepsis | 7 | 10 |
| Tachycardia | 5 | 6 |
| Headache | 0 | 4 |
| Rise in blood pressure | 4 | 0 |
| Vasovagal episode | 0 | 3 |
| Sublingual discomfort | 2 | 0 |
| Uncoagulated full blood count sample | 1 | 0 |
| Acute cholecystitis | 0 | 1 |
| Constipation | 1 | 0 |
| Feeling dizzy | 1 | 0 |
| Nausea | 0 | 1 |
| Perineal tear | 1 | 1 |
| Placenta accreta | 0 | 1 |
| Postnatal readmission for retained products | 0 | 1 |
| Potential deep-vein thrombosis | 1 | 0 |
| Self-discharge from hospital | 0 | 1 |
| Vomiting | 0 | 1 |
| Facial rash | 1 | 0 |
| General anaesthetic | 1 | 0 |
Chapter 6 Resource use, costs and cost-effectiveness
This chapter describes methods for conducting the economic analysis alongside the trial and presents the results in the context of the preceding clinical effectiveness and safety data. As stated in the trial protocol, the primary economic outcome is the net incremental cost (or cost saving) to the NHS of using GTN for the treatment of retained placenta compared with standard practice. Included in the analysis are the costs of drug acquisition and administration, monitoring prior to delivery of the placenta or transfer to theatre, placenta delivery and further health service resource use up to 6 weeks following delivery. The methods for measuring and valuing the relevant resource-use data are summarised in the following sections. Following this, results are presented by treatment allocation group alongside key primary and secondary clinical effectiveness and safety outcomes in a cost–consequences balance sheet.
Methods
Although the cost of GTN is low, there may be further costs associated with its administration and the monitoring and management of women thereafter. It is therefore important in the context of scarce maternity resources to explicitly quantify the net costs (or cost savings) associated with its use. Provision was therefore made in the GOT-IT protocol to carry out a simple cost analysis using clinical and resource-use data collected for individual participants recruited to the trial. This analysis explicitly quantifies the difference in mean costs between the active intervention and placebo groups.
Resource use associated with the alternative management strategies was estimated from the time of randomisation through to 6 weeks post childbirth. This included:
-
staff time for administration of the study drug and monitoring of the patient until time of placenta delivery or transfer to theatre for manual removal of placenta
-
resource use associated with complications arising following administration of the study drug or placebo
-
subsequent costs associated with delivery of the placenta (either spontaneously or operatively)
-
costs associated with the postnatal stay (to discharge)
-
subsequent health service contacts potentially relating to retained products of conception up to 6 weeks post discharge.
The time from administration of the study drug to spontaneous delivery of the placenta (or the decision to proceed with manual removal), and the incidence of complications following administration of the drug, were collected using the trial case report forms. Information about health service use in the 6 weeks following discharge was collected via a 6-week postnatal record check and several health service resource-use questions were included in the participant postnatal questionnaire (also at 6 weeks). Resource use was valued using routine sources of nationally relevant unit costs. 40–44
Mean costs are summarised by treatment allocation group following the principles of intention to treat, and the incremental cost (cost saving) associated with the use of GTN is estimated using linear regression with cluster robust CIs to adjust for potential centre effects. The cost data is presented alongside the primary and secondary outcome data in a cost–consequence balance sheet, indicating which strategy each outcome favours.
Measurement and valuation of resource-use data
The time from randomisation to delivery of placenta or transfer to theatre for manual removal of placenta was derived from the trial case report forms and costed using the unit cost per patient contact hour for someone on band 6. 40 This multiplier includes an overhead and capital apportionment. For those not requiring manual removal of placenta or transfer to theatre for other reasons, the remaining maternal stay was costed using a national average bed-day cost following vaginal deliveries, applied to actual duration of stay following delivery of the placenta. This unit cost was taken as relevant excess bed-day cost obtained from the NHS Reference Costs 2015 to 2016. 41 Because we did not have detailed data on mode of vaginal delivery for individual patients, we applied a weighted average (by national activity levels) of the excess bed-day costs for all health-care resource-use groups relating to normal and assisted deliveries: NZ30-34 and NZ40-44. Drug costs for GTN were also applied to those randomised to the GTN group (Table 14). 42
| Resource | How measured? | Source of measurement | Unit cost (£) | Source of valuation |
|---|---|---|---|---|
| aCoro Nitro® (glyceryl trinitrate) | Drug (2 × 400 µg/puff) administered or not | From case report form – randomisation | 3.44/200-dose unit | British National Formulary 42 |
| Placebo | Placebo administered or not | From case report form – randomisation | 0 | Not applicable |
Tables 15–17 summarise the data sources used for the costing of hospital resources (see Table 15), primary care (see Table 16) and secondary care (see Table 17). For women who were transferred to the theatre but did not subsequently require a manual removal of placenta, the cost of theatre resources was estimated by applying a running cost per hour of obstetric theatre time obtained from Scottish Health Service Costs (2016) data. 43 For women requiring a blood transfusion, a unit cost per red cell unit transfused was applied, assuming an average of two units per woman. 44 For women who were transferred to theatre and received manual removal of placenta, these episodes were costed using the NHS reference cost for such episodes. Manual removal of the placenta maps to health-care resource-use group code NZ27Z (postnatal therapeutic procedures). 41 The unit cost for this health-care resource-use group code reflects the average cost of these procedures across the NHS. The time and date of discharge were additionally used to estimate the overall health-care resource-use group code-based reference cost for each manual removal of placenta episode.
| Resource | How measured? | Source of measurement | Unit cost (£) | Source of valuation |
|---|---|---|---|---|
| Monitoring of patient by hospital | Length of time to placenta delivery/or time transfer to theatre | From case report form – clinical observation @ 5–15 minutes | 108/hour of patient contact | Curtis and Burns40 |
| Management by surgical team in theatre (for those with spontaneous delivery of placenta) | Length of time in theatre | From case report form – clinical observation @ 5–15 minutes | 667/hour | Information Services Division Scotland43 |
| Hospital stay (following spontaneous delivery of placenta) | Length of stay (number of days) | From case report forms – randomisation and pre-discharge | 454.86/day | Health resource group codes (NES/XSa for NZ27Zb)41 |
| Manual removal of placenta | Manual removal of placenta performed or not | From case report form – clinical observation @ 5–15 minutes | Day case, 983.38; non-elective short stay (1 day), 1149.59; non-elective (> 1 day), 1149.59 + (496.61 × number of days > 1 day) | Health resource group codes (NZ27Zb, OPCS R29.1c)41 |
| Resource | How measured? | Source of measurement | Unit cost (£) | Source of valuation |
|---|---|---|---|---|
| Calls | Number of calls made | From postnatal patient questionnaire | 7.33/10 minutes per call | Curtis and Burns40 |
| Visits | Number of visits | From postnatal patient questionnaire | 14.67/20 minutes per visit | Curtis and Burns40 |
| Health visitor calls | Number of calls made to health visitor | From postnatal patient questionnaire | 7.00/10 minutes per call | Curtis and Burns40 |
| Health visitor visits | Number of health visitor visits | From postnatal patient questionnaire | 14/20 minutes per visit | Curtis and Burns40 |
| GP visits | Number of visits to GP | From postnatal patient questionnaire | 36/9.22 minutes per visit | Curtis and Burns40 |
| GP calls | Number of telephone consultations with GP | From postnatal patient questionnaire | 36/9.22 minutes per call | Curtis and Burns40 |
| GP home visits | Number of home visits by GP | From postnatal patient questionnaire | 44.84/11.4 minutes per home visit | Curtis and Burns40 |
| Resource | How measured? | Source of measurement | Unit cost (£) | Source of valuation |
|---|---|---|---|---|
| Outpatient attendance | Number of outpatient appointments | From postnatal patient questionnaire | 136.79 per attendance | Curtis and Burns40 |
| Hospital readmission | Length of stay (number of days) | Case report form completed at 6 weeks post childbirth | Various | HRG-based reference costs39 |
The 6-week postnatal record check provided data on any readmissions to hospital deemed to be potentially relevant to retained placenta or study drug. All readmissions were costed using appropriate health-care resource-use group codes combined with length of stay. Finally, the postnatal patient questionnaire provided information on participants’ use of community midwifery, health visitor and primary care services up to 6 weeks post discharge. Reported use of these services was costed using national unit cost data. 40 In addition, the questionnaire collected data on any further outpatient appointments. Outpatient attendances were costed using the average unit price for outpatient appointments obtained from national sources. 40 The questionnaire was piloted with 15 women as part of the qualitative study at Edinburgh Royal Infirmary prior to finalisation (see Chapter 3). On the basis of these interviews, the wording of the questionnaire was refined slightly to avoid capturing routine primary care and health visitor contacts relating to the health of the baby. Tables 14–17 summarise the type of resource use captured and the associated sources of measurement and valuation. All costs were expressed for the 2015–16 financial year.
Analysis of cost data
Total costs were estimated for each woman enrolled in the trial as the sum of each cost element. These are summarised by treatment allocation group (by intention to treat) and broken down into the following categories: intervention and monitoring costs [until delivery of the placenta (non-manual removal of placenta) or transfer to theatre], theatre costs for non-manual removal of placenta procedures, manual removal of placenta costs, total episode costs (randomisation to discharge) and postdischarge costs (primary and secondary care). The mean differences in cost between groups was estimated using ordinary least squares regression. Cluster robust standard errors and CIs were implemented to account for possible within-centre correlation. All analyses were conducted in Stata 13® (StataCorp LP, College Station, TX, USA).
Further sensitivity analysis was conducted to assess the robustness of the findings to the costing approach. For the first sensitivity analysis [sensitivity analysis 1 (SA1)], we costed all manual removals using the running cost per hour of obstetric theatres in Scotland43 and costed the postnatal admission using the average bed-day cost following vaginal deliveries. For the second sensitivity analysis [sensitivity analysis 2 (SA2)], we costed manual removals using the difference between the NHS reference costs for vaginal deliveries with (NZ32, NZ33, NZ42 and NZ43) and without (NZ30, NZ31, NZ40 and NZ41) postnatal surgery. 41
The number of missing data was very low for major cost drivers associated with the initial delivery episode and readmissions to hospital, but substantial for postdischarge outpatient and primary care costs up to 6 weeks. Alternative assumptions about mechanisms for missing data were applied depending on the variable; missing elements of primary care use, where participants had responded to some resource-use questions but not others, were assumed missing not at random but because of irrelevance to the participant. In this case, zeros were assigned. Other resource-use variables were assumed missing at random. We originally planned to use multiple imputation to generate multiple plausible values for these missing data items, but because data was complete for over ≈90% of the major cost drivers, this was deemed unnecessary. The postdischarge outpatient and primary care costs up to 6 weeks formed only a very small component of the overall costs.
Differences in costs are presented alongside the statistical comparisons of primary and secondary efficacy and safety outcomes in the form of a cost–consequence balance sheet. This provides a summary of the strategy favoured on cost and all the other outcomes of interest. All effects on the clinical, patient-oriented and safety outcomes were derived as detailed in the statistical analysis section (see Chapter 5).
Results
Table 18 summarises the resource use associated with the index hospital episode, from time of randomisation to time of discharge. There were no obvious notable differences in elements of resource use associated with the hospital stay. Manual removal of placentae were slightly more frequent in the GTN group, as were blood transfusions.
| Type of hospital resources (initial episode) | Number of observations | Treatment group (N = 1104)a | |
|---|---|---|---|
| GTN (n = 541) | Placebo (n = 563) | ||
| Time to placenta delivery (hours), mean (SD) | 1087 | 1.31 (0.81) | 1.28 (0.77) |
| Time to theatre (hours), mean (SD) | 1065 | 0.79 (0.73) | 0.75 (0.66) |
| Time in theatre (hours), mean (SD) | 1000 | 0.82 (0.75) | 0.82 (0.71) |
| Method of placenta removal, n (%) | 1104 | ||
| Non-manual removal of placenta (spontaneous, controlled cord traction) | 134 (24.8) | 146 (25.9) | |
| Manual removal of placenta | 407 (75.2) | 417 (74.1) | |
| Blood transfusion, n (%) | 1084 | 61 (11.4) | 43 (7.8) |
| Length of stay (hours), mean (SD) | 1014 | 45.46 (35.33) | 42.74 (29.81) |
Table 19 summarises further secondary care resource use between discharge and 6 weeks post discharge. Again, there are no major notable differences between the groups.
| Outpatient resources | Number of observations | Treatment group (N = 1104) | |
|---|---|---|---|
| GTN | Placebo | ||
| Number of outpatient attendances, mean (SD) | 466 | 0.19 (0.72) | 0.14 (0.47) |
| Readmission, n (%) | 1098 | ||
| Yes | 16 (3.0) | 29 (5.2) | |
| No | 520 (97) | 533 (94.8) | |
| Average length of stay if readmitted (number of nights), mean (SD) | 45 | 3.94 (13.12) | 1.31 (1.85) |
Table 20 summarises primary care usage to 6 weeks post discharge. Again, there were no notable differences in elements of resource use by treatment allocation. Outpatient visits were slightly more common in the GTN group than in the placebo group, and readmissions were slightly more common in the placebo group than in the GTN group, but the length of stay among those readmitted was higher in the GTN group.
| Type of primary care resources | Number of observations | Treatment group, mean (SD) (N = 1104) | |
|---|---|---|---|
| GTN | Placebo | ||
| Number of calls | 460 | 0.30 (1.12) | 0.17 (0.46) |
| Number of visits | 457 | 0.24 (0.90) | 0.28 (0.93) |
| Number of calls made to health visitor | 452 | 0.04 (0.37) | 0.07 (0.39) |
| Number of health visitor visits | 452 | 0.10 (0.52) | 0.18 (0.80) |
| Number of visits to GP | 457 | 0.38 (0.68) | 0.49 (0.96) |
| Number of GP telephone consultations | 446 | 0.18 (0.50) | 0.17 (0.51) |
| Number of home visits by GP | 436 | 0.03 (0.19) | 0.004 (0.07) |
The costs associated with the initial delivery episode, from time of randomisation to time of discharge, are summarised in Table 21 by treatment allocation. Overall, the costs were similar between groups, with the total episode cost being slightly higher in the GTN group than in the placebo group. There were no obvious differences between the groups in terms of the postdischarge hospital costs (Table 22) or postdischarge primary care costs (Table 23). The estimates of postdischarge primary care costs and outpatient costs are subject to a substantial amount of missing data, because of reliance on participant responses to the 6-week postnatal questionnaire.
| Hospital resource (initial episode) | Number of observations | Treatment group, mean (SD) (N = 1104) | |
|---|---|---|---|
| GTN (£) | Placebo (£) | ||
| Cost of intervention | 1104 | 3.40 (0.36) | 0 (0) |
| Cost of monitoring by hospital | 1062 | 98.08 (72.42) | 92.51 (63.62) |
| Cost of surgical resource (non-manual removal of placenta in theatre) | 1095 | 18.15 (112.11) | 26.86 (212.83) |
| Cost of hospital stay (for non-manual removal of placenta, i.e. spontaneous, controlled cord traction) | 1085 | 154.00 (381.61) | 182.76 (429.66) |
| Cost of manual removal of placenta | 1033 | 1019.98 (823.80) | 949.91 (710.47) |
| Cost of blood transfusions | 1084 | 38.01 (105.84) | 25.92 (89.17) |
| Total episode cost | 966 | 1367 (734) | 1317 (642) |
| Total episode cost (SA1) | 908 | 1611 (970) | 1534 (894) |
| Total episode cost (SA2) | 969 | 1464 (841) | 1381 (713) |
| Outpatient attendance | Number of observations | Treatment group, mean (SD) (N = 1104) | |
|---|---|---|---|
| GTN (£) | Placebo (£) | ||
| Cost of outpatient appointments | 466 | 25.65 (98.42) | 18.86 (63.96) |
| Cost of hospital readmission | 1066 | 52.05 (858.84) | 43.32 (263.98) |
| Primary care resource | Number of observations | Treatment group, mean (SD) (N = 1104) | |
|---|---|---|---|
| GTN (£) | Placebo (£) | ||
| Cost of calls | 460 | 2.24 (8.19) | 1.24 (3.35) |
| Cost of visits | 457 | 3.52 (13.20) | 4.16 (13.66) |
| Cost of health visitor calls | 452 | 0.26 (2.58) | 0.48 (2.72) |
| Cost of health visitor visits | 452 | 1.42 (7.24) | 2.56 (11.18) |
| Cost of GP appointments | 457 | 13.80 (24.35) | 17.69 (34.64) |
| Cost of GP calls | 446 | 6.41 (17.92) | 6.03 (18.49) |
| Cost of GP home visits | 436 | 1.27 (8.64) | 0.20 (3.00) |
| Total primary care cost | 424 | 25.13 (52.29) | 28.40 (58.59) |
Incremental results
Table 24 summarises total costs by category and treatment allocation, and the estimated between-group difference. It can be noted that there are no significant between-group differences in any of the major cost categories. Directionally, there is a tendency towards slightly higher hospital episode costs in the GTN group than in the placebo group and this difference is consistent across both the approaches used to costing (SA1 and SA2).
| Category | Number of observations | Treatment group, mean (SD) (N = 1104) | Mean difference (95% CI)a | |
|---|---|---|---|---|
| GTN (£) | Placebo (£) | |||
| Total episode cost | 966 | 1366.62 (733.61) | 1317.12 (642.42) | 49.50 (–42.63 to 141.64) |
| Hospital episode cost (SA1) | 908 | 1610.98 (970.21) | 1534.46 (894.39) | 76.53 (–52.62 to 205.67) |
| Hospital episode cost (SA2) | 969 | 1464.35 (840.59) | 1381.53 (713.59) | 82.81 (–33.09 to 198.72) |
| Total primary care cost | 424 | 25.13 (52.29) | 28.40 (58.59) | –3.28 (–13.93 to 7.38) |
| Cost of outpatient appointment | 466 | 25.65 (98.42) | 18.86 (63.96) | 6.79 (–10.79 to 24.37) |
| Cost of hospital readmission | 1098 | 52.05 (858.84) | 43.32 (263.98) | 8.73 (–61.92 to 79.39) |
| Total NHS costb | 369 | 1514 (1732) | 1459 (779) | 55.30 (–199.20 to 309.79) |
Table 25 presents the main cost outcomes alongside the principal clinical, patient-oriented and safety outcomes, and secondary outcomes found to differ significantly between groups. Although the costs are directionally slightly higher in the GTN group than in the placebo group, there are no significant differences in the primary clinical, patient-oriented or safety outcomes. Furthermore, there is some evidence of a detrimental effect of GTN on several secondary outcomes, including the need for blood transfusions, side effects and blood pressure or heart rate.
| Cost/outcomes | Treatment group | Effect size | 95% CI | |
|---|---|---|---|---|
| GTN | Placebo | |||
| Cost of episode, mean (SD), mean difference | £1367 (734) | £1317 (642) | +£49.50 | –43 to 142 |
| Total costs to NHS, mean (SD), mean difference | £1514 (1732) | £1459 (779) | +£55.30 | –199 to 310 |
| Undelivered in 15 minutes, n (%), OR | 505 (93.3) | 518 (92.0) | 1.01 | 0.98 to 1.04 |
| Patient satisfaction (would recommend to a friend), n (%), OR | ||||
| Predischarge | 288/382 (75.4) | 303/388 (78.1) | 0.87 | 0.62 to 1.22 |
| 6 weeks | 166/221 (75.1) | 178/238 (74.8) | 1.02 | 0.66 to 1.56 |
| Palpitations/heart racing, n (%), OR | ||||
| Predischarge | 36/368 (9.8) | 15/375 (4.0) | 2.60** | 1.40 to 4.84 |
| 6 weeks | 14/200 (7.0) | 10/225 (4.4) | 1.62 | 0.70 to 3.73 |
| Blood loss (ml), n (%), OR | ||||
| Blood loss (ml) | ||||
| < 500 | 238/537 (44.3) | 249/560 (44.5) | ||
| 500–1000 | 180/537 (33.5) | 224/560 (40.0) | ||
| > 1000 | 119/537 (22.2) | 87/560 (15.5) | 1.14 | 0.88 to 1.48 |
| Blood transfusion | 61/533 (11.4) | 43/551 (7.8) | 1.53* | 1.04 to 2.25 |
| Fall in blood pressure/heart rate | 323/531 (60.8) | 131/544 (24.1) | 4.90*** | 3.73 to 6.42 |
Summary/discussion
The economic analysis is based on resource-use data that were collected for individual participants who were enrolled in the trial, combined with nationally relevant unit cost data. The strengths of the analysis include the secure randomisation, low loss to follow-up on the elements of resource use that are the most significant cost drivers and the generalisability of the unit costs applied. A potential limitation of the analysis is the reliance on reference costs as opposed to detailed bottom-up costing, because reference costs can lack the precision to capture the cost impact of finer differences in patterns of resource use between groups. To overcome this limitation we have presented more detailed data on resource use separately from costs, and these data show very similar levels and patterns between the treatment allocation groups. We also conducted further sensitivity analysis to assess the impact of utilising different methods to cost the delivery episode. A further limitation of the trial was the low level of follow-up on postdischarge resource use of primary care and hospital outpatient care. However, it can be noted from the available data that these costs were a relatively minor component of the overall costs and did not differ significantly between groups. In addition to the above, the economic analysis lacked an appropriate multidimensional measure of value by which to compare the alternatives. This is primarily attributable to the limitation of quality-adjusted life-years in this clinical context. Therefore, the analysis relied on a cost–consequence approach, which compared the different costs and multiple consequences of the alternative interventions without generating an incremental cost-effectiveness ratio or net benefit statistic.
However, considering the estimated costs in the context of the clinical effectiveness and safety findings, the data provide a clear message. The use of GTN for the treatment of retained placenta is not cost-effective compared with standard practice. Although there is a non-significant trend towards slightly higher costs in the GTN group, there are no significant differences in the primary clinical, patient-oriented or safety outcomes. Furthermore, directionally these primary outcomes favour placebo, and several secondary outcomes also point towards an increased side-effect profile for GTN over placebo, a possible safety concern with respect to blood pressure or heart rate and an increased number of blood transfusions required.
Chapter 7 Discussion and conclusions
Aim and overview
Non-surgical management options for retained placenta are limited and it is recognised that further research is required to examine medical management strategies for retained placenta. 1,8 Current management of manual removal of retained placenta poses a risk of bleeding and infection, and a new safer medical management that is more acceptable to women is urgently required.
Our large, pragmatic, randomised, double-blind trial was designed to determine the effectiveness of GTN spray as an alternative management for removal of retained placenta. It included an internal pilot study and the substantive trial measured four primary outcomes: clinical, safety, patient sided and economic. We also collected data for a number of secondary outcomes.
Summary of findings
This large, multicentre RCT demonstrated no difference between the GTN and placebo groups for the clinical outcome of the placenta remaining undelivered or the need for manual removal of placenta within 15 minutes of the study drug being administered. For the primary safety outcome, although numerically there appeared to be a greater number of bleeds of < 1000 ml in the GTN group, when compared with the placebo group this difference was not statistically significant. The only patient-sided difference noted was that the group that received the intervention of GTN reported an increase in the side effects of palpitations and tachycardia pre discharge. These side effects are consistent with previously reported side effects for GTN spray.
The main findings of the secondary outcomes were noted to be a decrease of 15 mmHg in systolic or diastolic blood pressure and/or increase in heart rate of > 20 b.p.m. noted for those in the GTN group. In addition, a greater number of participants in the GTN group also required a blood transfusion between the time of delivery and discharge from hospital.
The health economic analysis demonstrated no notable differences in elements of resource use associated with the hospital stay. Following discharge from hospital, there were no differences noted in the use of primary and secondary care services, but outpatient visits were marginally more frequent in the GTN group than in the placebo group. Readmission to hospital was slightly more common in the placebo group than in the GTN group, although the length of stay was noted to be higher in the GTN group than in the placebo group. The overall cost of the initial hospital stay was noted to be very similar for the two groups, with the GTN group being fractionally higher.
Strengths and limitations of the trial
We believe that the GOT-IT trial is the largest multicentre, randomised double-blind trial that has been undertaken to determine the effectiveness of GTN for retained placenta. The selection of a variety of hospitals has allowed us to produce results that are robust, reliable and generalisable. Furthermore, all sites received onsite monitoring visits from trained clinical trial monitors, which strengthened the validity of the data.
The group-sequential design of the trial provided the DMC opportunities for interim data evaluations. This approach allowed the maximum number of participants to be recruited into the trial, while also providing the opportunity for the trial to be stopped early if there had been either an overwhelming evidence of benefit or signs of futility. Hence, the use of a group-sequential approach confirmed that we were required to randomise the maximum number of participants to the trial to achieve the definitive result for our study. The inclusion of the internal pilot study was beneficial in obtaining the views of both women and staff to refine the consent pathway and optimise recruitment strategies for the substantive trial.
The trial completed the recruitment phase 2 months ahead of the projected recruitment completion date. As the primary clinical outcome was obtained within 15 minutes of study drug administration, we achieved 100% completeness. The majority of women who were invited to participate in the trial were willing to do so as they believed that the study medication (if they received the GTN intervention) could potentially prevent them from having a manual removal of their placenta. Thus, we are confident that our cohort accurately represented the demographic of women who sustain retained placenta.
We experienced problems in the early phase of recruitment with one component of the inclusion criteria. The term ‘haemodynamically stable’ was originally defined as a systolic blood pressure level of > 100 mmHg and a heart rate of < 110 b.p.m. However, in a minority of cases, clinicians were interpreting women as being ‘haemodynamically stable’ if their systolic blood pressure and heart rate were just outside these parameters. To clarify this, we redefined the definition of ‘haemodynamically stable’ in the protocol and submitted it as a substantial amendment to gain required approvals. The redefined definition read as follows.
Haemodynamically stable (must satisfy all three definitions):
-
haemodynamically stable
-
a heart rate of ≤ 119 b.p.m.
-
a systolic blood pressure level of > 100 mmHg.
This redefinition prevented any further protocol violations.
We anticipated that recruitment to the GOT-IT trial might be very challenging. The unpredictable nature of retained placenta meant that it could occur at any point within a 24-hour period. Successful delivery of this trial would therefore depend on clinicians (and not research staff) identifying, consenting and randomising women to trial entry alongside providing them with clinical care for an obstetric emergency. Very few of the trial sites had any experience of delivering a CTIMP in a busy labour ward setting, which posed logistical challenges particularly around safe storage of the investigational medicinal product in the labour ward setting. Finally, the good clinical practice requirements for such trials meant that all research and clinical staff were required to have either full good clinical practice training or study-specific training (with sponsor-approved good clinical practice training embedded within it) prior to consenting a participant into the trial. Of these, we believed that the last was likely to be the biggest barrier to clinician involvement in the trial. Working closely with our sponsor, we therefore developed study-specific training, including sponsor-approved good clinical practice training, to facilitate trial delivery. We believe that this was instrumental in the success of recruitment to this trial as this study-specific training allowed clinicians and clinical midwives who were not fully good clinical practice trained to consent eligible participants to the trial.
To further aid recruitment in an intrapartum setting, we chose to randomise trial participants using a ‘next off the shelf’ system rather than by a telephone or computing randomisation system. We were concerned that use of a telephone or computer system to randomise a participant would increase the time until the study drug was administered. We were therefore concerned that any possible delay in administering study medication, in the context of the life-threatening medical emergency of retained placenta, would influence the clinical teams’ decision to enrol participants and would subsequently affect study recruitment. Thus, the trial medication was signed out of pharmacy via a pharmacy accountability log and signed in to the labour ward via a labour ward accountability log. Once the study medication was on the labour ward, it was stored in a specific locked area subject to regular temperature monitoring. Locating the study drug on the labour ward allowed it to be administered rapidly with no delay. Use of the drug was recorded on a labour ward dispensing log, and return of the used vial to pharmacy was captured on the labour ward accountability log. The used vial was then signed back into pharmacy via the pharmacy accountability log before undergoing destruction as per local policy.
The main weakness of our trial was our underestimation of the anticipated return of the 6-week questionnaire sent out from the trial office. Despite sending reminders, the return rate of 6-week questionnaires for both the GTN group and the placebo group of the trial was initially only 20%. However, following the implementation of the thank-you cards, as a result of recommendations from the qualitative research, we saw the overall return rate increase to 43%. We believe that this increase in return rate was attributable to thanking the women for their participation in the trial by sending the cards. Nevertheless, an overall return rate of 43% was lower than anticipated and we attribute this to women finding it difficult to have the time to complete and return questionnaires via the post when they are looking after a young infant. We are aware that this low return rate may have some influence on the results of the economic evaluation. However, as the return rates were similar for both the GTN group and the placebo group, we believe that any similarities or minor differences noted between the two groups would have remained similar should the return rates have been higher.
Conclusions
-
No benefit was observed from use of GTN as the number of placentae that remained undelivered or required manual removal of placenta within 15 minutes was similar in both the GTN group and the placebo group (primary clinical outcome).
-
Measured blood loss was comparable for both groups (primary safety outcome).
-
Self-reported palpitations were reported more commonly prior to hospital discharge in participants who had received GTN compared with placebo. There were no other differences in side effect or satisfaction profile between groups (primary patient-sided outcome).
-
The health economic evaluation did not report any significant differences in the use of health resources between the two groups during the in-hospital stay, or the period from discharge to 6 weeks following hospital discharge (primary economic outcome).
-
Qualitative research with participants and staff helps to inform the recruitment and consent pathway, and inform study design for trials recruiting in emergency settings.
Acknowledgements
Acknowledgements of contributions to the project
We are extremely grateful to all women who participated in this trial.
We are grateful to members of the TSC [Andrew Shennan (chairperson), Deborah Bick, Kitty Bloemenkamp, Louise Brown and Claire Snowdon] and the DMC [Phillip Bennett (chairperson), Lucy Chappell and Amanda Farrin].
We are also grateful to the principal investigators: Vanessa Mackay (Queen Elizabeth University Hospital, Glasgow), Clare Tower (St Mary’s Hospital, Manchester), Paul Ayuk (Royal Victoria Hospital, Newcastle), Fiona Crosfill (Royal Preston Hospital), Rita Arya (Warrington Hospital), Janet Cresswell (Chesterfield Hospital), Shanthi Pinto (Leighton Hospital, Crewe), Louise Page (West Middlesex Hospital, London), Aparna Reddy (Stoke Mandeville Hospital), Joe Ogah (Furness Hospital, Morecambe Bay), Tadala Saukila (University Hospital of North Durham), Tadala Saukila (Darlington Memorial Hospital), Jen O’ Brien (Darlington Memorial Hospital), Priti Wuppalapati (Royal Bolton Hospital), Karen Brackley (Princess Anne Hospital, Southampton), Aarti Ullal (Royal Sunderland Hospital), Catherine Greenwood (The John Radcliffe Hospital, Oxford), George Bugg (Queen’s Medical Centre, Nottingham), Jim Thornton (Nottingham City Hospital), Elizabeth Martindale (Burnley General Hospital), Yashashri Choudari (Burnley General Hospital), Atul Nalawade (University Hospital of North Tees), Simon Wood (Countess of Chester Hospital), James Thomas (St Peter’s Hospital, Chertsey), Saikat Banerjee (St Peter’s Hospital, Chertsey), Deepika Meneni (The James Cook University Hospital, Middlesbrough), Claire Oxby (York District Hospital), Mr Patrick Bose and Mr Mark Selinger (Royal Berkshire Hospital, Reading), Sheena Hodgett (The Princess Royal Hospital, Telford), Premila Thampi (Milton Keynes University Hospital) and Maud Van de Venne (Frimley Park Hospital, Frimley); and the research midwives Aileen Jordan, Hayley Moir and Kirsten Cliff (all Edinburgh), Kirsteen Paterson, Therese McSorley and Lorna Mackay (all Glasgow), Linda Peacock (Manchester), Andrea Fenn (Newcastle), Katrina Rigby, Ann Docherty, Julie Earnshaw, Angela Philipson, and Stephanie Horridge (all Preston), Rachel Crone and Lindsay Roughley (both Warrington), Mary Kelly-Baxter (Chesterfield), Janet Brown and Caroline Dixon (both Crewe), Amy Barker and Marie O’ Connell (both West Middlesex), Julie Tebbutt (Stoke Mandeville), Karen Burns (Furness), Vicki Atkinson (Durham), Katrina Rhead, Christine Dawe and Claire Hopkins (all Bolton), Nicki Martin and Fiona Walbridge (Southampton), Gillian Campbell and Eileen Walton (both Sunderland), Fenella Roseman and Sarah Collins (both Oxford), Yvette Davis, Catriona Hussain, Gill Kirkwood and Carys Smith (all Nottingham), Hilary Jackson and Rebekah McCrimmon (both Burnley), Sharon Gowans (North Tees), Kerry Barker-Williams and Nichola Kearsley (both Chester), Beth Peers, Claire Atkinson and Hayley Tarft (all Chertsey), Hazel Alexander (South Tees), Jacqui Jennings (Darlington), Holly Alcock and Samantha Roche (both York), Sue Hallett and Sharon Westcar (both Reading), Helen Millward (Telford), Edel Clare (Milton Keynes) and Jay Weerasinghe (Frimley Park).
We are also grateful to all the study pharmacy teams for their input into this trial.
We are grateful to the trials team at the Centre for Healthcare Randomised Trials (ChaRT), Aberdeen, for providing support and expertise, especially Alison McDonald for providing expert trial management advice, and Mark Forrest for excellent design and database management.
We are also grateful to the study sponsor for sponsoring the trial and for providing advice and support throughout the entire duration. We are particularly grateful to Elizabeth Craig for providing monitoring oversight for the trial. We are also grateful to Richard Anderson, who served as an observational member of our TSC. We are also grateful to all staff working in NHS research and development offices who have supported this trial.
Finally, we are extremely grateful to Susan Morrow (previous Trial Manager), Tariq Derdeb (Assistant Trial Manager) and Gayle Beveridge, Hilary Guthrie and Amy Witherspoon (our Trial Administrators).
Contributions of authors
Fiona C Denison (Reader/Honorary Consultant in Maternal and Fetal Medicine) was the chief investigator. She contributed to the study design and protocol writing, study management, oversight of entire study and the writing and final editing of the report.
Kathryn F Carruthers (Trial Manager) supervised the management and day-to day running of the trial, supervised data collection and drafted the final report.
Jemma Hudson (Trial Statistician) provided statistical support, performed the statistical analysis of the study and contributed to the writing of the report.
Gladys McPherson was a co-applicant and was involved with the design of the database.
Graham Scotland (Health Economist) designed and planned the health economic analyses, provided health economic supervision and advice for the study and contributed to the final report.
Sheonagh Brook-Smith (Senior Clinical Midwife) was a co-applicant and helped with trial design and implementation of the pilot phase.
Cynthia Clarkson (layperson) contributed to the study design, provided comments and advice from a layperson’s perspective and contributed to the final report.
Mathilde Peace (layperson) contributed to the study design and provided comments and advice from a layperson’s perspective.
Jane Brewin (layperson) contributed to the study design, provided comments and advice from a layperson’s perspective and contributed to the final report.
Gin Nie Chua (Health Economist) contributed to the health economic analysis and the writing of Chapter 6.
Nina Hallowell (Associated Professor at Nuffield Department of Population Health, University of Oxford) contributed to the design and implementation of the qualitative trial and the qualitative chapter of the report (see Chapter 3).
Jane E Norman (Professor of Maternal and Fetal Health) was a co-applicant and contributed to the study design, writing the protocol, study management as a member of the Trial Management Group and final editing of the report.
Julia Lawton (Professor of Health and Social Science) was a co-applicant and contributed to the design and implementation of the qualitative trial and the writing of the qualitative chapter of the final report (see Chapter 3).
John Norrie (Professor of Medical Statistics and Trial Methodology) was a co-applicant and was responsible for the design of the study, provided statistical supervision and advice as a member of the Trial Monitoring Group and contributed to the writing and final editing of the report.
Publications
Hallowell N, Snowdon C, Morrow S, Norman JE, Denison FC and Lawton J. The role of therapeutic optimism in recruitment to a clinical trial in a peripartum settings: balancing hope and uncertainty. Trials 2016;17:267.
Lawton J, Snowdon C, Morrow S, Norman JE, Denison FC and Hallowell N. Recruiting and consenting into a peripartum trial in an emergency setting: a qualitative study of the experiences and views of women and healthcare professionals. Trials 2016;17:195.
Denison FC, Norrie J, Lawton J, Norman JE, Scotland G, McPherson G, et al. A pragmatic group sequential, placebo-controlled, randomised trial to determine the effectiveness of glyceryl trinitrate for retained placenta (GOT-IT): a study protocol. BMJ Open 2017;7:e017134.
Lawton J, Hallowell N, Snowdon C, Norman JE, Carruthers K and Denison FC. Written versus verbal consent: a qualitative study of stakeholder views of consent procedures used at the time of recruitment into a peripartum trial conducted in an emergency setting. BMC Med Ethics 2017;18:36.
Denison FC, Carruthers KF, Hudson J, McPherson G, Chua GN, Peace M, et al. Nitroglycerin for treatment of retained placenta: a randomized, placebo-controlled, multicentre double blind trial in the UK. PLOS Med in press; 2019.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly, Everyone should be able to find out about how patient data are used. #datasavelives You can find out more about the background to this citation here: understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- National Institute for Health and Care Excellence (NICE) . NICE Clinical Guideline 190: Intrapartum Care for Healthy Women and Babies 2014. www.nice.org.uk/guidance/cg190 (accessed 1 February 2018).
- Cheung WM, Hawkes A, Ibish S, Weeks AD. The retained placenta: historical and geographical rate variations. J Obstet Gynaecol 2011;31:37-42. https://doi.org/10.3109/01443615.2010.531301.
- Stones RW, Paterson CM, Saunders NJ. Risk factors for major obstetric haemorrhage. Eur J Obstet Gynecol Reprod Biol 1993;48:15-8. https://doi.org/10.1016/0028-2243(93)90047-G.
- Combs CA, Murphy EL, Laros RK. Factors associated with postpartum hemorrhage with vaginal birth. Obstet Gynecol 1991;77:69-76.
- Lennox C, Marr J. Scottish Confidential Audit of Severe Maternal Morbidity: Reducing Avoidable Harm. 10th Annual Report 2014. www.healthcareimprovementscotland.org/our_work/reproductive,_maternal_child/programme_resources/scasmm.aspx (accessed 1 February 2018).
- Combs CA, Laros RK. Prolonged third stage of labor: morbidity and risk factors. Obstet Gynecol 1991;77:863-7.
- Ely JW, Rijhsinghani A, Bowdler NC, Dawson JD. The association between manual removal of the placenta and postpartum endometritis following vaginal delivery. Obstet Gynecol 1995;86:1002-6. https://doi.org/10.1016/0029-7844(95)00327-N.
- Abdel-Aleem H, Abdel-Aleem MA, Shaaban OM. Nitroglycerin for management of retained placenta. Cochrane Database Syst Rev 2015;11. https://doi.org/10.1002/14651858.CD007708.pub3.
- Barnes F. Hour-glass contraction of the uterus treated with nitrite of amyl. Br Med J 1882;1. https://doi.org/10.1136/bmj.1.1107.377.
- Dufour P, Vinatier D, Puech F. The use of intravenous nitroglycerin for cervico-uterine relaxation: a review of the literature. Arch Gynecol Obstet 1997;261:1-7. https://doi.org/10.1007/s004040050189.
- Chedraui PA, Insuasti DF. Intravenous nitroglycerin in the management of retained placenta. Gynecol Obstet Invest 2003;56:61-4. https://doi.org/10.1159/000072734.
- DeSimone CA, Norris MC, Leighton BL. Intravenous nitroglycerin aids manual extraction of a retained placenta. Anesthesiology 1990;73. https://doi.org/10.1097/00000542-199010000-00031.
- Lowenwirt IP, Zauk RM, Handwerker SM. Safety of intravenous glyceryl trinitrate in management of retained placenta. Aust N Z J Obstet Gynaecol 1997;37:20-4. https://doi.org/10.1111/j.1479-828X.1997.tb02212.x.
- Peng AT, Gorman RS, Shulman SM, DeMarchis E, Nyunt K, Blancato LS. Intravenous nitroglycerin for uterine relaxation in the postpartum patient with retained placenta. Anesthesiology 1989;71:172-3. https://doi.org/10.1097/00000542-198907000-00039.
- Rolbin SH, Hew EM, Bernstein A. Uterine relaxation can be life saving. Can J Anaesth 1991;38:939-40. https://doi.org/10.1007/BF03036985.
- Bullarbo M, Tjugum J, Ekerhovd E. Sublingual nitroglycerin for management of retained placenta. Int J Gynaecol Obstet 2005;91:228-32. https://doi.org/10.1016/j.ijgo.2005.08.020.
- Ekerhovd E, Bullarbo M. Sublingual nitroglycerin seems to be effective in the management of retained placenta. Acta Obstet Gynecol Scand 2008;87:222-5. https://doi.org/10.1080/00016340701855654.
- Redick LF, Livingston E. A new preparation of nitroglycerin for uterine relaxation. Int J Obstet Anesth 1995;4:14-6. https://doi.org/10.1016/0959-289X(95)82102-G.
- Laslett LJ, Baker L. Sublingual nitroglycerin administered by spray versus tablet: comparative timing of hemodynamic effects. Cardiology 1990;77:303-10. https://doi.org/10.1159/000174612.
- Marmor A. Comparative evaluation of a new formulation of isosorbide dinitrate oral spray and sublingual nitroglycerin tablets. Am J Cardiol 1990;65:43J-45J. https://doi.org/10.1016/0002-9149(90)91311-S.
- Curry SH, Lopez LM, Lambert CR, Kwon HR, Stack RK. Plasma concentrations and hemodynamic effects of intravenous, sublingual, and aerosolized nitroglycerin in patients undergoing cardiac catheterization. Biopharm Drug Dispos 1993;14:107-18. https://doi.org/10.1002/bdd.2510140203.
- Wight LJ, VandenBurg MJ, Potter CE, Freeth CJ. A large scale comparative study in general practice with nitroglycerin spray and tablet formulations in elderly patients with angina pectoris. Eur J Clin Pharmacol 1992;42:341-2. https://doi.org/10.1007/BF00266360.
- Farley AE, Graham CH, Smith GN. Contractile properties of human placental anchoring villi. Am J Physiol Regul Integr Comp Physiol 2004;287:R680-5. https://doi.org/10.1152/ajpregu.00222.2004.
- Royal College of Obstetricians and Gynaecologists (RCOG) . Obtaining Valid Consent to Participate in Perinatal Research Where Consent Is Time Critical (Clinical Governance Advice No. 6a) 2016.
- Hallowell N, Snowdon C, Morrow S, Norman JE, Denison FC, Lawton J. The role of therapeutic optimism in recruitment to a clinical trial in a peripartum setting: balancing hope and uncertainty. Trials 2016;17. https://doi.org/10.1186/s13063-016-1394-1.
- Lawton J, Snowdon C, Morrow S, Norman JE, Denison FC, Hallowell N. Recruiting and consenting into a peripartum trial in an emergency setting: a qualitative study of the experiences and views of women and healthcare professionals. Trials 2016;17. https://doi.org/10.1186/s13063-016-1323-3.
- Lawton J, Hallowell N, Snowdon C, Norman JE, Carruthers K, Denison FC. Written versus verbal consent: a qualitative study of stakeholder views of consent procedures used at the time of recruitment into a peripartum trial conducted in an emergency setting. BMC Med Ethics 2017;18. https://doi.org/10.1186/s12910-017-0196-7.
- Coviello EM, Grantz KL, Huang CC, Kelly TE, Landy HJ. Risk factors for retained placenta. Am J Obstet Gynecol 2015;213:864.e1-864.e11. https://doi.org/10.1016/j.ajog.2015.07.039.
- Great Britain . Data Protection Act 1998 1998. www.legislation.gov.uk/ukpga/1998/29/contents (accessed 1 February 2018).
- Aspire Pharma Ltd . Glytrin, 400 Micrograms Per Metered Dose, Sublingual Spray n.d. www.medicines.org.uk/emc/medicine/10739 (accessed 1 February 2018).
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70:659-63. https://doi.org/10.2307/2336502.
- O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics 1979;35:549-56. https://doi.org/10.2307/2530245.
- Denison F, McPherson G, Scotland G, Brook-Smith S, Clarkson C, Peace M, et al. A pragmatic adaptive sequential placebo controlled randomised trial to determine the effectiveness of Glyceryl trinitrate for retained placenta (Got-it trial). Southampton: National Institute for Health Research; n.d.
- Royal College of Obstetricians and Gynaecologists (RCOG) . Obtaining Valid Consent to Participate in Research While in Labour (Clinical Governance Advice No. 6a) 2010.
- Vernon G, Alfirevic Z, Weeks A. Issues of informed consent for intrapartum trials: a suggested consent pathway from the experience of the release trial [ISRCTN13204258]. Trials 2006;7. https://doi.org/10.1186/1745-6215-7-13.
- Britten N. Qualitative interviews in medical research. BMJ 1995;311:251-3. https://doi.org/10.1136/bmj.311.6999.251.
- Pope C, Mays N. Reaching the parts other methods cannot reach: an introduction to qualitative methods in health and health services research. BMJ 1995;311:42-5. https://doi.org/10.1136/bmj.311.6996.42.
- Glaser BG, Strauss AL. The Discovery of Grounded Theory. New York, NY: Aldine; 1967.
- Strauss A, Corbin JM. Basics of Qualitative Research: Grounded Theory Procedures and Techniques. London: Sage Publications, Ltd; 1990.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2016. Canterbury: Personal Social Services Research Unit, University of Kent; 2016.
- Department of Health and Social Care . NHS Reference Costs 2015 to 2016 n.d. www.gov.uk/government/publications/nhs-reference-costs-2015-to-2016.
- Joint Formulary Committee . British National Formulary n.d. www.medicinescomplete.com (accessed 5 April 2019).
- Information Services Division Scotland . Scottish Health Service Costs (2016) n.d. www.isdscotland.org/Health-Topics/Finance/Costs/File-Listings-2016.asp (accessed 5 April 2019).
- National Institute for Health and Care Excellence (NICE) . Costing Statement: Blood Transfusion Implementing the NICE Guideline on Blood Transfusion (NG24) 2015. www.nice.org.uk/guidance/ng24/resources/ (accessed 5 April 2019).
- NHS . OPCS Classification of Interventions and Procedures 2019. www.datadictionary.nhs.uk/web_site_content/supporting_information/clinical_coding/opcs_classification_of_interventions_and_procedures.asp (accessed 20 July 2018).
List of abbreviations
- AE
- adverse event
- b.p.m.
- beats per minute
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CTIMP
- Clinical Trial of an Investigational Medicinal Product
- DMC
- Data Monitoring Committee
- GOT-IT
- Glyceryl trinitrate fOr reTaIned placenTa
- GP
- general practitioner
- GTN
- glyceryl trinitrate
- HTA
- Health Technology Assessment
- ISRCTN
- International Standard Randomised Controlled Trial Number
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- OR
- odds ratio
- RCT
- randomised controlled trial
- SA1
- sensitivity analysis 1
- SA2
- sensitivity analysis 2
- SAE
- serious adverse event
- TSC
- Trial Steering Committee