

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 12/127/134. The contractual start date was in June 2014. The draft report began editorial review in November 2019 and was accepted for publication in July 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Disclaimer
This report contains transcripts of interviews conducted in the course of the research and contains language that may offend some readers.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2020. This work was produced by Appleton et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2020 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Parts of this chapter have been reused from Lyttle et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Parts of this chapter have been reused from Lyttle et al. 2 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
In addition, the exclusion criteria have been reused from the study ISRCTN registry. 3 This article is available under the terms of the Creative Commons Attribution License http://creativecommons.org/licenses/by/4.0/.
Scientific background
Convulsive status epilepticus (CSE) is the most common life-threatening neurological emergency in children, with an incidence of 20 per 100,000 children per year. 4,5 It is the second most common reason for unplanned admission to paediatric intensive care units (PICUs) in the UK, accounting for 5.6% of all PICU admissions. 6 Mortality is low, but morbidity, including neurodisability, learning difficulties and de novo and drug-resistant epilepsy, may be as high as 22%. 7–10 Predictably, these may result in major long-term demands on acute and chronic health and social care resources. 4 The longer the duration of CSE, the more difficult it is to terminate and the greater the morbidity risk. 4,9,10
The current UK emergency care pathway for the management of childhood CSE is the stepwise algorithm that is advocated in advanced paediatric life support (APLS) guidance. 11 First-line treatment is two doses of a benzodiazepine given 10 minutes apart. A second-line anticonvulsant is administered if the child continues to fit 10 minutes after the second dose of benzodiazepine. APLS guidance recommends phenytoin (Epanutin, Pfizer Inc., New York, NY, USA) as the first-choice second-line anticonvulsant. Phenobarbital (AAH Pharmaceuticals, Coventry, UK) is recommended if the child is allergic to phenytoin, has previously not responded to it or has experienced a serious adverse event (SAE). Failure to stop CSE necessitates rapid sequence induction (RSI), intubation and admission to PICU, with consequent potential for iatrogenic consequences, including pneumonia, hospital-acquired infections and prolonged admission.
There is reasonable randomised controlled trial (RCT) evidence to support the use of benzodiazepines as first-line anticonvulsants,12 but there is a dearth of evidence for second-line drug treatment and no high-quality RCT evidence to support any second-line treatment. 13 There is an absence of randomised evidence to support the use of phenytoin as the second-line anticonvulsant, despite its use as a standard intravenous (i.v.) anticonvulsant for the treatment of CSE since the 1940s. A retrospective case note review, in which 87% (331/381) of children administered a second-line anticonvulsant received phenytoin, reported seizure cessation in 190 cases (50%). 14 There is considerably more literature on phenytoin’s potential adverse effects, including potentially fatal cardiac arrhythmias and Stevens–Johnson syndrome (Tables 1 and 2). 15–18 The risk of a cardiac arrhythmia is related to the rate of infusion and, therefore, phenytoin must be infused over a period of at least 20 minutes. 18
| Eligibility criteria | Reason for ineligibilitya | Number of patients |
|---|---|---|
| Inclusion | ||
| 1 | Outside age range | 23 |
| 2 | Patient does not present with generalised tonic–clonic, generalised clonic or focal clonic status epilepticus that requires second-line treatment to terminate the seizureb | 656 |
| 3 | First-line treatment not administered in accordance with APLS guidelines or personalised rescue care plan to try and terminate the seizure | 133 |
| Exclusion | ||
| 1 | Absence, myoclonic or non-CSE, or infantile spasms | 100 |
| 2 | Known or suspected pregnancy | 0 |
| 3 | Contraindication or allergy to either trial treatment | 45 |
| 4 | Known renal failure (i.e. patients on peritoneal or haemodialysis, or with renal function that is < 50% expected for age) | 3 |
| 5 | Previous administration of a second-line antiepileptic drug prior to arrival in the ED | 7 |
| 6 | Had previously entered the EcLiPSE trial | 38 |
| Data recorded | Assumption | Number of patients |
|---|---|---|
| Patient recorded as ‘no’ to inclusion criterion 2 and ‘yes’ to exclusion criterion 1, but their seizure continued after benzodiazepines | Patient had incorrect seizure type | 21 |
| Patient recorded as ‘no’ to inclusion criterion 2 and ‘no’ to exclusion criterion 1 and their seizure stopped after benzodiazepines | No second-line treatment was required (seizure stopped), but patient had correct seizure type | 533 |
| Patient recorded as ‘no’ to inclusion criterion 2 and ‘yes’ to exclusion criterion 1 and their seizure stopped after benzodiazepines | Patient had incorrect seizure type and no second-line treatment was required | 57 |
| Missing response for exclusion criterion 1 | No assumptions made: data confirmed as unobtainable | 3 |
| Patient recorded ‘no’ to inclusion criterion 2 and ‘no’ to exclusion criterion 1 and their seizure continued after benzodiazepines | No assumptions made: data suggested correct seizure type and seizure continuing | 11 |
| Patient has no benzodiazepines recorded | No assumptions made | 19 |
| Patient has missing benzodiazepine administration outcome | No assumptions made: data confirmed as unobtainable | 3 |
| Patient has other outcome recorded for benzodiazepines | No assumptions made | 9 |
| Total | 656 |
Levetiracetam (Keppra, UCB Pharma, Brussels, Belgium) is a broad-spectrum anticonvulsant that effectively treats focal and generalised tonic–clonic and myoclonic seizures. A growing body of evidence, predominantly, but not exclusively, anecdotal, suggests that i.v. levetiracetam is safe and effective in the treatment of acute repetitive seizures and both CSE and non-CSE, with reported seizure cessation rates between 76% and 100%. 19–27 There is limited evidence that i.v. levetiracetam may also be as effective as i.v. lorazepam (Ativan, Pfizer Inc.), which is the current first-choice first-line anticonvulsant in the treatment of CSE. Levetiracetam and lorazepam administered intravenously to 79 patients (the majority of whom were adults) were equally effective in terminating CSE {levetiracetam terminated CSE in 76.3% of patients and lorazepam in 75.6% of patients [relative risk 0.97, 95% confidence interval (CI) 0.44 to 2.13]}. 28 A systematic review of levetiracetam published in 201229 indicated that efficacy ranged from 44% to 94%, with reported higher rates in retrospective studies. Two RCTs, published in 2015, involving predominantly adults, directly compared i.v. levetiracetam with either i.v. phenytoin30 or i.v. phenytoin plus i.v. sodium valproate (Epilim, Sanofi, Paris, France)31 and showed no difference between the comparators. Chakravarthi et al. 30 reported on 44 patients who presented with ‘consecutive status’ and were randomised to treatment with phenytoin (20 mg/kg) or levetiracetam (20 mg/kg). Both drugs showed a similar efficacy in status termination within 30 minutes of commencement of drug infusion. Phenytoin achieved control in 15 out of 22 (68.2%) patients and levetiracetam in 13 out of 22 (59.1%) patients (p = 0.53). 30 In the study reported by Mundlamuri et al. ,31 the presenting seizure was controlled with lorazepam plus phenytoin infusion in 34 out of 50 (68%) patients, lorazepam plus valproate infusion in 34 out of 50 (68%) patients and with lorazepam plus levetiracetam infusion in 39 out of 50 (78%) patients. There was no statistically significant difference between the subgroups (p = 0.44). 31 Reported i.v. levetiracetam doses range from 20 to 60 mg/kg. Chakravarthi et al. 30 and Mundlamuri et al. 31 used doses of 20 mg/kg and 25 mg/kg, respectively. Adverse reactions (ARs) with levetiracetam seem to be infrequent and mild, even at high doses. These include dizziness, somnolence, headache and transient agitation, but there have been no reports of cardiac arrhythmias, hypotension, tissue extravasation reactions, Stevens–Johnson syndrome or hepatotoxicity. 32–34 Levetiracetam can be infused over 5–10 minutes, which suggests that, theoretically, CSE may be terminated more rapidly than with phenytoin. Consequently, a reasonable hypothesis is that levetiracetam may be more effective and safer than i.v. phenytoin in terminating CSE. 13,35,36
Convulsive status epilepticus management was identified as a key priority area for research by a number of sources, including the Paediatric Emergency Research in the UK and Ireland (PERUKI)37,38 in its inaugural prioritisation exercise,38 and the National Institute for Health and Care Excellence in its update of national epilepsy guidelines published in January 2012. 39 A high-quality RCT is, therefore, essential to determine whether phenytoin or levetiracetam is the better drug in managing CSE, as highlighted in a recent systematic review. 13 A meta-analysis published in 2014 concluded with the following statement:
The evidence does not support the first-line use of phenytoin. There is not enough evidence to support the routine use of lacosamide. Randomized controlled trials are urgently needed. 36
One RCT has recently been completed that evaluated the efficacy and safety of i.v. levetiracetam and phenytoin in the management of CSE in children aged 3 months to 16 years. 40,41 A second RCT recently evaluated i.v. fosphenytoin, levetiracetam and sodium valproate in the management of CSE in children (aged > 2 years) and adults. 42
The Emergency treatment with Levetiracetam or Phenytoin in Status Epilepticus in children (EcLiPSE) trial was a Phase IV, multicentre, parallel-group, randomised controlled open-label superiority trial comparing i.v. levetiracetam with i.v. phenytoin.
Rationale for research
Convulsive status epilepticus is the most common life-threatening neurological emergency in childhood. It can result in significant morbidity, with acute and chronic impacts on the family and health and social care systems. The current recommended first-choice second-line treatment in children aged ≥ 6 months is i.v. phenytoin (fosphenytoin, which is a pro-drug of phenytoin in the USA). However, this is not based on any robust RCT evidence and the drug is associated with significant and potentially serious adverse reactions (SARs). Emerging evidence suggests that i.v. levetiracetam may be effective as a second-line agent for CSE, and fewer ARs have been described. This trial was designed to determine whether i.v. phenytoin or i.v. levetiracetam is the more effective and safer drug in the treatment of childhood CSE.
Intervention
The EcLiPSE trial was an open-label trial using investigational medicinal products (IMPs) with marketing authorisation in the UK. These became IMPs only when the packaging was opened in the setting of this study. IMP provision was the responsibility of each site in accordance with standard clinical practice. Both IMPs were stored in line with local requirements for general medicine supplies.
A single dose of the randomly allocated treatment was administered by i.v. infusion. The levetiracetam dose was 40 mg/kg (with a maximum dose of 2500 mg) over 5 minutes, diluted to a maximum of 50 mg/ml with 0.9% sodium chloride. The dose of 40 mg/kg was based on the available published data at the time of the full study application to the Health Technology Assessment programme in 2013. The phenytoin dose was 20 mg/kg (with a maximum dose of 2000 mg) at a rate not exceeding 1 mg/kg/minute (or > 20 minutes for doses of > 1 g), diluted with 0.9% sodium chloride to a maximum concentration of 10 mg/ml. This dose was based on national guidelines available in 2013. 11
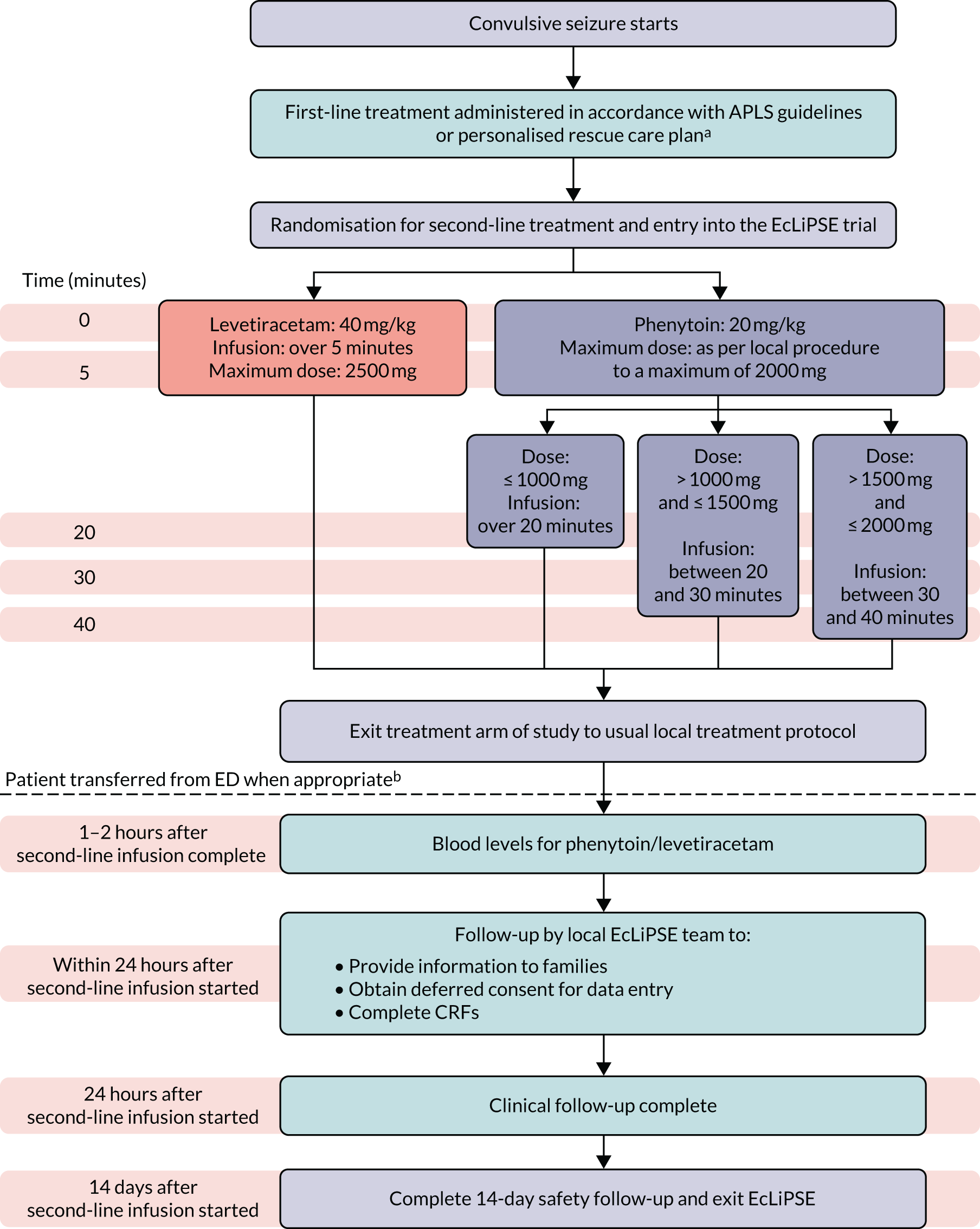
The allocated treatment was prepared and administered in accordance with standard clinical care, with independent checking performed by two trained personnel. Trial-specific labelling was not required, rather an approved ‘i.v. additive label’ was used. If the randomised treatment was discontinued prior to administration of the full dose this was recorded. If CSE persisted at the end of the IMP infusion, further medical management was decided by the local clinical team independent of the trial protocol (Figure 1).
FIGURE 1.
Study design. a, Administration of the first-line treatment may have occurred prior to arrival in the ED. b, If a patient was randomised but not treated with a second-line anticonvulsant, follow-up would end at this point. ED, emergency department.
Objectives
The study objectives were to determine:
-
whether i.v. phenytoin or i.v. levetiracetam is the more efficacious second-line anticonvulsant for the emergency management of CSE in children
-
whether or not i.v. levetiracetam is associated with fewer ARs or adverse events (AEs) than i.v. phenytoin
-
the potential barriers and solutions to recruitment and consent in the EcLiPSE trial to inform future trials with regard to recruiter training and trial conduct in this clinical setting (i.e. a nested consent study).
Chapter 2 Trial design and methods
Trial registration and ethics
The trial was approved by the National Research Ethics Service Committee North West – Liverpool Central on 3 March 2016 (reference 15/NW/0090). The trial is registered as ISRCTN22567894 (registered 27 August 2015) and EudraCT identifier 2014-002188-13 (registered on 21 May 2014).
Participant inclusion and exclusion criteria
The full study protocol was published in 2017. 2
Inclusion criteria
-
Males and females aged 6 months to 17 years 11 months (inclusive).
-
The presenting seizure was generalised tonic–clonic, generalised clonic or focal clonic status epilepticus that requires second-line treatment to terminate the seizure.
-
First-line treatment was administered in accordance with APLS guidelines or the child’s personalised rescue care plan to try and terminate the presenting seizure.
Eligibility notes
Patients with the following features were eligible for inclusion in the trial, assuming that all other inclusion and exclusion criteria were met.
-
Patients administered more than two doses of benzodiazepines, which is above the recommended dose in APLS guidelines.
-
Patients whose personalised rescue care plan included rectal paraldehyde as the first-line treatment.
-
Patients receiving oral phenytoin or levetiracetam as part of their regular oral antiepileptic drug regime.
Exclusion criteria
-
Absence, myoclonic or non-CSE, or infantile spasms.
-
Patients with a known or suspected pregnancy.
-
Patients with known contraindication or allergy to levetiracetam or phenytoin. This included when the child’s personalised rescue care plan stated that the child never responded to, or had previously experienced a SAR to, phenytoin, levetiracetam or both.
-
Patients with known renal failure (i.e. patients on peritoneal or haemodialysis, or with renal function that is < 50% expected for age).
-
Previous administration of a second-line antiepileptic drug prior to arrival in the emergency department (ED).
-
Patients known to have previously been treated as part of the EcLiPSE trial.
Recruitment
Thirty EDs (‘sites’) throughout Great Britain and Northern Ireland participated in the study. The sites were selected from the membership of PERUKI [a collaborative paediatric emergency medicine (PEM) research network]. 37 Participating sites included tertiary or district general hospitals with EDs that treat children only, or children and adults. A full list of participating centres is included in Appendix 3. Centres were selected based on factors such as membership of PERUKI, site research infrastructure, projected number of recruits based on the local population and proposed training strategy.
Screening commenced once a child arrived in the ED and received their first-line treatment for CSE. A unique participant screening form was used and included an eligibility assessment and reasons for non-randomisation where appropriate. If eligible, the participant was randomised to the trial.
Informed consent
The trial used research without prior consent (RWPC), also known as ‘deferred consent’, because of the time-critical management of CSE, in accordance with regulatory requirements, RWPC guidance and pre-trial research. 43,44 Parents/legal representatives/patients (hereafter termed ‘participants’) were approached once the child’s clinical condition was stable. This was ideally within 24 hours of randomisation and prior to discharge from hospital, at which point written informed consent was sought to continue data collection and use data already collected.
If consent was not sought prior to discharge the participant would be contacted within 5 working days of randomisation by a delegated member of the research team and informed of the participant’s involvement and details of the trial. Written information and a consent form were posted to the family. The covering letter asked participants to return the enclosed form, indicating their consent for use of the data already collected and continued participation in trial follow-up, within 4 weeks of the date of the letter. If no response was received within 4 weeks, the covering letter verified that the participant was included within the trial.
If the participant died before consent was sought, the site research team obtained information from colleagues and bereavement counsellors to establish the most appropriate time and the most appropriate practitioner to notify the parents/legal representative of their child’s involvement in the research study.
When it was considered inappropriate to seek consent prior to the parent/legal representative’s departure from hospital, the parent/legal representative was notified by a personalised letter and written information about the trial from the most appropriate practitioner 4 weeks after randomisation. Wherever possible, this practitioner would already be known to the family. The letter explained the EcLiPSE trial, reasons for deferred consent, how to opt in or out of the trial and provided contact details if parents wished to discuss the trial with a member of the research team (either in person or by telephone).
A second letter was sent to the bereaved family if there had been no response within 4 weeks of the initial letter. The second letter included information about the EcLiPSE trial, reasons for deferred consent and how to opt in or out of the trial. It also provided contact details if parents wished to discuss the EcLiPSE trial with a member of the research team, either in person or by telephone. Finally, it informed the family that the participant’s data would be included in the trial if no consent form was returned within 4 weeks of the letter being sent, unless the family first notified the site team.
Finally, there was also a qualitative mixed-method study involving participants to explore approaches to recruitment and deferred consent. This is outlined in Chapter 4.
Randomisation
Participants were randomised to levetiracetam or phenytoin in a ratio of 1 : 1 using random variable block sizes of two and four. A computer-generated randomisation schedule was produced by an independent statistician who had no further involvement in the trial. Randomisation was stratified by centre for logistical purposes. Centres were provided with sequentially numbered and EcLiPSE trial-labelled randomisation packs. These were stored in an appropriate secure location within the ED for ready access on presentation of eligible patients.
Randomisation packs were opaque brown cardboard tamper-proof A4 envelopes. The construction was resistant to accidental damage or tampering and contents could not be viewed without fully opening the envelope. Each pack was sequentially numbered and during randomisation the clinician/nurse used the next sequentially numbered pack.
The case report form (CRF) that was completed at the patient’s bedside while the patient was in the ED was included in the randomisation pack. The CRF was prepopulated with the centre code, participant randomisation number and the randomly allocated treatment.
The randomisation pack should have been opened only once eligibility was confirmed on the screening form. However, because of the time required to prepare the randomised treatment for infusion, this was undertaken prior to when the infusion was required to avoid trial participation creating a delay in treatment. This meant that some randomised participants would experience seizure cessation while the infusion was being prepared.
Once randomised, the patient was administered the randomly allocated treatment, as required clinically, to terminate seizure activity.
If the participant was randomised but the seizure terminated prior to infusion of the randomly allocated treatment, the patient could subsequently be treated with the randomised treatment allocation if the patient’s seizure restarted while still in the ED. However, if the patient’s seizure restarted after leaving the ED, the randomised treatment could not be given.
If the randomised treatment was not administered while the patient was in the ED and instead the comparator second-line was administered in error, the participant was still considered as recruited and the randomisation number applied. The treatment administered was recorded, along with reasons why it had not been possible to treat as per allocation.
If the patient had been given a RSI prior to administration of any second-line treatment the decision to administer a second-line treatment was outside the EcLiPSE trial and the patient was treated as per the site’s routine care.
Blinding
It was not possible to blind the trial interventions in the EcLiPSE trial because of the different times required for their infusion. Although a double-dummy approach was considered, this would have increased complexity during a paediatric emergency situation. Therefore, this was an open-label study.
Outcome measures
Primary outcome
The primary outcome was time from randomisation to cessation of all visible signs of convulsive seizure activity. All ‘visible signs of convulsive seizure activity’ was defined by cessation of all continuous rhythmic clonic activity.
Secondary outcomes
The secondary outcomes were as follows:
-
the need for further anticonvulsants to manage CSE after administration of the trial treatment
-
the need for RSI because of ongoing CSE
-
the need for admission to a critical care unit (i.e. a high-dependency unit or a PICU)
-
the occurrence of SARs, including death, airway complications, cardiovascular instability (e.g. cardiac arrest, arrhythmia and hypotension requiring intervention), extravasation injury (e.g. ‘purple glove syndrome’) and extreme agitation.
Data collection
Follow-up
There were three time points for data collection in the EcLiPSE trial (see Figure 1). The first was in the ED during the acute CSE treatment phase. The second was at 24 hours following the administration of the randomised treatment, wherein data collected included further seizures, concomitant anticonvulsants that may have been required to treat other acute seizures and ARs. The third and final time point was undertaken 14 days after administration of the randomised treatment through review of hospital notes and a single-sheet four-question questionnaire completed by the child’s parents. The questionnaire included information on further hospital admissions and organ failure.
Blood samples
Samples were taken 1–2 hours after completion of the randomised treatment to measure drug levels, a common practice when giving phenytoin. 45 Levetiracetam levels were measured as part of trial conduct by an accredited central laboratory. Measurement of phenytoin levels was undertaken in the laboratory of each participating site as part of routine care.
Sample size
The sample size was calculated on the basis of published seizure cessation rates for phenytoin (50–60%)14 and levetiracetam (76–100%). 19–27 A sample size of 140 randomised and consented participants per group, with a total of 183 events of CSE cessation, was required for a 0.05-level two-sided log-rank test for equality of survival curves to detect an increase in seizure cessation rates from 60% to 75% [a constant hazard ratio (HR) of 0.661] at 80% power. The sample size was increased to 308 participants to allow for 10% loss to follow-up, which proved unnecessary. The final sample size was 286 participants and the Independent Data and Safety and Monitoring Committee (IDSMC) and Trial Steering Committee (TSC) were consulted before the decision to stop recruitment because of low attrition and completeness of data. The difference to detect was influenced by the size of difference deemed to be clinically important and convincing to change clinical practice. Although smaller differences could also have been considered important, they needed to be balanced against the costs of the medicinal products and the experience of delivering them within emergency care setting.
Trial management and oversight
Trial Management Group
The Trial Management Group (TMG) was responsible for the day-to-day practical and clinical aspects of the trial. The team was multidisciplinary (see Appendix 2) and included the chief investigator, several co-investigators, sponsor representatives, a patient and public involvement (PPI) contributor (see Appendix 5) and members of the clinical trials unit.
Independent Data and Safety Monitoring Committee
The IDSMC was responsible for safeguarding the interests of the EcLiPSE trial participants, assessing the efficacy and safety of the interventions throughout the trial and monitoring the overall progress and conduct of the trial. The IDSMC comprised an independent paediatrician, an independent professor of neurology and an independent statistician (see Appendix 1). The IDSMC met annually during the course of the trial and provided recommendations to the TSC. The Haybittle–Peto approach was used by the IDSMC as a guide to consider stopping the trial within interim reports with 99.9% CIs.
Trial Steering Committee
The TSC was responsible for providing overall oversight of the trial. The TSC comprised an independent paediatrician, an independent consultant in PEM, an independent statistician and a representative from the TMG (see Appendix 1). Co-sponsor representatives were invited to meetings as observers. The TSC met annually throughout the study and remained masked to accumulating data until the end of the trial. The TSC remained happy with trial progress and received monthly updates of recruitment and progress. The TSC met in December 2017.
Both chairpersons of the TSC and the IDSMC supported a joint final meeting that was arranged to coincide with the final results meeting. This meeting took place in Manchester on 9 July 2018 and the results of the study were presented to all EcLiPSE trial team members of each participating site.
Internal pilot
The EcLiPSE trial included an 18-month internal pilot and involved five centres.
The 18-month period was chosen to allow five centres to be opened, be fully up to speed with trial procedures and be achieving the optimal recruitment rate (assumed to be achieved 3 months after opening). This time frame also allowed each site a minimum of 6 months of active recruitment at the optimal level to demonstrate their recruitment rates and support prediction of trial activity into the main phase of the trial.
Success criteria of the pilot were based on the below.
Recruitment
-
If the predicted recruitment period is ≤ 36 months, then proceed to main trial.
-
If the predicted recruitment period is between 36 and 48 months, then consider, and introduce, ways to reduce this (e.g. increase the number of centres, address training needs or determine if new evidence suggests that eligibility criteria could be widened) then proceed to main trial with amendments.
-
If the predicted recruitment period is > 36 months and no obvious solutions exist, then abandon the plan for the main trial.
Deferred consent
-
If the deferred consent rate is ≥ 80%, then proceed to the main trial.
-
If the deferred consent rate is between 60% and 80%, and there is no clear association between provision of deferred consent and the child’s outcome, then analyse reasons why patients/guardians do not want to participate to identify any aspects amenable to change and proceed to the main trial, as amended.
-
If deferred consent is < 60%, then analyse reasons why patients/guardians do not want to participate. If consent declination is associated with poor patient outcome (e.g. death), abandon the main trial.
Completeness of primary outcome data
-
If primary outcome data are available for > 90% of randomised and consented participants, then proceed to the main trial.
-
If primary outcome data are available for between 70% and 90% of randomised and consented participants, then analyse reasons for missing data and identify whether or not any aspects are amenable to change and proceed to the main trial, as amended.
-
If primary outcome data are available for < 70% of participants randomised and consented, then abandon the plan for the main trial.
Statistical methods
A detailed statistical analysis plan is available online [see NIHR Journals Library project web page URL: www.journalslibrary.nihr.ac.uk/programmes/hta/12127134/# (accessed 25 September 2020)]. All analyses were undertaken with SAS® software, version 9.4 (SAS Institute Inc., Cary, NC, USA).
The primary analysis was based on a modified intention-to-treat principle. All randomised and consented participants who received a second-line treatment were included in the analysis according to their allocated treatment. Children who were randomised but whose CSE stopped without requiring second-line treatment (and did not restart in the ED) were excluded. The safety analysis included the same participants, grouped according to the actual treatment received. To avoid double counting, SAEs were reported separately from AEs.
Statistical tests were two-sided at a 5% significance level and results are presented with 95% CIs. The primary outcome was analysed using the log-rank test and is presented as a Kaplan–Meier curve. All participants were followed up to cessation of CSE, with censoring used in the event of RSI or death. If RSI was administered, time was censored at RSI plus 12 hours (i.e. 720 minutes). In patients who died before cessation of CSE, time was censored at the time of death plus 48 hours (i.e. 2880 minutes). RSI and death represent informative censoring and, therefore, the censoring times were inflated to signify the negative outcome for the child with further sensitivity analyses. The sensitivity analyses considered the robustness of the results to the primary analysis approach and included Gray’s test, treating RSI as a competing risk, calculating time to cessation of CSE from start of infusion instead of randomisation and censoring participants at the time of an additional second-line treatment after no response to the allocated treatment. Additional analysis using a Cox proportional hazards model adjusted for baseline characteristics of weight (i.e. < 12 kg, 12–36 kg or > 36 kg), sex and whether or not this was the child’s first seizure. Two covariates (i.e. site of infusion and additional anticonvulsants given in parallel) specified in the analysis plan were not included because they were measured after randomisation. Additionally, centre (i.e. the site) could not be included as a factor in the Cox model because of the lack of convergence. Schoenfeld residual plots were used to check the assumption of proportionality. The binary secondary outcomes of need for further anticonvulsants, RSI and admission to critical care were analysed using the chi-square test and presented with relative risks. Logistic regression models were fitted as additional analyses to the primary chi-square tests, with adjustments as per the Cox proportional hazards model. No adjustment was made for multiplicity for the secondary outcomes. Baseline categorical data and AE data are summarised using numbers and percentages, and continuous data are summarised as medians and interquartile ranges (IQRs). A post hoc analysis was undertaken for the reasons underlying the further management of the presenting episode of CSE, the assessment of which was carried out without knowledge of the allocated intervention.
Role of the funding source
The trial funder monitored trial progress and approved oversight committee membership, but had no role in trial design, data collection, data analysis, data interpretation or writing of the report. The corresponding author had full access to all data in the trial.
Changes to the protocol
The EcLiPSE trial opened to recruitment on version 3.0 of the protocol and closed on version 5.0. Changes to the protocol are summarised in Appendix 4. In summary, the key changes from version 1.0 to version 2.0 included increasing the follow-up period to 14 days to collect longer-term safety data, adding information that the primary outcome would be calculated from the time of randomisation and providing additional clarifications on eligibility criteria. Amendments between subsequent versions included clarification on dose, safety reporting, questionnaire administration and consent process.
Chapter 3 Clinical effectiveness results
Parts of this chapter have been reused from Lyttle et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Recruitment
Participants were recruited from a total of 30 sites, all EDs. Although 30 sites were formally opened, two subsequently closed with neither site having recruited any participants. The first participant was recruited from Alder Hey Children’s Hospital, Liverpool, on 22 July 2015 under version 3.0 of the protocol and the last participant was recruited from Southampton General Hospital, Southampton, on 7 April 2018 under version 5.0 of the protocol.
The five top-recruiting sites recruited 124 out of the 286 (43.4%) participants.
The observed site opening and participant recruitment rates closely followed those predicted (Figure 2).
FIGURE 2.
Recruitment graph.
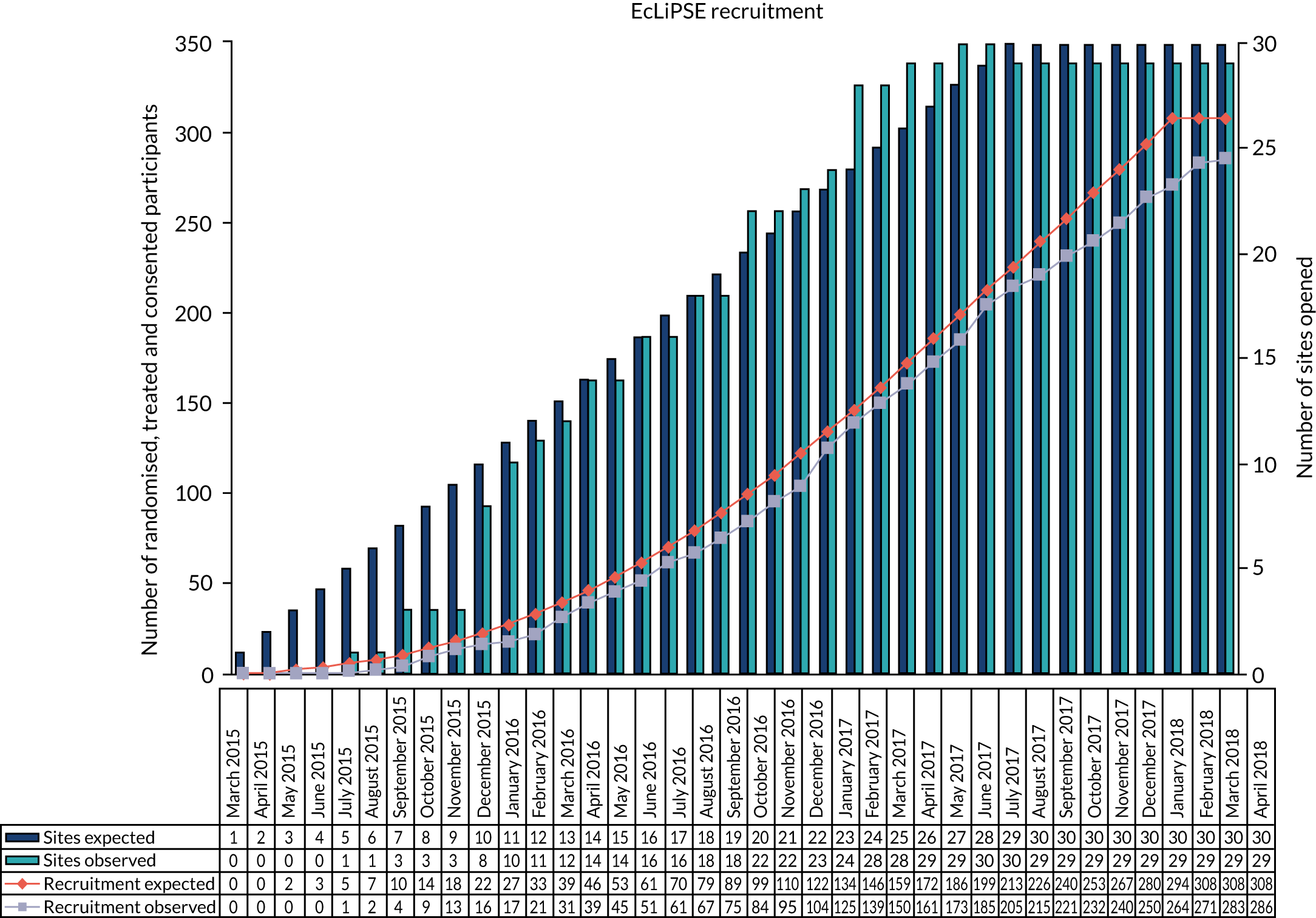
Study commencement was delayed by approximately 6 weeks because of contractual issues between the two co-sponsors of the study and, therefore, recruitment closed 6 weeks later than planned, on 10 April 2018.
Participant screening and throughput are summarised in Figure 3.
FIGURE 3.
Consolidated Standards of Reporting Trials (CONSORT) flow diagram. ITT, intention to treat.
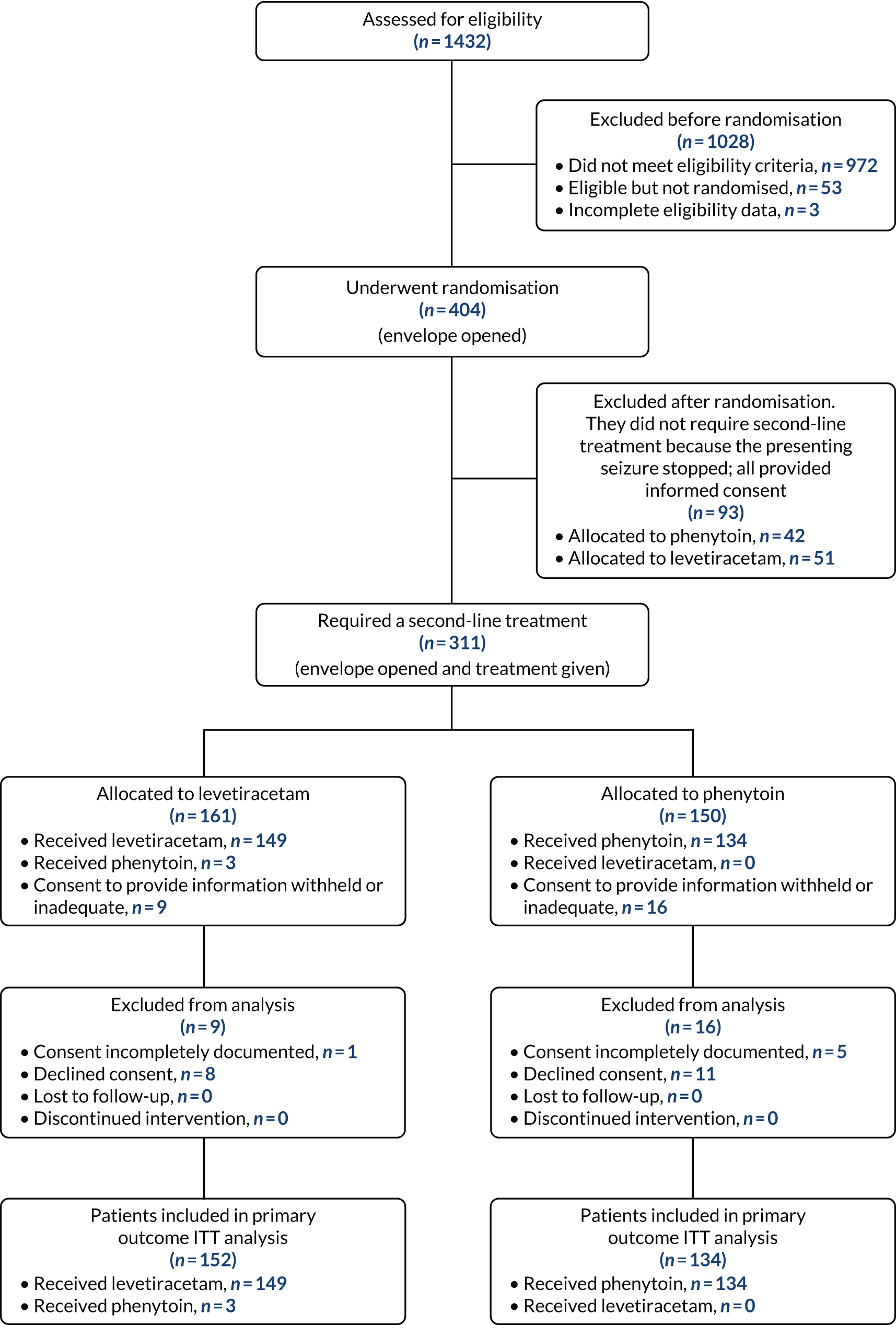
Of the 1432 children screened for eligibility, 404 were randomised (i.e. the randomisation pack was opened). A total of 1028 children were excluded before randomisation, including 972 children who did not meet the eligibility criteria (see Tables 1 and 2). Reasons for not randomising 53 children who were considered eligible included no trial-trained doctor available, loss of, or failure to, achieve i.v. access, clinical judgement (e.g. child too sick) and treatment given before random allocation.
Inclusion criterion 2 (see Table 1) combined seizure type with the need for second-line treatment. Table 2 looks at this inclusion criteria in greater detail to distinguish eligibility owing to seizure type and ongoing need for second-line treatment.
Of the 404 children randomised, 93 did not require a second-line treatment. Of those children randomised who did require a second-line treatment, 25 were excluded from the analysis as consent was either incompletely documented (n = 6) or declined (n = 19). Table 3 provides the reasons why consent was declined.
| Reason consent declined | Group | Total | |
|---|---|---|---|
| Levetiracetam | Phenytoin | ||
| Dad did not like the idea of being interviewed for the consent study. When it was explained again that this was optional dad said he did not like the idea | 1 | 0 | 1 |
| Dad felt that he had been deceived and that consent should have been asked first | 1 | 0 | 1 |
| Father did not want information to be used | 1 | 0 | 1 |
| Mum did not want sponsors or regulatory authorities to have access to the child’s medical records | 1 | 0 | 1 |
| Not happy with DOB or gender to be used or have mum’s name written on consent form | 0 | 1 | 1 |
| Parents felt patient had been through enough | 1 | 0 | 1 |
| Parents would like to put upsetting time in past | 1 | 0 | 1 |
| Too much going on and does not want any of their details collected | 0 | 1 | 1 |
| Language barrier – mum given information and understood brief concepts but not enough to give informed consent | 0 | 1 | 1 |
| Not interested | 0 | 1 | 1 |
| Unclear reason. Parents have been given lot of information and patient was diagnosed with a new condition since admission. Information overload | 0 | 1 | 1 |
| Stressed, patient very unwell | 1 | 0 | 1 |
| Do not like signing things | 0 | 1 | 1 |
| Social worker explained that this is a complex social care case – currently with courts and they are unable to consent without court approval | 0 | 1 | 1 |
| Taken advice from solicitor due to ongoing litigation in respect to birth injury | 0 | 1 | 1 |
| No reason given | 1 | 3 | 4 |
| Total | 8 | 11 | 19 |
Compliance with the intervention
Data on compliance (adherence) with treatment are shown in Tables 4 and 5.
| Patients randomised and consented | Group, n (%) | Total (N = 379), n (%) | |
|---|---|---|---|
| Levetiracetam (N = 203) | Phenytoin (N = 176) | ||
| Received no second-line treatment | 51 (25.1) | 42 (23.9) | 93 (24.5) |
| Received allocated treatment | 149 (73.4) | 134 (76.1) | 283 (74.7) |
| Received other second-line treatment to that allocated | 3 (1.5) | 0 (0.0) | 3 (0.8) |
| Received treatment followed by a second second-line treatment | 22a (14.5) | 13a (9.7) | 35a (12.2) |
| Compliance data | Group, n (%) | Total (N = 286), n (%) | |
|---|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | ||
| Patient given lower dose of trial treatment | 8 (5.3) | 4 (3.0) | 12 (4.2) |
| Patient given higher dose of trial treatment | 2 (1.3) | 1 (0.8) | 3 (1.1) |
| Dose administration shorter than expected | 0 (0.0) | 1 (0.8) | 1 (0.4) |
| Dose administration longer than expected | 27 (17.8) | 34 (25.4) | 61 (21.3) |
| Treatment prematurely discontinued | 0 (0.0) | 2 (1.5) | 2 (0.7) |
| Unauthorised route of administration (intraosseous) | 6 (4.0) | 0 (0.0) | 6 (2.1) |
| Received initial second-line treatment other than that allocated | 3 (1.5) | 0 (0.0) | 3 (0.8) |
| Received further second-line treatmenta | 22 (14.5) | 13 (9.7) | 35 (13.6) |
Baseline characteristics
The baseline characteristics of the study participants were comparable (Table 6). However, the levetiracetam-treated group comprised more participants in whom the presenting episode of CSE [n = 69 (45%) vs. n = 49 (37%)] represented their first convulsive seizure ever, with a lower proportion of participants with a chronic epilepsy, as evidenced by participants taking oral maintenance antiepileptic drugs [n = 51 (34%) vs. n = 55 (41%)].
| Demographic | Group | |
|---|---|---|
| Levetiracetam (N = 152, 53%) | Phenytoin (N = 134, 47%) | |
| Sex, n (%) | ||
| Male | 75 (49) | 72 (54) |
| Female | 77 (51) | 62 (46) |
| Age | ||
| 6 months to < 2 years, n (%) | 65 (43) | 53 (40) |
| 2–11 years, n (%) | 81 (53) | 74 (55) |
| 12–17 years, n (%) | 6 (4) | 7 (5) |
| Median (years) (IQR) | 2.7 (1.3–5.9) | 2.7 (1.6–5.6) |
| Range (years) | 0.6–16.1 | 0.6–17.9 |
| Weight (kg) | ||
| < 12, n (%) | 52 (34) | 42 (31) |
| 12–36, n (%) | 86 (57) | 80 (60) |
| > 36, n (%) | 14 (9) | 12 (9) |
| Median (IQR) | 12.1 (10.0–19.0) | 12.0 (10.0–18.0) |
| Range | 7.5–70.0 | 6.0–66.0 |
| Participant’s first seizure, n (%) | 69 (45) | 49 (37) |
| Presenting seizure type, n (%) | ||
| Generalised tonic–clonic | 107 (70) | 105 (78) |
| Generalised clonic | 12 (8) | 7 (5) |
| Focal clonic | 33 (22) | 22 (16) |
| Seizure cause, n (%)a | ||
| Febrile convulsion | 63 (41) | 58 (43) |
| Seizure (pre-existing epilepsy) | 46 (30) | 46 (34) |
| First afebrile seizure | 16 (11) | 12 (9) |
| Central nervous system infection | 6 (4) | 7 (5) |
| Intracranial vascular event (bleed/stroke) | 2 (1) | 2 (1) |
| Traumatic brain injury | 0 | 0 |
| Substance misuse | 1 (< 1) | 0 |
| Indeterminate | 10 (7) | 7 (5) |
| Other | 27 (18) | 26 (19) |
| Maintenance antiepileptic drugs at presentation, n (%)a | ||
| Levetiracetam | 29 (15.9) | 26 (19.4) |
| Sodium valproate | 16 (10.5) | 19 (14.2) |
| Carbamazepineb | 12 (7.9) | 10 (7.5) |
| Clobazamc | 9 (5.9) | 9 (6.7) |
| Topiramated | 4 (2.6) | 8 (6.0) |
| Phenytoin | 0 (0.0) | 1 (0.7) |
| Other | 11 (7.2) | 18 (13.4) |
Protocol deviations
Protocol deviations were comparable across both treatment groups. Major and minor deviations are shown in Table 7.
| Protocol deviation | Group, n (%) | Total (N = 286), n (%) | |
|---|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | ||
| Any protocol deviation | 52 (34.2) | 51 (38.1) | 103 (36) |
| At least one major deviation | 10 (6.6) | 8 (6) | 18 (6.3) |
| At least one minor deviation | 43 (28.3) | 45 (33.6) | 88 (30.8) |
Major protocol deviations comprised:
-
premature discontinuation of randomised treatment (none in the levetiracetam-treated group and two in the phenytoin-treated group)
-
duration of infusion shorter than expected (none in the levetiracetam-treated group and one in the phenytoin-treated group)
-
lower dose of the intervention administered (eight in the levetiracetam-treated group and four in the phenytoin-treated group)
-
missing data for primary outcome (two in both the levetiracetam- and phenytoin-treated groups).
Primary outcome
Seizure cessation was achieved in 106 out of the 152 (70%) levetiracetam-treated participants and in 86 out of the 134 (64%) phenytoin-treated participants.
Table 8 provides the median time to seizure cessation from randomisation and Figure 4 shows the Kaplan–Meier curve and log-rank test. As the event of interest (i.e. seizure cessation) is positive, the lower curve indicates a shorter time to seizure cessation; however, there is no statistically significant difference between the treatment arms (log-rank p-value > 0.05).
| Event | Group | |
|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | |
| Number of events (seizure cessation), n (%) | 106 (69.7) | 86 (64.2) |
| Number of censored times (RSI), n (%) | 46a (30.3) | 48b (35.8) |
| Median time (minutes) to cessation of seizure from randomisation (IQR) | 35 (20–NAc) | 45 (24–NAc) |
FIGURE 4.
Kaplan–Meier plot time from randomisation to seizure cessation. Product-limit survival estimates with number of subjects at risk. This figure been reused from Lyttle et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
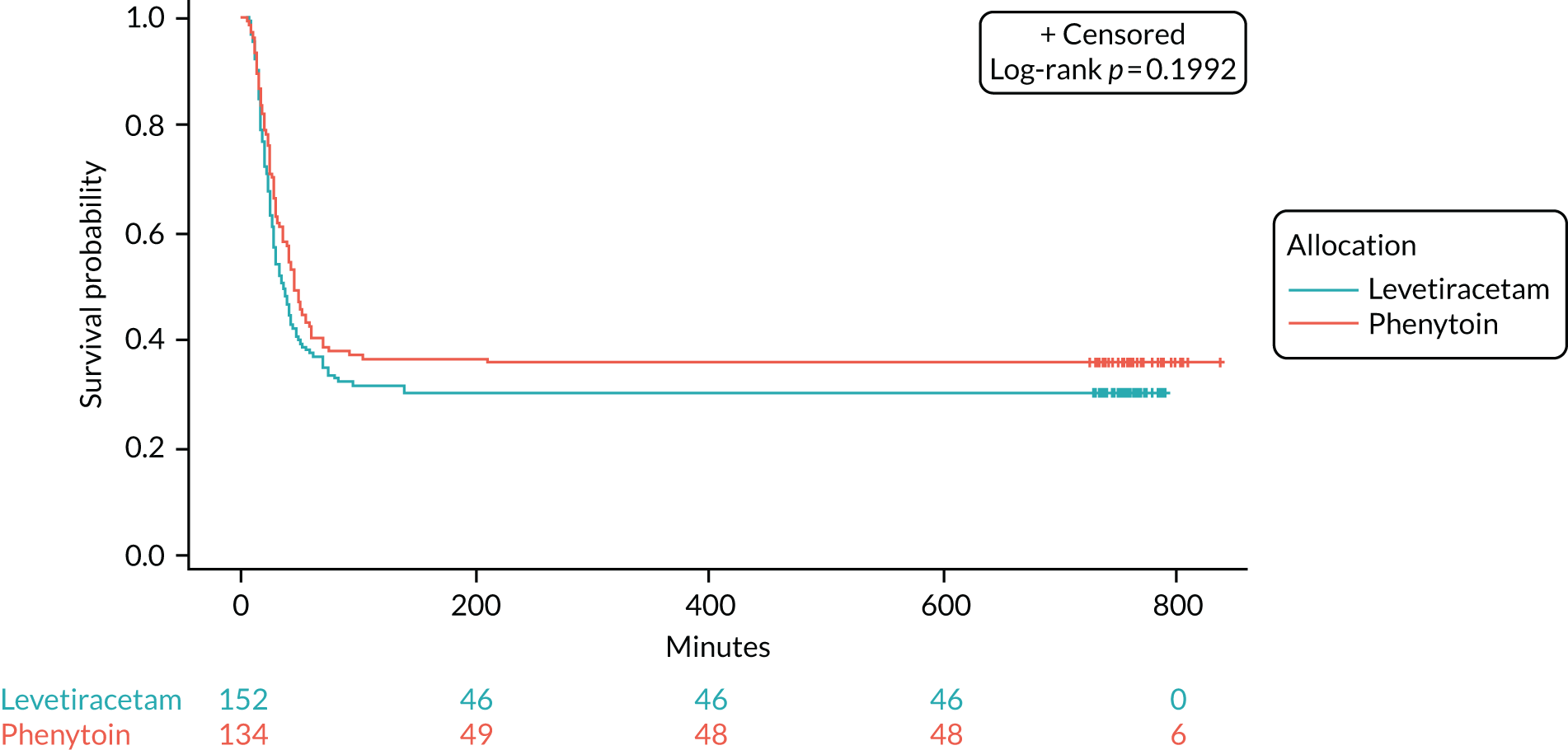
The unadjusted HR was 1.2 (95% CI 0.91 to 1.6; p = 0.2) in favour of levetiracetam. The Schoenfeld residuals for the unadjusted model (p = 0.72) indicated the independency of time and the validity of the proportionality assumption. The Schoenfeld residuals for the adjusted model indicated that the assumption of proportionality for weight was not met (p = 0.05, p-value ranged from 0.27 to 0.71 for other variables). The data were subgrouped according to weight category as per the baseline table (i.e. < 12 kg, 12–36 kg and > 36 kg) and estimates from the adjusted model calculated (see Appendix 6, Tables 15–29). The proportionality assumption within each subgroup of data was supported by the Schoenfeld residuals. Direction of treatment effect was consistent across subgroups, CIs were wide and results were not statistically significant. The treatment effect was increased for children in the > 36 kg subgroup, but remained non-significant and numbers within this group are small.
A range of sensitivity analyses were considered to assess seizure cessation from the start of the infusion, rather than from randomisation (see Appendix 7, Table 32) and the impact censoring regarding RSI and death. The results demonstrated robustness of conclusions and are presented in Appendices 6 and 7.
Secondary outcomes
The study comprised four secondary outcomes. These assessed the efficacy and safety of the two trial interventions (Table 9).
| Secondary outcome | Group, n (%) | Unadjusted RR (95% CI) | p-value | |
|---|---|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | |||
| Need for further anticonvulsantsa | 57 (37.5) | 50 (37.3) | 1.01 (0.74 to 1.36) | 0.97 |
| Need for further anticonvulsants for the presenting CSEb | 24 (15.8) | 20 (14.9) | 1.06 (0.61 to 1.83) | 0.84 |
| Need for further anticonvulsants for a subsequent seizure (within 24 hours)b,c | 14 (9.2) | 17 (12.7) | 0.72 (0.37 to 1.4) | 0.33 |
| RSI to terminate an ongoing seizure | 44 (30.0) | 47 (35.1) | 0.83 (0.59 to 1.16) | 0.27 |
| Admission to critical care | 97 (63.8) | 72 (53.7) | 1.19 (0.97 to 1.45) | 0.08 |
| SAR | 0 | 2d | ||
| 14-day follow-up | ||||
| Discharged from hospital | 145 (95.4) | 130 (97.0) | ||
| Readmitted to hospital | 12 (7.9) | 10 (7.5) | ||
| Patient died | 1 (0.7) | 1 (0.8) | ||
| Organ failure | 1 (0.7) | 0 (0.0) | ||
One participant who was allocated and received phenytoin experienced a SAR. This was profound hypotension, which was considered to be immediately life-threatening and responded to emergency treatment. This participant also experienced a severe unexpected serious adverse reaction (SUSAR), which manifested as a large increase in seizure frequency and marked sedation within 24 hours of receiving phenytoin. The SUSAR was considered medically significant and the participant required admission to the intensive care unit. The SUSAR resolved without complication.
Safety, tolerability and compliance
Two patients died in the study, but neither death was considered related to the randomised treatment. One participant presented to the ED in a generalised tonic–clonic seizure and unconscious. Resuscitation was immediate. The participant received levetiracetam (the randomised treatment) and then phenytoin, followed by RSI with thiopentone (Archimedes, Reading, UK) because of abnormal posturing. The participant died 36 hours following admission and a post-mortem examination revealed severe brain oedema secondary to encephalitis. Consent for recruitment into the study was subsequently obtained from the participant’s carers. The death was considered to be unrelated to the randomised treatment by the principal investigator and chief investigator.
The second participant received phenytoin. The participant died and the results of the post-mortem examination were not available prior to closure to recruitment to the study. The principal investigator and chief investigator considered the death to be unrelated to the randomised treatment. Consent was sought from the participant’s carers but had not been obtained by the time recruitment to the study closed on 10 April 2018 and, therefore, this participant’s data are not included in the analysis.
Adverse events and serious adverse events
A total of 51 AEs were reported in 39 out of the 286 participants. Some participants experienced more than one AE. Forty-one of the AEs were classified as mild, nine as moderate and one as severe. Sixteen out of 130 levetiracetam-treated participants, 18 out of 132 phenytoin-treated participants and four out of 24 participants who received both drugs experienced at least one AE. Each individual AE had a prevalence of < 10%. In the levetiracetam-treated group (20 AEs in 16 participants), a psychiatric AE was reported in 12 participants (agitation in 11 and hallucinations in one). In the phenytoin-treated group (23 AEs in 18 participants), a cardiovascular AE was reported in eight participants, an extravasation/administration site reaction AE in seven (severe in one) and an agitation AE in four. In the group that received levetiracetam and phenytoin (eight AEs in four participants), an extravasation/administration site reaction was reported in three. The full list of reported AEs is shown in Table 10.
| AE | Group | Total (N = 286) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levetiracetam (N = 132) | Phenytoin (N = 130) | Both drugs (N = 24) | ||||||
| Events, n | Patients, n (%) | Events, n | Patients, n (%) | Events, n | Patients, n (%) | Events, n | Patients, n (%) | |
| Agitation | 11 | 11 (8.3) | 4 | 4 (3.1) | 0 | 0 (0.0) | 15 | 15 (5.2) |
| Hypotension | 2 | 2 (1.5) | 3 | 3 (2.3) | 1 | 1 (4.2) | 6 | 6 (2.1) |
| Catheter site related | 1 | 1 (0.8) | 1 | 1 (0.8) | 3 | 2 (8.3) | 5 | 4 (1.4) |
| Extravasation | 0 | 0 (0.0) | 4 | 4 (3.1) | 1 | 1 (4.2) | 5 | 5 (1.75) |
| Tachycardia | 1 | 1 (0.8) | 3 | 3 (2.3) | 1 | 1 (4.2) | 5 | 5 (1.75) |
| Rash | 2 | 2 (1.5) | 1 | 1 (0.8) | 0 | 0 (0.0) | 3 | 3 (1.1) |
| Hypertension | 0 | 0 (0.0) | 2 | 2 (1.5) | 0 | 0 (0.0) | 2 | 2 (0.7) |
| Reaction to ceftriaxonea | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (4.2) | 1 | 1 (0.4) |
| Confused | 1 | 1 (0.8) | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Decreased consciousness | 0 | 0 (0.0) | 1 | 1 (0.8) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Hallucination | 1 | 1 (0.8) | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Infusion site erythema | 0 | 0 (0.0) | 1 | 1 (0.8) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Mechanical ventilation complication | 0 | 0 (0.0) | 1 | 1 (0.8) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Pallor | 0 | 0 (0.0) | 1 | 1 (0.8) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Stridor | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (4.2) | 1 | 1 (0.4) |
| Vomiting | 0 | 0 (0.0) | 1 | 1 (0.8) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Wheezing | 1 | 1 (0.8) | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (0.4) |
| Total | 20 | 16 (12.1) | 23 | 18 (13.9) | 8 | 4 (16.7) | 51 | 38 (13.3) |
Five SAEs were reported in four participants [including one participant who experienced two SAEs (participant 00133027)]. In three participants, the SAE was considered unrelated to the intervention, and one each was considered to be possible and probable (Table 11).
| SAE number | Description | Preferred term (System Organ Class) | Treatment received | Seriousness | Severity | Expectedness | Relationship assessment | Outcome | |
|---|---|---|---|---|---|---|---|---|---|
| Principal investigator | Chief investigator | ||||||||
| 00133007-001 | Attended ED fitting. Randomised to levetiracetam, but given phenytoin. Stopped self-ventilating and required RSI and was intubated. CT showed fractured VP shunt and required urgent shunt revision. Taken to theatre 25 November 2015 at 02.45. Shunt revision complete. Admitted to PICU for < 24 hours. Admitted to neurosurgical ward until 30 November 2015 | Device malfunction (general disorders and administration site conditions) | Phenytoin | Prolonged existing hospitalisation | Moderate | Unexpected | Unrelated | Unrelated | Resolved |
| 00133027-005 | Patient weaned off sedation on 22 March 2017. At 13.55 patient started having seizure episodes. Documented in notes that the patient had approximately 40 seizures until phenobarbital given at 18.10, 22 March 2017. No further seizures but conscious level decreased until 18.00, 26 March 2017 | Seizure (nervous system disorders) | Phenytoin | Medically significant or important | Moderate | Unexpected | Unlikely | Possibly | Resolved |
| 00133027-006 | Patient became hypotensive at 01.45, 22 March 2017. Patient given fluid boluses as documented on concomitant medication form. Patient remained hypotensive and so given adrenaline infusion. Hypotension resolved completely (26 March 2017). No further problems | Hypotension (vascular disorders) | Phenytoin | Immediately life-threatening | Moderate | Expected | Possibly | Probably | Resolved |
| 00133029-008 | Patient randomised and given levetiracetam. Noted to have only slight twitching after. Patient then started having decorticate/decerebrate posturing. RSI. Desaturated and bradycardia owing to ET tube problems. No ETC02 reading, reintubated but ventilation difficult causing desaturation and bradycardia. Arrest CPR commenced | Cardiac arrest (cardiac disorders) | Levetiracetam |
Immediately life-threatening Prolonged existing hospitalisation |
Severe | Unexpected | Unrelated | Unrelated | Resolved |
| 00243026-002 | Attended the ED fitting and unconscious. Pupils equal and reactive but sluggish. Tolerating airway. CT showed massive raised intracranial pressure. Pupils become fixed and dilated just before being taken to emergency theatre. Oral secretions positive for mycoplasma pneumoniae | Intracranial pressure increased (nervous system disorders) | Phenytoin and levetiracetam | Immediately life-threatening | Severe | Unexpected | Unrelated | Unrelated | Fatal |
An additional follow-up questionnaire was completed by sites and the families of participants who had been randomised, treated and consented to take part in the study. The questionnaire was completed 2 weeks following randomisation. Results are shown in Table 12.
| Follow-up | Allocation | Yes, n (%) | No, n (%) | Information not provided in patient notes, n (%) | Unknown or missing, n (%) | Total, n (%) |
|---|---|---|---|---|---|---|
| Discharged from hospital | Levetiracetam | 145 (95.4) | 7 (4.6) | 0 (0) | 0 (0) | 152 (53.15) |
| Phenytoin | 130 (97) | 4 (3) | 0 (0) | 0 (0) | 134 (46.9) | |
| Total | 275 (96.1) | 11 (3.8) | 0 (0) | 0 (0) | 286 (100) | |
| Readmitted to hospital | Levetiracetam | 12 (7.9) | 81 (53.3) | 25 (16.4) | 34 (22.4) | 152 (53.1) |
| Phenytoin | 10 (7.5) | 64 (47.8) | 19 (14.2) | 41 (30.6) | 134 (46.9) | |
| Total | 22 (7.7) | 145 (50.7) | 44 (15.4) | 75 (26.2) | 286 (100 | |
| Patient died | Levetiracetam | 1 (0.7) | 111 (73) | 22 (14.5) | 18 (11.8) | 152 (53.1) |
| Phenytoin | 1 (0.7) | 93 (69.4) | 20 (14.3) | 20 (14.9) | 134 (46.9) | |
| Total | 2 (0.7) | 204 (71.3) | 42 (14.7) | 38 (13.3) | 286 (100) | |
| Organ failure | Levetiracetam | 1a (0.7) | 110 (72.4) | 20 (13.2) | 21 (13.8) | 152 (53.1) |
| Phenytoin | 0 (0) | 98 (73.1) | 16 (11.9) | 20 (14.9) | 134 (46.9) | |
| Total | 1 (0.3) | 208 (72.7) | 36 (12.6) | 41 (14.3) | 286 (100) |
Only 74 (25.9%) families completed their 14-day follow-up questionnaire. As documented in the internal meeting minutes, 8 May 2018, details for these questionnaires are not presented within this report because of the low response rate.
Laboratory parameters (haematological, biochemical analysis and urinalysis)
Laboratory analyses in the trial protocol were limited to measurements of blood levels of levetiracetam and phenytoin in the randomised, treated and consented participants. Blood samples were obtained between 1 and 2 hours after completion of infusion of the two treatments. Biochemistry laboratories in each site processed the samples in accordance with their standard operating procedures. The reference ranges for blood levels of levetiracetam were provided by a single central laboratory that analysed all of the samples of levetiracetam-treated participants. The reference ranges for blood levels of phenytoin were provided by the biochemistry department of each participating site that analysed samples of its phenytoin-treated participants.
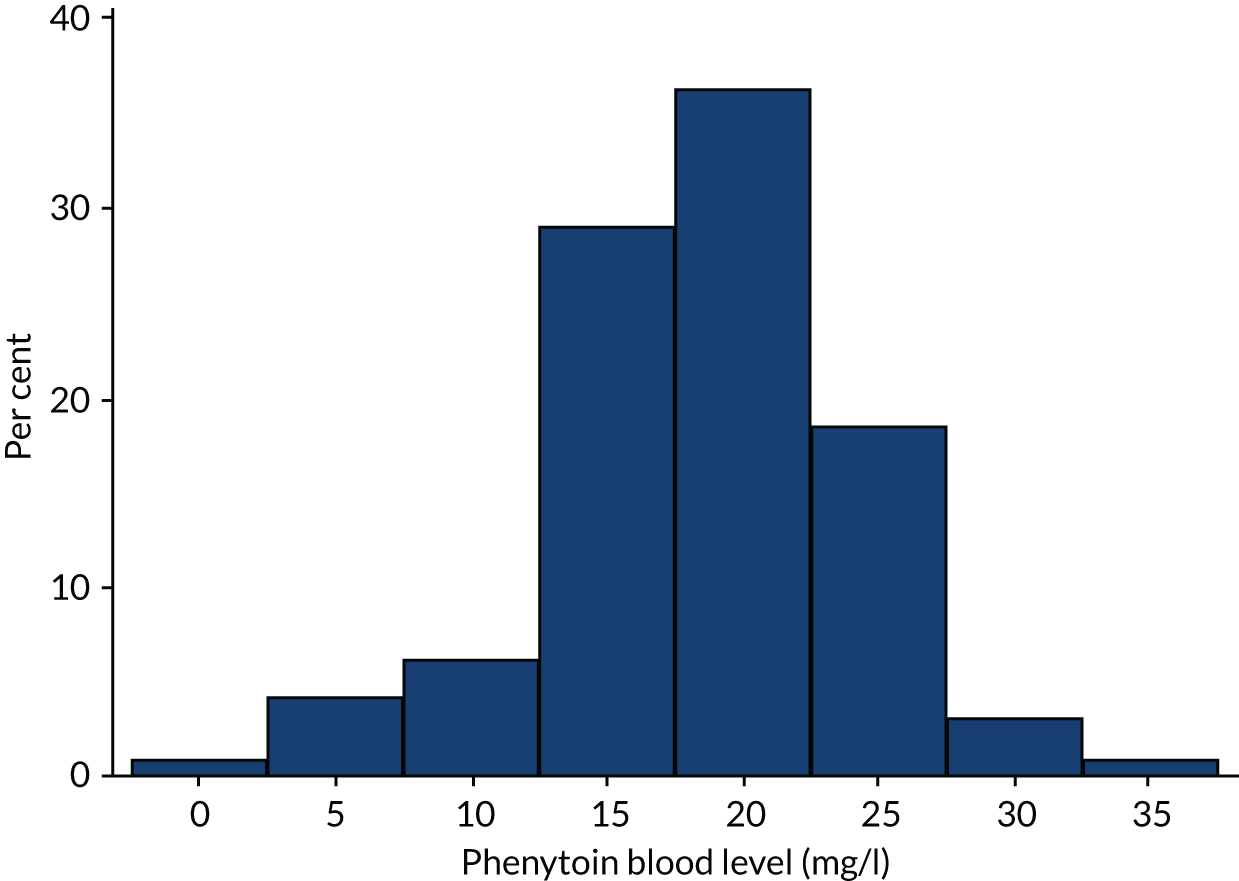
A total of 192 participants underwent measurement of a blood level [96 participants in the levetiracetam-treated group (63.2%) and 96 participants in the phenytoin-treated group (77.7%)] and the results are shown in Figures 5 and 6.
FIGURE 5.
Levetiracetam blood levels.
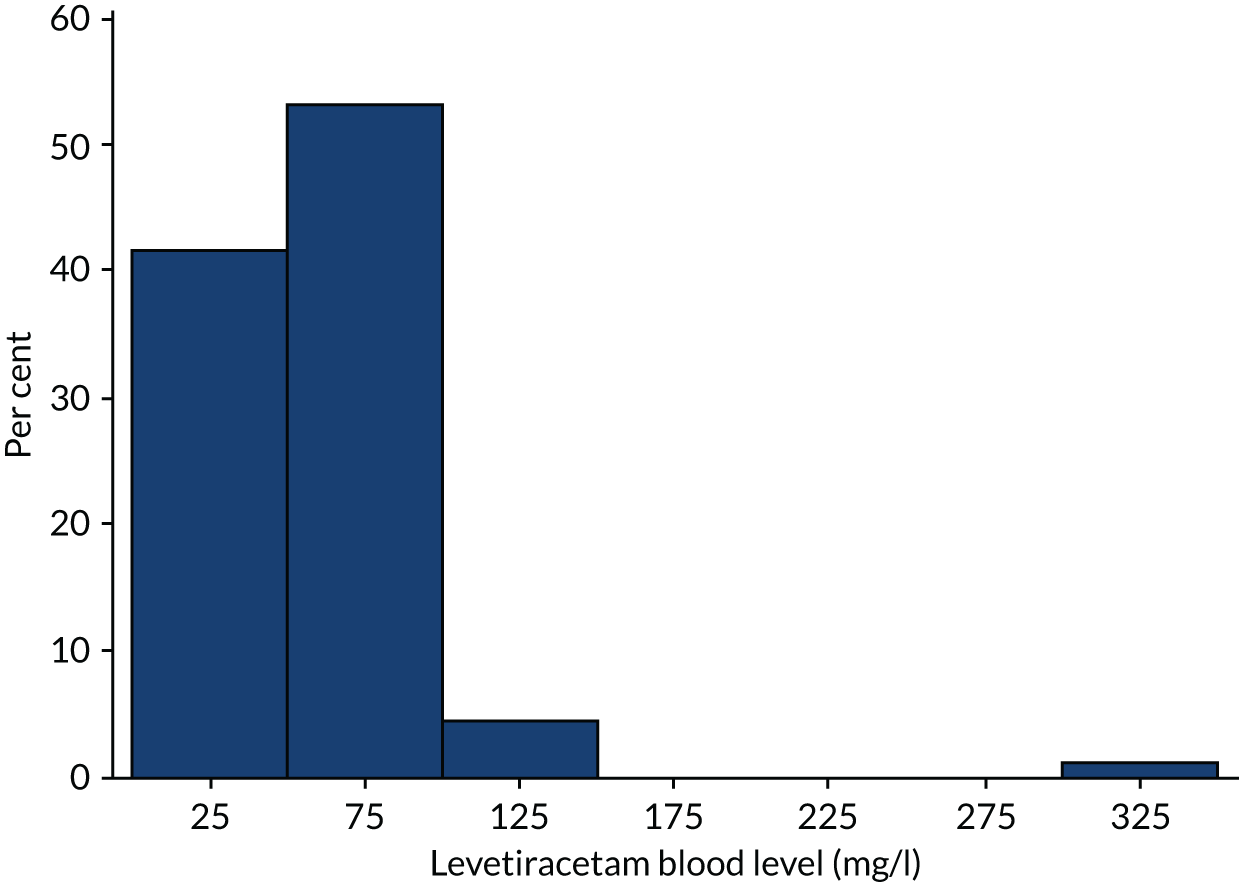
FIGURE 6.
Phenytoin blood levels.
Chapter 4 Nested consent study
Background
Convulsive status epilepticus is a medical emergency that has insufficient time to obtain informed consent within the therapeutic window. The use of RWPC (also known as deferred consent) in the EcLiPSE trial was supported by parents who took part in trial feasibility work. 43
A nested study was designed to identify potential barriers and solutions to recruitment and consent in the EcLiPSE trial, and to inform recruiter training on recruitment, consent, trial conduct and future trials in this setting.
Objectives
Consent study objectives were to explore:
-
how information about the trial and RWPC was exchanged during recruitment discussions
-
parents’ and practitioners’ views and experiences of RWPC and consent decision-making
-
the impact of an unblinded trial design.
We included a strand of work that aimed to develop trial recruiter training on RWPC using recommendations made by parents in the feasibility study and related guidance. 43,46–49 Our objective was to evaluate the effectiveness of the EcLiPSE trial site initiation visit (SIV) training on practitioners’ confidence in recruitment, consent and trial conduct.
Methods
Study design
The consent study used a mixed-methods approach,50,51 involving parents of randomised participants and EcLiPSE trial practitioners. The design and development of the consent study, including recruitment strategy, questionnaires and topic guides, were informed by previous work43,46 in paediatric emergency and critical care in the NHS. EcLiPSE trial feasibility work43 was used to develop participant information and practitioner training materials.
All EcLiPSE trial sites were eligible for inclusion in the consent study, which involved the following research methods:
-
recorded trial recruitment and consent discussions between parents and EcLiPSE trial practitioners
-
parent questionnaires completed after EcLiPSE trial consent discussions (including those who decline consent)
-
telephone interviews with parents approximately 1 month after hospital discharge
-
telephone interviews with the site principal investigator or research nurse within the first 12 months of site opening
-
focus groups with practitioners at the end of the first year
-
a semistructured questionnaire with practitioners administered before and after SIV training
-
an online questionnaire of practitioners in final phase of the trial (approximately 8 months before trial closure).
Participants
We aimed to collect parent questionnaires at all sites throughout trial recruitment. Based on previous research,43,44 we anticipated interviewing approximately 15–25 parents/legal representatives, conducting 6–10 focus groups and 10 additional practitioner interviews to reach data saturation (i.e. the point where no new major themes are discovered in analysis). We planned to collect recorded trial discussions at all sites for the first 4 months of the trial or until data saturation point. We aimed to include practitioners at all sites in the online survey and all staff attending each SIV in the training evaluation.
Eligibility
Parents
Parents (including legal representatives) who did and did not consent to their child’s participation in the trial were eligible to take part in the consent study, unless they were unable to speak or read English.
Practitioners
All practitioners involved in screening, recruiting, randomising and consenting parents/legal representatives during the trial were eligible to take part in the consent study. Those not intending to stay for the full SIV training were excluded from the SIV questionnaire element.
Recruitment
Recruitment to recorded trial discussions, parent questionnaires and interviews
As described in Chapter 2, the principal investigator, research nurse or other designated member of the site research team approached the parent/legal representative to discuss the trial and seek consent as soon as possible after completion of trial treatment (ideally within 24 hours of randomisation). During this discussion, the principal investigator/research nurse briefly explained the aims of the consent study and sought verbal permission for audio-recording of trial discussions. If permission was declined the recruitment discussion was not recorded. If permission was given the recruiter activated an audio-recorder. Written consent was then sought for all consent study elements as part of the EcLiPSE trial consent process. This included written consent for the use of recorded trial discussion data, as well as consent to complete a questionnaire before their child was discharged from hospital and to take part in an interview approximately 1 month later.
Parents were asked to place the completed questionnaires in a sealed, stamped, addressed envelope and return it to the EcLiPSE trial practitioner to post to the consent study team. There was also a link to an online version of the questionnaire at the top of the paper questionnaire, enabling parents to complete the survey online if preferred. Recruitment for questionnaires took place throughout the active trial recruitment phase. In the rare instance that consent was not sought prior to discharge or before the participant was transferred to another hospital, the questionnaire was sent to parents/legal representatives along with the parent/legal representative information sheet and consent form to complete. We asked trial recruiters to record all trial discussions (e.g. an initial discussion followed by a full trial discussion after the family had considered the trial information), which were then uploaded to a secure website for transcription. LR made contact with families to arrange telephone interviews within 1 month of consent. Initially, parents’ expressions of interest to participate were responded to in sequential order. As trial recruitment progressed, we stopped interviewing parents from high-recruiting sites and purposively sampled across all recruiting sites to help ensure sample variance.
Recruitment to practitioner focus groups and interviews
LR e-mailed selected sites and invited practitioners to participate in a telephone interview or focus group. Selection of sites was based on accrual rates (i.e. high and low rates) and recruitment issues identified in the ongoing analysis of recorded trial discussion, parent questionnaires and interviews. Verbal consent was sought, including consent for recorded trial discussions. All focus groups were facilitated by LR who sought audio-recorded verbal consent from participants before each focus group began.
Recruitment to the site initiation visit evaluation
The trial co-ordinator (AH) liaised with the site principal investigator or research nurse to invite all relevant staff to the SIV. KW, LR or AH provided a brief description of the evaluation before the opening presentation and invited practitioners who intended to stay for the full training to participate by completing part A of the questionnaire before training and part B at the end of training. Questionnaire completion was taken as indication of consent. Personal details were not requested to ensure anonymity.
The trial co-ordinator (AH) e-mailed all eligible sites (28 out of 30 sites because two had closed) 8 months before the scheduled trial end date, and invited staff involved in the EcLiPSE trial to complete an online questionnaire [see appendix on the NIHR Journals Library project web page: URL www.journalslibrary.nihr.ac.uk/programmes/hta/12127134/#/ (accessed 28 September 2020)]. MDL sent e-mail reminders on behalf of the trial team and PERUKI. It was anticipated that some of the same staff who took part in a telephone interview or focus groups would also complete the online questionnaire.
Conduct of interviews and focus groups
LR began interviews and focus groups with parents and practitioners with a description of the consent study aims. The interview commenced using an interview topic guide (see Appendix 8). Respondent validation was used to add unanticipated topics to the topic guide as interviewing and analysis progressed. After the interview, participants were thanked for their time.
Any distress during the parent interviews was managed with care and compassion and participants were free to decline to answer any questions that they did not wish to answer or to stop the interviews at any point.
Transcription
Digitally recorded trial discussions were transcribed verbatim by a professional transcription company (VoiceScript Ltd, Bristol, UK). Transcripts were anonymised and checked for accuracy. All identifiable information, such as names (e.g. of patients, family members or the hospital their child had been admitted to), were removed.
Data analysis
Qualitative data
LR (a psychologist) led the analysis with assistance from KW (a sociologist). Qualitative focus group and interview data analysis was interpretive, and iterative analysis was based on thematic analysis, a method for identifying, analysing and reporting patterns (or themes) within data. Utilising a thematic analysis approach, the aim was to provide accurate representation of parent and practitioner views to address the study aims and objectives. This approach allows for themes to be identified at a semantic level (i.e. surface meanings or summaries) or at a latent level (i.e. interpretive, theorising the significance of the patterns and their broader meanings and implications). 50 NVivo 10 software (QSR International, Warrington, UK) was used to assist in the organisation and coding of data.
Quantitative data
LR entered all parent and practitioner online questionnaire and SIV training questionnaire data into SPSS version 26 (SPSS Inc., Chicago, IL, USA). Descriptive statistics are presented with percentages and the chi-squared test for trend.
For the SIV training questionnaire analysis, we used paired samples t-test and Wilcoxon signed-ranks test (95% CI), as appropriate. Questionnaires with recruitment and consent-related data missing were excluded from the analysis. To investigate the presence of informative missing data, the results of those who completed only part A of the questionnaire were compared with those who completed parts A and B of the questionnaire. This was also undertaken for those who completed only the ‘after’ questionnaire.
Data synthesis
Our approach to synthesising qualitative and quantitative data52 drew on the constant comparative method. 53,54 As part of an iterative process, KW and LR used early findings during trial conduct to create updates via newsletters for EcLiPSE trial recruiters and brief feedback sessions at the end of each focus group. These outputs contained recommendations to assist ongoing approaches to recruitment and consent in the EcLiPSE trial.
Results
Participants: parents
Two hundred and eighteen parents of the 289 (75%) children randomised and treated in the EcLiPSE trial consented to participate in some aspect of the consent study. A total of 212 out of 218 (97%) parents consented to complete a questionnaire and in 13 instances two parents completed a questionnaire for one child. A total of 143 out of 212 (67%) paper-version questionnaires were received (Figure 7). No parents chose to complete the questionnaire using the online version.
FIGURE 7.
Parent characteristics by method.

Recorded trial discussions and interview elements closed in August 2017 (with 193 patients treated and randomised at that stage of the trial) as we reached the data saturation point. 55 Just under half (95/193, 49%) of eligible parents gave consent for the recorded trial discussion. Of these recorded discussions, 76 out of 95 (80%) were received and analysed. Of 114 (59%) of the 193 eligible parents who agreed to be approached for interview, 59 (51%) were invited to participate in an interview by telephone or e-mail. Of these, eight (13%) had incorrect contact details, 10 (16%) did not respond and 11 (18%) declined to interview on telephone contact. Interview and recorded trial discussions data were obtained from 17 out of 25 (68%) sites. Of these, 36 out of 76 (47%) recorded trial discussions were led by doctors and 19 out of 76 (25%) conversations were led by nurses or research nurses. Most (45/76, 60%) practitioners recorded one trial discussion, 24 out of 76 (31%) recorded two parts of a trial discussion and 7 out of 76 (9%) recorded three or more parts. In 37 (49%) cases, it was clear that only the second part of the conversation had been recorded. We obtained a full data set (i.e. the questionnaire, recorded trial discussions and parental interview) for 19 families.
Participants: practitioners
We interviewed EcLiPSE trial principal investigators (n = 4) and lead research nurses (n = 6) who were the main staff involved in recruitment discussions with parents within their first year of site opening. Telephone interviews took place 8–18 months post SIV training (mean 11 months, 345 days; range 260–577 days) and 5–16 months after site opening (mean 8 months, 265 days; range 168–490 days).
A total of 36 practitioners (i.e. 20 nurses and 16 doctors) took part in one of six focus groups held 13–18 months post SIV (mean 14 months, 455 days; range 400–574 days) and a mean of 12 months (mean 374 days; range 329–420 days) after site opening.
The SIV evaluation involved 26 out of 30 (87%) sites. A total of 149 out of 312 (47%) staff were eligible for inclusion, as they anticipated staying for the full site initiation meeting and completed a questionnaire. Clinical commitments had an impact on staff’s ability to attend the entire SIV. Consequently, 24 out of 149 (16%) questionnaires were partially completed and excluded from the analysis because of missing data.
A total of 199 practitioners from 29 out of the 30 (97%) sites completed the online questionnaire approximately 8 months before the end of the trial.
See Appendix 10 for consent study participant characteristics.
We present consent study findings under key themes identified in the analysis of parent and practitioner data.
Trial acceptability and challenges: practitioner perspectives
For many hospitals, the EcLiPSE trial was the first clinical trial to be led by their ED. In the early stages of the design of the EcLiPSE trial, the trial team recognised practitioner concerns about the acceptability of RWPC. A training package was, therefore, developed using parents’ perspectives from the EcLiPSE feasibility study40 and CONseNt methods in paediatric Emergency and urgent Care Trials (CONNECT) guidance. 48 The aim was to help provide practitioners with confidence in recruitment and RWPC in the EcLiPSE trial.
Practitioner perspectives before trial recruitment
Before site initiation training, 33 out of 118 (28%) practitioners who completed a questionnaire indicated that they had concerns about recruiting participants to the trial. A higher proportion of practitioners (48/120, 40%) was concerned about seeking RWPC (Table 13). During interviews and focus groups practitioners described how their concern arose from lack of knowledge and experience of RWPC. Some staff were apprehensive about the acceptability of RWPC and ‘quite concerned how parents would take that [RWPC]’ (focus group 4, female, nurse, P1) and about ‘how to approach it with the parents’ (practitioner telephone interview, female, lead research nurse, P3) after their child had been entered into the trial.
| Question | Yes, n (%)a | No, n (%)a | Odds ratio | 95% CI | p-value |
|---|---|---|---|---|---|
| Do you have any concerns about recruiting to the EcLiPSE trial? | 33 (28) | 85 (72) | |||
| Experienced in RWPC | 4 (19) | 17 (71) | 0.54 | 1.17 to 1.74 | 0.296 |
| Not experienced in RWPC | 28 (30) | 64 (70) | |||
| Do you have any concerns about seeking consent for the EcLiPSE trial? | 48 (40) | 72 (60) | |||
| Experienced in RWPC | 6 (26) | 17 (74) | 0.46 | 0.17 to 1.30 | 0.128 |
| Not experienced in RWPC | 40 (43) | 52 (57) |
Interestingly, previous experience with RWPC was not associated with concerns about recruitment or seeking consent, suggesting that concerns may have arisen from issues other than the consent process. Sixty-nine out of 115 (60%) practitioners who completed the questionnaire anticipated that there would be practical or logistical difficulties in conducting the EcLiPSE trial. This included concerns about adequate research support to conduct consent discussions with families, particularly ‘over the weekend’ (SIV questionnaire part A, female, doctor, P55), as well as the challenge of training all relevant staff across departments. In focus groups, doctors discussed their concerns about using a trial protocol in an emergency resuscitation situation, while not compromising the clinical care of critically ill children:
I thought, oh God, this sounds horrendous. Literally I was thinking, how is this going to work? This is going to be a nightmare.
Focus group 3, female, doctor, P3
As the number of eligible patients per site was expected to be fairly small (i.e. approximately 0.5 patients per month), practitioners referred to the anticipated challenge of maintaining trial awareness to ensure that eligible patients were not missed. Principal investigators were most concerned about engaging and motivating all staff to maximise trial success:
Difficult to control for other people who may be less interested in our department being involved.
SIV questionnaire part A, female, doctor, P121
Practitioner perspectives after training and experience of recruitment
As shown in Appendix 9, Tables 38 and 39, improved levels of confidence were observed for all four questionnaire statements, regardless of whether or not practitioners had prior experience of RWPC. It was notable that following training 82 (66%) practitioners felt that their confidence in explaining the study to families had improved, whereas 90 (72%) practitioners felt more confident in explaining RWPC to families. Approximately half of the practitioners also indicated that their confidence in explaining randomisation (47%) and addressing parents’ objections to randomisation (51%) had improved.
Questionnaire part B (after training) free-text responses, as well as interview and focus group discussions, indicated that the EcLiPSE trial training had addressed many of the practitioners’ concerns about recruitment and RWPC. After training, many described how the trial and its approach to consent seemed more ‘feasible’ (SIV questionnaire part B, female, doctor, P128) and ‘logical and straightforward’ (SIV questionnaire part B, female, nurse, P30). Research nurses, in particular, valued the examples of tailored communication, such as the ‘terminology used to explain this to families’ (SIV questionnaire part B, female, nurse, P109), as well as ‘see how nurse handled difficult questions’ from parents (SIV questionnaire part B, female, nurse, P27). During telephone interviews, research nurses also commented on the training:
There are some good sort of one-line quotes that you can take from it.
Practitioner telephone interview, female, lead research nurse, P4
Although fewer logistical concerns were expressed after training, some practitioners restated concerns about staff availability to cover consent discussions with families 7 days per week. Questionnaire, interview and focus group participants also highlighted the challenge of disseminating training to relevant ED staff, including new doctors at the trainee rotational changeover, and having sufficient staff trained to cover consent-seeking during evenings, weekends and over busy periods.
Despite these challenges, principal investigators and research nurses took the lead in providing regular or bespoke training sessions to help ensure that all relevant staff were able to screen, recruit or seek consent:
I’ve trained new people who’ve joined the team . . . then just the ongoing simulation training . . . and making sure people are still familiar with the protocol.
Practitioner telephone interview, male, doctor, P2
In focus groups, practitioners suggested that the trial management team could provide recruitment and consent support through study updates, advice (when required) and recruitment training tips from the ongoing consent study. This support was provided through regular contact and newsletter updates, which included access to training materials online and on a USB (universal serial bus) provided to each site principal investigator. Practitioners described how trial team support and access to training materials facilitated dissemination of training to new staff. In the online questionnaire, 8 months before the end of trial recruitment, practitioners involved in any element of the EcLiPSE trial were asked to select the statements that they felt were relevant to their site. The majority of practitioners indicated that the trial was running well, which was supported by trial recruitment data (i.e. recruiting to target with a 95% consent rate). Only two practitioners (2/199, 1%) reported that anxieties about RWPC were a barrier to recruitment. Some practitioners reported that staff shortages had led to patients being missed and some (6%) indicated that training was not frequent enough.
Trial acceptability: parents’ perspectives
Recorded trial discussion and interview data showed that the majority of parents were first aware of their child’s involvement in the study after the emergency situation had passed. At an early point in trial discussions, practitioners provided a brief explanation of the reasons why informed consent had not been sought. These explanations were often consistent with CONNECT guidance48 and related SIV training provided to practitioners during site training.
The majority of parents found the EcLiPSE trial to be acceptable and parents were ‘happy’ for their child ‘to participate’ in the trial (recorded trial discussion 6, parent). These findings are supported by all sources of data collected, including practitioners’ descriptions of consent discussions with parents and the high consent rate for the EcLiPSE trial.
Consistent with previous studies in an emergency setting,47,56 just over one-third of parents who completed the questionnaire were initially surprised to find out that their child had already been entered into the trial without their prior consent (Table 14).
| Statement number | Statement | Response, n (%)a | ||
|---|---|---|---|---|
| Agree | Neither agree nor disagree | Disagree | ||
| 1 | The practitioner checked that it was a convenient time to discuss research before discussing the EcLiPSE trial | 129 (90) | 9 (6) | 5 (4) |
| 2 | I was initially surprised to find out that my child had already been entered into the EcLiPSE trial | 56 (39) | 49 (34) | 38 (27) |
| 3 | The information I received about the EcLiPSE trial was clear and straightforward to understand | 139 (97) | 4 (3) | 0 (0) |
| 4 | I understood why consent for my child’s participation in the EcLiPSE trial was sought after the treatment had been given | 139 (97) | 2 (1) | 2 (1) |
| 5 | I had enough opportunity to ask questions about the EcLiPSE trial | 134 (94) | 7 (5) | 2 (1) |
| 6 | I was satisfied with the deferred consent process for the EcLiPSE trial | 129 (90) | 9 (6) | 5 (4) |
| 7 | It was difficult to take in the information I was given about the EcLiPSE trial | 20 (14) | 17 (12) | 105 (73) |
| 8 | It was difficult to make a decision about the EcLiPSE trial | 15 (11) | 15 (11) | 112 (78) |
| 9 | I made this decision | 122 (85) | 14 (10) | 7 (5) |
| 10 | Someone took this decision away from me | 8 (6) | 15 (11) | 119 (83) |
| 11 | I was not in control of this decision | 18 (13) | 20 (14) | 102 (71) |
| 12 | The decision about the research was inappropriately influenced by others | 8 (6) | 15 (11) | 115 (80) |
This experience was also described by parents who were interviewed. However, these parents described their surprise was momentary, as practitioner explanations about why informed consent could not be sought in an emergency situation had addressed any initial surprise or concern:
I think the only thing I really found surprising was the whole informing you about it afterwards, but he explained all that and I do understand why that was done . . . when he explained it all, I mean it makes sense. It’s not the situation to start having discussions, it’s an emergency situation.
Parent interview, mother, P14
The majority (129/143, 90%) of parents who completed a questionnaire indicated that they were satisfied with the consent process. In addition, they indicated that the timing of the consent discussion was appropriate [statement 1: 129/143 (90%)], that trial information was straight forward and easy to understand [statement 3: 139/143 (97%)], and that they understood the reasons why consent had been sought after treatment had been given [statement 4: 139/143 (97%)]. Moreover, responses to statements 8–12 suggest that parents felt that they made voluntary consent decisions, which were not inappropriately influenced by others (see Table 14).
However, there was one exception. One mother (parent interview, mother, P11) described how she had adequate capacity to make a decision about a trial when her child was being treated:
I was asking questions and stuff while she was having the seizure so it could’ve been mentioned to me and I would’ve taken it in and I wouldn’t have questioned it after.
Parent interview, mother, P11
This mother described her sense of loss of control over the situation as ‘they’d done it without telling us’. However, this parent felt able to consent for the use of her child’s data in the EcLiPSE trial as ‘they use these two medications all the time’, which she found reassuring.
During trial discussions, parents often voiced their gratitude, thanking doctors and nurses for conducting research into a condition that threatened their child’s life.
As shown in Table 14, despite parental satisfaction with the RWPC process, some parents (20/143, 14%) reported finding it difficult to take in study information at the point that they were approached about the trial, whereas others (15/143, 10%) reported finding it difficult to make a decision. During interviews, some parents described their child’s recent hospital episode as being ‘like a big blur’ (parent interview, mother, P1) because of the stressful situation.
Why parents agreed to the use of their child’s information in the EcLiPSE trial
During interviews, parents were asked for the reasons why they had provided consent for use of their child’s data in the EcLiPSE trial. The questionnaire also included statements to identify the reasons why parents had consented for their child’s information to be used in the trial. Parents commonly indicated multiple reasons for providing consent. The majority (135/143, 94%) of parents indicated that a reason they provided consent for the use of their child’s information in the trial was to help other children in the future, whereas 46 (32%) parents indicated that this was their main reason.
In total, 124 out of 143 parents (87%) also indicated that they provided consent in the belief that medical research, such as the EcLiPSE trial, is important, and 11 out of 94 (12%) parents indicated that this was their main reason for giving consent. Interestingly, just under half of parents stated that they had consented because their child had recovered. However, this was not a common reason described during interviews, with only two parents mentioning that they were happy to participate in the trial ‘because it worked’ and one parent suggesting that ‘if it didn’t work then it might’ve been different’ (parent interview, mother, P24).
The nature of the trial interventions appeared to influence parental decision-making. Although parents spoke of how it is ‘important that research happens’ (parent interview, mother, P28), many also stated that they had found the EcLiPSE trial acceptable because they did not believe that the interventions would harm or pose any risk to their child, as both were used in the treatment of seizures and were likely to be effective in stopping a seizure:
It didn’t quite seem like a drastic choice anyway, two commonly used medicines. So it wasn’t like a risk at all.
Recorded trial discussion 27, parent
Others spoke of how their child may have benefited from involvement in the trial if the treatment had been effective in stopping the seizure more quickly. In addition, some parents also acknowledged that, as a result of the trial, there was potential for their child to benefit from evidence-based medicine in the future:
Well I guess potentially if it happened again there might be benefit for [child’s name], and it may have benefited her at the time . . . It might have stopped the fitting.
Parent interview, mother, P28
Importantly, throughout interviews and during the EcLiPSE trial recruitment discussions, parents cited their trust in practitioners as acting in the best interests of their child, with 89 out of 143 (62%) questionnaire participants indicating that a reason they consented was ‘because I trust the doctor or nurse who explained EcLiPSE’:
I was happy for them, as experts, to take it into their own hands and do what they felt was best for my child, and for me to sit on the sidelines and just hope that everything was going to be OK.
Recorded trial discussion 70, parent
Finally, parents valued practitioners’ explanation of the trial, including the time they had taken out of their clinical duties to explain it to them. Some described how they appreciated having an in-depth conversation with a consultant about the trial and how clear explanations helped them in making their decision on consent:
They just made the effort to ring and talk about it, so can’t remember her name either, but she was incredible. She just took me through everything very slowly and explained everything.
Parent interview, mother, P6
Parental awareness and responses to the EcLiPSE trial at the point of randomisation
During SIVs, practitioners frequently asked the trial team how best to respond to parents who noticed the trial randomisation envelope being opened during resuscitation. In the site training evaluation questionnaire, fewer than half (60/125, 48%) of practitioners responded positively to the statement ‘I feel confident in dealing with parents who object to their child being randomised’. Prior experience of RWPC was not associated with increased confidence in dealing with parents’ objections at the point of randomisation (p = 0.288). Practitioners were concerned that parents would have questions that would lead to discussions that would potentially delay the administration of time-critical treatments. Practitioners were also worried that parents might object to or be upset about RWPC, creating friction in an already highly emotive situation:
You’re like oh my God, what are they [parent] going to say, what are they going to do.
Practitioner telephone interview, female, lead research nurse, P8
Specific training was developed to help address such concerns, including how to respond to parents who asked about the study at the point of randomisation, and to direct parents to posters and leaflets in the ED if further information was requested.
Parents were asked during interviews about the first time that they had heard about the trial and explored their knowledge of trial processes taking place during resuscitation. The majority of parents stated that they ‘didn’t know anything about the study at that point’ (parent interview, mother, P1). This view was echoed by practitioners who stated that, in most cases, ‘I don’t think they specifically asked the question [about the study]’ (focus group 6, male, doctor, P1). Doctors and nurses who had randomised patients stated how the process worked well and without the anticipated problems. This was attributed to how ‘parents are focussed on their child’ and the opening of a randomisation envelope not being ‘explicitly done in front of them’ (focus group 5, female, doctor, P1).
A few parents stated that they had noticed trial posters in the ED but had not given them any further consideration because of the emergency situation. Nevertheless, some parents, particularly those with previous experience of their child being admitted to hospital with tonic–clonic seizures, did notice the randomisation envelope. In these cases, a brief discussion about the trial, including what drug had been allocated, appeared to make parents feel that practitioners ‘were keeping me involved’ (parent interview, mother, P15) and prevented concerns or negative responses during resuscitation. Practitioners described how the provision of brief information about the EcLiPSE trial when parents noticed the opening of an envelope, or ‘something different’ was happening, was important to maintain parental trust by being ‘really transparent’ (focus group 2, female, nurse, participant 6). However, when no questions were asked, it was not always possible for practitioners to know whether or not parents had noticed trial processes were taking place. As the below quotation from a recorded trial discussion shows, one parent had noticed the randomisation process, had not asked questions and had not noticed any the EcLiPSE trial information in the ED. This lack of knowledge about what was going on made this father feel uncomfortable and suspicious that something ‘dodgy’ was going on:
I’m like what’s all this whispering about? What’s the dodgy envelope? My kid is unconscious; tell me what’s going on. So it definitely made me uncomfortable. I don’t have the answer but yes, I definitely felt that.
Recorded trial discussion 52, parent
How information about the trial was exchanged during discussions on recruitment into the EcLiPSE trial
Previous studies have shown that how a clinical trial is explained is a key determinant of recruitment and how patients experience and understand a trial. 57–59 To the best of our knowledge, there are no published reports on how practitioners explained to parents that their child had been included in a trial in the ED, and whether or not they understood this explanation. Nineteen matched recorded trial discussions and parent interviews were analysed to explore how EcLiPSE trial information, including discussion of RWPC, was exchanged between practitioners and parents. This included exploring questions parents had about the trial and how they were addressed, as well as how parents recalled and understood aspects of the study when questioned during interviews approximately 1 month later. We also drew on wider data (i.e. 76 recorded trial discussions and 30 parent interviews) to corroborate our findings.
Our analysis was informed by the Realpe et al. 57 six-step model, which provides a framework for successful recruitment to RCTs. The model steps are (1) explain the condition, (2) reassure patients about receiving treatment, (3) establish uncertainty, (4) explain the study purpose, (5) give a balanced view of treatments and (6) explain study procedures (see Appendix 10, Figure 11). There are also two elements used throughout the trial discussion: (1) responding to patients’ concerns and (2) showing confidence and a relaxed manner. Owing to the nature of the audio-recorded data, we were unable to fully assess whether or not the practitioners showed confidence and a relaxed manner.
Discussion of trial information and parental understanding
The sequence of how trial information was presented by EcLiPSE trial practitioners differed to the Realpe et al. 57 six-step model (see Appendix 10, Figure 13). This difference reflects the RWPC approach, as well as parents’ prior knowledge of their child’s condition, which meant that a discussion of seizures (i.e. step 1 in Realpe et al. ’s57 model) was often not part of the trial discussion.
We found that parental understanding of the EcLiPSE trial was enhanced when practitioners provided a comprehensive description of trial aims; explained the reasons for RWPC; discussed uncertainty about which intervention was best; provided a balanced description of trial intervention; provided a clear explanation about randomisation; and provided an opportunity for questions. Interestingly, this common sequence of information provision did not match the order in the participant information sheets, suggesting that practitioners had tailored verbal information for parents and had not simply read out the information sheet (see Appendix 10, Figure 14 for a new seven-step model to optimise recruitment in future trials that involve RWPC).
How checking understanding and providing opportunities for questions led to patient-centred trial discussions
In the majority of conversations about recruitment, practitioners checked parental understanding and provided opportunities for them to ask questions, and 38 out of 76 (50%) parents asked questions during these discussions. Previous experience of their child having a seizure did not appear to have an impact on whether or not parents asked questions.
Practitioners tailored discussions to specific parental questions. This meant that, although the six steps were often covered in discussions, practitioners would not strictly follow the sequence or they would revisit certain steps (e.g. description of treatments) to provide additional information. Questions typically focused on treatment information, such as which drug their child received, and the safety, effectiveness and side effects.
Parents spoke of how their questions about the trial had been sufficiently addressed during the trial discussion:
The doctor explained everything and he was really good.
Parent interview, mother, P1
Parental understanding of the EcLiPSE trial was influenced by the level of verbal information provided
Analysis of recorded trial discussions indicated that most practitioners clearly described the trial’s aims and objectives. Initial descriptions of the trial focused on its size, including the number of hospitals involved, the trial interventions, the trial’s aims and why the child was eligible and had been recruited. Consequently, during interviews, most parents were able provide a reasonably clear description of the aims of the trial:
Basically that there’s two medicines that they can give that they know stops seizures but they didn’t know which was the best one or the preferred one to use and that’s why you’re doing this study, to see how it affects different people.
Parent interview, mother, P15
Practitioners sometimes described an equivalence, ‘The study is looking at is it [Keppra] as effective as phenytoin’ (recorded trial discussion 27, practitioner), rather than a superiority hypotheses, ‘Whether actually is Keppra more efficient at stopping a seizure more quickly than phenytoin’ (recorded trial discussion 4). This seemed to influence parents’ understanding of the trial design and is not necessarily surprising, as a fairly technical understanding would be needed to understand clinical trials methodology.
Matched audio-recorded discussions and interview analysis showed that more detailed trial discussions, covering all six steps, led to improved parental understanding and retained information. For example, when some parents were asked to recall what they had understood about the trial during the recruitment conversation, as well as to the researcher in an interview 1 month later, they were able to clearly explain the differences in how the two interventions were used in hospital departments and the rationale of the trial:
What I have understood is that you have been using this new drug for years, but what you still want to know, is the drug still capable of working or is there another way you can control kids who have fits like [child] has for such a long time, and without the research how are you going to know what is the best thing for the kids without causing them too much distress than what they are already going through.
Recorded trial discussion 6, parent
What they explained was even though there were two drugs for children with epilepsy, to try and control it, it did say one of the drugs had only just been recently used in A&E [accident and emergency]. He said they had both been used for years, but one of them was kept out of A&E. Now he said they were introducing it into A&E, and they just wanted to know which drug would be best suited to control children with epilepsy.
Parent interview 3, matched with recorded trial discussion 6
By contrast, practitioners who had brief discussions with parents, covering only limited aspects of the trial, resulted in parents being unable to recall it in any detail when interviewed. For example, in one very brief trial discussion (i.e. 3 minutes and 27 seconds in length) that covered the trial aims, the two drugs involved and reassurance that their child received ‘the normal treatment she would normally have anyway’ (recorded trial discussion 2, trial recruiter), the parent was subsequently unable to recall any details of the conversation, including which drug her child had received:
I can’t remember any names of them.
Parent interview, mother, P4
Despite all parents in this sample being provided with, and confirming that they had read and understood, a written information sheet, it was clear that verbal information provided during trial discussions was prioritised, understood and recalled. Parents appeared to value the manner in which information was delivered by practitioners. During interviews, parents often referred to how the trial was ‘really well explained’ (parent interview, mother, P15) by doctors or nurses who were ‘sympathetic’ and ‘absolutely lovely’ (parent interview, mother, P13).
Descriptions and understanding of randomisation was assisted by the use of an envelope
Previous studies have shown that patients often struggle to understand trial processes, such as randomisation,60 and may not believe that chance was involved in treatment allocation. 61 Interestingly, analysis of recorded trial discussions and interview data indicated that the majority of practitioners had explained the EcLiPSE trial randomisation process clearly and parents had understood the process. Practitioners often provided confident, yet simple, descriptions of randomisation. These explanations may have been assisted by the tangible nature of the EcLiPSE trial randomisation process itself, which involved the opening of a pre-filled envelope. However, there were some examples of when parents were unable to recall details of the randomisation process, despite there being a description in the parent information sheet. We found that in these examples practitioners had not explained this to parents during trial discussions:
Do you know, I can’t but I just know I know what randomised selection and stuff means anyway. I couldn’t really tell you how it was explained to me.
Parent interview, mother, P3 matched with recorded trial discussion 2
Parental capacity to understand trial information was influenced by previous experience of child seizures
There were examples of practitioners giving a comprehensive explanation of the trial, yet parents were unable to recall any of the conversation. This lack of recall was attributed to the highly emotive situation and sleep deprivation. We found that parents of children who had experienced their first seizure were often unable to recall the recruitment conversation or describe any key aspects of the trial, including the trial drug administered to their child. This finding was also apparent in the subset of parent interviews for which we had a matched recorded trial discussion. In these examples, their first experience of their child having a seizure understandably had an impact on parental capacity, despite recruitment conversations taking place after their child’s seizure had stopped and the life-threatening situation had passed:
It was really traumatic. The only thing I can remember is going in where the ambulance took us and there were loads and loads of doctors around getting nurses and stuff and she was just lying on the bed. Then they came in and said, I can’t remember, but I later remembered the first thing is that they said they have an envelope and they opened the envelope.
Parent interview, mother, P4
In addition, a few parents continued to be distressed and upset when their child had fully recovered from their seizure and lacked the capacity at the time to understand the recruitment conversation:
I do remember the consultant kept saying to me, are you OK, do you understand what was just . . . and I was just crying the whole time I think.
Parent interview, mother, P18
By contrast, parents of children who had previously been admitted to hospital with a tonic–clonic seizure were often able to provide detailed and accurate descriptions of the trial, such as the trial aims, treatment interventions and why informed consent could not be sought in the emergency situation:
Basically that you were doing which medicine, either Keppra or phenytoin, worked best for stopping seizures . . . It wasn’t really putting her in any danger because it’s what they use anyway for children.
Parent interview, mother, P11
The impact of an unblinded trial design
Parental knowledge of intervention allocation
As the EcLiPSE trial was unblinded, trial recruiters commonly informed parents about which trial intervention their child had received. In total, 23 out of 30 (77%) parents interviewed recalled which drug their child had received. One parent stated that both drugs had been given to their child, but was unsure which one was the trial intervention. Six out of 30 (20%) parents could not recall which drug their child had received, but this did not appear to concern parents because their child had recovered:
Gosh, I don’t know. Basically we were just glad he got through it. I didn’t really care what he was given.
Parent interview, mother, P6
Equipoise and uncertainty about which drug is best
A few of the staff interviewed in focus groups at the end of the first year of the trial described how they had presented the intervention allocation in a positive light to parents, regardless of which drug the child had received:
So they’ve either had the, you’ve got the standard drug, great news. Or, you’ve got the new wonder drug, great news.
Focus group 6, male, doctor, P1
Analysis of the first 34 recorded trial discussions that were received indicated that seven practitioners (21%) favoured the allocated intervention. Most practitioners (6/7, 86%) spoke more positively about levetiracetam and did not describe the uncertainty about whether or not it is more effective at stopping long-lasting seizures than phenytoin. This positive presentation of one intervention is likely to result from the unblinded trial design, lack of equipoise and perhaps practitioners wishing to reassure parents that their child’s safety had not been compromised by participation in the trial.
As part of the iterative design, the consent team added a section to the November 2016 EcLiPSE trial newsletter for sites, which highlighted the importance of providing a balanced description of both trial interventions. Analysis of the recorded discussions that took place after the newsletter had been distributed indicated that this simple intervention was effective because subsequent discussions were equipoised. In total, only 7 out of the 76 (9%) recorded trial discussions were not in equipoise following the newsletter. Overall, trial practitioners provided parents with a brief but balanced description of the two interventions. Importantly, when practitioners described uncertainty as to which drug was better during discussions, parents often provided very similar descriptions of the uncertainly of effectiveness during an interview:
There are these two drugs and one of them – we have used both of them for a long time but we have only used one of them in the emergency setting but we are wondering whether the other one might work well, better or worse. That is what we are trying to find out.
Recorded trial discussion 6, trial recruiter
What they explained was even though there were two drugs for children with epilepsy, to try and control it, it did say one of the drugs had only just been recently used in A&E [accident and emergency]. He said they had both been used for years, but one of them was kept out of A&E. Now he said they were introducing it into A&E, and they just wanted to know which drug would be best suited to control children with epilepsy.
Parent interview, mother, P3, matched with recorded trial discussion 6
Successfully conducting an emergency department-led paediatric trial
During telephone interviews and focus groups, the researcher sought practitioners’ views on perceived barriers to and facilitators of conducting an ED-led trial. The EcLiPSE TMG was particularly interested in what facilitated or hindered ‘buy-in’ at the participating sites, with ‘buy-in’ being defined as when all those involved in trial conduct are supportive of the trial and working to ensure that the trial is a success.
Motivation and leadership
In addition to the training issues described above, motivation and leadership within and across sites were identified as important factors in the successful conduct of the EcLiPSE trial.
Lead research nurses or principal investigators commonly described their enthusiasm for taking part in the study from the very early stages of the trial. This enthusiasm was evident, despite some initial concerns about training staff for the trial. Site leads acted as advocates for the study, raising awareness and ensuring that all relevant staff were trained in delivering the protocol:
I think that we had taken on the study so we were very keen to do it. So myself as the lead and the research nurses, so even though we internally had our concerns, we were very strong at promoting it after we had that initial site visit. In our education with all the juniors, we very strongly advocated it.
Practitioner telephone interview, female, doctor, P3
A strong motivated leader, such as the principal investigator or lead research nurse, was seen as important to engage staff and ensure that eligible patients were not missed. Practitioners emphasised the importance of principal investigators taking responsibility for promoting the trial. This included making time to disseminate training and motivating the wider team to take ownership in recruiting patients 24 hours per day, 7 days per week. This was often challenging in busy EDs. Strong leadership was not evident at a few sites, which had a negative impact on staff engagement and ultimately the number of patients recruited at these hospitals. This was rare but highlighted the importance of identifying a motivated and committed principal investigator throughout the trial:
There were three consultants and then there were two ANPs [advanced nurse practitioners], and they’re just not interested. It’s not just this study, we’ve had it with a couple of others, there’s just a bit of a lack of interest with research . . . So I think nobody is taking ownership really.
Practitioner telephone interview, female, lead research nurse, P1
The importance of a clinically relevant research question and simple trial design
Practitioners described how the EcLiPSE trial research question was important and clinically relevant, which was a crucial factor in engendering interest and ‘buy-in’ from the site teams, as well as support from parents who also wanted evidence-based treatments for their child:
It’s a really important question that people want answered, and I think that’s one of the reasons . . . I think for parents it’s something that they want. They want answers. If you’ve got a child in status, I want the best drug which I think is different to some of the other studies that are around.
Focus group 4, female, doctor, P2
Many ED practitioners cited the value of the simple and pragmatic trial design, which meant that the protocol was easy to follow in an emergency situation. It also facilitated engagement with the trial. During focus groups, they spoke of the benefits of having a trial protocol that aligned very closely to their usual practice and did not involve much additional work and documentation:
The great thing about EcLiPSE we have had was the pragmatism was just really primary for practice and, unlike commercial studies that I’ve been involved in where they try and bolt on a thousand and one extra things for you to do.
Focus group 3, female, nurse, P3
Collective engagement through site initiation visits
During interviews, practitioners spoke of how the involvement of a number of the EcLiPSE trial team members, including the chief investigator, in delivering the SIV training had helped to create a sense of importance of the research question and the trial. It also helped engage practitioners. They also valued the invitation that as many people could attend the SIV as possible so that the trial could be explained and questions answered:
I think people were very impressed that the chief investigator had arrived . . . they saw that as a really, really good sign that people were taking this very seriously.
Practitioner telephone interview, male, doctor, P2
Teamwork
Many spoke about how working together as a team, both with the central EcLiPSE trial team and at the site, was important to assist engagement on site initiation and to facilitate recruitment:
Trial teams don’t generally come back and ask all these questions. We don’t usually get this much support.
Focus group 4, female, nurse, P3
Teleconferences involving all sites were viewed as a useful method of sharing good practice and raising any concerns. Individual sites produced trial posters visible in their ED and developed their own incentives to help maintain awareness of the trial to assist recruitment. These were agreed by the central EcLiPSE trial team and, when appropriate, the Research Ethics Committee. Finally, despite best efforts, a minority of sites described how they struggled to work as a team because of insufficient research nurse support or lack of engagement by key staff:
I think not having [nurse] working in research, well that hasn’t helped . . . But even when he was working with us, I don’t think we were necessarily always completing a log screen for every single case that came in.
Practitioner telephone interview, male, lead research nurse, P9
Discussion
Our findings show how interactive site training, developed using pre-trial research and RWPC guidance, can significantly alleviate practitioner concerns about recruitment and consent in a challenging PEM trial. For many participating hospitals, the EcLiPSE trial was the first time that teams worked together to ensure that critically ill children were randomised to a trial during resuscitation without prior informed consent. Prior to site training, many practitioners expressed concerns about conducting the trial and lacked confidence in how to communicate to parents some of its elements, including RWPC. Practitioners rated highly the clarity and content of SIV training. Significant improvements were observed in practitioners’ confidence in explaining the study, randomisation and RWPC to families, as well as how to respond to parents who might object to their child being randomised during an emergency resuscitation. Site training provided an opportunity to discuss and learn about the potential challenges and solutions to trial recruitment and conduct. 62,63 Our findings suggest that the use of training videos significantly aided this process and helped practitioners to visualise potentially difficult trial processes, including screening in a resuscitation situation. Practitioners particularly valued the RWPC video, which had been informed by data on parents’ views and priorities from pre-trial research43 and CONNECT study guidance on RWPC. 48,49 The video provided practitioners with examples of how to communicate RWPC to parents,48,49 as well as preparing them for a range of questions parents might ask about the trial. 43
Recorded trial discussions and interviews conducted with parents and practitioners throughout the EcLiPSE trial provided insights into how information about the trial and RWPC had been exchanged and understood. Overall, trial practitioners provided parents with a brief but balanced description of the two interventions to parents. Parental understanding of the trial was enhanced when practitioners clearly described trial aims, the reason for RWPC, uncertainty about which intervention was better, a balanced description of both interventions and the randomisation process, and welcomed questions. We provide a bespoke seven-step framework to optimise PEM trial RWPC discussions. Further testing of this framework in future PEM trials is required, as are parent and practitioners views and experiences of RWPC.
Multiple factors, including trial design, logistics and leadership, were found to both challenge and contribute to trial recruitment and conduct. Importantly, both parents and practitioners wanted to know the answer to the research question, which appeared to underpin many of the decisions and behaviours identified in our study. Our findings also suggest that practitioners were engaged and invested in the EcLiPSE trial because of its pragmatic design, which aimed to answer an important clinical question that they believed would quickly inform their daily practice. This engagement was apparent across the majority of EcLiPSE trial sites, despite initial concerns about whether or not the trial was possible, which was often confounded by inexperience of conducting an ED-led trial. Our findings highlight the important role of researchers and funding panels when the clinical relevance of the research questions and how challenging trials are more likely to succeed, if these are relevant and important to all key stakeholders, including patients, family members and clinicians. 64,65
As has been shown in recent pilot studies56,66 exploring treatments for paediatric suspected infection, parents and practitioners found RWPC to be acceptable. They understood its principle of avoiding unethical delays in the delivery of potentially life-saving treatments. Practitioner concerns that parents would notice the randomisation envelope being opened in the ED and would object to the trial or RWPC, or both, were mostly unfounded. Parents of children who had a prior diagnosis of epilepsy seemed to be more likely to notice something different was occurring, such as the opening of the randomisation envelope. These parents valued how practitioners provided them with brief description of the study, including what drug had been allocated. As shown in other studies57,67,68 that have explored trial recruitment and decision-making, patients and family members often prioritise verbal over written information provision, while the highly stressful and time-critical ED context is likely to impact on parental capacity and, indeed, desire to read even short written study information. 47 Future trials would benefit from considering how their study and RWPC could be briefly communicated to parents of children who are regular ED attenders, if deemed appropriate.
Strengths and limitations
Although most sites (87%) took part in the evaluation, only 47% of eligible practitioners anticipated that they would be able to stay for the full consent training, and completed the questionnaires. Of these practitioners, 16% were excluded from analysis because of incomplete questionnaires. This attrition was because practitioners had to leave training early because of clinical commitments. However, our study was strengthened by the conduct of interviews, focus groups and an online questionnaire with staff who had, but also who had not, attended the SIV. This mixed-methods approach provided insight into multiple perspectives to assist understanding of the longer-term uptake and impact of training on practitioner confidence in recruitment and RWPC,50,51 and potential barriers to trial success. All sites participated in the consent study and practitioners were purposively sampled for focus groups and interviews to ensure sample variance (e.g. low- and high-recruiting sites). The majority of parents (75%) of children randomised and treated in the EcLiPSE trial consented to participate in some aspect of the consent study and qualitative recruitment stopped when data saturation was reached. 55,69 However, none of the 19 (4.7%) parents who declined their child’s involvement in the EcLiPSE trial consented to take part in the consent study and, therefore, their views have not been able to be represented.
Chapter 5 Discussion
Parts of this chapter have been reused from Lyttle et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
To the best of our knowledge, the EcLiPSE trial is the largest and most clinically pragmatic RCT to compare levetiracetam with phenytoin in the treatment of paediatric CSE unresponsive to first-line treatment. This trial, powered for superiority, did not detect a statistically significant difference in any of the primary and secondary outcomes. The direction of effect favoured levetiracetam across the primary outcome (i.e. time from randomisation to CSE cessation) and secondary outcomes (e.g. the need for RSI and SARs), other than for the secondary outcome of admission to critical care, for which the direction of effect favoured phenytoin. These findings were robust in all sensitivity analyses.
The results of the EcLiPSE trial were published in May 2019. 1 A similar RCT, the Convulsive Status Epilepticus Paediatric Trial (ConSEPT), which was undertaken in 13 EDs in New Zealand and Australia, of 233 children aged 2 months to 16 years and used a similar protocol, including identical doses and rates of administration of the two drugs, was published simultaneously in the same journal. 41 The primary outcome of the ConSEPT trial was clinical cessation of seizure activity 5 minutes after the completion of infusion of the study drug, with levetiracetam infused over 5 minutes and phenytoin infused over 20 minutes. Analysis was by intention to treat. Results showed that clinical cessation of seizure activity 5 minutes after completion of infusion of the randomised drug occurred in 68 (60%) patients in the phenytoin group and 60 (50%) patients in the levetiracetam group (risk difference –9.2%, 95% CI –21.9% to 3.5%; p = 0.16). The authors41 concluded that levetiracetam was not shown to be superior to phenytoin for the second-line management of paediatric CSE.
More recently, the results of the Established Status Epilepticus Treatment Trial (ESETT) from the USA showed no statistical significance difference in its primary outcome (i.e. cessation of status and improvement in consciousness at 60 minutes without the use of additional antiseizure medication) between levetiracetam (at a dose of 60 mg/kg), sodium valproate and fosphenytoin (a pro-drug of phenytoin and at a dose of 20 mg/kg). The primary outcome was achieved in approximately 50% of patients and the frequency of AEs was similar in the three drugs. 70,71
Prior to the publication of the ConSEPT trial, reported CSE cessation rates for levetiracetam and phenytoin were broadly similar to a number of previously reported observational retrospective, and predominantly adult, studies. 14,30,31 However, CSE cessation rates as high as 85–95% have been reported, although these studies display significant heterogeneity in design and outcomes. 72–74 A recent prospective RCT74 of only 50 children recruited over a 6-month period reported that levetiracetam (at a dose of 30 mg/kg) terminated CSE in 92% of children and fosphenytoin (at a dose of 20 mg/kg) terminated CSE in 84% of children (p = 0.66). 74 These rates are considerably higher than those found in the EcLiPSE trial, ConSEPT trial41 and ESETT. 70,71 This also applied to the time to terminate CSE from the time of commencement of the infusion. Fosphenytoin terminated CSE earlier (2.5 ± 1.4 minutes) than levetiracetam (3.3 ± 1.2 minutes; p = 0.03). 74 The equivalent median times in the EcLiPSE trial, from time of commencement of infusion, were 17.5 and 24.5 minutes for levetiracetam and phenytoin, respectively, and in the ConSEPT trial these were 17 and 22 minutes, respectively. The methodology of the study by Senthilkumar et al. 74 was limited and each treatment group comprised only 25 patients (this may explain their markedly discrepant findings with the EcLiPSE trial for both CSE cessation and the speed with which this was achieved). Finally, although fosphenytoin can be administered more rapidly than phenytoin, it can still not be administered as quickly as levetiracetam. Consequently, the findings of Senthilkumar et al. 74 remain difficult to understand and it is uncertain if they would be generalisable.
One RCT,28 undertaken in adults with CSE, compared the efficacy of i.v. phenytoin (20 mg/kg), sodium valproate (30 mg/kg) and levetiracetam (25 mg/kg) in 150 patients unresponsive to i.v. lorazepam. CSE stopped in 34 (68%) patients treated with phenytoin, 34 (68%) patients treated with valproate and 39 (78%) patients treated with levetiracetam (p = 0.44). A paediatric RCT,72 published in 2018, evaluated 100 children aged 3–12 years receiving levetiracetam (30 mg/kg) or phenytoin (20 mg/kg) if CSE continued after one dose of i.v. diazepam. 72 Efficacy was high and almost identical in both groups. A lower diastolic blood pressure was recorded in phenytoin-treated patients (p = 0.023). It is difficult to translate these findings to clinical practice because of the trial’s design, including its many exclusion criteria and its primary outcome,71 which was ‘absence of seizure activity within 24 hours’. This is an unusual and very rarely used primary outcome in other studies of CSE. It is also not a practical and ‘real-life’ clinical goal, as the emphasis should be on the termination of the presenting seizure as soon as possible after treatment has been given, and not the child’s condition after 24 hours. Consequently, it would be difficult, and probably inappropriate, to use the same primary outcome in routine clinical practice in the UK and elsewhere. Childhood CSE management in the UK follows the APLS algorithm,11 which is applicable to the vast majority of children presenting to an ED. Our study design used eligibility criteria that were as inclusive as possible, and followed a well-recognised treatment pathway that reflected routine clinical practice.
Children with focal and generalised CSE were included because their management is the same in the APLS algorithm. In addition, it may be difficult to accurately distinguish focal and generalised convulsive seizures in infants and children aged < 3 years.
We did not detect a statistically significant difference between levetiracetam and phenytoin in time to CSE cessation. A superiority design was selected for three reasons: (1) the reported CSE cessation rates for each drug, hypothesising that levetiracetam would be more effective; (2) the absence of any RCT data comparing the effectiveness of either phenytoin or levetiracetam with placebo and (3) the shorter infusion time of levetiracetam (i.e. 5 minutes vs. at least 20 minutes for phenytoin). We selected time from randomisation, and instructed sites to undertake randomisation at the latest possible point that would allow reconstitution of the allocated treatment to provide scientific and clinical rigour. As the median time to commencement of infusion exceeded 10 minutes in each arm, we also undertook a sensitivity analysis, using time to cessation of CSE from commencement of the infusion. This supported our primary analysis findings and demonstrated that the median time from commencement of the infusion was similar to the median time from randomisation in both treatment groups. This is interesting, as it might have been expected that CSE would have been terminated more quickly with levetiracetam (with infusion time of 5 minutes) than phenytoin (with infusion time of at least 20 minutes). One explanation for this observation could be that the anticonvulsant effect of phenytoin may be achieved prior to completion of its infusion. As far as we are aware, there is no literature that has specifically evaluated how rapidly an infusion of phenytoin might terminate CSE once it has been commenced. The ConSEPT trial41 also showed no significant difference between levetiracetam and phenytoin in time to CSE-cessation.
Progression to RSI in CSE may be required for one or a combination of reasons, including continuing CSE, respiratory depression, clinical deterioration and stability for transfer, or to safely undertake investigations, specifically neuroimaging. However, RSI abolishes visible CSE activity and may, therefore, prevent an assessment of CSE cessation directly related to trial treatment. Participants were, therefore, censored at the time of RSI, but the censoring time was increased to allow for this to be a negative and potentially informative outcome. This may have artificially inflated the time to CSE cessation. However, sensitivity analyses that censored patients at the time of RSI, and defined RSI as a competing risk, did not change our findings.
Safety profiles were similar across both treatments. Owing to their relative infrequency in relation to the trial population size, together with good clinical management in participating sites, the trial showed low rates of SARs. Only 4 out of 286 (1.4%) patients experienced a total of five SARs (three in the phenytoin-treated group and one in the levetiracetam-treated group) and in only one of these patients was the reaction (i.e. marked hypotension) considered as being ‘probably’ related to the study medication (phenytoin). One SAR occurred in a patient who received both anticonvulsants. One phenytoin-treated patient experienced a SUSAR, which manifested as a large increase in seizure frequency and marked sedation within 24 hours of receiving phenytoin. An equally good safety profile was also reported by the ConSEPT trial team,41 with one death in the phenytoin group 27 days after randomisation because of haemorrhagic encephalitis that was considered to be unrelated to the study drug. The authors reported no other SAEs or SARs. The safety profile of the 255 children (aged 2–17 years) who participated in the ESETT70,71 was also good. Two deaths occurred, one in the levetiracetam-treated group and one in the sodium valproate-treated group. Life-threatening hypotension and cardiac arrhythmias were rare and did not differ by treatment group in any age. The only significant safety outcome was seen in children requiring intubation more frequently in the fosphenytoin-treated group, an observation that could not be readily explained by the authors. The authors reported no other differences in safety outcomes. 71
The good safety profile of both anticonvulsants is encouraging, particularly for phenytoin, in view of its well-recognised serious potential adverse effects of hypotension, cardiac arrhythmias and severe extravasation reactions, including the ‘purple glove syndrome’. 15,16 Rarely, the arrhythmia may be fatal caused by non-resuscitatable cardiac asystole. 18 In the USA, fosphenytoin, a pro-drug of phenytoin, replaced phenytoin as the preferred second-line management of CSE, primarily because of its slightly faster rate of infusion but also because of its perceived better safety profile. However, it may also cause SARs and this, together with its relative cost to phenytoin, has precluded its use in the UK.
The literature on the safety of levetiracetam is less extensive because of the comparative short period that it has been used in the treatment of CSE. However, despite the fact that this period spans < 20 years, in comparison with the 50 years with phenytoin, it is important to note that, to date, there have been no reports of severe Stevens–Johnson syndrome, purple glove syndrome or fatal cardiac arrhythmias associated with the use of i.v. levetiracetam.
In the EcLiPSE trial, levetiracetam was well tolerated at an infusion rate of 5 minutes and this was more rapid than previously reported (i.e. 10–15 minutes). 27,30,31 Agitation was the most commonly reported AE in the levetiracetam-treated group, as reported previously. 26 There were no new or unexpected SARs with levetiracetam. Sedation, somnolence and dizziness are rare side effects in adults, but these may in part reflect the prior use of benzodiazepines or craniotomy in these study populations. 20,74 Anxiety has also been reported in adults, but was not reported as an AE or AR in this or other paediatric studies. It is possible that anxiety in the adult may equate to agitation in the child. Clearly, in view of the fact that 90% of our study population was aged ≤ 10 years (and 41% aged ≤ 2 years), anxiety might be difficult, if not impossible, for them or their carers to describe, and instead used the terms ‘agitated’ or ‘irritable’.
More participants in the levetiracetam-treated group required admission to critical care (either a high-dependency unit or a PICU), but this did not reach statistical significance. This finding is difficult to explain. It was not explained by the demography of the two treatment groups, ongoing CSE, the need for RSI, additional anticonvulsants, or the frequency of AEs or AEs. Potential explanations could include a different type or nature (e.g. more severe) of epilepsy in the levetiracetam-treated group or that the attending clinicians had a lower threshold of transferring participants to critical care, as levetiracetam was a relatively new anticonvulsant. There did not appear to be a difference in the two treatment groups regarding the type of epilepsy or additional comorbid conditions, or the numbers that were still on critical care 24 hours after randomisation following post hoc analysis.
Children who were already receiving levetiracetam and phenytoin as oral maintenance antiepileptic drugs were included in the study because this reflects real life. In most emergency situations and, in particular, in the absence of information from carers, children will be treated in accordance with the APLS algorithm. In addition, a recognised cause of CSE in patients with epilepsy at all ages is poor adherence to antiepileptic medication and consequent low blood levels of the medication. The relatively high proportion of children receiving levetiracetam was partly expected and reflects current clinical practice and perception that this drug has a broad spectrum of action (in treating different seizure types) and is safe. The very small number of children receiving phenytoin was also predictable, as it is perceived as having a very narrow spectrum of action, numerous drug interactions, significant long-term side effects and is difficult to monitor. We consider it unlikely that the inclusion of children on levetiracetam and phenytoin significantly affected our findings. Theoretically, it might have been predicted that those children already receiving levetiracetam would not have responded as well to i.v. levetiracetam or have shown more adverse side effects, or both, although this was not reflected in the overall results. However, the small number of patients receiving levetiracetam across both treatment arms precluded any formal subgroup analysis.
The EcLiPSE trial is a unique trial for many reasons. First, to the best of our knowledge, it is the first adequately powered RCT to compare the efficacy and safety of two anticonvulsants as second-line treatments for CSE. Second, to the best of our knowledge, it is the first adequately powered RCT to evaluate phenytoin as a second-line treatment for CSE, despite this drug’s position as the first-choice second-line treatment for > 50 years. Third, the trial incorporated a nested consent study that evaluated the process of RWPC in a PEM trial. 43,44 Last, to the best of our knowledge, it was the first multicentre RCT to be supported by and delivered across the then emerging PERUKI collaborative. 37,38
This trial has a number of strengths. First, it evaluated a specific step (i.e. second-line treatment) in a UK clinical algorithm for the management of childhood CSE. 11 A similar trial75 that assessed the first-line non-i.v. treatment of CSE in the same algorithm led to a change in national clinical practice. Second, it demonstrated that RWPC is acceptable and successful, with 385 out of 404 (95%) randomised participants providing consent. Likewise, in those who were randomised and treated, 286 out of 311 (92%) participants provided consent. RWPC is essential for the successful delivery of paediatric emergency care trials. The high consent rate mirrors that found in a previous trial75 of first-line CSE management (consent rate 97%) and a pilot RCT56 that compared fluid boluses in shock (consent rate 100%). Third, it was a pragmatic trial and recorded only key primary and secondary outcomes in the resuscitation room. This approach, supported by focused data-collection materials and simple allocation and enrolment methods, facilitated successful delivery of the study across all sites, as shown by the small numbers of missed patients, high protocol adherence and accurate data capture for key outcomes. Finally, the trial was conducted in EDs in secondary and tertiary institutions throughout PERUKI, thereby facilitating dissemination and increasing generalisability of our findings.
This trial has some limitations. First, it was open label. A double-blind design was considered too complex for most participating sites, in part because of the markedly different infusion rates of the two drugs, and within the context of the life-threatening and time-critical nature of CSE. Second, the number of participants in one arm of the trial fell below the sample size calculation requirement (134 vs. 140), whereas in the other arm it surpassed requirements (152 vs. 140); however, the effect of this on power is considered unimportant. Third, there was probably subjectivity in the assessment of ‘cessation of all signs of continuous, rhythmic clonic activity’ as the clinical event for our primary outcome, rather than fixed time points to assess CSE cessation. Clearly, these three limitations may collectively increase the risk of bias. However, continual assessment of a child’s condition reflects ‘real-life’ practice in a dynamic situation, in which clinicians constantly evaluate and prepare for the next step in the treatment algorithm. Site training included a simulated demonstration of the end point to ensure an understanding of the key outcome measure for the trial. In the ConSEPT trial,41 the primary outcome assessment (i.e. CSE cessation 5 minutes after completion of the randomised infusion) was video-recorded. This was to explore possible observer bias owing to the unblinded nature of the study design. Recordings were obtained for 84 (71%) participants in the levetiracetam-treated group and 71 (62%) participants in the phenytoin-treated group. Although this might have been feasible in a research setting in a few sites, it would not be easily applied to routine clinical practice. It would not have been feasible or pragmatic for each participant to undergo a video-recording or an electroencephalogram (EEG) to determine CSE cessation time more precisely. It is not possible to state definitively and without EEG whether or not any patients may have developed non-CSE following either treatment. However, treatment algorithms for non-CSE generally follow the same flow as CSE, and there was no difference between treatment groups in the number of additional anticonvulsants given after trial treatment. EEGs were not used to determine or confirm seizure cessation in the ConSEPT trial41 or ESTT. 70,71 Third, the time point of randomisation resulted in CSE terminating prior to administration of trial treatment in a number of cases; however, this affected both treatment arms equally, and was essential to maintain high standards of clinical care and avoid treatment delays. Fourth, we included safety measures as key secondary outcomes because of previous reports of harm. However, this trial was not powered to demonstrate difference in SARs (a secondary outcome) between treatment groups, given their low incidence rate. Finally, we considered a superiority design was more appropriate for reasons given above.
Added value of this study
This is an adequately powered RCT that pragmatically and directly compares two anticonvulsants in the second-line treatment of paediatric CSE in an emergency setting. It is also, to the best of our knowledge, the first scientifically-robust clinical trial to compare the efficacy and safety of levetiracetam with phenytoin in this common paediatric neurological emergency. We found no significant differences between the two anticonvulsants in any primary or secondary outcomes, including time to seizure cessation, need for additional anticonvulsants and progression to RSI. The safety profile was similar between both treatments (note that this is in contrast to existing observational evidence that phenytoin appears to have a worse safety profile, including causing a potentially fatal cardiac arrhythmia).
The study comprised a number of challenges, but also opportunities, that might have adversely affected recruitment, including research within a paediatric emergency situation that involved RWPC, which was a new concept to many participating centres. Early input from parents was obtained through pre-trial feasibility43 and CONNECT guidance,46–48 which informed site training to maximise recruitment into the EcLiPSE trial. The nested consent study provides valuable insight from parents and practitioners to inform the design and conduct of future trials in this setting, including a bespoke seven-step framework to optimise PEM trial recruitment discussions. Multiple factors, including trial design, organisation and leadership, were found to both challenge and contribute to trial recruitment and conduct. Early engagement with PERUKI optimised success of the trial through collaboration with clinicians and researchers in the development and delivery of the study, together with the selection of the most appropriate sites in which to recruit patients.
Implications of all the available evidence
The results of the EcLiPSE trial indicate that levetiracetam could be considered as an alternative treatment to phenytoin for the second-line management of paediatric CSE. Recently published RCT data from the ConSEPT trial41 and ESTT70,71 seem to confirm our results and our conclusion.
Additional treatment-related factors may be important to consider and are likely to be relevant to the wider interpretation of our findings. First, levetiracetam is widely used as oral maintenance therapy for many childhood epilepsies because of its broad-spectrum activity and safety profile; this was clearly reflected in the EcLiPSE trial, with levetiracetam being the most commonly used oral antiepileptic drug in all participants on presentation to the ED. By contrast, phenytoin is a rarely used maintenance antiepileptic drug because of its complex pharmacokinetics and potential toxicity. However, despite its rare use, anecdotally, many ED clinicians are reluctant to give a loading dose of phenytoin in CSE to children on oral maintenance phenytoin because of the risk of potential overdosing and the risk of potential cardiovascular toxicity, including a fatal arrhythmia. There seemed to be no similar concerns for levetiracetam, and there was no observed increase in AEs following the i.v. administration of a dose of 40 mg/kg to children already receiving maintenance levetiracetam. In addition, in the levetiracetam-treated participants, blood levels of the drug showed no obvious difference between those who were and were not receiving it as a maintenance oral antiepileptic drug on presentation. Second, a significant minority of children who present in CSE for the first time will be commenced on maintenance therapy prior to discharge. This is more likely to be with levetiracetam than phenytoin because of the latter’s adverse safety profile and unreliable pharmacokinetics. One observational study in adults showed that 8% of patients treated with i.v. fosphenytoin for CSE were subsequently commenced on oral phenytoin, compared with 78% of patients treated with i.v. levetiracetam who were subsequently commenced on oral levetiracetam. 72 Third, ease of drug preparation and administration is also important in the management of CSE.
Throughout the EcLiPSE trial, levetiracetam was reported by all clinical teams to be easier to prepare and administer than phenytoin because of the latter’s calculations performed in reconstituting the drug, the number of vials required and procedures needed for its administration (these observations are supported by the literature20,74).
The majority of participants in the EcLiPSE trial were managed in accordance with the national APLS algorithm, unless the clinical team considered otherwise. For children in whom the second-line anticonvulsant fails to terminate CSE, RSI with thiopentone or another agent is the next step in the algorithm. However, treatment strategies in the management of CSE are evolving. This includes the emerging use of two, rarely three, second-line anticonvulsants in preference to the traditional practice of immediate progression to RSI after failure of the first second-line drug. Clinicians might consider the risks of RSI, and its potential iatrogenic consequences, to be greater than the administration and assessment of a second second-line treatment. In total, 24 out of the 286 participants (8.4%) in the EcLiPSE trial received both treatments sequentially (17 of these were randomised to, and received, levetiracetam first). This could reflect the acceptance of a second second-line treatment being conditional on the amount of time elapsed since CSE-onset.
In the ConSEPT trial,41 if CSE continued following administration of the randomised drug then the alternative drug was given, with further assessment of seizure activity performed 5 minutes after the infusion of the second trial drug was completed. Persisting CSE after the administration of both drugs was subsequently managed by local protocols, all of which advised RSI and intubation. Consequently, 42 participants received phenytoin followed by levetiracetam and 48 participants received levetiracetam followed by phenytoin. Clinical cessation of seizure activity at 2 hours following the administration of the randomised drug only was seen in 62 (54%) participants in the phenytoin-treated group and 61 (51%) participants in the levetiracetam-treated group. However, seizure cessation at 2 hours having received one or both study drugs increased this to 89 (78%) participants in the phenytoin-treated group and 86 (72%) participants in the levetiracetam-treated group. The authors41 concluded that although both drugs failed to terminate CSE in a significant number of patients when given alone, treatment with one drug and followed by the other reduced the failure rate by > 50% at the expense of only an additional 10 minutes (compared with giving phenytoin alone). The authors41 argued that clinicians should, therefore, consider the sequential use of either medication first, before progressing to RSI and intubation. It is understandable that clinicians might consider the risks of RSI and intubation to be greater than the risks of administration and assessment of an additional second-line treatment. However, the administration of two second-line treatments might substantially delay the use of RSI. The ConSEPT trial team suggest that any delay would be < 10 minutes. 41 However, in practice, the preparation and administration of levetiracetam is likely to take > 10 minutes and closer to 15 minutes and phenytoin is likely to take closer to 20 or 25 minutes because of its more complicated preparation. Such a delay would significantly add to the overall period of CSE since its onset and would increase the potential risk of neurological and cognitive impairment.
Chapter 6 Conclusions
To the best of our knowledge, this was the first adequately powered RCT that has directly compared any anticonvulsant in the second-line treatment of paediatric CSE in an emergency setting.
There was no statistical difference between the two anticonvulsants in any primary or secondary outcome. Although the safety profile in the EcLiPSE trial was similar between the two treatments, observational evidence over the past 50 years has indicated that phenytoin may be associated with a worse safety profile, including death caused by cardiac arrhythmias and Stevens–Johnson syndrome. Clearly, the observational data for levetiracetam are small and span approximately 20 years, but no deaths from any cause attributed to this anticonvulsant have been reported during this period.
The results of the EcLiPSE trial suggest that levetiracetam may be considered as an alternative first-choice treatment to phenytoin in the second-line management of paediatric CSE. Additional benefits for levetiracetam compared with phenytoin are its ease of preparation and administration. Recent results from two additional RCTs,41,70,71 in a total of 617 children and adults, would support its potential first-choice position in the second-line management in children with benzodiazepine-resistant CSE.
The possible sequential use of phenytoin followed by levetiracetam, or levetiracetam followed by phenytoin, is important; however, it requires debate and ideally by a multispecialty group that would include PEM, paediatric neurology, paediatric intensive care and general paediatrics.
The EcLiPSE trial has provided new and robust evidence that, together with other recently published RCT data,41,70,71 will help to inform clinicians in their management of paediatric CSE. This evidence will also be assessed by National Institute for Health and Care Excellence in its revised epilepsy guideline that is due to be published in 2022.
Finally, the nested consent study has provided valuable insight into factors that have helped to facilitate the successful conduct of, and recruitment to, a challenging ED-led trial. These factors are relevant for both the families of the trial participants and the practitioners who conducted the trial. Its results will hopefully facilitate future research in paediatric EDs.
Acknowledgements
The EcLiPSE TMG is very grateful to all of the families and staff at the participating sites who agreed to take part in the EcLiPSE trial, and all of the members of the TSC and IDSMC.
The EcLiPSE TMG is grateful to Ms Rachael Kelly, managing editor, and the Cochrane Epilepsy Group, University of Liverpool, Liverpool, UK, for assistance with the literature searches for this report.
Contributions of authors
Richard E Appleton (https://orcid.org/0000-0002-0742-2113) (Consultant Paediatric Neurologist) conceived the study, designed the study and protocol, analysed the data, and prepared and reviewed the manuscript.
Naomi EA Rainford (https://orcid.org/0000-0002-5876-3946) (Trial Statistician, Clinical Trials Research Centre) designed the study and protocol, analysed the data, and prepared and reviewed the manuscript.
Carrol Gamble (https://orcid.org/0000-0002-3021-1955) (Lead Statistician and Head of Biostatistics, Clinical Trials Research Centre) designed the study and protocol, analysed the data, and prepared and reviewed the manuscript.
Shrouk Messahel (https://orcid.org/0000-0003-0645-3070) (Consultant in PEM) designed the study and protocol, and prepared and reviewed the manuscript.
Amy Humphreys (https://orcid.org/0000-0002-5996-2613) (Trial Manager, Clinical Trials Centre) designed the study and protocol, was responsible for trial management, and prepared and reviewed the manuscript.
Helen Hickey (https://orcid.org/0000-0003-0467-0362) (Head of Trials Management, Clinical Trials Research Centre) designed the study and protocol, was responsible for trial management, and prepared and reviewed the manuscript.
Kerry Woolfall (https://orcid.org/0000-0002-5726-5304) (Senior Lecturer, Department of Health Services Research) designed the study and protocol, analysed the data for the nested consent study, and prepared and reviewed the manuscript.
Louise Roper (https://orcid.org/0000-0002-2918-7628) (Chartered Health Psychologist) was responsible for data analysis of the nested consent study, and prepared and reviewed the manuscript.
Joanne Noblet (https://orcid.org/0000-0001-5232-8495) (Senior Sister in Paediatric Emergency Care) designed the study and protocol, and reviewed the manuscript.
Elizabeth Lee (https://orcid.org/0000-0002-2846-2448) (Research Nurse in Paediatric Emergency Care) designed the study and protocol, and reviewed the manuscript.
Sarah Potter (https://orcid.org/0000-0003-1463-279X) (Senior Sister in Paediatric Emergency Care) designed the study and protocol, and reviewed the manuscript.
Paul Tate (https://orcid.org/0000-0002-4246-1409) (Data Manager) was responsible for data collation and data management.
Nadia Al Najjar (https://orcid.org/0000-0002-2851-0913) (Trial Manager) was responsible for trial management.
Anand Iyer (https://orcid.org/0000-0002-3932-4729) (Consultant Paediatric Neurologist) designed the study and protocol, and prepared and reviewed the manuscript.
Vicki Evans (Parent and PPI representative) designed the study and protocol, and reviewed the manuscript.
Mark D Lyttle (https://orcid.org/0000-0002-8634-7210) (Consultant in PEM) designed the study and protocol, and prepared and reviewed the manuscript.
Publications
EcLiPSE Study. Convulsive status epilepticus: an update. Epilepsy Professional 2017;47:14–19.
Lyttle MD, Gamble C, Shrouk M, Hickey H, Iyer A, Woolfall K, et al. Emergency treatment with levetiracetam or phenytoin in status epilepticus in children-the EcLiPSE study: study protocol for a randomised controlled trial. Trials 2017;18:283.
Cook R, Davidson P, Martin R, NIHR Dissemination Centre. Levetiracetam is a useful alternative to phenytoin for epileptic seizures in children. BMJ 2019;367:l5464.
Lyttle MD, Rainford NEA, Gamble C, Messahel S, Humphreys A, Hickey H, et al. Levetiracetam versus phenytoin for second-line treatment of paediatric convulsive status epilepticus (EcLiPSE): a multicentre, open-label, randomised trial. Lancet 2019;393:2125–34.
Woolfall K, Roper L, Humphreys A, Lyttle MD, Messahel S, Lee E, et al. Enhancing practitioners’ confidence in recruitment and consent in the EcLiPSE trial: a mixed-method evaluation of site training – a Paediatric Emergency Research in the United Kingdom and Ireland (PERUKI) study. Trials 2019;20:181.
Roper L, Lyttle MD, Gamble C, Humphreys A, Messahel S, Lee ED, et al. Planning for success: overcoming challenges to recruitment and conduct of an open-label emergency department–led paediatric trial. Emerg Med J 2020;0:1–7.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Lyttle MD, Rainford NEA, Gamble C, Messahel S, Humphreys A, Hickey H, et al. Levetiracetam versus phenytoin for second-line treatment of paediatric convulsive status epilepticus (EcLiPSE): a multicentre, open-label, randomised trial. Lancet 2019;393:2125-34. https://doi.org/10.1016/S0140-6736(19)30724-X.
- Lyttle MD, Gamble C, Messahel S, Hickey H, Iyer A, Woolfall K, et al. Emergency treatment with levetiracetam or phenytoin in status epilepticus in children-the EcLiPSE study: study protocol for a randomised controlled trial. Trials 2017;18. https://doi.org/10.1186/s13063-017-2010-8.
- ISRCTN registry . Emergency Treatment With Levetiracetam or Phenytoin in Status Epilepticus n.d. www.isrctn.com/ISRCTN22567894 (accessed 24 September 2020).
- Novorol CL, Chin RF, Scott RC. Outcome of convulsive status epilepticus: a review. Arch Dis Child 2007;92:948-51. https://doi.org/10.1136/adc.2006.107516.
- Chin RF, Neville BG, Peckham C, Bedford H, Wade A, Scott RC. NLSTEPSS Collaborative Group . Incidence, cause, and short-term outcome of convulsive status epilepticus in childhood: prospective population-based study. Lancet 2006;368:222-9. https://doi.org/10.1016/S0140-6736(06)69043-0.
- PICANet . Annual Report 2010 n.d. www.picanet.org.uk/wp-content/uploads/sites/25/2018/05/PICANet_Annual_Report_2010-1.pdf (accessed 18 October 2020).
- Metsäranta P, Koivikko M, Peltola J, Eriksson K. Outcome after prolonged convulsive seizures in 186 children: low morbidity, no mortality. Dev Med Child Neurol 2004;46:4-8. https://doi.org/10.1017/s0012162204000027.
- Chin RF, Verhulst L, Neville BG, Peters MJ, Scott RC. Inappropriate emergency management of status epilepticus in children contributes to need for intensive care. J Neurol Neurosurg Psychiatry 2004;75:1584-8. https://doi.org/10.1136/jnnp.2003.032797.
- Hussain N, Appleton R, Thorburn K. Aetiology, course and outcome of children admitted to paediatric intensive care with convulsive status epilepticus: a retrospective 5-year review. Seizure 2007;16:305-12. https://doi.org/10.1016/j.seizure.2007.01.007.
- Eriksson K, Metsäranta P, Huhtala H, Auvinen A, Kuusela AL, Koivikko M. Treatment delay and the risk of prolonged status epilepticus. Neurology 2005;65:1316-18. https://doi.org/10.1212/01.wnl.0000180959.31355.92.
- Advanced Life Support Group . A Practical Approach to Emergencies (APLS) 6th Edition n.d. www.alsg.org/home/mod/data/view.php?d=12%26rid=143 (accessed 30 October 2019).
- McTague A, Martland T, Appleton R. Drug management for acute tonic–clonic convulsions including convulsive status epilepticus in children. Cochrane Database Syst Rev 2018;1. https://doi.org/10.1002/14651858.CD001905.pub3.
- Brigo F, Bragazzi N, Nardone R, Trinka E. Direct and indirect comparison meta-analysis of levetiracetam versus phenytoin or valproate for convulsive status epilepticus. Epilepsy Behav 2016;64:110-15. https://doi.org/10.1016/j.yebeh.2016.09.030.
- Lewena S, Pennington V, Acworth J, Thornton S, Ngo P, McIntyre S, et al. Emergency management of pediatric convulsive status epilepticus: a multicenter study of 542 patients. Pediatr Emerg Care 2009;25:83-7. https://doi.org/10.1097/PEC.0b013e318196ea6e.
- Appleton RE, Gill A. Adverse events associated with intravenous phenytoin in children: a prospective study. Seizure 2003;12:369-72. https://doi.org/10.1016/S1059-1311(02)00338-2.
- Craig S. Phenytoin poisoning. Neurocrit Care 2005;3:161-70. https://doi.org/10.1385/NCC:3:2:161.
- Gallop K. Review article: phenytoin use and efficacy in the ED. Emerg Med Australas 2010;22:108-18. https://doi.org/10.1111/j.1742-6723.2010.01269.x.
- NHS Improvement . Risk of Death and Severe Harm From Error With Injectable Phenytoin n.d. https://improvement.nhs.uk/news-alerts/risk-death-and-severe-harm-error-injectable-phenytoin/ (accessed 30 October 2019).
- Berning S, Boesebeck F, van Baalen A, Kellinghaus C. Intravenous levetiracetam as treatment for status epilepticus. J Neurol 2009;256:1634-42. https://doi.org/10.1007/s00415-009-5166-7.
- Trinka E, Dobesberger J. New treatment options in status epilepticus: a critical review on intravenous levetiracetam. Ther Adv Neurol Disord 2009;2:79-91. https://doi.org/10.1177/1756285608100460.
- Knake S, Gruener J, Hattemer K, Klein KM, Bauer S, Oertel WH, et al. Intravenous levetiracetam in the treatment of benzodiazepine refractory status epilepticus. J Neurol Neurosurg Psychiatry 2008;79:588-9. https://doi.org/10.1136/jnnp.2007.130260.
- Michaelides C, Thibert RL, Shapiro MJ, Kinirons P, John T, Manchharam D, et al. Tolerability and dosing experience of intravenous levetiracetam in children and infants. Epilepsy Res 2008;81:143-7. https://doi.org/10.1016/j.eplepsyres.2008.05.004.
- Rüegg S, Naegelin Y, Hardmeier M, Winkler DT, Marsch S, Fuhr P. Intravenous levetiracetam: treatment experience with the first 50 critically ill patients. Epilepsy Behav 2008;12:477-80. https://doi.org/10.1016/j.yebeh.2008.01.004.
- Wheless JW, Clarke D, Hovinga CA, Ellis M, Durmeier M, McGregor A, et al. Rapid infusion of a loading dose of intravenous levetiracetam with minimal dilution: a safety study. J Child Neurol 2009;24:946-51. https://doi.org/10.1177/0883073808331351.
- Kirmani BF, Crisp ED, Kayani S, Rajab H. Role of intravenous levetiracetam in acute seizure management of children. Pediatr Neurol 2009;41:37-9. https://doi.org/10.1016/j.pediatrneurol.2009.02.016.
- McTague A, Kneen R, Kumar R, Spinty S, Appleton R. Intravenous levetiracetam in acute repetitive seizures and status epilepticus in children: experience from a children’s hospital. Seizure 2012;21:529-34. https://doi.org/10.1016/j.seizure.2012.05.010.
- Prasad K, Al-Roomi K, Krishnan PR, Sequeira R. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev 2005;4. https://doi.org/10.1002/14651858.CD003723.pub2.
- Misra UK, Kalita J, Maurya PK. Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. J Neurol 2012;259:645-8. https://doi.org/10.1007/s00415-011-6227-2.
- Zelano J, Kumlien E. Levetiracetam as alternative stage two antiepileptic drug in status epilepticus: a systematic review. Seizure 2012;21:233-6. https://doi.org/10.1016/j.seizure.2012.01.008.
- Chakravarthi S, Goyal MK, Modi M, Bhalla A, Singh P. Levetiracetam versus phenytoin in management of status epilepticus. J Clin Neurosci 2015;22:959-63. https://doi.org/10.1016/j.jocn.2014.12.013.
- Mundlamuri RC, Sinha S, Subbakrishna DK, Prathyusha PV, Nagappa M, Bindu PS, et al. Management of generalised convulsive status epilepticus (SE): a prospective randomised controlled study of combined treatment with intravenous lorazepam with either phenytoin, sodium valproate or levetiracetam – pilot study. Epilepsy Res 2015;114:52-8. https://doi.org/10.1016/j.eplepsyres.2015.04.013.
- Wright C, Downing J, Mungall D, Khan O, Williams A, Fonkem E, et al. Clinical pharmacology and pharmacokinetics of levetiracetam. Front Neurol 2013;4. https://doi.org/10.3389/fneur.2013.00192.
- Ramael S, Daoust A, Otoul C, Toublanc N, Troenaru M, Lu ZS, et al. Levetiracetam intravenous infusion: a randomized, placebo-controlled safety and pharmacokinetic study. Epilepsia 2006;47:1128-35. https://doi.org/10.1111/j.1528-1167.2006.00586.x.
- Szaflarski JP, Sangha KS, Lindsell CJ, Shutter LA. Prospective, randomized, single-blinded comparative trial of intravenous levetiracetam versus phenytoin for seizure prophylaxis. Neurocrit Care 2010;12:165-72. https://doi.org/10.1007/s12028-009-9304-y.
- Hirsch LJ. Levitating levetiracetam’s status for status epilepticus. Epilepsy Curr 2008;8:125-6. https://doi.org/10.1111/j.1535-7511.2008.00266.x.
- Yasiry Z, Shorvon SD. The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: a meta-analysis of published studies. Seizure 2014;23:167-74. https://doi.org/10.1016/j.seizure.2013.12.007.
- Lyttle MD, O’Sullivan R, Hartshorn S, Bevan C, Cleugh F, Maconochie I. PERUKI . Pediatric Emergency Research in the UK and Ireland (PERUKI): developing a collaborative for multicentre research. Arch Dis Child 2014;99:602-3. https://doi.org/10.1136/archdischild-2013-304998.
- Hartshorn S, O’Sullivan R, Maconochie IK, Bevan C, Cleugh F, Lyttle MD. Paediatric Emergency Research in the UK and Ireland (PERUKI) . Establishing the research priorities of paediatric emergency medicine clinicians in the UK and Ireland. Emerg Med J 2015;32:864-8. https://doi.org/10.1136/emermed-2014-204484.
- National Institute for Health and Care Excellence . Epilepsies: Diagnosis and Management n.d. www.nice.org.uk/guidance/cg137 (accessed 30 October 2019).
- Dalziel SR, Furyk J, Bonisch M, Oakley E, Borland M, Neutze J, et al. A multicentre randomised controlled trial of levetiracetam versus phenytoin for convulsive status epilepticus in children (protocol): Convulsive Status Epilepticus Paediatric Trial (ConSEPT) – a PREDICT study. BMC Pediatr 2017;17. https://doi.org/10.1186/s12887-017-0887-8.
- Dalziel SR, Borland ML, Furyk J, Bonisch M, Neutze J, Donath S, et al. Levetiracetam versus phenytoin for second-line treatment of convulsive status epilepticus in children (ConSEPT): an open-label, multicentre, randomised controlled trial. Lancet 2019;393:2135-45. https://doi.org/10.1016/S0140-6736(19)30722-6.
- Bleck T, Cock H, Chamberlain J, Cloyd J, Connor J, Elm J, et al. The established status epilepticus trial 2013. Epilepsia 2013;54:89-92. https://doi.org/10.1111/epi.12288.
- Woolfall K, Young B, Frith L, Appleton R, Iyer A, Messahel S, et al. Doing challenging research studies in a patient-centred way: a qualitative study to inform a randomised controlled trial in the paediatric emergency care setting. BMJ Open 2014;4. https://doi.org/10.1136/bmjopen-2014-005045.
- Woolfall K, Roper L, Humphreys A, Lyttle MD, Messahel S, Lee E, et al. Enhancing practitioners’ confidence in recruitment and consent in the EcLiPSE trial: a mixed-method evaluation of site training – a Paediatric Emergency Research in the United Kingdom and Ireland (PERUKI) study. Trials 2019;20. https://doi.org/10.1186/s13063-019-3273-z.
- Piper JD, Hawcutt DB, Verghese GK, Spinty S, Newland P, Appleton R. Phenytoin dosing and serum concentrations in paediatric patients requiring 20 mg/kg intravenous loading. Arch Dis Child 2014;99:585-6. https://doi.org/10.1136/archdischild-2013-305093.
- Woolfall K, Gamble C, Frith L, Young B. the CONNECT Advisory Group . Research Without Prior Consent (Deferred Consent) in Trials Investigating the Emergency Treatment of Critically Ill Children: CONNECT Study Guidance Version 2.0 n.d. www.liverpool.ac.uk/psychology-health-and-society/research/connect/ (accessed 14 December 2018).
- Woolfall K, Frith L, Gamble C, Gilbert R, Mok Q, Young B. CONNECT Advisory Group . How parents and practitioners experience research without prior consent (deferred consent) for emergency research involving children with life threatening conditions: a mixed method study. BMJ Open 2015;5. https://doi.org/10.1136/bmjopen-2015-008522.
- Woolfall K, Frith L, Dawson A, Gamble C, Lyttle MD, . the CONNECT Advisory Group . 15 minute consultation: an evidence-based approach to research without prior consent (deferred consent) in neonatal and paediatric critical care trials. Arch Dis Child Educ Pract Ed 2015;101:49-53. https://doi.org/10.1136/archdischild-2015-309245.
- Woolfall K, Frith L, Dawson A, Gamble C, Young B. Research Without Prior Consent (Deferred Consent) in Trials Investigating the Emergency Treatment of Critically Ill Children: CONNECT Study Guidance. Liverpool: University of Liverpool; 2015.
- Cresswell JW, Plano Clark VL. Designing and Conducting Mixed Methods Research. London: SAGE Publications Ltd; 2007.
- Johnstone PL. Mixed methods, mixed methodology health services research in practice. Qual Health Res 2004;14:259-71. https://doi.org/10.1177/1049732303260610.
- O’Brien BC, Harris IB, Beckman TJ, Reed DA, Cook DA. Standards for reporting qualitative research: a synthesis of recommendations. Acad Med 2014;89:1245-51. https://doi.org/10.1097/ACM.0000000000000388.
- Strauss A, Corbin J. Basics of Qualitative Research: Techniques and Procedures for Developing Grounded Theory. Thousand Oaks, CA: SAGE Publications Ltd; 1998.
- Glaser B. The constant comparative method of qualitative analysis. Soc Probl 1965;12:436-45. https://doi.org/10.2307/798843.
- Saunders B, Sim J, Kingstone T, Baker S, Waterfield J, Bartlam B, et al. Saturation in qualitative research: exploring its conceptualization and operationalization. Qual Quant 2018;52:1893-907. https://doi.org/10.1007/s11135-017-0574-8.
- Inwald DP, Canter R, Woolfall K, Mouncy P, Zenasni Z, O’Hara C, et al. Restricted fluid bolus volume in early septic shock: results of the Fluids in Shock pilot trial. Arch Dis Child 2019;104:426-31. https://doi.org/10.1136/archdischild-2018-314924.
- Realpe A, Adams A, Wall P, Griffin D, Donovan JL. A new simple six-step model to promote recruitment to RCTs was developed and successfully implemented. J Clin Epidemiol 2016;76:166-74. https://doi.org/10.1016/j.jclinepi.2016.02.002.
- Shilling V, Williamson PR, Hickey H, Sowden E, Beresford MW, Smyth RL, et al. Communication about children’s clinical trials as observed and experienced: qualitative study of parents and practitioners. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0021604.
- Behrendt C, Gölz T, Roesler C, Bertz H, Wünsch A. What do our patients understand about their trial participation? Assessing patients’ understanding of their informed consent consultation about randomised clinical trials. J Med Ethics 2011;37:74-80. https://doi.org/10.1136/jme.2010.035485.
- Featherstone K, Donovan JL. ‘Why don’t they just tell me straight, why allocate it?’ The struggle to make sense of participating in a randomised controlled trial. Soc Sci Med 2002;55:709-19. https://doi.org/10.1016/s0277-9536(01)00197-6.
- Appelbaum PS, Roth LH, Lidz CW, Benson P, Winslade W. False hopes and best data: consent to research and the therapeutic misconception. Hastings Cent Rep 1987;17:20-4. https://doi.org/10.2307/3562038.
- Fallowfield L, Langridge C, Jenkins V. Communication skills training for breast cancer teams talking about trials. Breast 2014;23:193-7. https://doi.org/10.1016/j.breast.2013.11.009.
- Jenkins V, Fallowfield L, Solis-Trapala I, Langridge C, Farewell V. Discussing randomised clinical trials of cancer therapy: evaluation of a Cancer Research UK training programme. BMJ 2005;330. https://doi.org/10.1136/bmj.38366.562685.8F.
- Reay H, Arulkumaran N, Brett SJ. Priorities for future intensive care research in the UK: results of a James Lind Alliance priority setting partnership. J Intensive Care Soc 2014;15:288-96. https://doi.org/10.1177/175114371401500405.
- Petit-Zeman S, Firkins L, Scadding JW. The James Lind Alliance: tackling research mismatches. Lancet 2010;376:667-9. https://doi.org/10.1016/S0140-6736(10)60712-X.
- Peters MJ, Woolfall K, Khan I, Deja E, Mouncey PR, Wulff J, et al. Permissive versus restrictive temperature thresholds in critically ill children with fever and infection: a multicentre randomized clinical pilot trial. Crit Care 2019;23. https://doi.org/10.1186/s13054-019-2354-4.
- Wade J, Donovan JL, Athene Lane J, Neal DE, Hamdy FC. It’s not just what you say, it’s also how you say it: opening the ‘black box’ of informed consent appointments in randomised controlled trials. Soc Sci Med 2009;68:2018-28. https://doi.org/10.1016/j.socscimed.2009.02.023.
- Mills N, Donovan JL, Wade J, Hamdy FC, Neal DE, Lane JA. Exploring treatment preferences facilitated recruitment to randomized controlled trials. J Clin Epidemiol 2011;64:1127-36. https://doi.org/10.1016/j.jclinepi.2010.12.017.
- Tong A, Sainsbury P, Craig J. Consolidated criteria for reporting qualitative research (COREQ): a 32-item checklist for interviews and focus groups. Int J Qual Health Care 2007;19:349-57. https://doi.org/10.1093/intqhc/mzm042.
- Kapur J, Elm J, Chamberlain JM, Barsan W, Cloyd J, Lowenstein D, et al. Randomized trial of three anticonvulsant medications for status epilepticus. N Engl J Med 2019;381:2103-13. https://doi.org/10.1056/NEJMoa1905795.
- Chamberlain JM, Kapur J, Shinnar S, Elm J, Holsti M, Babcock L, et al. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): a double-blind, responsive-adaptive, randomised controlled trial. Lancet 2020;395:1217-24. https://doi.org/10.1016/S0140-6736(20)30611-5.
- Singh K, Aggarwal A, Faridi MMA, Sharma S. IV Levetiracetam versus IV phenytoin in childhood seizures: a randomized controlled trial. J Pediatr Neurosci 2018;13:158-64. https://doi.org/10.4103/jpn.JPN_126_17.
- Nakamura K, Inokuchi R, Daidoji H, Naraba H, Sonoo T, Hashimoto H, et al. Efficacy of levetiracetam versus fosphenytoin for the recurrence of seizures after status epilepticus. Medicine 2017;96. https://doi.org/10.1097/MD.0000000000007206.
- Senthilkumar CS, Selvakumar P, Kowsik M. Randomized controlled trial of levetiracetam versus fosphenytoin for convulsive status epilepticus in children. Int J Pediatr Res 2018;5:237-42. https://doi.org/10.17511/ijpr.2018.i04.13.
- McIntyre J, Robertson S, Norris E, Appleton R, Whitehouse WP, Phillips B, et al. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet 2005;366:205-10. https://doi.org/10.1016/S0140-6736(05)66909-7.
Appendix 1 Trial oversight committees
Trial Steering Committee
The independent members of the TSC were as follows: Professor Imti Choonara (chairperson), Professor Caroline Doré, Dr Michael Barrett, Dr Ronan O’Sullivan, Gail Miller, Jane Morgan and Denise Bustansy.
The non-independent member of the TSC was Professor (Honours) Richard Appleton.
Independent Data and Safety Monitoring Committee
Professor Steff Lewis (chairperson), Dr Julie Ellison and Professor Tony Marson.
Trial Management Group
Dr Richard Appleton, Dr Mark Lyttle, Professor Carrol Gamble, Helen Hickey, Amy Humphreys, Nadia Al-Najjar, Naomi Rainford (née: Bacon), Dr Kerry Woolfall, Louise Roper, Victoria Evans, Dr Shrouk Messahel, Dr Anand Iyer, Joanna Noblet, Elizabeth Lee, Rachel Greenwood-Bibby, Tracey Bingham, Alice Smith, Sarah Potter, Phoebe Moulsdale, Holly Lavigne-Smith, Lucy Cooper, Emma Johnston and Dr Mary Ryan.
Appendix 2 Trial management team
The Liverpool Clinical Trials Centre (previously known as the Clinical Trials Research Centre), University of Liverpool, conducted all trial management.
Statistical lead
Professor Carrol Gamble.
Senior trial manager
Helen Hickey.
Senior data managers
Clare Jackson and Sue Howlin.
Trial co-ordinators
Amy Humphreys and Nadia Al-Najjar.
Statisticians, data managers and database developers
Statisticians
Naomi Rainford (née Bacon: trial statistician).
Randomisation team
Ashley Jones and Anna Rosala-Hallas.
Quality control
Ashley Best.
Database development and Information Systems quality control
Janet Harrison and Linda Kane.
Data manager
Paul Tate.
Appendix 3 Recruiting centres in centre number order
| Participating trust | Investigator |
|---|---|
| Royal Devon and Exeter NHS Foundation Trust |
Dr Rachel Howells, Consultant Paediatrician (PI) Dr Andy Appelboam, Consultant EM (co-PI) Su Wilkins, Clinical RN |
| South Tees Hospitals NHS Foundation Trust |
Dr Ramesh Kumar, Consultant Paediatrician (PI) Dr Alex Scott, Consultant EM (co-investigator) |
| Cambridge University Hospitals NHS Foundation Trust |
Dr Matthew Pereira, Consultant EM (PI) Dr Khurram Iftikhar, Consultant EM (previous PI) Susie Hardwick, ED RN |
| University Hospitals of Leicester NHS Trust | Dr Damian Roland, Consultant PEM (PI) |
| University Hospitals Bristol and Weston NHS Foundation Trust |
Dr Mark Lyttle, Consultant PEM (PI) Sarah Potter, RN Phoebe Moulsdale, RN Holly Lavigne-Smith, RN Alice Smith, Clinical RN Pauline Jackson, Clinical Paediatric RN |
| Birmingham Children’s Hospital NHS Foundation Trust |
Dr Stuart Hartshorn, Consultant PEM (PI) Louise Rogers, Clinical Research Sister Juliet Hopkins, Junior Sister Clinical Research |
| The Royal London Hospital |
Dr Ami Parikh, Consultant PEM (PI) Olivia Boulton, ED RN |
| Western Sussex Hospitals NHS Foundation Trust |
Dr Mike Linney, Consultant Paediatrician (PI) Katia Vamvakiti, Consultant Paediatrician (co-PI) Sharon Floyd, Paediatric and Neonatal RN Gillian Hobden, Paediatric RN |
| St George’s University Hospitals NHS Foundation Trust |
Dr Yasser Iqbal, ED Consultant (PI) Sarah Rounding, Senior Clinical RN |
| King’s College Hospital NHS Foundation Trust |
Dr Emer Sutherland, Consultant Emergency Physician (PI) Sinead Helyar, Senior ACET RN |
| City Hospitals Sunderland NHS Foundation Trust |
Dr Niall Mullen, Consultant PEM (PI) Dr Madhuri Dasarathi, Consultant Paediatrician (co-PI) Paul Corrigan, RN |
| Nottingham University Hospitals NHS Trust |
Dr Christopher Gough, Consultant EM (PI) Sonya Finucane, RN |
| Alder Hey Children’s NHS Foundation Trust |
Dr Shrouk Messahel, Consultant PEM (PI) Jo Noblet, Senior AED Sister Liz Lee, RN Rachel Greenwood-Bibby, RN |
| Central Manchester University Hospitals NHS Foundation Trust | Dr Katherine Potier, Consultant EM (PI) |
| Sheffield Children’s NHS Foundation Trust |
Dr Derek Burke, Consultant PEM (PI) Dr Shammi Ramlakhan, Consultant PEM (previous PI) Jayne Evans, RN Julie Morcombe, RN |
| University Hospital Lewisham |
Dr Tina Sajjanhar, Consultant PEM (PI) Dr Maggie Nyirenda Nyang’wa, Paediatrician (co-PI) |
| Watford General Hospital |
Dr Chaniyil Ramesh, Consultant Paediatrician (PI) Solomon Kamal-Uddin, Consultant PEM (co-PI) |
| University Hospital of Wales |
Dr Jeff Morgan, Consultant PEM (PI) Dr Sara Edwards, Consultant PEM (previous PI) |
| NHS Ayrshire and Arran |
Dr Joanna Mulligan, Consultant EM (PI) Claire Bell, Paediatric RN |
| NHS Greater Glasgow and Clyde |
Dr Vincent Choudhery, Consultant PEM (PI) Stewart MacLeod, Consultant Paediatric Neurologist (co-PI) Ashleigh Neil, Senior RN |
| NHS Lothian |
Dr Jen Browning, Consultant EM (PI) Julia Grahamslaw, RN |
| Royal Belfast Hospital for Sick Children |
Dr Julie-Ann Maney, Consultant PEM (PI) Dr Elizabeth Dalzell, Consultant PEM (co-PI) Muriel Millar, Children’s RN |
| Derby Teaching Hospitals NHS Foundation Trust |
Dr Hani Faza, Consultant Paediatrician (PI) Dr Gisela Robinson, Consultant PEM (co-PI) Dr Rachel Sunley, Senior Trainee PEM (previous Co-PI) Coral Smith, RN |
| Guy’s & St Thomas’ NHS Foundation Trust | Dr John Criddle, Consultant PEM (PI) |
| University Hospital Southampton NHS Foundation Trust |
Dr Jason Barling, Consultant PEM (PI) Ruth Ensom, Senior Paediatric RN |
| Brighton and Sussex University Hospitals NHS Trust |
Dr Catherine Bevan, Paediatric Consultant (PI) Rebecca Ramsay, Senior RN |
| Great North Children’s Hospital |
Dr Mark Anderson, Consultant Paediatrician (PI) Kirsty Devine, Paediatric RN |
| Wirral University Teaching Hospital NHS Foundation Trust |
Dr Mark Buchanan, Consultant EM (PI) Sharon Hughes, RN |
| Chelsea and Westminster Healthcare NHS Foundation Trust |
Dr James Ross, Consultant PEM (PI) Dr Jo Hacking, Consultant PEM (previous PI) Dr Sara Edwards, Senior Registrar PEM (previous PI) Natasha Ramsay, Paediatric Nurse Practitioner |
| Leeds General Infirmary |
Dr Alice Downes, Consultant PEM (PI) Dr Helen Mollard, Consultant EM (previous PI) Nicola Balatoni, Research Sister |
Appendix 4 Changes to the protocol
Summary of amendments from protocol v1.0 (13 January 2015) to protocol v2.0 (23 April 2015)
| Protocol section | Summary of changes |
|---|---|
| Protocol summary |
|
| Primary end point |
|
| Internal pilot |
|
| Inclusion criteria |
|
| Exclusion criteria |
|
| Screening |
|
| Randomisation |
|
| Patients randomised but not treated with a second-line treatment in the ED |
|
| Trial treatments introduction |
|
| Preparation, dosage and administration of levetiracetam |
|
| Accountability procedures for study treatments |
|
| Assessment of compliance with study treatments |
|
| Co-enrolment guidelines |
|
| Schedule for follow-up |
|
| Trial assessments |
|
| Seizure activity |
|
| Procedures for assessing safety |
|
| Blood samples |
|
| Consent study |
|
| Part A: audio-recordings |
|
| Sample size estimate |
|
| Notes on AE recording timelines |
|
| Flow chart for reporting requirements of AEs |
|
| Reporting of overdose |
|
| CRFs |
|
| N/A |
|
Summary of amendments from protocol v2.0 (23 April 2015) to protocol v3.0 (17 June 2015)
| Protocol section | Summary of changes |
|---|---|
| Trial treatments introduction |
|
| Preparation, dosage and administration of levetiracetam |
|
| Preparation, dosage and administration of phenytoin |
|
Summary of amendments from protocol v3.0 (17 June 2015) to protocol v4.0 (27 august 2015)
| Protocol section | Summary of changes |
|---|---|
| N/A |
|
| Protocol summary: schematic study design |
|
| Trial treatments introduction |
|
| Preparation, dosage and administration of levetiracetam |
|
| Preparation, dosage and administration of phenytoin |
|
| Part B: parent/legal representative questionnaires |
|
| Part C: interviews |
|
| N/A |
|
Summary of amendments from protocol v4.0 (27 August 2015) to protocol v5.0 (5 April 2017)
| Protocol section | Summary of changes |
|---|---|
| Contact details |
|
| Protocol summary |
|
| Randomisation |
|
| Consent/assent form completion |
|
| Definitions of legal representatives |
|
| Death prior to deferred consent being sought |
|
| Discharge/transfer prior to deferred consent being sought |
|
| Patients randomised but not treated with second-line treatment in the ED or second-line treatment administered after RSI/seizure stop time |
|
| Deferred consent declined |
|
| Participant transfers |
|
| Trial treatments: introduction |
|
| Arm A: levetiracetam – formulation |
|
| Arm B: phenytoin – formulation |
|
| Schedule for follow-up |
|
| 14-day follow-up questionnaire |
|
| Blood samples |
|
| Consent study |
|
| Urgent safety measures |
|
| Protocol deviation and serious breaches |
|
| Records retention |
|
| N/A |
|
Appendix 5 Patient and public involvement
The lead PPI contributor was a mother of a child with a severe form of epilepsy caused by a genetic disorder. In the first 6 years of her life, her daughter had experienced frequent episodes of CSE that had resulted in numerous attendances in the ED or hospital admissions. It was considered that she would be a valuable member of the team and was involved throughout the development of the grant application and included as a co-applicant.
Aims
The key aims of her involvement were to comment on the clinical relevance of the subject of the trial and to provide input on those methodologies that related to patient-recruitment and RWPC, including the patient and participation information leaflets and the process for obtaining consent post discharge.
Methodology
The methodology focused on the following.
-
Providing input on the terminology and practice of ‘deferred consent’ (i.e. RWPC). This included advice on the content and structure of site training for the simulated interview of taking informed deferred consent from families.
-
A review of the parent/patient information leaflets and covering letters.
-
Attendance at the TMG meetings, which were predominantly undertaken by telephone conference.
Study results
The lead PPI attended TMG meetings at which the results of the EcLiPSE trial and the nested consent study were discussed.
Reflections
The PPI agreed that the trial was important and felt that the primary and secondary outcomes were appropriate and relevant. She felt that she had a specific value in guiding other members of the TMG in the potential consequences of seeking deferred consent in an emergency and emotionally sensitive situation. This included invaluable comments in how to best communicate with the parents of a child who had died and had been recruited into the EcLiPSE trial. She was concerned about two issues over her involvement. First, that she was unable to participate in many of the regular TMG meetings, primarily because of their dates and times, which clashed with her work responsibilities and childcare. Second, because of the specialised pharmacological aspects of the trial, which at times she felt were somewhat ‘over her head’, despite explanations provided by the chief investigator and clinical members of the TMG.
Report writing, academic paper preparation and dissemination
The PPI was not actively involved in the writing of the protocol (published in the online journal Trials2), but did comment on the final draft prior to its publication. Her input was similar in the definitive results paper (published online in The Lancet1). She also contributed to the final report to the Health Technology Assessment programme, with her focus being on the Plain English summary and preparation of the end-of-study results information sheet for all participating sites and their teams. She will continue to be involved in dissemination activities and preparation of academic papers.
Appendix 6 Primary outcome proportionality assumption
All children
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.2 (0.91 to 1.6) | 0.2 |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.6100 | 1000 | 0.7200 |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.6207 | 1000 | 0.7100 |
| Sex: female vs. male | 0.9470 | 1000 | 0.2520 |
| Weight | 1.8151 | 1000 | 0.0050 |
| First seizure: no vs. yes | 0.9665 | 1000 | 0.2670 |
Children with a weight of < 12 kg
FIGURE 8.
Kaplan–Meier for children with a weight of < 12 kg. Product-limit survival estimates with number of subjects at risk.
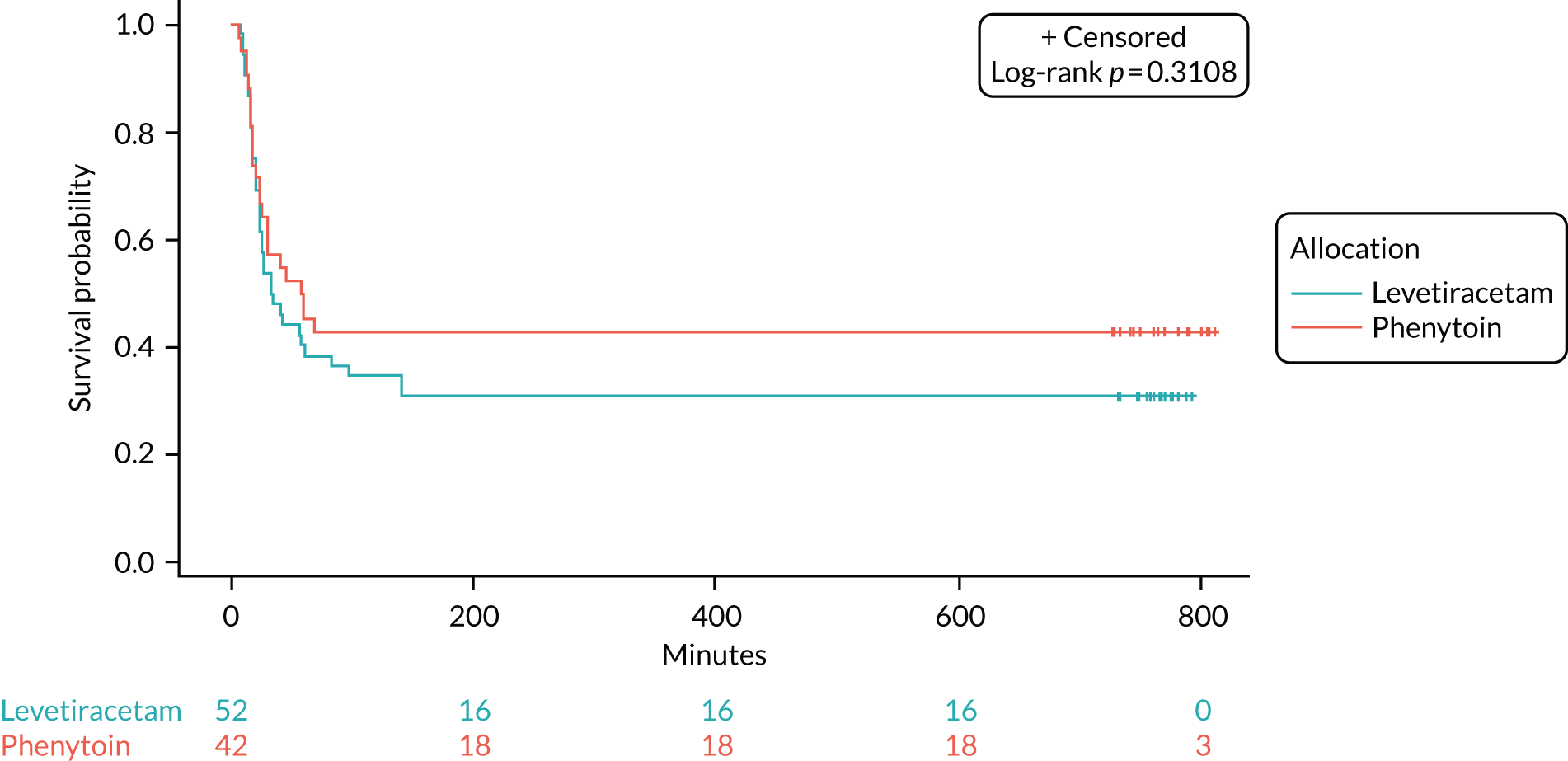
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.4659 | 1000 | 0.8870 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.301 (0.776 to 2.181) | 0.3185 |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.4595 | 1000 | 0.9040 |
| Sex: female vs. male | 1.0542 | 1000 | 0.1410 |
| Weight | 1.2529 | 1000 | 0.1400 |
| First seizure: no vs. yes | 0.8390 | 1000 | 0.2930 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.250 (0.737 to 2.120) | 0.4087 |
| Sex: female vs. male | 1.020 (0.599 to 1.736) | 0.9418 |
| Weight | 1.172 (0.873 to 1.573) | 0.2910 |
| First seizure: no vs. yes | 1.495 (0.862 to 2.593) | 0.1526 |
Children with a weight between 12 and 36 kg
FIGURE 9.
Kaplan–Meier for children with a weight between 12 and 36 kg. Product-limit survival estimates with number of subjects at risk.
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.7902 | 1000 | 0.3950 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.081 (0.748 to 1.561) | 0.6791 |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.8037 | 1000 | 0.3870 |
| Sex: female vs. male | 0.7263 | 1000 | 0.5030 |
| Weight | 0.9563 | 1000 | 0.2280 |
| First seizure: no vs. yes | 0.7400 | 1000 | 0.5240 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.080 (0.742 to 1.571) | 0.6886 |
| Sex: female vs. male | 1.036 (0.710 to 1.511) | 0.8540 |
| Weight | 0.998 (0.968 to 1.029) | 0.9047 |
| First seizure: no vs. yes | 0.960 (0.629 to 1.465) | 0.8497 |
FIGURE 10.
Kaplan–Meier for children with a weight of > 36 kg. Product-limit survival estimates with number of subjects at risk.
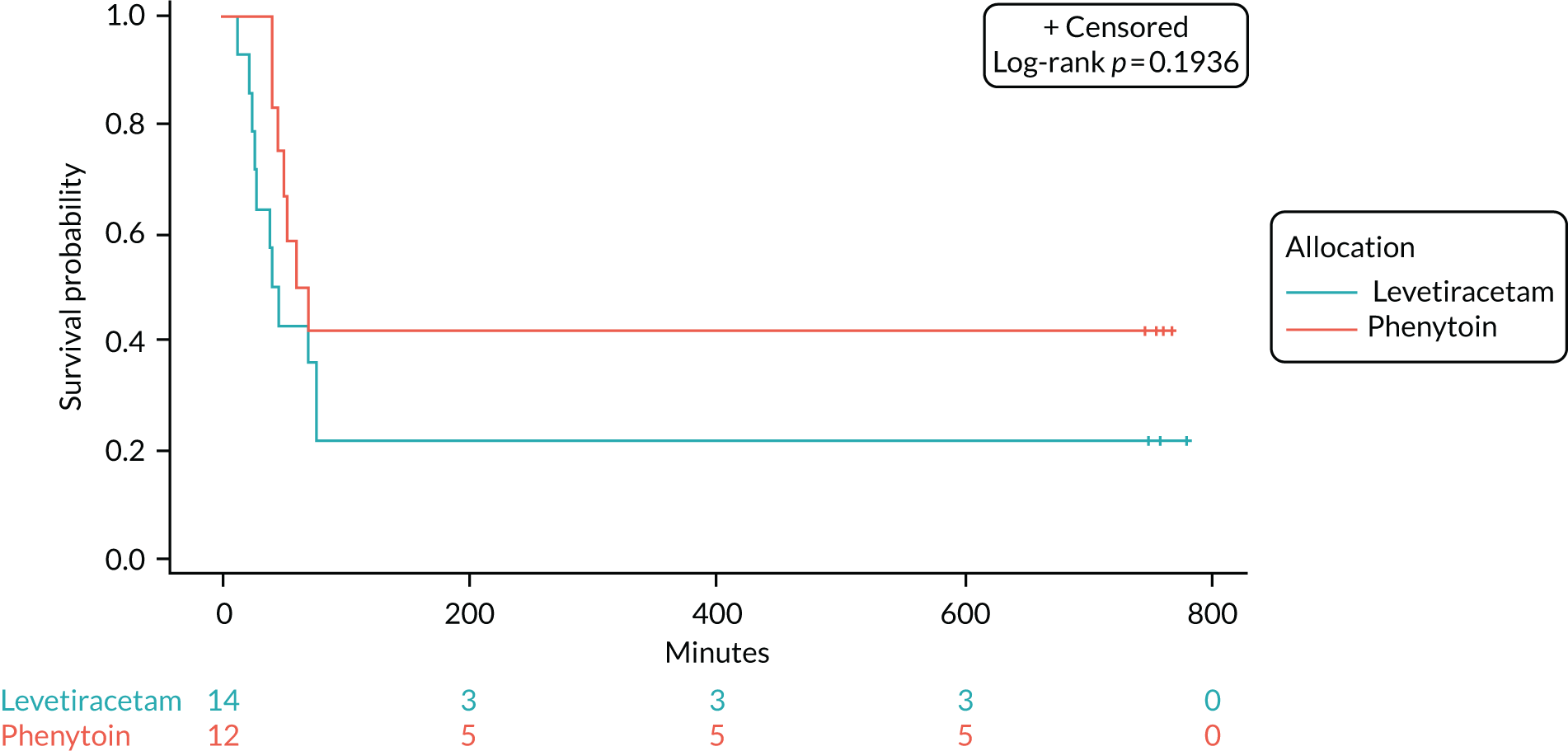
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 1.0623 | 1000 | 0.0910 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.845 (0.712 to 4.781) | 0.2072 |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 1.1107 | 1000 | 0.0860 |
| Sex: female vs. male | 0.8301 | 1000 | 0.5380 |
| Weight | 0.7626 | 1000 | 0.6220 |
| First seizure: no vs. yes | 0.8002 | 1000 | 0.2320 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.690 (0.621 to 4.599) | 0.3043 |
| Sex: female vs. male | 0.934 (0.276 to 3.162) | 0.9128 |
| Weight | 1.039 (0.977 to 1.104) | 0.2201 |
| First seizure: no vs. yes | 2.084 (0.455 to 9.538) | 0.3443 |
Appendix 7 Primary outcome sensitivity analyses
Time to seizure cessation using Gray’s test for competing risks
| Event | Group, n (%) | |
|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | |
| Number of events of interest (seizure cessation) | 106 (69.74) | 86 (64.18) |
| Number of competing events (RSI) | 46 (30.26) | 48 (35.82) |
FIGURE 11.
Cumulative incidence plot.
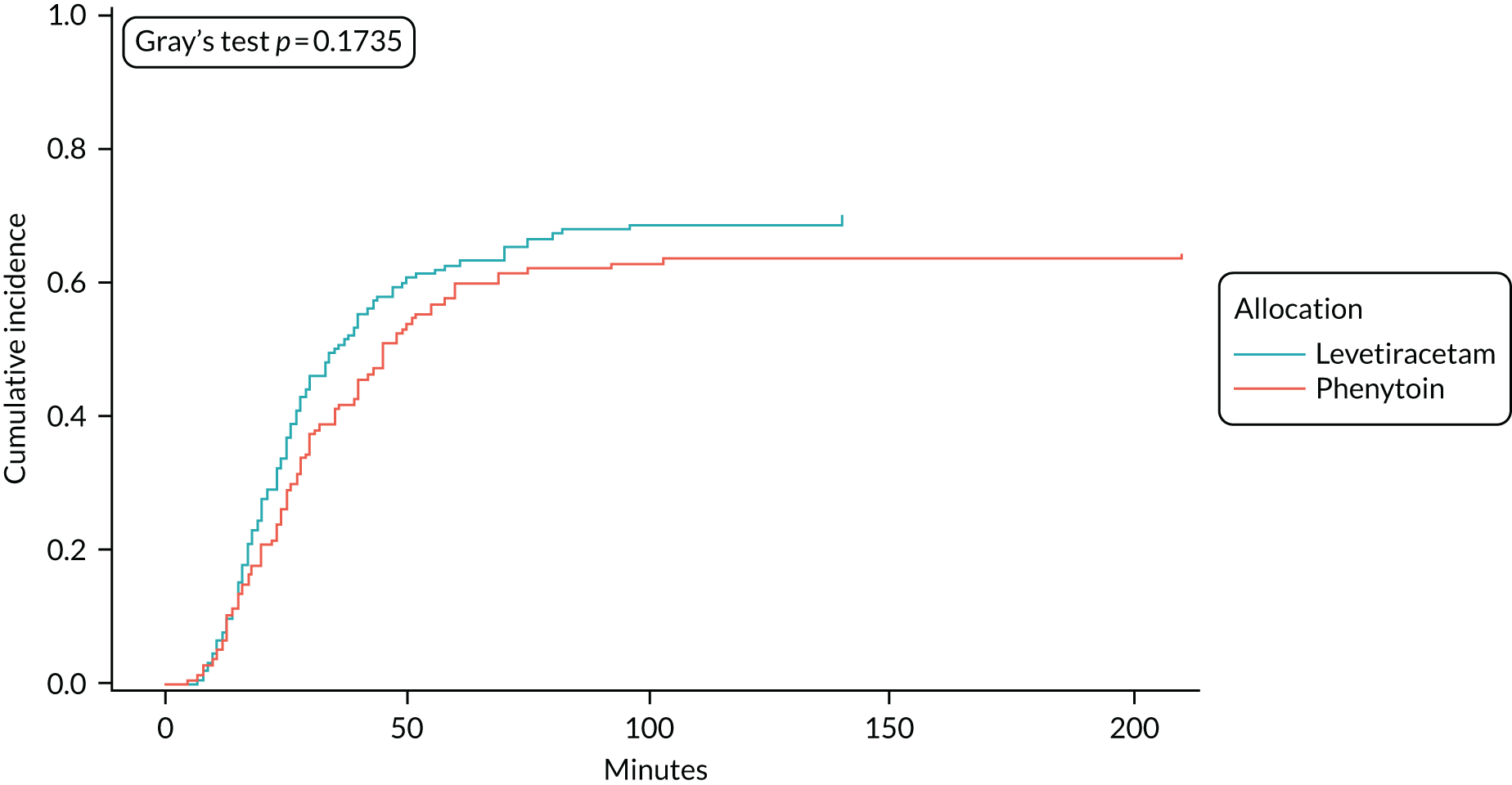
| Variable | Sub-distribution hazard ratio (95% confidence interval) | p-value | |
|---|---|---|---|
| Levetiracetam vs. phenytoin | HR for seizure cessation | 1.202 (0.908 to 1.592) | 0.1976 |
| HR for RSI | 0.837 (0.560 to 1.250) | 0.3843 | |
Time to seizure cessation from infusion
| Variable | Group | |
|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | |
| Number of events (seizure cessation), n (%) | 106 (69.74) | 86 (64.18) |
| Number of censored times (RSI), n (%) | 46 (30.26) | 48 (35.82) |
| Median time (minutes) to cessation of seizure from start of infusion (IQR) | 17.50 (8.00–NA) | 24.50 (8.00–NA) |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.6704 | 1000 | 0.5850 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.153 (0.867 to 1.532) | 0.3279 |
Time to seizure cessation censoring at time of second second-line treatment
| Variable | Group | |
|---|---|---|
| Levetiracetam (N = 152) | Phenytoin (N = 134) | |
| Number of events (seizure cessation), n (%) | 98 (64.47) | 85 (63.43) |
| Number of censored times (RSI), n (%) | 41 (26.97) | 45 (33.58) |
| Number of censored times (second second-line treatment), n (%) | 13 (8.55) | 4 (2.99) |
| Median time (minutes) to cessation of censoring at time of second second-line treatment (IQR) | 35 (20–NA) | 45 (24–NA) |
| Supremum test for proportional hazards assumption | |||
|---|---|---|---|
| Variable | Maximum absolute value | Replications | Pr > MaxAbsVal |
| Levetiracetam vs. phenytoin | 0.8734 | 1000 | 0.3200 |
| Variable | HR (95% CI) | p-value |
|---|---|---|
| Levetiracetam vs. phenytoin | 1.152 (0.861 to 1.540) | 0.3412 |
Appendix 8 Example telephone interview and focus group questions
| Interview topic | Example question |
|---|---|
| Practitioners | |
| Knowledge and experience pre training |
Please define your role in the EcLiPSE trial Prior to the EcLiPSE trial did you have any previous experiences of trial recruitment? If yes, how long have you worked in trials? Have you been in any trials that used RWPC (deferred consent)? How many children have you personally recruited to the EcLiPSE trial? How many children has your site recruited to the EcLiPSE trial? |
| Thoughts about the EcLiPSE trial before training and practitioner engagement with the trial |
What were your thoughts about the EcLiPSE trial before the SIV? Prompt: did you have any concerns? What was the feeling among your site colleagues about the EcLiPSE trial before the meeting? Do you think there was support for the trial? Prompt: was there ‘buy-in’ for this study before the meeting? What do you think helps with getting ‘buy-in’ from the site team? |
| Experience and of training |
Is there anything that stands out about the training? Prompt: anything you found particularly useful/anything not useful? What did you think about the randomisation video? Prompts: how useful was it? Anything you did not find useful? Did you watch it again after the training? What did you think about the consent video? Prompts: how useful was it? Anything you did not find useful? Did you watch it again after the training? Did you have a simulation? Prompts: how useful was it? How could it have been improved? |
| Consent in emergency care |
When you first heard that the EcLiPSE trial was to use a RWPC (deferred consent) approach, what were your initial thoughts? Did you have any concerns? If yes, what were they? Have your views about deferred consent changed over time? If yes, could you tell me a bit more about that? At what point did they change before/after SIV? After experience of RWPC? |
| Consent process: randomisation |
Who is responsible for doing what? How do you think the randomisation process has been going so far? Do you open the envelope in front of parents? If no, why? Do you have posters/leaflets up on the wall in resus? Have any parents asked about the EcLiPSe trial in resus? What questions have parents asked in resus? Have there been any awkward parent/doctor conversations? Has a parent declined to participate in resus, what happened? Have you ever given any brief information about the trial during resus or soon afterwards? (Explore) Is the randomisation/resuscitation process smooth? Do you think it could be improved? (Discuss) |
| Parents | |
| Consent process |
Would you mind if I start by getting an overall picture of what happened when you first heard about the EcLiPSE trial . . . could you tell me a bit about that? (Explore any knowledge about the trial before admission) During the actual treatment in the room where your child was having the seizure, did you ask any questions about the study? What were you told? Did you see any leaflets or posters about the trial? Could you tell me what they explained about the EcLiPSE trial? Could you tell me what the EcLiPSE trial was looking at? What do you think about the use of deferred consent in an emergency situation (e.g. when a child has entered hospital via accident and emergency or born very early)? |
| Decision-making |
Did you feel that your child may benefit from taking part in the trial? Could you describe the possible benefits you expected your child to gain from taking part in the EcLiPSE trial? Did this influence your decision in any way? |
| Assent | Did the nurse or doctor explain the EcLiPSE trial to your child and give them a leaflet to seek their permission to take part? If yes:
|
| Improving the trial and research discussion in the future |
When do you think is the best time to approach parents to obtain consent in an emergency situation? When do you think parents should be consulted about their child’s involvement in an emergency trial? Prompt: what if the trial involved a new drug? Could you tell me a bit more about your reasons for this? Is deferred consent acceptable for that type of research? Who do you think should approach the parents about a trial? Prompt: do you think it should be a doctor or nurse involved in a child’s care who approaches parents about a trial? Do you think it should be someone separate from the care team? Could you tell me why you think this? |
Appendix 9 Consent study participant characteristics
Characteristics: parents
All parents who took part in the consent study had consented to the use of their child’s information in the EcLiPSE trial. Despite attempts to involve decliners, none of the 19 parents who declined the use of their child’s data in the EcLiPSE trial consented to participate in the consent study. None of the parents included in the nested consent study lost a child, although fortunately very few children involved in the EcLiPSE trial died. No individuals identified themselves as legal representatives and therefore this term is not used in the remainder of this appendix.
Of the 143 parents who returned a questionnaire, 93 (65%) were mothers and 39 (27%) were fathers. Ten (7%) had missing parent information.
One parent was present in approximately half (41/76, 54%) of the recorded trial discussions, whereas two parents (or family members) were present in 35 (46%) discussions. Details of who (e.g. mother, father or relative) took part in recorded trial discussions could not be fully established, as not all individuals recorded completed the consent form. Of the 76 recorded trial discussions, 37 (49%) children had received phenytoin and 35 (46%) children had received levetiracetam. In four discussions it was unclear which study drug the child had received. Recorded trial discussion data suggested that 31 out of 76 (40%) children in this subsample had experienced previous seizures, whereas 17 (22%) had not. In 28 (36%) children it was not clear whether or not the child had experienced a previous seizure. In total, 21 out of the 76 (21%) parents indicated that their child had previously taken, or was currently taking, levetiracetam as a preventative treatment.
LR interviewed 30 parents (i.e. 25 mothers and five fathers) over the telephone at a mean of 6 weeks (range 1–8 weeks) after their child had been discharged from hospital. Interviews ranged from 18 to 63 minutes (mean 34 minutes). Eighteen parents reported that their child had experienced previous seizures prior to their child’s involvement in the EcLiPSE trial. For 12 parents, this was their child’s first seizure. Children of parents involved in the consent study were aged between 9 months and 8.5 years at the time of recruitment to the EcLiPSE trial (with a median of 2.8 years) compared with a range of 6 months to 17 years 11 months (with a median of 2.8 years) for the EcLiPSE trial sample (379 children).
Thirteen parents reported their child being randomised to levetiracetam and 10 parents said that their child was randomised to phenytoin. In six cases, the parent reported that they could not remember what trial drug was given and one knew both had been given, but was unsure of the order in which they were given.
Characteristics: practitioners
As shown in Figure 12, SIV questionnaire respondents comprised 45 (36%) nurses and 57 (46%) doctors. Those who defined themselves as ‘other’ had specific roles (e.g. ‘nurse specialist hospice’). Six nurses and four doctors took part in a telephone interview. All had a lead role (e.g. principal investigator, lead research nurse) in the EcLiPSE trial recruitment. Telephone interviews lasted, on average, 37 (range 21–47) minutes. Of the 36 practitioners who took part in one of the six focus groups, 20 (56%) were nurses and 16 (44%) were doctors. On average, focus groups lasted 61 (range 37–88) minutes. Of the 199 practitioners who took part in the online questionnaire, 39% (n = 78) were nurses, 59% (n = 117) were doctors and 5% (n = 3) were classified as ‘other’ (e.g. clinical research lead). It is likely that some of the practitioners who took part in a telephone interview, focus group or SIV questionnaire also completed the online questionnaire.
FIGURE 12.
Practitioner characteristics by method.
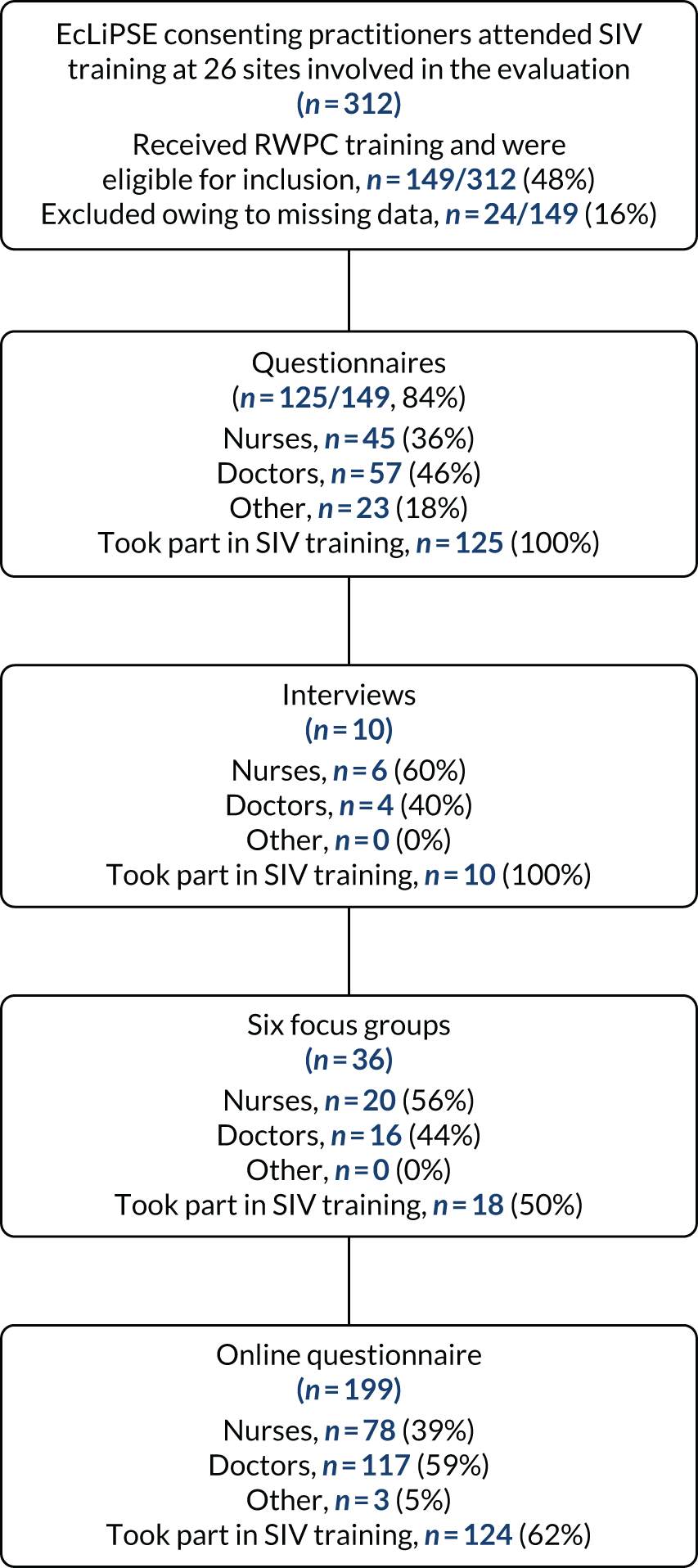
| I feel confident in . . . | Before training, n (%) | After training, n (%) | Changes in confidence after training, n (%) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strongly disagree | Mildly disagree | Neither agree nor disagree (neutral) | Mildly agree | Strongly agree | Mean confidence score | Strongly disagree | Mildly disagree | Neither agree nor disagree (neutral) | Mildly agree | Strongly agree | Mean confidence score | Improved | Decreased | No change | Mean difference (95% CI) | p-value | |
| 1. Explaining the study to families | 11 (9) | 27 (22) | 30 (24) | 30 (24) | 27 (22) | 3.28 | 0 (0) | 0 (0) | 4 (3) | 52 (42) | 69 (55) | 4.52 | 82 (66) | 4 (3) | 39 (31) | –1.24 (–1.46 to –1.02) | 0.000a |
| 2. Explaining randomisation to families | 8 (6) | 7 (6) | 20 (16) | 49 (39) | 41 (33) | 3.86 | 0 (0) | 0 (0) | 2 (2) | 42 (34) | 81 (65) | 4.63 | 58 (47) | 3 (2) | 64 (51) | –0.77 (–0.96 to –0.57) | 0.002b |
| 3. Explaining RWPC to familiesc | 20 (16) | 24 (19) | 40 (32) | 21 (17) | 20 (16) | 2.98 | 0 (0) | 1 (1) | 12 (10) | 48 (39) | 61 (50) | 4.39 | 90 (72) | 2 (2) | 32 (26) | –1.40 (–1.62 to –1.17) | 0.000a |
| 4. Dealing with parents who object to their child being randomised | 8 (6) | 28 (22) | 29 (23) | 30 (24) | 30 (24) | 3.37 | 0 (0) | 2 (2) | 16 (13) | 56 (45) | 51 (41) | 4.25 | 64 (51) | 5 (4) | 56 (45) | –0.88 (–1.08 to –0.68) | 0.000b |
| Statement | n (%) |
|---|---|
| The trial is running well and we have no issues | 125 (63) |
| The trial is running well but we do have some issues | 40 (20) |
| The trial is not running well as we have some issues | 5 (3) |
| Staff shortages have led to patients being missed | 13 (7) |
| There is a lack of support from the central EcLiPSE trial team | 1 (1) |
| Site training is not frequent enough | 11 (6) |
| It is difficult to find staff to cover consent-seeking | 5 (3) |
| There is a lack of support for the EcLiPSE trial at site | 2 (1) |
| Anxieties about research without consent are a barrier to recruitment | 2 (1) |
Appendix 10 How trial information about the trial was exchanged during discussion about recruitment to the EcLiPSE trial
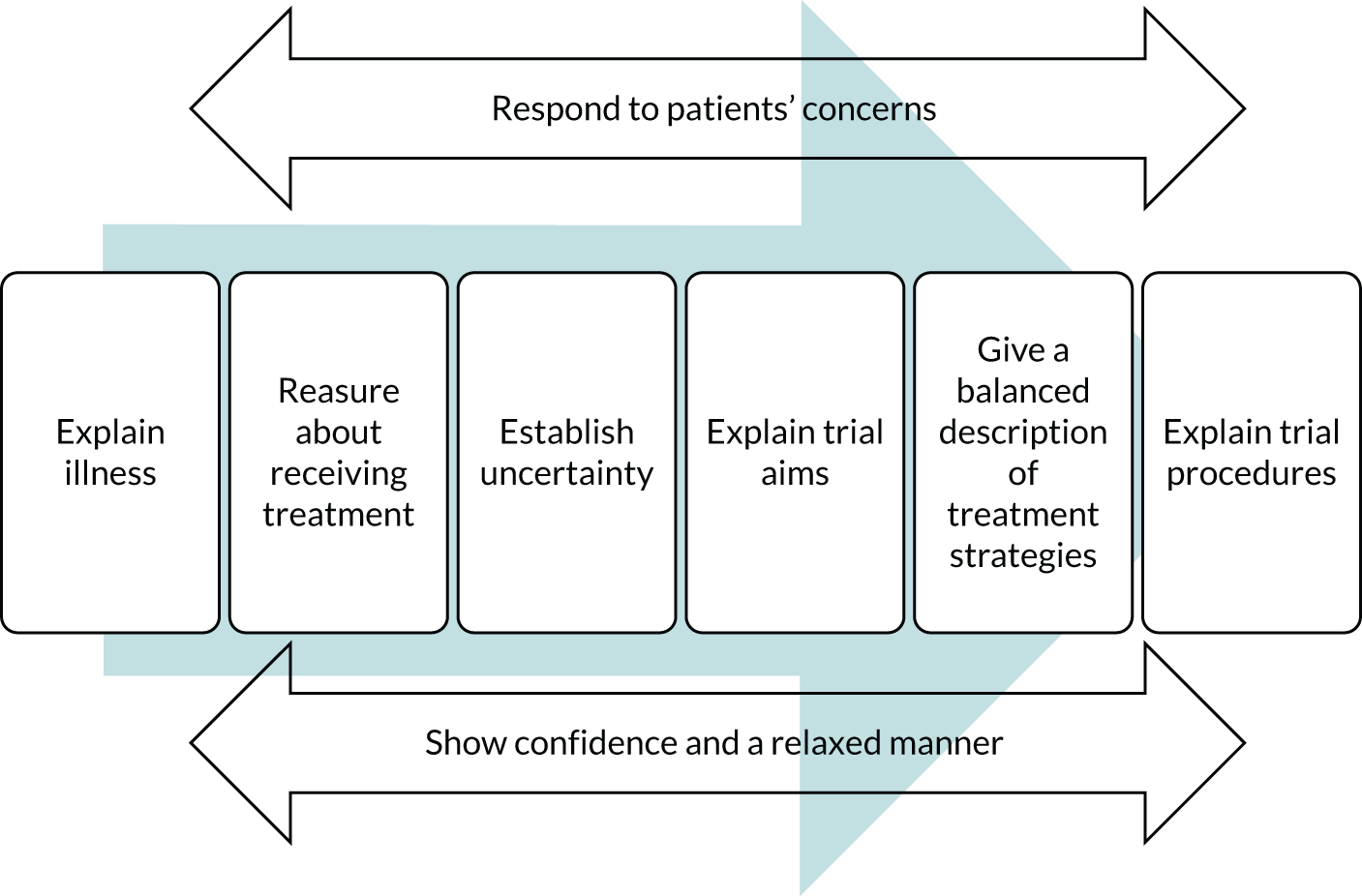
FIGURE 14.
A seven-step framework to assist recruitment in trials that involve RWPC.
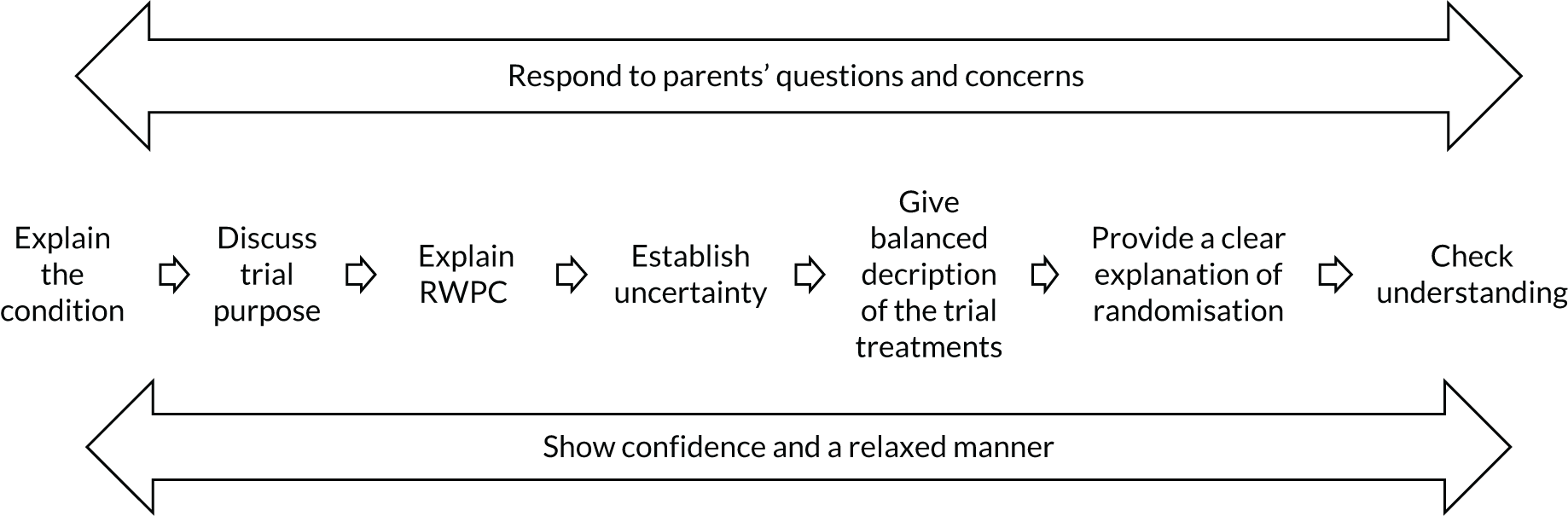
List of abbreviations
- AE
- adverse event
- APLS
- advanced paediatric life support
- AR
- adverse reaction
- CI
- confidence interval
- CONNECT
- CONseNt methods in paediatric Emergency and urgent Care Trials
- CRF
- case report form
- CSE
- convulsive status epilepticus
- EcLiPSE
- Emergency treatment with Levetiracetam or Phenytoin in Status Epilepticus in children
- ED
- emergency department
- EEG
- electroencephalogram
- ESETT
- Established Status Epilepticus Treatment Trial
- HR
- hazard ratio
- IDSMC
- Independent Data and Safety and Monitoring Committee
- IMP
- investigational medicinal product
- IQR
- interquartile range
- i.v.
- intravenous
- PEM
- paediatric emergency medicine
- PERUKI
- Paediatric Emergency Research in the UK and Ireland
- PICU
- paediatric intensive care unit
- PPI
- patient and public involvement
- RCT
- randomised controlled trial
- RSI
- rapid sequence induction
- RWPC
- research without prior consent
- SAE
- serious adverse event
- SAR
- serious adverse reaction
- SIV
- site initiation visit
- SUSAR
- suspected unexpected serious adverse reaction
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee