

Notes
Article history
The research reported in this issue of the journal was funded by the HS&DR programme or one of its preceding programmes as project number 15/80/39. The contractual start date was in March 2017. The final report began editorial review in February 2020 and was accepted for publication in November 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HS&DR editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2021. This work was produced by Mouncey et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2021 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background and rationale
Vasopressors are life-sustaining drugs that are administered to patients in critical care to avoid hypotension, which is associated with myocardial injury, kidney injury and death. 1,2 They work by causing vasoconstriction, which may reduce blood flow and cause other secondary effects on cardiac, metabolic, microbiome and immune function. 3 Balancing the risks of hypotension with the risks of vasopressors is a daily challenge when managing patients in critical care units.
National clinical audit data indicate that close to half (44%) of all patients admitted to adult general critical care units across England, Wales and Northern Ireland have hypotension and receive vasopressors. It is estimated that 40–50% of these cases will represent vasodilatory hypotension. This is similar in other health-care systems. 4,5
To guide vasopressor administration, doctors typically prescribe a mean arterial pressure (MAP) target and bedside nurses adjust the dose/rate of vasopressor infusions to achieve the target MAP. The 2012 Surviving Sepsis Campaign guidelines6 recommended maintaining a MAP of > 65 mmHg; however, the guidelines were based on low-quality evidence and did not provide guidance for an upper limit. The guidelines also suggested a higher MAP target for older patients and those with chronic hypertension, recommendations that were later removed in 2016. 7,8 Studies suggest that, as the optimal MAP target is not well established, clinicians tend to err on the side of targeting higher MAPs, potentially exposing patients to greater doses and durations of vasopressors than may be necessary. 9,10
An individual patient data meta-analysis11 of two randomised clinical trials (RCTs) that evaluated different MAP targets12,13 suggested that increased exposure to vasopressors, through targeting higher MAPs, may be associated with a greater risk of death in older critically ill patients. 11,14
The 65 trial tests the hypothesis that, in critically ill patients aged ≥ 65 years who receive vasopressors for vasodilatory hypotension, reducing vasopressor exposure through permissive hypotension (i.e. a MAP target of 60–65 mmHg) compared with usual vasopressor exposure reduces 90-day mortality and is cost-effective. This follows similar strategies to minimise the intensity of other critical care interventions, including oxygen therapy,15 enteral feeding,16 mechanical ventilation,17 blood transfusions,18 intravenous fluids for patients after trauma19 and severe febrile illness in children. 20
Efficient design
The 65 trial was funded through the National Institute for Health Research (NIHR) Health Technology Assessment programme Efficient Study Designs call and designed in such a way to maximise efficiency and minimise the additional workload that research can create for critical care unit teams. The trial took a data-enabled approach, maximising the use of routinely collected and available data. The trial was nested in an existing network of research-active critical care units participating in the Case Mix Programme (CMP). The CMP (i.e. the national clinical audit for adult critical care in England, Wales and Northern Ireland) is a source of high-quality, robust and representative data. 21 The trial additionally utilised national death registration data held by NHS Digital. Primary data collection was largely limited to protocol adherence and patient safety data.
Aim and objective
Aim
The aim was to evaluate the clinical effectiveness and cost-effectiveness of reducing vasopressor exposure through permissive hypotension (using a MAP target of 60–65 mmHg) in critically ill patients aged ≥ 65 years with vasodilatory hypotension.
Objective
The objective was to estimate the clinical effectiveness and cost-effectiveness of reducing vasopressor exposure through permissive hypotension when compared with usual care.
Chapter 2 Methods
Reproduced with permission from Richards-Belle et al. 22
Design
The 65 trial was a pragmatic, multicentre, parallel-group, open-label RCT of reduced exposure to vasopressors through permissive hypotension (i.e. a lower MAP target of 60–65 mmHg) in older critically ill patients with vasodilatory hypotension, with an integrated economic evaluation. 22
Setting
The trial was carried out in NHS adult general critical care units in England, Wales and Northern Ireland.
Sites
The trial aimed to recruit eligible patients from a representative sample of 65 NHS adult general critical care units. Adult general critical care units were defined as intensive care units (ICUs) or combined ICU and high-dependency units. Standalone high-dependency units and specialist critical care units (e.g. cardiothoracic) were excluded.
Eligibility/requirements
Sites were considered for participation if they were active participants in the CMP and able to:
-
identify two local joint-principal investigators (PIs) – one critical care consultant and one senior critical care nurse – to lead the 65 trial locally
-
identify a 65 trial research nurse who would be responsible for day-to-day local trial co-ordination
-
incorporate the 65 trial into routine critical care clinical practice
-
agree to randomise eligible patients and adhere to individual patient randomisation allocations
-
agree to data collection requirements and to maintain a screening and enrolment log
-
continue active participation in the CMP
-
comply with all responsibilities as stated in the 65 trial Clinical Trial Site Agreement and all requirements of the protocol
-
comply with the UK Policy Framework for Health and Social Care Research23 and the International Conference on Harmonisation guidelines on good clinical practice. 24
Site identification, initiation and activation
A call for expressions of interest was sent via e-mail to all adult general critical care units actively participating in the CMP by the Intensive Care National Audit & Research Centre (ICNARC; London, UK) Clinical Trials Unit (CTU). Advertisements were also placed on ICNARC’s website (URL: www.icnarc.org) and Twitter feed (URL: www.twitter.com; Twitter, Inc., San Francisco, CA, USA).
Site initiation visits were planned for each site, facilitated by the chief investigator (PRM), lead clinical investigator (FL) and/or trial manager (ARB). During the visits, the following were covered: trial background/rationale and procedures for screening and randomising patients, seeking informed deferred consent/opinion, data collection and safety monitoring.
Investigator site files (ISFs) containing all essential documents [e.g. trial protocol, standard operating procedures (covering screening, randomisation, delivery of the interventions, consent procedures, safety monitoring, etc.), relevant approvals, information sheets and consent forms] were provided.
Sites were activated and authorised to commence screening and recruitment once the following were in place:
-
a completed site initiation visit
-
all relevant institutional approvals (e.g. local confirmation of capacity and capability)
-
a 65 trial Clinical Trial Site Agreement signed by the local NHS trust/health board and the sponsor (i.e. ICNARC)
-
a signed delegation log had been submitted to the ICNARC CTU.
Once the ICNARC CTU confirmed that all necessary documentation was in place, a site activation e-mail was issued to the joint PIs.
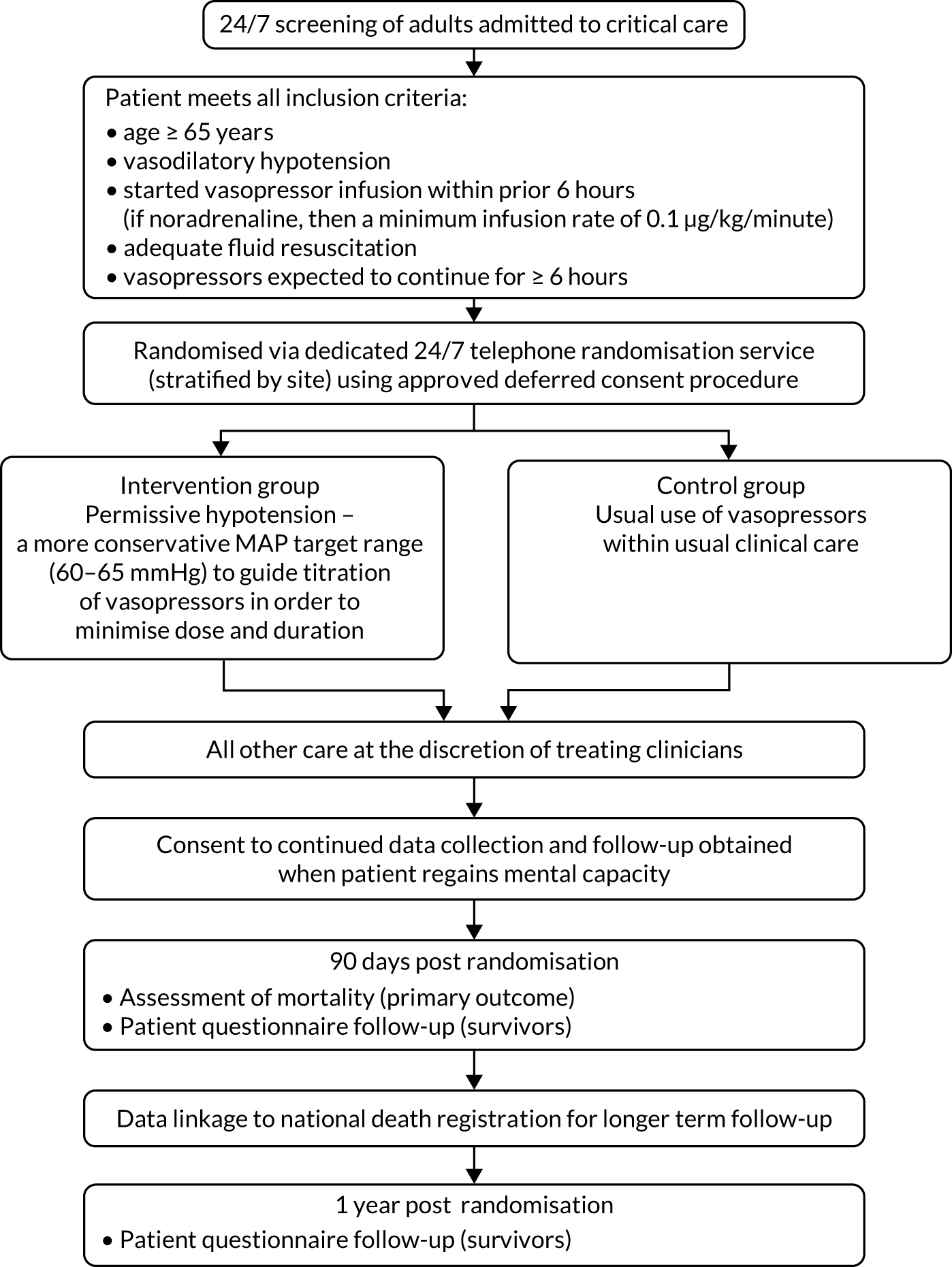
Patients
The full flow of eligible patients through the 65 trial is shown in Figure 1.
FIGURE 1.
Patient flow through the 65 trial. Reproduced with permission from Richards-Belle et al. 22
Eligibility
The target population was critically ill patients aged ≥ 65 years receiving vasopressors for vasodilatory hypotension. Patients were eligible if they met all inclusion criteria and none of the exclusion criteria.
Inclusion criteria
-
Patients were aged ≥ 65 years.
-
Patients had vasodilatory hypotension (assessed by treating clinician).
-
Patients had started an infusion (for at least 1 hour) of vasopressors within the prior 6 hours (if noradrenaline, then a minimum infusion rate of 0.1 µg/kg/minute was required).
-
Patients had adequate fluid resuscitation completed or ongoing.
-
Vasopressors were expected to be continued for ≥ 6 further hours.
In the original approved protocol, patients were eligible if a decision to start vasopressors (at any dose) had been made. The inclusion criteria were updated in December 2017 after routine central monitoring of available trial data for 159 control group patients had identified a group of patients who received only a relatively short duration (and often low doses) of vasopressors. The inclusion criteria were therefore refined to specify that, at the time of randomisation, patients must have been on a vasopressor infusion for at least 1 hour and if receiving noradrenaline then they must have been on an infusion of at least 0.1 µg/kg/minute.
Exclusion criteria
-
Vasopressors being used solely as therapy for bleeding, acute ventricular failure (left or right) or post-cardiopulmonary bypass vasoplegia.
-
Ongoing treatment for brain injury or spinal cord injury.
-
Death perceived as imminent.
-
Previous enrolment to the 65 trial.
Screening
Potentially eligible patients admitted (or accepted for admission) to the participating adult general critical care unit were screened against the eligibility criteria by local clinical/research teams. Site research teams maintained a screening and enrolment log of patients aged ≥ 65 years with vasodilatory hypotension and who received vasopressors. The log detailed randomised patients, reasons for exclusion and non-enrolment.
Randomisation
Patients were randomised in a 1 : 1 ratio to either the permissive hypotension group or to the usual-care group, using a dedicated 24 hours per day, 7 days per week, telephone or web-based randomisation service hosted by Sealed Envelope Ltd (URL: www.sealedenvelope.com/, London, UK). Allocation was concealed, used randomised permuted blocks of variable lengths (of 4, 6 and 8) and stratified by recruiting site. As the trial was large, the risk of chance imbalance in prognostic factors was low and the need to randomise patients during a very short time frame mandated that the randomisation process was as simple as possible. For these reasons, we elected not to stratify the randomisation process on any baseline covariates.
Treatment groups
Permissive hypotension (mean arterial pressure target 60–65 mmHg)
‘Permissive hypotension’ aimed to reduce exposure (i.e. dose and duration) to vasopressors through use of a lower MAP target range (i.e. 60–65 mmHg) to guide vasopressor administration.
The choice of vasopressor was at the discretion of the treating clinician, with administration (aside from the MAP target) as per local practice and guidelines. The following were considered as vasopressors: noradrenaline, vasopressin, terlipressin, phenylephrine, adrenaline, dopamine and metaraminol.
The decision to discontinue vasopressors depended on the patients’ ability to maintain the MAP target stipulated by the protocol without vasopressors. Clinical teams were actively reminded to consider discontinuing vasopressor if the patient was able to maintain a MAP value of at least 60 mmHg. The MAP target applied to any point during the critical care unit admission that the patient was deemed to require vasopressors.
If a patient developed an exclusion criterion (see Exclusion criteria) after randomisation, it was at the discretion of the treating clinical team whether or not the MAP target continued, with patient safety guiding this decision.
Usual care
Patients randomised to the usual-care group received usual vasopressor exposure (including the MAP target) at the discretion of the treating clinician and as per local practice and guidelines.
Treating clinician(s) were aware of the treatment allocation. All other usual care was provided at the discretion of the treating clinical team and as per local practice.
Co-interventions
As per standard care of patients receiving vasopressors, central venous catheters (to avoid extravasation) and arterial catheters (for close MAP monitoring) would usually be in place. The use of pure inotropes {i.e. dobutamine, milrinone or levosimendan [Leximda, Orion Pharma (UK) Ltd]} fluids and corticosteroids [e.g. hydrocortisone, methylprednisolone (Solu-Medrone, Pfizer Ltd), dexamethasone, prednisone/prednisolone] was recorded, but left to the discretion of the treating team.
Consent procedures
Patients in critical care requiring vasopressors often need this treatment started as a life-saving measure during an emergency, time-sensitive situation. The majority of patients lack mental capacity because of their medical condition and the effects of concomitant treatments administered as part of standard care (e.g. mechanical ventilation, sedative and analgesic drugs). Any delay in commencing treatment could be detrimental to the patient and to the scientific validity of the trial. This, alongside the potential distress of the emergency situation, rendered attempts to obtain either prospective informed consent from the patient or the opinion of their personal consultee (i.e. relative or close friend) prior to starting the trial treatment inappropriate.
Considering these reasons, eligible patients were enrolled and randomised to receive the allocated treatment immediately. This method is known as ‘deferred consent’ or ‘research without prior consent’ and was covered by an emergency waiver of consent under the Mental Capacity Act25 and approved by the South Central – Oxford C Research Ethics Committee (REC) (reference 17/SC/0142).
In the situation where a patient was deemed by the treating clinical team to have full mental capacity and was able to provide prospective informed consent at the point of randomisation, they were approached prior to randomisation for verbal consent. If providing verbal consent, they were then followed up for full written informed consent, in line with the procedures outlined below.
Patient informed deferred consent
Following randomisation, patients were approached by an authorised, trained member of the site research team when deemed to have full mental capacity to provide informed deferred consent. A patient information sheet (PIS) (see Report Supplementary Material 1), providing information on the aim of the trial, what participation would mean for the patient, confidentiality and data security, and the future availability of the trial results, was provided to the patient. Patients were given time to read the PIS and to ask any questions they had prior to confirming their consent decision. If in agreement, a consent form (see Report Supplementary Material 2) was then provided, indicating that the information, given orally and in writing, had been read and understood, and that participation was voluntary and could be withdrawn at any time without consequence. The consent form covered options for continuing participation, access to the medical records for ongoing data collection, questionnaire follow-up (i.e. at 90 days and 1 year) and the sharing of anonymised data.
After verifying that the PIS and consent form were understood, the trained member of the site research team invited the patient to sign the consent form. A copy was given to the patient, a copy placed in the patient’s medical records and the original kept in the local ISF. If the patient was unable to physically sign the consent form (e.g. because of weakness or reduced dexterity), an independent witness signed on their behalf and in their presence.
The patient’s general practitioner (GP) was then sent a letter by the site research team to inform them of their patient’s participation in the trial (provided consent had been given for this).
Personal consultee opinion
For the reasons outlined in Consent procedures, it was usually not possible to involve patients in the consent process early on. In the interim, once notified of the randomisation of a patient into the 65 trial, an authorised and trained member of the site research team approached the patient’s personal consultee (i.e. relative or close friend) as soon as appropriate and practicably possible to discuss the trial and to seek their opinion as to the patient’s likely wishes and feelings regarding participation. Ideally, this approach would take place within 24–48 hours of randomisation, once the patient’s medical situation was no longer considered an emergency (but the specific timing would vary according to each patient’s clinical scenario).
The personal consultee was provided with a personal consultee information sheet (see Report Supplementary Material 3), which contained all the information provided on the PIS, supplemented by information about why the personal consultee was being approached at this stage. A personal consultee opinion form (see Report Supplementary Material 4) was provided, which indicated that the information, given orally and in writing, had been read and understood, the patient’s participation was voluntary and could be withdrawn at any time without consequence, and that, in the personal consultee’s opinion, the patient would not object to taking part.
Personal consultees were given time to read the personal consultee information sheet and were invited to ask any questions they had about the patient’s participation in the 65 trial. After verifying that the personal consultee information sheet and opinion form were understood, the trained member of the site research team then invited the personal consultee to sign the personal consultee opinion form. If a personal consultee advised that, in their opinion, the patient would not choose to participate, then the trial treatment was stopped (if ongoing) and the personal consultee was asked if, in their opinion, the patient would be willing to continue with ongoing data collection and/or to be followed up at 90 days and 1 year.
On patient recovery, patients were approached directly for informed deferred consent (see Patient informed deferred consent). The patient’s decision was final and superseded that of the personal consultee when there was disagreement.
Nominated consultee opinion
In the case of a patient who had died, a nominated consultee was appointed. The nominated consultee could include an independent mental capacity advocate appointed by the NHS trust or an independent doctor (i.e. not associated with the conduct of the trial). The opinion of the nominated consultee was sought in the same manner as for the personal consultee.
A nominated consultee was also approached if no personal consultee was available or if one was available but did not want to be consulted. On patient recovery, the patient was approached directly for informed deferred consent (see Patient informed deferred consent). The patient’s decision was final and superseded that of the nominated consultee when there was disagreement.
Discharge prior to consent/opinion being sought
If a patient was discharged from hospital with mental capacity prior to consent/opinion being confirmed, the most appropriate member of the site research team followed up the patient by telephone and post to obtain informed consent. If there was no response 4 weeks after contacting the patient by post for a second time, the participant’s data were included in the trial unless the patient notified the site research team otherwise.
If the patient was discharged without mental capacity, then the opinion of the personal consultee was sought in line with the above process (i.e. telephone call then postal approach).
If the participant was transferred to another hospital participating in the trial before the consent procedures were complete, then the local site research team contacted the site research team at the receiving hospital to hand over the consenting procedures.
Refusal or withdrawals of consent/opinion
If patient-informed deferred consent (or consultee opinion) was refused or withdrawn, this decision was respected and abided by, and no further contact made. All data up to the point of this decision were retained in the trial records, unless the patient or consultee requested otherwise.
Safety monitoring
All patients eligible for the 65 trial were critically ill and, because of the complexity of their condition, at increased risk of experiencing adverse events and serious adverse events (SAEs). 26 In the 65 trial, the labelling of a SAE was limited to serious events that might reasonably occur as a consequence of either sustaining lower MAP values and/or higher doses of vasopressors required to maintain higher MAP values. In addition to reporting any unexpected and possibly related SAEs, research teams were asked to screen for, and record occurrences of, the following events (up to critical care unit discharge):
-
supraventricular cardiac arrhythmia
-
ventricular cardiac arrhythmia
-
myocardial injury
-
extremity necrosis
-
mesenteric ischaemia
-
severe acute renal failure.
If occurring, each event was assessed for its severity, using the scale below:
-
‘None’ – no event or complication.
-
‘Mild’ – complications result in only temporary harm and do not require clinical treatment.
-
‘Moderate’ – complications require clinical treatment, but do not result in significant prolongation of hospital stay. Does not usually result in permanent harm and, where this does occur, the harm does not cause functional limitations to the patient.
-
‘Severe’ – complications require clinical treatment and result in significant prolongation of hospital stay and/or permanent functional limitation.
-
‘Life-threatening’ – complications may lead to death.
-
‘Fatal’ – patient died as a direct result of the complication/adverse events.
A reportable event with the severity assessed as ‘severe’, ‘life-threatening’ or ‘fatal’ was considered a SAE in the 65 trial and this was reported on the 65 trial SAE reporting form. On receipt, a member of the ICNARC CTU trial team reviewed the form for completeness and internal consistency, which was then followed by review by a clinical member of the 65 Trial Management Group (TMG) to evaluate whether or not the event qualified for expedited reporting to the REC. If the event was judged to be unexpected and potentially related to the trial intervention(s), the ICNARC CTU trial team submitted a report to the REC within 15 calendar days.
Questionnaire follow-up
Health-related quality of life (HRQoL) [assessed using the EuroQol-5 Dimensions, five-level version (EQ-5D-5L) questionnaire],27 cognitive decline [assessed using the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) short form]28 and health service and resource use were collected via self-report follow-up questionnaires administered to surviving patients at 90 days and 1-year post randomisation. Follow-up for patient-reported 1-year outcomes was truncated and completed when the last patient reached 90 days.
Allowing time for administrative procedures, the questionnaire follow-up process started at 82 days post randomisation for the 90-day follow-up time point and at 358 days post randomisation for the 1-year follow-up time point. Patients who had died since leaving hospital were logged and the follow-up process ended. Questionnaire packs were sent to participants by post and included a self-addressed stamped envelope and pen for ease of return. Participants could indicate if they no longer wished to complete the questionnaire.
Non-responders were telephoned 3 weeks later by a trained member of the 65 trial team from the ICNARC CTU to check whether or not they had received the questionnaire and were given the option to complete the questionnaire over the telephone or to receive another copy, either by post or by e-mail.
For patients identified as current hospital inpatients, or residents in a care home or rehabilitation centre, the relevant institution was contacted to establish the most appropriate way to proceed with follow-up. If possible, staff members at the relevant institution assisted the patient with completion and return to the ICNARC CTU.
Outcomes
Primary clinical outcome: 90-day mortality
The primary clinical outcome was 90-day mortality, defined as death due to any cause within 90 days following the date of randomisation.
Secondary clinical outcomes
Mortality at discharge from the critical care unit and acute hospital
Mortality at discharge from the critical care unit was defined as death due to any cause before discharge to any location providing a level of care less than level 2 (i.e. high-dependency care). Mortality at discharge from acute hospital was defined as death due to any cause before discharge from acute hospital. Patients transferred from the original acute hospital to another acute hospital were followed up until they left the acute hospital.
Duration of survival to longest available follow-up
Duration of survival was calculated as the duration (in days) from the date of randomisation to the date of death. Patients were censored at the last date on which they were known to be alive.
Duration of advanced respiratory and renal support during the critical care unit stay
Advanced respiratory support and renal support were defined in accordance with the UK Department of Health and Social Care’s Critical Care Minimum Data Set. 29 Advanced respiratory support was defined as receiving one or more of the following: invasive mechanical ventilatory support applied via a translaryngeal tube or via a tracheostomy; bilevel positive airway pressure applied via a translaryngeal tracheal tube or via a tracheostomy; continuous positive airway pressure via a translaryngeal tracheal tube; or extracorporeal respiratory support. Note that mask/hood continuous positive airway pressure, mask/hood bilevel positive airway pressure and high-flow nasal cannula were not considered advanced respiratory support. Renal support was defined as receiving either acute renal replacement therapy (e.g. haemodialysis, haemofiltration) or renal replacement therapy for chronic renal failure.
The duration of organ support was defined as the number of calendar days (00.00 to 23.59) on which the organ support was received at any time. Any days outside the critical care unit were assumed to be free of organ support.
Days alive and free of advanced respiratory and renal support within first 28 days
For patients surviving to 28 days following randomisation, the number of days alive and free of advanced respiratory and renal support to day 28 was defined as the number of calendar days (00.00 to 23.59) on which neither advanced respiratory support nor renal support was received at any time. Patients dying between randomisation and day 28 were assigned a value of zero.
Duration of critical care unit and acute hospital stay
Duration of critical care unit stay was calculated as the sum of the duration (in days) from the date and time of randomisation to the date and time of first discharge from, or death in, the critical care unit plus the duration of any subsequent admissions to the critical care unit within the same acute hospital stay. Duration of acute hospital stay was calculated as the duration (in days) from the date of randomisation to the date of discharge from, or death in, acute hospital.
Cognitive decline at 90 days and 1 year
Cognitive decline was assessed using the IQCODE short version,28 with the total score calculated as the mean of the scores (from 1 to 5) on 16 items.
Primary economic outcome: incremental net monetary benefit at 90 days
The incremental net monetary benefit (INMB) of permissive hypotension compared with usual care at 90 days was calculated by multiplying the mean gain or loss in quality-adjusted life-years (QALYs) by the National Institute for Health and Care Excellence (NICE)-recommended threshold in the UK (of £20,000) and subtracting the incremental cost. 30,31
Secondary economic outcomes
Health-related quality of life at 90 days and 1 year
Health-related quality of life at 90 days and 1 year was measured using the EQ-5D-5L. 27 The EQ-5D-5L requires patients to describe their health on five dimensions: (1) mobility, (2) self-care, (3) usual activities, (4) pain/discomfort and (5) anxiety/depression. The mean [standard deviation (SD)] was reported among survivors at the relevant time point. EQ-5D-5L responses were used to report each patient’s described health, which was then valued using the EQ-5D-5L value set for England 201832 according to health state preferences from the general population to calculate EQ-5D-5L utility scores, anchored on a scale from 0 (death) to 1 (perfect health).
Life-years and quality-adjusted life-years at 90 days and 1 year
The 65 trial data were linked with national death registrations held by NHS Digital. Information on the date and time of deaths was used to calculate the survival time and life-years up to 90 days and up to 1 year for each randomised patient. QALYs at 90 days were calculated by valuing each patient’s survival time by their HRQoL at 90 days, according to the ‘area under the curve’ approach. 33 For survivors at 90 days, QALYs were calculated using the 90-day EQ-5D-5L scores, assuming an EQ-5D-5L score of zero at randomisation and a linear interpolation between randomisation and 90 days. For decedents between randomisation and 90 days, a zero QALY gain was assumed. The same approach was taken to calculate the QALYs at 1 year.
Resource use and costs at 90 days and 1 year
Overview
Resource use categories considered were chosen a priori and according to those where differences between the treatment groups were deemed likely to drive incremental costs. The categories were resource use associated with the trial interventions, hospital admissions (index admission and readmissions) and visits to outpatients and community health-care services. Vasopressor use (duration and infusion rate) was judged as the key element of the trial interventions. Total costs at 90 days were calculated by combining resource use data with unit costs at 2017/18 prices (GBP).
Intervention
The costs of vasopressors associated with delivering the trial interventions for both treatment groups (i.e. permissive hypotension and usual care) were included. Detailed patient-level data on the volume of vasopressor use were collected in the case report form (CRF). The costing of vasopressors was informed by expert clinical opinion and based on standard dose/units of vasopressors (i.e. noradrenaline, adrenaline, dopamine, phenylephrine, vasopressin, metaraminol, terlipressin) that are routinely used in critical care units.
Hospital stay
The use of hospital resources from the index hospital admission (i.e. the hospital stay following randomisation) and any readmission(s) to hospital was extracted from the CRF and CMP database. 21 For each patient, location and duration of the index hospital admission (including time spent in critical care and on general medical wards) were recorded up to 90 days. Length of stay in critical care was calculated as total duration (in days), including fractions of days, from the date and time of randomisation for the critical care unit stay during which the patient was randomised until the time of discharge from, or death in, critical care. Within the index admission, the total duration of critical care stay included all time spent in critical care between randomisation and discharge from acute hospital (including any transfers to critical care units in other hospitals). Data on the number of organs supported on each day of critical care were extracted from the CMP database and each critical care episode then assigned a Healthcare Resource Group (HRG), applying a standard HRG grouper algorithm. 29 For the index admission, total length of stay was calculated as the total duration (in days) from the date of randomisation to the date of ultimate discharge from, or death in, hospital.
A hospital readmission was defined as a further hospital admission after ultimate acute hospital discharge from the index admission. Readmission data were collected from two sources: (1) the CMP database21 and (2) health services questionnaires administered to patients surviving to 90 days. The former provided information on duration of critical care unit stay and total hospital stay, including subsequent transfer to other care areas (e.g. general medical wards) within the same hospital and to other hospitals. The latter provided information on readmissions that did not include a further stay in critical care.
The resource use items included the total number of hospital outpatient visits and community service use following discharge from the index admission but before 90 days post randomisation. Visits to outpatient and community health-care services [e.g. GP visits, nurses (i.e. from the GP clinic, hospital or a psychiatric nurse), health visitor, occupational therapist, speech and language therapist, counsellor, physiotherapist, psychiatrist and psychologist] were collected via the health service questionnaire. Resource use items included usage for reasons both related and unrelated to the initial critical care admission in which the patient was randomised.
Unit costs
The unit costs required for valuing the resource use data were taken from national unit cost databases (Table 1). Vasopressor unit costs were taken from British National Formulary. 34 The costs per critical care bed-day by HRG and general medical bed-day were taken from the Payment by Results database. 35 Unit costs for hospital outpatient visits and community service use were obtained from Unit Costs of Health and Social Care. 36 All unit costs were reported in 2017–18 prices.
| Item | Unit cost (£) | Source |
|---|---|---|
| Vasopressor | ||
| Noradrenaline (4 mg) | 5.8 | BNF34 |
| Dopamine (200 mg) | 2 | BNF34 |
| Phenylephrine (10 mg) | 5 | BNF34 |
| Terlipressin (1 mg) | 17.9 | BNF34 |
| Adrenaline (1 mg) | 0.6 | BNF34 |
| Vasopressin (1unit) | 85 | BNF34 |
| Metaraminol (20 mg) | 8.8 | BNF34 |
| Hospital costs (bed-day) | ||
| Critical care bed-day: zero organs supported | 838 | NHS reference costs35 |
| Critical care bed-day: one organ supported | 1136 | NHS reference costs35 |
| Critical care bed-day: two organs supported | 1524 | NHS reference costs35 |
| Critical care bed-day: three organs supported | 1710 | NHS reference costs35 |
| Critical care bed-day: four organs supported | 1893 | NHS reference costs35 |
| Critical care bed-day: five organs supported | 2038 | NHS reference costs35 |
| Critical care bed-day: six or more organs supported | 2075 | NHS reference costs35 |
| General medical bed-day | 337 | NHS reference costs35 |
| Outpatient and community health services | ||
| Hospital outpatient | 134 | PSSRU36 |
| GP general practice visit (per visit) | 37 | PSSRU36 |
| GP home visit (per visit) | 86 | PSSRU36 |
| GP nurse visita | 11 | PSSRU36 |
| GP nurse home visita | 19 | PSSRU36 |
| Hospital nursea | 9 | PSSRU36 |
| Health visitora | 7 | PSSRU36 |
| Health visitor home visita | 12 | PSSRU36 |
| Occupational therapista | 9 | PSSRU36 |
| Physiotherapista | 9 | PSSRU36 |
| Psychiatrista | 27 | PSSRU36 |
| Psychiatric nursea | 9 | PSSRU36 |
| Psychologista | 14 | PSSRU36 |
| Counsellora | 9 | PSSRU36 |
| Speech and language therapista | 9 | PSSRU36 |
Data collection and management
Case report forms/electronic case report forms
The data set was restricted to those fields required to confirm eligibility, to describe the patient population, to describe and monitor protocol adherence, to assess primary and secondary outcomes and to enable linkage to the CMP, NHS Digital and patient follow-up (Table 2).
A dedicated, secure electronic CRF was developed to enable site research staff to enter trial data. Access to the trial database was restricted to authorised site research staff recorded on the local delegation log and centrally approved by the data manager (NH or MS). Each individual was assigned a unique username and had access to data only for patients recruited at their site.
Data management
To ensure completeness and accuracy, and to enable effective trial monitoring, data entered by authorised site research staff into the electronic CRF were regularly checked and validated by the data manager. This was carried out on an ongoing basis so that any issues could be identified by the trial team in a timely manner. The electronic CRF had built-in functionality to prevent erroneous data from being entered and checks for unusual and missing data. Other data queries were programmed using either Structured Query Language or Stata® coding (StataCorp LP, College Station, TX, USA) and fed back to site research teams via e-mail.
Data received from completed follow-up questionnaires were entered into a secure database at the ICNARC CTU, following a standard operating procedure. Identifiable data (where present) were removed and anonymised at the point of entry. Queries relating to the entry of follow-up questionnaires were reviewed by the trial manager (ARB) and data manager (NH or MS), with disagreement reviewed by a third member of the team, either the chief investigator (PRM) or health economist (ZS). To ensure accuracy, 10% of questionnaires entered by each data entry personnel were cross-checked by a second member of the ICNARC CTU team. In addition, all questionnaires with any missing EQ-5D-5L values or three or more missing IQCODE item responses were checked and verified. Any errors found were logged and corrected on the database.
Data linkage: Case Mix Programme and NHS Digital
65 trial data were linked to the CMP database by the trial statistician (KT) and, following the signing of a data-sharing agreement, to national death registrations in the Medical Research Information Service Database Administration Service, held by NHS Digital.
| Data collection | Baseline (at point of randomisation) | During critical care unit stay | End of critical care unit stay | At hospital discharge | At 90 days | At 1 yeara |
|---|---|---|---|---|---|---|
| Collected in-hospital | ||||||
| Patient details | ✓ | |||||
| Clinical/baseline data | ✓ | |||||
| MAP/vasopressors data | ✓ | ✓ | ✓ | |||
| Co-interventions data | ✓ | ✓ | ||||
| Safety monitoring data | ✓ | ✓ | ||||
| Discharge data | ✓ | |||||
| At follow-up | ||||||
| EQ-5D-5L27 | ✓ | ✓ | ||||
| IQCODE short version28 | ✓ | ✓ | ||||
| Health services/resource use | ✓ | ✓ | ||||
Governance, management and oversight
Health Research Authority and research ethics application
An application for approval by the Health Research Authority (London, UK) and ethics opinion from the South Central – Oxford C REC (Oxford, UK) was submitted on 7 March 2017. Following a meeting of the REC on 31 March 2017, Health Research Authority approval and favourable ethics opinion were both confirmed on 24 April 2017 (Integrated Research Application System number 215503 and REC reference 17/SC/0142).
Substantial amendments
Following initial favourable ethics opinion, two substantial amendments were submitted, both of which received favourable ethics opinion from the South Central – Oxford C REC. These were are follows:
-
To refine the inclusion criteria by specifying that participants must (a) have already commenced on a vasopressor infusion prior to randomisation (for at least 1 hour) and (b) if receiving noradrenaline (the most commonly used vasopressor), currently be receiving a dose of at least 0.1 µg/kg/minute (see Inclusion criteria for further details). In addition, to meet NHS Digital consent materials requirements, information sheets and consent/opinion forms were amended to include greater detail on data processing.
-
To revise the trial power calculation following a recommendation by the Trial Steering Committee (TSC) (see Final power calculation).
Local governance
Confirmation of capacity and capability was obtained from each participating NHS trust/health board prior to commencement of recruitment. In addition, a clinical trial site agreement, based on the model agreement for non-commercial research in the NHS, was signed by each participating NHS trust/health board and the sponsor (i.e. ICNARC).
Trial registration
To ensure transparency, the 65 trial was prospectively registered with the ISRCTN registry on 10 April 2017, with registration confirmed the following day (reference ISRCTN10580502).
Patient and public involvement
Two patient and public involvement (PPI) representatives (CW and DH) were trial co-investigators and members of the TMG, and both were involved in the development and management of the trial through to completion and dissemination. In addition, independent PPI representation was sought for membership of the TSC.
Monitoring
The monitoring plan followed a risk-based strategy and was guided by recruitment and protocol adherence data, with a view to visiting ≈ 25% of sites. On-site routine monitoring visits were carried out at a total of 18 (28%) sites. At these visits, barriers to or difficulties in delivering the trial were discussed and trial data/materials were reviewed. The latter included the ISF being checked for completeness, patient consent (or consultee opinion) forms were checked for all patients randomised at the time of the visit and source data verification was conducted on a random sample of patient CRFs. After the visit, a report was provided by the trial monitor to the site PIs, which summarised findings and actions required following the visit. The site PIs were responsible for resolving outstanding actions and reporting back to the ICNARC CTU. Follow-up teleconferences were arranged to resolve residual issues, if required.
Trial Management Group
The TMG was responsible for management of the trial and was led by the chief investigator (PRM) and lead clinical investigator (FL), both of whom took overall responsibility for trial delivery and oversaw progress against timelines and milestones. The TMG also comprised methodological, clinical and PPI co-investigators and members of the ICNARC CTU trial team, and met regularly throughout the trial period. The trial manager (ARB) was responsible for day-to-day management of the trial, with support from the research assistant (RD), data manager (NH or MS) and trial statistician (KT).
Trial Steering Committee
The NIHR convened an independently chaired and majority (≥ 75%) independent TSC to provide overall supervision of the trial on behalf of the funder and sponsor. The TSC was independently chaired by Professor Tim Walsh (University of Edinburgh, Edinburgh, UK) and included experienced clinicians, methodologists and a PPI representative.
Data Monitoring and Ethics Committee
The NIHR convened an independent Data Monitoring and Ethics Committee (DMEC), chaired by Professor John Norrie (University of Edinburgh), to monitor recruitment, protocol adherence and patient safety. The DMEC included experienced methodologists and clinicians.
Sponsorship
The 65 trial was sponsored by ICNARC and managed by the ICNARC CTU.
Network support
The 65 trial was adopted onto the NIHR central portfolio management system on 17 March 2017 (CPMS ID 34223) and supported by the NIHR Clinical Research Network (CRN) (division six). CRN-supported research nurses were in place across most participating sites. The NIHR CRN Critical Care Specialty Group monitored progress and presentations were held at local CRN meetings to maintain awareness. In addition, the UK Critical Care Research Group supported the trial.
Statistical analysis
Analysis principles
All analyses adhered to the intention-to-treat principle. Patients were analysed according to the initial treatment assignment, irrespective of whether or not the allocated treatment was received. All patients for whom the primary outcome was known were included in the primary analysis, regardless of protocol adherence. All statistical tests were performed for superiority and were two sided with significance set at p < 0.05. Effect estimates are reported with 95% confidence intervals (CIs). There was no adjustment for multiple testing. The results of subgroup analyses were interpreted, taking into account accepted criteria for credible subgroup effects. 37,38
Power calculations
Original power calculation
In the original approved protocol, the sample size was calculated as follows: assuming 90-day mortality of 35% in the usual-care group (based on CMP data for patients aged ≥ 65 years admitted to critical care and receiving advanced cardiovascular support), a sample size of 1402 patients provided 90% power to detect as statistically significant (p < 0.05) an 8% absolute risk reduction to 27%. Allowing for 2.5% withdrawal/loss to follow-up, we aimed to recruit a total of 1440 patients.
Final power calculation
In a substantial amendment to the protocol (from version 2.0 to 3.0), the expected absolute risk reduction was changed from 8% to 6% (i.e. an expected 90-day mortality of 29% in the intervention group, with all other parameters remaining unchanged), leading to a revised sample size of 2600 patients (1300 patients per group).
This change was recommended by the TSC after the internal pilot feasibility assessment noted that the recorded duration of vasopressors in the usual-care group was lower than expected, suggesting that the difference in treatment (and hence outcome) between arms may be smaller than initially anticipated.
Internal pilot
A feasibility assessment was conducted after the end of the internal pilot (i.e. at the first 6 months of the trial recruitment period) against the following progression criteria:
-
separation between groups of 10 mg (noradrenaline equivalent) in mean total vasopressor dose and/or a separation of 5 mmHg in peak MAP while receiving vasopressors
-
a minimum of 50 sites open to recruitment
-
the recruitment rate in open sites is at least 80% of the level anticipated.
Interim analyses
A single interim analysis of 90-day mortality was performed following the recruitment and follow-up to 90 days of 500 patients and reviewed by the DMEC. The interim analysis was conducted using a Haybittle–Peto stopping rule (p < 0.001) to guide recommendations for early termination due to either effectiveness or harm. The trial statistician, senior statistician and DMEC were not blinded to treatment allocation. All other investigators remained unaware of the results of the interim analysis, other than the recommendation of the DMEC to continue or to terminate recruitment.
Clinical effectiveness analysis
Timing of final analysis
The end of the trial was when the final patient recruited had completed their 90-day follow-up questionnaire. Following the end of the trial, any patients remaining in follow-up were censored, the trial database was locked and the final analysis was conducted.
Timing of outcome assessments
The timings of all outcome assessments were taken relative to the date of randomisation.
Screening data
Based on data from screening and enrolment logs, the following summaries are presented:
-
the total number of days of screening (calculated as the sum of the number of days of screening at each site)
-
the number of screened patients
-
the number of eligible patients (and per cent of patients screened)
-
the number of recruited patients (and per cent of patients eligible) and reasons for non-recruitment, where known.
The recruitment rate per site per month, defined as number of recruited patients/(total number of days screening × 12/365), was calculated both overall and by site and summarised across sites by the median [interquartile range (IQR)].
Recruitment and consent data
A CONSORT (Consolidated Standards of Reporting Trials) flow diagram39 was used to summarise the patient flow as follows:
-
The number of patients aged ≥ 65 years with vasodilatory hypotension and receiving vasopressors screened, including the number of patients who:
-
did not meet inclusion criteria (with reasons)
-
met an exclusion criterion (with reasons)
-
were eligible but did not undergo randomisation (with reasons).
-
-
The number of patients randomised.
-
The number of patients included in the primary outcome analysis (with reasons for those not included).
-
The numbers of patients returning a complete follow-up questionnaire at 90 days and 1 year.
The number and percentage of patients who had capacity at randomisation and gave consent was reported for each treatment group. Subsequent consent procedures were summarised in a flow diagram that included the following information for each treatment group.
-
For all patients:
-
whether or not a consultee (personal or nominated) was approached
-
whether or not the patient regained capacity prior to a consultee being approached.
-
-
For those where a consultee was approached:
-
whether or not the consultee gave agreement to continue trial participation, for access to medical records for ongoing data collection and to receive follow-up questionnaires or any other outcome of the approach
-
whether or not the patient regained capacity before hospital discharge.
-
-
For those who regained capacity:
-
whether or not the patient gave consent to continue trial participation, for access to medical records for ongoing data collection and to receive follow-up questionnaires or any other outcome of the approach.
-
-
For those discharged prior to consent/opinion being confirmed in hospital, the telephone/postal approach for consent/opinion was summarised.
Patients for whom consent was not given for continued trial participation (e.g. trial treatment) were included in the analysis of the primary outcome and all other secondary end points (unless otherwise specified).
Patients for whom consent was not given to receive follow-up questionnaires had missing data imputed so that they could be included in the analysis of cognitive decline and HRQoL at 90 days and 1 year (if known to be alive at these time points). These patients were also included in the analysis of all other end points (unless otherwise specified).
Patients for whom consent was not given for accessing their medical records for ongoing data collection were included in the reporting of baseline characteristics and trial treatment (as these data were gathered directly from source on the CRF), but not included in those end points that were collected using data retrospectively obtained from linked data sets (i.e. duration and days free from organ support, duration of unit and hospital stay) and were not included in the analysis of patient-reported outcomes (as these outcomes are reported only for patients known to be alive from medical records). The patients were censored for mortality end points on the date their consent was withheld.
Baseline patient characteristics
The following baseline demographic and clinical data were summarised for each treatment group, but not subjected to statistical testing:
-
demographics –
-
age [mean (SD)]
-
sex (male, female) [number (%)].
-
-
comorbidities [number (%)] –
-
chronic hypertension (yes, no)
-
chronic heart failure (yes, no)
-
atherosclerotic disease (yes, no)
-
chronic renal replacement therapy at ICU admission (yes, no).
-
-
dependency prior to admission to acute hospital (e.g. able to live without assistance in daily activities, minor/major assistance with daily activities, total assistance with all daily activities) [number (%)]
-
location prior to admission to critical care and urgency of surgery (e.g. emergency department/not in hospital, theatre elective/scheduled surgery, theatre emergency/urgent surgery, other critical care unit, ward or intermediate care area) [number (%)]
-
acute severity of illness from first 24 hours following admission to the unit –
-
Acute Physiology and Chronic Health Evaluation II (APACHE II) score40 [mean (SD)]
-
ICNARC physiology score41 [mean (SD)]
-
ICNARCH–2015 model predicted risk of death42 [median (IQR)]
-
Sepsis-343,44 (no sepsis, sepsis, septic shock) [number (%)] (note that Sepsis-3 criteria specify that there must be evidence of infection and two or more points on the Sequential Organ Failure Assessment score and this categorisation is based on data from the first 24 hours following admission to the ICU).
-
-
MAP (mmHg) at randomisation [mean (SD)]
-
vasopressor infusions received at randomisation [number (%)] –
-
none [patients in this category were eligible for recruitment prior to version 2.0 of the protocol if a decision had been taken to start vasopressors or if they had received vasopressors in the form of metaraminol or terlipressin boluses (see Inclusion criteria)]
-
noradrenaline equivalent < 0.1 µg/kg/minute
-
noradrenaline equivalent ≥ 0.1 µg/kg/minute
-
metaraminol
-
other/combination.
-
-
duration of vasopressor infusion prior to randomisation (minutes) [median (IQR)].
Protocol adherence
Exposure
Exposure to the intervention was assessed by the following parameters, calculated for each treatment group:
-
MAP – mean (SD) and median (IQR) of the (1) highest and (2) mean MAP for each patient while receiving vasopressors, and difference in means with 95% CI
-
receipt of vasopressors – the number and percentage of patients receiving each vasopressor either as a continuous infusion or bolus (noradrenaline, adrenaline, dopamine, phenylephrine, vasopressin, metaraminol, terlipressin)
-
duration of vasopressors – mean (SD) and median (IQR) of the total duration (hours) from the later of the time of randomisation or time of initiation of vasopressors to the end of the first episode of vasopressors (defined as the start of a 24-hour period during which the patient received no vasopressors), critical care discharge or death (whichever comes first), and difference in means with 95% CI
-
dose/rate of vasopressors when given as a continuous infusion – mean (SD) and median (IQR) of the (1) highest and (2) mean rate of noradrenaline equivalents (µg/kg/minute) and metaraminol (mg/hour), and difference in means with 95% CI
-
total dose of vasopressors (from either infusion or bolus) – the median (IQR) among patients receiving the relevant vasopressor(s) and mean (SD) among all patients (including those not receiving the vasopressors with a value of zero) of the total dose (mg) of vasopressors for (1) noradrenaline, adrenaline, dopamine, phenylephrine and vasopressin combined, expressed as noradrenaline equivalent (see below), (2) metaraminol and (3) terlipressin, and difference in means with 95% CI
-
total number of episodes of vasopressor treatment (recommencing vasopressors after 24 hours without vasopressor treatment defines the start of a new episode) – mean (SD) and median (IQR) of the number of vasopressor treatment episodes at critical care discharge, and difference in means with 95% CI
-
total number of days on vasopressors at critical care discharge – mean (SD) and median (IQR) of the total number of days on vasopressors, and difference in means with 95% CI
-
fluid balance – mean (SD) and median (IQR) of fluid balance (ml), measured as the cumulative sum of daily fluid balance during the first episode of vasopressor treatment
-
urine output – mean (SD) and median (IQR) of the mean daily urine output (ml/kg/hour) during the first episode of vasopressor treatment.
The distribution across patients of the daily values of the following parameters in each group was presented in the form of box and whisker plots for days 1–7 following randomisation among all patients receiving vasopressors on that day:
-
MAP – (1) highest and (2) mean MAP for each patient while receiving vasopressors
-
dose/rate of vasopressor infusion – (1) highest and (2) mean rate of noradrenaline equivalents (µg/kg/minute) and metaraminol (mg/hour)
-
daily fluid balance (ml)
-
daily urine output (ml/kg/hour).
The numbers of patients included on each day was reported at the foot of the figure. Patients were included in these summaries only if they had recorded treatment data up to discontinuation or death on treatment. Time to discontinuation of vasopressors was illustrated using Kaplan–Meier curves by group, with time measured in hourly intervals from randomisation (rounded down to the nearest whole hour). Time of discontinuation was defined as the start of the first period of 24 consecutive hours not on vasopressors, or the time of death for patients who died on treatment without having achieved 24 continuous hours free of vasopressors. Patients without recorded treatment data up to discontinuation or death on treatment were censored at the time of last recorded treatment.
Noradrenaline-equivalent doses were calculated using the two alternate conversion methods (Table 3).
| Vasopressor | Unit | Conversion factor for noradrenaline equivalent |
|---|---|---|
| Method 145 | ||
| Adrenaline | µg/kg/minute | × 1 |
| Dopamine | µg/kg/minute | /150 |
| Phenylephrine | µg/kg/minute | × 0.1 |
| Vasopressin | U/minute | × 2.5 |
| Method 246 | ||
| Adrenaline | µg/kg/minute | × 1 |
| Dopamine | µg/kg/minute | × 0.01 |
| Phenylephrine | µg/kg/minute | × 0.45 |
| Vasopressin | U/minute | × 5 × 100/weight (kg) |
Data on vasopressor infusions were collected on an hourly basis. Accordingly, to calculate total dose, each recorded infusion episode was assumed to last for exactly 1 hour. Analysis using the calculations from method 145 of Table 3 were used for the main results paper for this trial and corresponding analysis using calculations from method 246 was used in a sensitivity analysis.
A number of different exploratory graphical approaches were used to further visually summarise treatment pathways by arm. These did not incorporate any formal statistical comparisons beyond those specified in this statistical analysis plan.
Protocol deviations
Failure to discontinue vasopressors or reduce the dose/rate once MAP was above the upper limit of the MAP target range (i.e. 65 mmHg) for at least 3 consecutive hours in the permissive hypotension group defined a potential protocol deviation (with no treatment protocol deviation defined in the usual care group). Potential protocol deviations, identified from the trial data, triggered a query to the participating site, which had the opportunity to provide a justification. In some cases, the TMG determined that the event did not constitute a protocol deviation (e.g. MAP values may have been above range only transiently on the hour but within range between the hourly recordings in the trial data). The total number of such events that were decided not to constitute a deviation is reported.
Likewise, the number and percentage of patients with at least one protocol deviation in the permissive hypotension group is reported. Adherence was defined at the patient level as not having experienced any protocol deviation.
For each patient in the permissive hypotension group, the following measures of protocol adherence were also calculated: total time on vasopressors with recorded MAP within target range, total time on vasopressors with recorded MAP above target range, total time on vasopressors with recorded MAP of > 5 mmHg above upper limit of target and total time on vasopressors with recorded MAP below target range. These measures were summarised as mean (SD) or median (IQR) in patients with recorded treatment data up to discontinuation or death on treatment.
Co-interventions
The following parameters were calculated for each treatment group in patients with recorded treatment data up to discontinuation or death on treatment:
-
receipt of inotropes – the number and percentage of patients receiving inotropes (i.e. any of dobutamine, milrinone or levosimendan) at any time during the first recorded episode of vasopressor treatment
-
receipt of corticosteroids – the number and percentage of patients receiving corticosteroids at any time during the first recorded episode of vasopressor treatment.
Serious adverse events
The numbers of SAEs and number and percentage of patients experiencing each SAE following randomisation until critical care discharge is reported in each treatment group. The total number of patients experiencing one or more SAE is compared between groups using Fisher’s exact test.
Withdrawal/follow-up
The number and percentage of patients withdrawing consent (or consultees withdrawing agreement) to trial participation is reported in each group, with reasons provided. Data collected up until the point of withdrawal are included in the analysis, but no further data after the date of withdrawal were collected for that patient.
The number and percentage of patients lost to follow-up for mortality at 90 days (as a percentage of all randomised patients) and for questionnaire outcomes (as a percentage of survivors) at 90 days and 1 year are reported for each group. The total lost to follow-up for mortality includes consented patients for whom data are unavailable (i.e. true loss to follow-up), those who withdrew before 90 days and those for whom consent to access medical records for ongoing data collection was refused before 90 days.
The total lost to follow-up for 90-day questionnaire follow-up includes consented patients for whom data are unavailable (i.e. true loss to follow-up), those who withdrew and those for whom consent to receive questionnaires was never given. The baseline characteristics (as described in Baseline patient characteristics) of patients completing a follow-up questionnaire at each time point were compared with those of patients who did not complete a follow-up questionnaire who were known to be alive at that time point. The same approach was taken for 1-year questionnaires (note that 1-year follow-up is truncated).
Analysis methods
The primary outcome of number and percentage of deaths by 90 days following randomisation is reported. The primary effect estimate is the absolute risk reduction, reported with a 95% CI. The relative risk is also reported. Deaths by 90 days following randomisation are compared between the groups, unadjusted and using Fisher’s exact test. Owing to the anticipated low amount of clustering, unadjusted analyses did not take account of site-level effects.
An analysis, adjusted for baseline data, was also conducted using multilevel logistic regression with a random effect of site. Baseline variables adjusted for in the multilevel logistic regression model were:
-
age (linear)
-
sex
-
comorbidities
-
dependency prior to admission to acute hospital
-
location prior to admission to critical care and urgency of surgery
-
ICNARC physiology score (linear)41
-
Sepsis-344
-
vasopressors received as a continuous infusion at randomisation
-
duration of vasopressors prior to randomisation (linear).
(All categorical variables are defined and grouped as previously described under Baseline patient characteristics.)
Baseline variables were selected for inclusion in the adjusted analysis according to anticipated relationship with outcome. The results of the multilevel logistic regression model are reported as an adjusted odds ratio with 95% CI. The unadjusted odds ratio is presented for comparison.
The primary outcome (i.e. 90-day mortality) is analysed by the following prespecified patient subgroups:
-
age (linear)
-
chronic hypertension (yes, no)
-
chronic heart failure (yes, no)
-
atherosclerotic disease (yes, no)
-
predicted log-odds of acute hospital mortality from the ICNARCH–2015 risk prediction model (linear)42
-
Sepsis-344
-
vasopressors received at randomisation (see Baseline patient characteristics).
These analyses tested for an interaction between the subgroup categories (or subgroup variable for linear interactions) and the treatment group in a multilevel logistic regression model, adjusted for the same baseline variables as the primary analysis. For linear interactions, the interaction effect was illustrated by calculating the adjusted odds ratio within five categories at quintiles of the continuous variable. 48
The primary analysis was repeated, adjusting for adherence to allocated intervention (i.e. binary variable equal to zero for all patients allocated permissive hypotension with one or more recorded protocol deviation, and 1 for all other patients) and using a structural mean model with an instrumental variable of allocated treatment to estimate the complier-average causal effect of treatment. 49
An additional sensitivity analysis was performed, repeating the primary analysis in the subset of patients who would have been eligible for the trial following the inclusion criteria as defined in the protocol amendment to version 2.0 (i.e. patients restricted to those who had started vasopressors between 1 and 6 hours prior to randomisation, and excluding any patients who were receiving only noradrenaline at randomisation at a dose level of < 0.1 µg/kg/minute).
Secondary outcomes are reported by treatment group. Continuous outcomes are reported using either mean and SDs (for duration of advanced respiratory support for all patients, duration of renal support for all patients, number of days alive and free of advanced respiratory support to day 28, number of days alive and free of renal support to day 28, and IQCODE scores at 90 days and at 1 year) or median and IQR (for duration of advanced respiratory support in patients who received it, duration of renal support in patients who received it, and duration of critical care and acute hospital stay). Unadjusted comparisons of continuous outcomes are made using t-tests or Wilcoxon rank-sum tests (comparisons for duration of stay were stratified by survival status at discharge). Adjusted comparisons (for all continuous variables excluding duration of stay) are made using multilevel linear regression, adjusted for the same baseline variables as the adjusted analysis of the primary outcome, using bootstrapping to account for anticipated non-normality in the distribution. 50
Binary outcomes (i.e. mortality at discharge from critical care unit and acute hospital) are reported using numbers and percentages. Unadjusted comparisons were made using Fisher’s exact test and adjusted comparisons using multilevel logistic regression (adjusted for the same baseline variables as the adjusted analysis of the primary outcome).
Time-to-event outcomes (i.e. duration of survival to longest available follow-up) are reported using Kaplan–Meier curves and compared using the log-rank test. An adjusted comparison was performed using a Cox proportional hazards model adjusted for the same baseline variables as the primary analysis, with shared frailty at the site level.
A subgroup analysis of the in-hospital secondary outcomes was performed to compare unadjusted and adjusted secondary outcomes in those patients who did/did not have chronic hypertension at baseline.
Handling of missing data
The number of missing clinical primary outcome data was anticipated to be small, but is accounted for in a sensitivity analysis. The primary analysis was repeated once, assuming that all patients in the intervention group with missing outcomes survived and all patients in the usual-care group with missing outcomes did not survive. The analysis was then repeated with the opposite assumptions. This gives the absolute range of how much the results could change if the data were complete.
Analysis of cognitive decline at 90 days and 1 year was carried out once, using only patients with non-missing data (defined as having no more than three missing items from the 16-item IQCODE) and then repeating with missing data imputed among patients known to be alive at those time points, excluding those who did not consent to access of their medical records. Where necessary, missing data in baseline variables included in the adjusted models were also imputed.
Multiple imputation was undertaken using the multivariate imputation using chained equations algorithm, with the model including all baseline variables included in the adjusted models and all outcome variables. Twenty multiply imputed data sets were generated. Models were fitted in each imputed data set and results combined using Rubin’s rules. 51
Expert elicitation methods
The 65 trial primary analysis uses the assumption that missing HRQoL outcomes are ‘missing at random’ (MAR). This assumes that the probability that a patient’s outcome is missing is not dependent on the outcome of the patient after conditioning using observed variables, for example the patient’s characteristics at baseline. Sensitivity analysis that allows patients’ predicted health state to affect the probability that they return their questionnaire was carried out. For example, we may expect that patients who are in a relatively good state of health may be more likely to complete and return the HRQoL questionnaire, and this would mean that these outcome data may be ‘missing not at random’ (MNAR). The steps in the expert elicitation framework52 were followed for this additional analysis. These steps included (1) scoping a 65 trial-specific elicitation exercise, (2) development of an elicitation tool (including questions about the HRQoL outcomes), (3) eliciting expert opinion, (4) evaluating the elicitation results and (5) carrying out the sensitivity analysis, incorporating the elicited expert information.
To model the MNAR data53 fully, Bayesian pattern-mixture models were used, which allow calculation of a patient’s outcome differently, depending on whether the outcome is observed (pattern 1) or missing (pattern 2):
-
The observed data were used to calculate the outcome using the same statistical model as specified for the primary analysis.
-
A specified offset (sensitivity parameter) from the mean of the observed data was used to adjust this model. This sensitivity parameter is able to alter by treatment. 54
As some patients did not complete and return a HRQoL questionnaire, which lead to missing outcomes, we can interpret an offset as the HRQoL difference between these two groups of patients (i.e. those who did and did not return a questionnaire). As we cannot estimate the offsets from the observed data, expert opinion about patients’ likely HRQoL values is needed to inform the prior distributions of these parameters. For all other unknown parameters specified in the model, minimally informative priors were used.
Pattern-mixture models for the HRQoL and the cost-effectiveness analysis (CEA) were fitted using WinBUGS version 1.4.3 (MRC Biostatistics Unit, Cambridge, UK) (HRQoL) and JAGS software version 4.3.0. 55 In the HRQoL models, the random effects were not hierarchically centred, which improved the mixing of the Markov chain Monte Carlo chains. For the CEA, there were no random effects and the QALYs and costs were modelled jointly, consistent with the primary analysis. The costs include intervention and hospital costs, but exclude health services questionnaire costs. There are no missing costs.
Shiny, a web application framework within the statistical software R (The R Foundation for Statistical Computing, Vienna, Austria), was used to create a 65 trial-specific elicitation tool. This tool was based on the tool that was developed for the POPPI [Psychological Outcomes following a nurse-led Preventative Psychological Intervention for critically ill patients (POPPI)] trial. 56 The tool was updated iteratively during a piloting phase.
The experts’ opinions were able to be represented as a split normal distribution (truncated) by utlising three individual sliders that indicate the following: (1) which they thought the most likely value (mode), and (2 and 3) their uncertainty about the ‘most likely’ value (left and right SD). Experts were asked to imagine a group consisting of 100 patients included in the 65 trial, each patient had the same defined baseline characteristics (i.e. male, an APACHE II score equal to 19 and aged 74 years), was allocated to the usual-care group and had completed/returned a questionnaire. The experts were then shown a scale that indicated for this group of 100 patients, based on early trial data, our best estimate of the average (indicated with an arrow). The experts were then prompted to indicate their thoughts about the possible average score for an additional five groups that consisted of 100 patients who were similar to the index group except:
-
Group 1 – patients aged ≥ 10 years.
-
Group 2 – patients who were female.
-
Group 3 – patients who had an APACHE II score of 26.
-
Group 4 – patients who failed to complete/return a questionnaire.
-
Group 5 – patients allocated to the permissive hypotension group who failed to complete/return a questionnaire.
Groups 1–3 allowed assessment of how closely an expert’s elicited values are calibrated with outcomes that are known empirically. Groups 4 and 5 informed sensitivity parameter priors.
Once this step was completed, graphical feedback was provided that indicated any differences/overlap in their opinions about groups 4 and 5. If this indicated unreasonable answers, the expert was then given the option of revising their answers. Experts were then given additional new information about group 4 (i.e. usual-care group patients) and asked to reconsider the answers they had given for group 5 (i.e. permissive hypotension group patients). This allowed for the prospect that the elicited values for usual-care group patients who failed to complete/return a questionnaire were related to the permissive hypotension group patients who also failed to complete/return a questionnaire. This enabled construction of a joint distribution for each groups’ sensitivity parameters, allowing correlation between the groups. Important context to understand experts’ reasons for their views was collected using free-text question fields. This also enabled us to assess the reliability of experts’ responses to the previous questions.
The HRQoL scores were shown on a numeric scale from –28 to 100 (original EQ-5D-5L utility score scale multiplied by 100), anchored at 0 for ‘death’ and 100 for ‘perfect health’. A HRQoL score is calculated from a patient’s answers to five questions about (1) mobility, (2) self-care, (3) usual activities, (4) pain or discomfort and (5) anxiety or depression. Initially, an arrow shows the point on the scale linked to all the answers being ‘no problems’ (100). Using drop-down menus with the five available levels of severity, the expert may select other combinations of answers. The arrow then moves to show how the HRQoL score changes.
The 65 trial chief investigator (PRM) e-mailed contacts at the participating sites, inviting them to identify individuals who are involved in long-term patient follow-up. ICNARC then sent these individuals a PIS and a link to the elicitation tool. A maximum of two reminders to complete the questionnaire were sent via e-mail, with consent taken electronically as part of the elicitation tool.
Two statisticians (AJM and DH) independently examined the elicited information using prespecified criteria to identify those experts whose responses were ‘usable’. Of the usable responses from experts, responses were further categorised as ‘high’ and ‘very high’ in confidence (separated into two distinct subgroups). This was designed to identify any expert who had clearly misunderstood the exercise. Consistency in both quantitative and qualitative responses was required to be included within the high-confidence group. When agreement was observed between the expert’s views about groups 1–3 and the ‘truth’ (based on trial data), the expert was included within the very high-confidence group. The categorisations of the two statisticians were compared and, through discussion, discrepancies were resolved.
Following the approach discussed in Mason et al. ,52 we used a combination of pooled and individual priors to fully explore the sensitivity of the trial results to a range of expert opinion. The individual priors were selected from the very high-confidence subgroup.
We ran all models using two chains initialised to diffuse starting values to produce a sample of 100,000 after convergence for posterior inference. Convergence was assumed if the Gelman–Rubin convergence statistic57 for individual parameters was < 1.05 and a visual inspection of the trace plot for each parameter was satisfactory. The results from the Bayesian MNAR sensitivity analyses are compared with Bayesian MAR and complete-case analysis.
Statistical software
The analyses were conducted in Stata/SE version 14.2. Other packages, such as R, were used for specific analyses.
Health economics analysis
Statistical analysis of cost-effectiveness analysis at 90 days
Following recent NICE recommendations, the CEAs adopted a NHS and Personal Social Services perspective, and reported costs and QALYs. The EQ-5D-5L measure has been used for assessing the health status of each trial participant, which was valued using the valuation set for England32 to calculate the EQ-5D-5L index score. The QALY gain was calculated by combining survival data with EQ-5D-5L index scores. The corresponding INMB was estimated by valuing incremental QALYs at the NICE-recommended threshold (i.e. £20,000) for a QALY gain and subtracting incremental costs.
In line with best practice for the analysis of RCTs, the CEA followed the intention-to-treat principle58 and reported incremental costs, QALYs and cost-effectiveness up to 90 days, according to randomised group. Missing data are a common occurrence within RCTs, but often health economic analyses alongside RCTs fail to apply appropriate methods to address missing data. In this analysis, missing data in baseline covariates, resource use and outcomes (Table 4) were handled with multivariate imputation by chained equation. 60 Under this approach, each missing variable was imputed conditional on fully observed baseline variables (such as age, sex, chronic hypertension, chronic heart failure, atherosclerotic disease, dependency prior to admission to acute hospital, source of admission, ICNARC model physiology score, sepsis, septic shock, vasopressors received at randomisation, duration of vasopressor infusion prior to randomisation) and all other imputed variables. Missing data from follow-up EQ-5D-5L and health services questionnaires were imputed from patients who were eligible and who had fully completed those questionnaires. Missing/incomplete EQ-5D-5L scores of patients who did not return or fully complete the EQ-5D-5L questionnaire administered at 90 days were imputed from those survivors who did fully complete the questionnaire. Similarly, for those eligible patients who did not return the health services questionnaire, information on the use of health services up to 90 days post randomisation was imputed from those patients who completed this questionnaire. The CEA used bivariate seemingly unrelated regression model, which is a system of unrelated regression equations on the costs and effectiveness (QALY) component of the analysis, allowing for correlation between costs and QALYs. The economic analysis adjusted for same baseline covariates as for the clinical analysis to adjust for baseline imbalances between the randomised arms (see Clinical effectiveness analysis).
| Variable | Missing values in patients with known primary outcome, n (%) | Imputation model |
|---|---|---|
| Patient-level covariatesa | ||
| Allocated treatment group | 0 (0) | None required |
| Age | 0 (0) | None required |
| Sex | 9 (< 0.1) | Logistic regression |
| Chronic hypertension | 0 (0) | None required |
| Chronic heart failure | 1 (< 0.1) | Logistic regression |
| Atherosclerotic disease | 0 (0) | None required |
| Dependency prior to admission to acute hospital | 23 (< 0.1) | Multinomial logistic regression |
| Source of admission | 6 (< 0.1) | Multinomial logistic regression |
| ICNARC physiology score | 12 (< 0.1) | Predictive mean matching |
| Sepsis/septic shock | 9 (< 0.1) | Logistic regression |
| Vasopressors received at randomisation | 35 (1.4) | Multinomial logistic regression |
| Duration of vasopressor infusion prior to randomisation | 71 (2.9) | Predictive mean matching |
| Outcomes and resource use at 90 daysb | ||
| IQCODE score | 458 (18.6) | Predictive mean matching |
| EQ-5D-5L health utility | 444 (18.0) | Predictive mean matching |
| Health services questionnaire costs | 766 (31.2) | Predictive mean matching |
| Outcomes and resource use at 1 yearc | ||
| IQCODE score | 302 (15.1) | Predictive mean matching |
| EQ-5D-5L health utility | 309 (15.4) | Predictive mean matching |
| Health services questionnaire costs | 445 (22.3) | Predictive mean matching |
Analysis of uncertainty and sensitivity in cost-effectiveness
The incremental costs and QALYs were estimated using a single-level bivariate seemingly unrelated regression model. To express the uncertainty in the estimation of the incremental costs and QALYs, we generated 800 estimates of incremental costs and QALYs from the joint distribution of estimated incremental costs and QALYs from single-level regression model, assuming asymptotic normality. These incremental costs and QALYs were then plotted on the cost-effectiveness plane to summarise the joint uncertainties in incremental costs and QALYs. 61 From the joint distribution of incremental costs and QALYs, we have derived the cost-effectiveness acceptability curves by calculating the probability that, compared with usual care, the permissive hypotension intervention is cost-effective, given the data, at alternative levels of willingness to pay for a QALY gain.
The main assumptions made in the base-case scenario and how each was relaxed in sensitivity analyses are detailed below and summarised in Table 5.
| Assumption | Base case | Sensitivity analysis |
|---|---|---|
| HRQoL valuation | EQ-5D-5L value set was applied | Crosswalk (between EQ-5D-3L and EQ-5D-5L) value set was applied |
| Readmissions from the health services questionnaire | Included in the analysis | Excluded from the analysis |
| Distributional assumptions | Costs and QALYs normally distributed | Costs and QALYs gamma distributed |
Health-related quality of life from crosswalk value set
The base-case HRQoL was valued using EQ-5D-5L value sets for England defined by the EQ-5D-5L descriptive system. 32 An alternative approach to using the EQ-5D-5L value sets is to use validated mapping function to derive utility values for the EQ-5D-5L from the more traditional EuroQol-5 Dimensions, three-level version, value set. 62 To examine the sensitivity of the results to the choice of approach in the sensitivity analysis, HRQoL was estimated by mapping the five-level descriptive system onto the three-level valuation system using the mapping function developed by van Hout et al. 30,63
Readmissions from the health services questionnaire
The base-case analysis included readmissions to critical care recorded on the CMP database and also those recorded from responses to the health services questionnaire. The readmission costs to critical care could thereby be double counted. To consider the possible impact of double counting the same readmissions across both data sources, we have included only readmissions from the CMP database in the sensitivity analysis.
Distributional assumptions for costs and quality-adjusted life-years
The base-case regression model assumed that costs and QALYs were normally distributed. In sensitivity analyses, we assessed the robustness of the cost-effectiveness results to alternative distributional assumptions about both cost-effectiveness end points. Following methodological guidance,30,58 a gamma distribution for costs and QALYs was considered in the sensitivity analysis. Gamma distribution was considered in the sensitivity analysis because costs had a right-skewed distribution64 and QALYs had anticipated large proportion of decedents with zero QALYs, and the remainder of the distribution was right skewed.
The results of the sensitivity analysis were reported as mean INMBs with corresponding 95% CIs.
Cost-effectiveness analysis by subgroups at 90 days
Prespecified subgroup analyses were conducted for the same subgroups that were considered in the clinical effectiveness evaluation (see Analysis methods). The above seemingly unrelated regression models were expanded to include subgroup by randomised arm interaction terms65 and used to report INMB at 90 days by subgroups.
Cost-effectiveness outcomes at 1 year
Use of health-care resources (e.g. critical care, general medical length of stay, outpatient and community care) between 90 days and 1 year was measured using readmission information from the CMP and health services questionnaires at 1 year. Total costs at 1 year were estimated by valuing resource with appropriate unit costs. Life-years up to 1 year was reported using the follow-up survival data. HRQoL data up to 1 year were combined with survival data to report QALYs at 1 year. For patients surviving up to 1 year, we used EQ-5D-5L responses at 1 year, assuming a linear interpolation between the EQ-5D-5L scores at 90 days and 1 year. For decedents between 90 days and 1 year, where an EQ-5D-5L score at 90 days was available, a linear interpolation was applied between the 90-day EQ-5D-5L and the date of death (when a zero EQ-5D-5L score was applied).
As not all randomised patients were followed up to 1 year, their survival, resource use and HRQoL data were censored. Any administrative censoring at 1 year of resource use, survival and HRQoL was assumed at random. The statistical analysis of CEA end points at 1 year followed the same approaches that are outlined for the 90 days end point.
Chapter 3 Results
Sites and patients
Site selection
Relative to the target of 65 sites, expressions of interest to participate were received from 124 NHS adult general critical care units across England, Wales and Northern Ireland. Of these, 101 completed a site feasibility questionnaire and were considered for participation by the ICNARC CTU. A total of 67 sites were invited to participate and were chosen based on a variety of factors, including geography, good research track record in previous multicentre RCTs, display of enthusiasm for the trial and adequate research nurse support. Two selected sites ultimately did not open (one had two critical care units within their trust and focused resources on delivering the trial in one and the other had a change in local priorities, limiting research participation).
Site set-up
The full site target was reached, with all 65 sites recruiting patients into the 65 trial. The first sites opened in July 2017, 2 months ahead of schedule, with the final site opening in September 2018 (Figure 2). By the end of the internal pilot, 59 sites were open to recruitment.
FIGURE 2.
Actual vs. expected number of sites opening to recruitment. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
From a research governance perspective, the median time from provision of the final local information pack to the issuing of local confirmation of capacity and capability was 70 (IQR 44–129) days. The median time from local confirmation of capacity and capability to the start of patient screening at sites was 20 (IQR 5–49) days. The median time from the start of patient screening to the first patient recruited at sites was 9 (IQR 3–21) days. Overall, the whole process from site selection to first patient recruited took a median of 132 (IQR 83–169) days.
Each site participated in the 65 trial for a median of 18 (IQR 17–19) months. Of the 65 sites that opened, seven were closed early. Reasons for closing early included lack of equipoise (n = 2), lack of research nurse resources (n = 2), low recruitment (n = 2) and reaching the contractual recruitment target and choosing to discontinue screening (n = 1).
Patient screening, randomisation and consent
Screening and randomisation
Between 3 July 2017 and 16 March 2019, a total of 10,755 patients aged ≥ 65 years receiving vasopressors for vasodilatory hypotension were screened across the 65 sites (Figures 3 and 4). Of these patients, just under 40% (n = 4271) did not meet inclusion criteria. Of those patients meeting inclusion criteria, just under 50% (n = 3066) met one or more exclusion criteria [the most common being ongoing treatment for brain injury (n = 1033) and death perceived as imminent (n = 690)]. Around 5% (n = 330) of eligible patients did not undergo randomisation. Reasons for not randomising eligible patients ranged mainly from clinician decisions (e.g. some decided that certain patients required a higher MAP target) to very few patient decisions (e.g. a small number of patients with capacity prospectively declined participation). As a result, 2600 patients were randomised (Table 6) and after excluding two duplicates (two patients were randomised twice and only the first randomisation was kept for analysis) there were 1291 patients allocated to the permissive hypotension group and 1307 patients allocated to the usual-care group.
FIGURE 3.
A CONSORT (Consolidated Standards of Reporting Trials) flow diagram of screening, randomisation and follow-up through the 65 trial. a, Patients are included in more than one category if they met more than one exclusion criteria. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
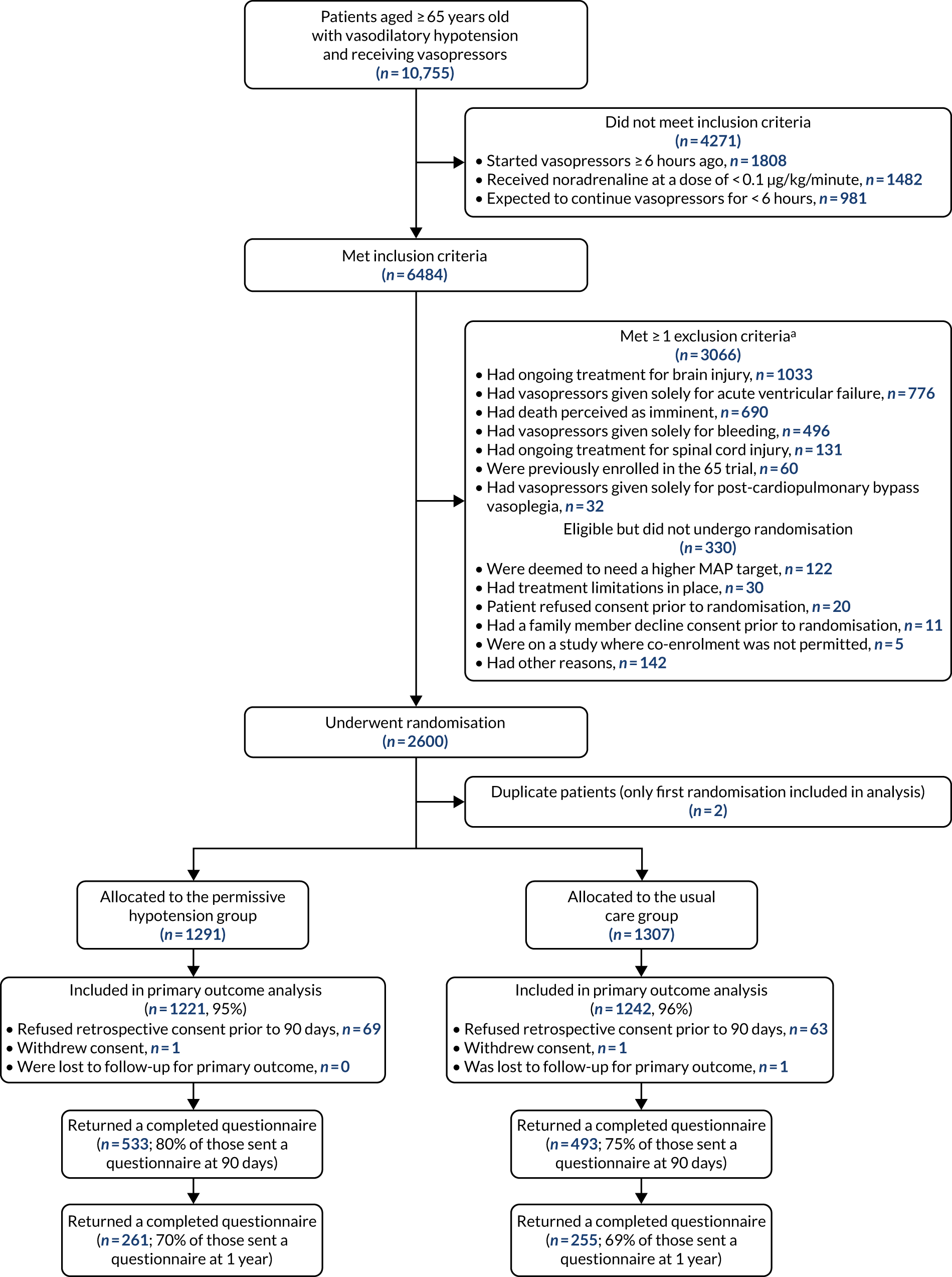
FIGURE 4.
Recruitment over time: actual and pre-trial estimate. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
| Site | Month opened | Annual critical care unit admissions, na | Overall recruitment (N = 2600), n |
|---|---|---|---|
| Dorset County Hospital (Dorset, UK) | July 2017 | 668 | 32 |
| Musgrove Park Hospital (Taunton, UK) | July 2017 | 721 | 70 |
| Peterborough City Hospital (Peterborough, UK) | July 2017 | 609 | 50 |
| Poole Hospital (Poole, UK) | July 2017 | 635 | 72 |
| Salford Royal Hospital (Salford, UK) | July 2017 | 1914 | 20 |
| King’s College Hospital (London, UK) | July 2017 | 1524 | 21 |
| Russells Hall Hospital (Dudley, UK) | July 2017 | 839 | 96 |
| The York Hospital (York, UK) | July 2017 | 976 | 15 |
| University Hospital Lewisham (London, UK) | July 2017 | 684 | 66 |
| Queen Alexandra Hospital (Portsmouth, UK) | July 2017 | 1115 | 80 |
| Royal Cornwall Hospital (Cornwall, UK) | July 2017 | 795 | 2 |
| Warwick Hospital (Warwick, UK) | July 2017 | 374 | 114 |
| William Harvey Hospital (Ashford, UK) | July 2017 | 849 | 56 |
| Basingstoke and North Hampshire Hospital (Basingstoke, UK) | July 2017 | 934 | 41 |
| Princess Royal University Hospital (Orpington, UK) | July 2017 | 599 | 47 |
| Ipswich Hospital (Ipswich, UK) | July 2017 | 754 | 58 |
| Bristol Royal Infirmary (Bristol, UK) | July 2017 | 1073 | 38 |
| Medway Maritime Hospital (Kent, UK) | August 2017 | 472 | 56 |
| Manchester Royal Infirmary (Manchester, UK) | August 2017 | 2303 | 23 |
| Morriston Hospital (Swansea, UK) | August 2017 | 1211 | 37 |
| Broomfield Hospital (Chelmsford, UK) | August 2017 | 722 | 46 |
| Tunbridge Wells Hospital (Tunbridge Wells, UK) | August 2017 | 485 | 84 |
| Addenbrooke’s Hospital (Cambridge, UK) | August 2017 | 830 | 38 |
| Royal Stoke University Hospital (Stoke-on-Trent, UK) | August 2017 | 1319 | 40 |
| Countess of Chester Hospital (Chester, UK) | August 2017 | 689 | 64 |
| Royal Gwent Hospital (Newport, UK) | August 2017 | 895 | 24 |
| Worthing Hospital (Worthing, UK) | September 2017 | 606 | 53 |
| Northampton General Hospital (Northampton, UK) | September 2017 | 828 | 33 |
| Derriford Hospital (Plymouth, UK) | September 2017 | 1307 | 10 |
| Darent Valley Hospital (Dartford, UK) | September 2017 | 635 | 40 |
| Royal Berkshire Hospital (Reading, UK) | September 2017 | 744 | 76 |
| Aintree University Hospital (Liverpool, UK) | September 2017 | 1144 | 67 |
| Royal Glamorgan Hospital (Pontyclun, UK) | September 2017 | 484 | 35 |
| James Cook University Hospital (Middlesbrough, UK) | September 2017 | 1842 | 20 |
| Queen Elizabeth Hospital (Gateshead, UK) | September 2017 | 863 | 6 |
| Royal Oldham Hospital (Oldham, UK) | September 2017 | 639 | 24 |
| Royal Blackburn Hospital (Blackburn, UK) | September 2017 | 1617 | 43 |
| Charing Cross Hospital (London, UK) | September 2017 | 1092 | 29 |
| St Mary’s Hospital (London, UK) | September 2017 | 610 | 15 |
| Southmead Hospital (Bristol, UK) | September 2017 | 2354 | 95 |
| Royal Devon and Exeter Hospital (Exeter, UK) | October 2017 | 916 | 23 |
| Altnagelvin Hospital (Londonderry, UK) | October 2017 | 393 | 6 |
| Darlington Memorial Hospital (Darlington, UK) | October 2017 | 646 | 50 |
| Glangwili General Hospital (Carmarthen, UK) | October 2017 | 649 | 106 |
| Hammersmith Hospital (London, UK) | October 2017 | 539 | 6 |
| Arrowe Park Hospital (Wirral, UK) | October 2017 | 754 | 11 |
| University Hospital Coventry (Coventry, UK) | October 2017 | 1122 | 31 |
| Antrim Area Hospital (Antrim, UK) | October 2017 | 359 | 16 |
| Leicester Royal Infirmary (Leicester, UK) | October 2017 | 1163 | 46 |
| Pinderfields Hospital (Wakefield, UK) | October 2017 | 799 | 24 |
| Queen Elizabeth Hospital, Woolwich (London, UK) | October 2017 | 774 | 142 |
| Queens Medical Centre (Nottingham, UK) | October 2017 | 1072 | 10 |
| Royal Liverpool University Hospital (Liverpool, UK) | October 2017 | 1603 | 17 |
| Royal Victoria Infirmary (Newcastle-upon-Tyne, UK) | October 2017 | 1281 | 55 |
| Torbay Hospital (Torquay, UK) | November 2017 | 533 | 51 |
| Lister Hospital (Stevenage, UK) | November 2017 | 1003 | 12 |
| Gloucestershire Royal Hospital (Gloucester, UK) | November 2017 | 886 | 47 |
| Royal Preston Hospital (Preston, UK) | December 2017 | 1401 | 9 |
| Norfolk and Norwich Hospital (Norwich, UK) | January 2018 | 1569 | 33 |
| North Devon District Hospital (Barnstaple, UK) | January 2018 | 440 | 6 |
| St Thomas’ Hospital (London, UK) | January 2018 | 1239 | 9 |
| Blackpool Victoria Hospital (Blackpool, UK) | February 2018 | 733 | 27 |
| Yeovil District Hospital (Yeovil, UK) | February 2018 | 585 | 8 |
| Northern General Hospital (Sheffield, UK) | March 2018 | 2324 | 6 |
| University Hospital of North Tees (Stockton-on-Tees, UK) | September 2018 | 684 | 13 |
The overall site recruitment rate was 2.4 patients per month with a median recruitment rate of 2.1 (IQR 1.0–3.1) patients across sites. Many were able to embed 65 trial screening and randomisation into routine clinical practice, leading to a high proportion of randomisations occurring outside standard office working days and hours (Figures 5 and 6).
FIGURE 5.
Randomisation by day of week. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
FIGURE 6.
Randomisation by time of day. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
Consent
Of the 2598 unique patients, 15 (permissive hypotension, n = 8; usual care, n = 7) requested that all data be removed from the trial and are not included in further reporting or analysis. For the remaining 2583 patients, consent for ongoing data collection and linkage was obtained for 2461 (95%). A further five patients (or their consultees) declined consent after 90 days and were included in the analysis up to that point. By 90 days, a further two patients had withdrawn consent, leaving 2464 eligible for inclusion in the analysis of the primary outcome. Consent and withdrawal rates by 90 days did not differ by randomised group. The full flow of the consent procedures, according to randomised groups, is shown in Figures 7 and 8.
FIGURE 7.
Delivery of consent procedures for patients randomised to the permissive hypotension group. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
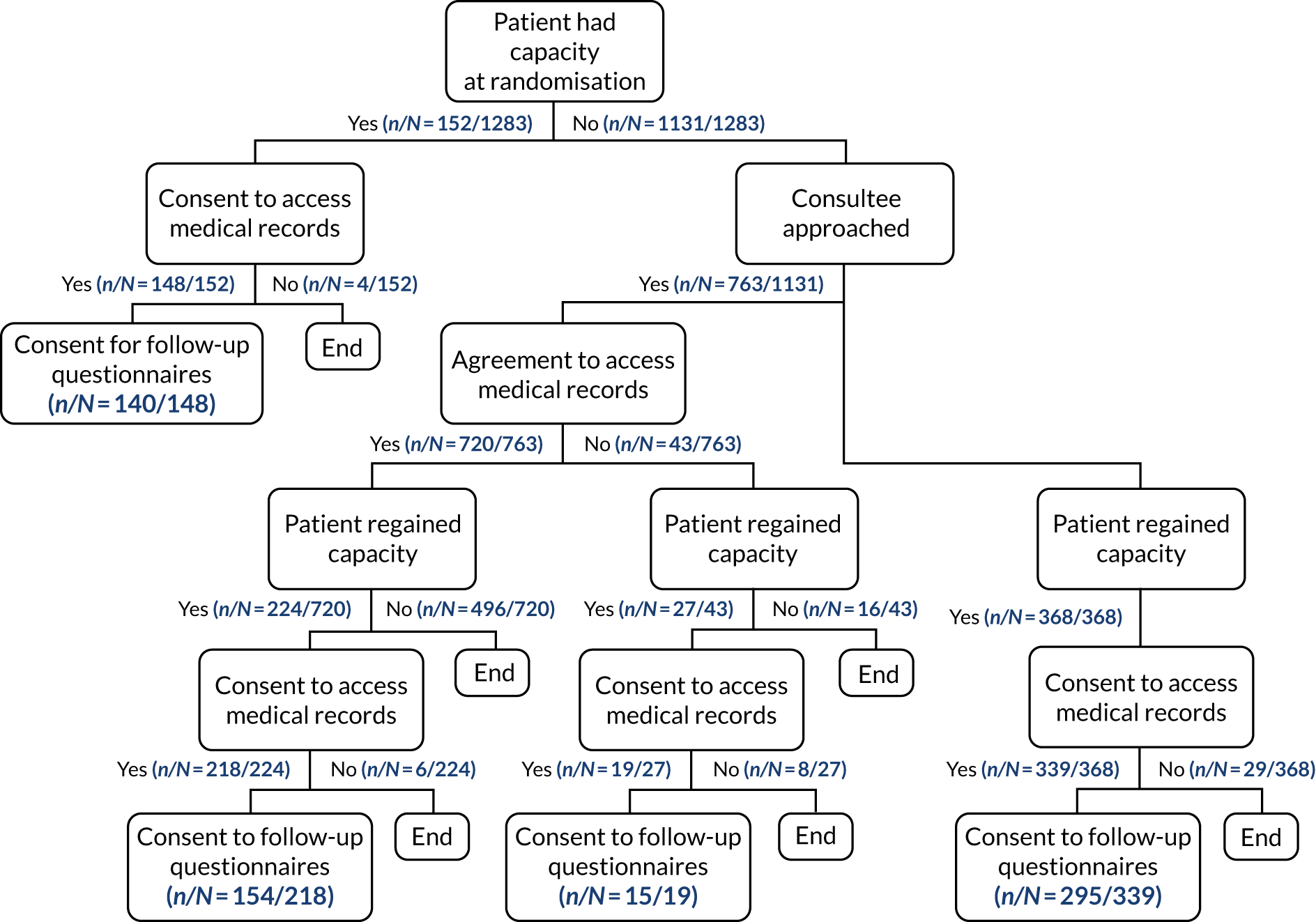
FIGURE 8.
Delivery of consent procedures for patients randomised to the usual-care group. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
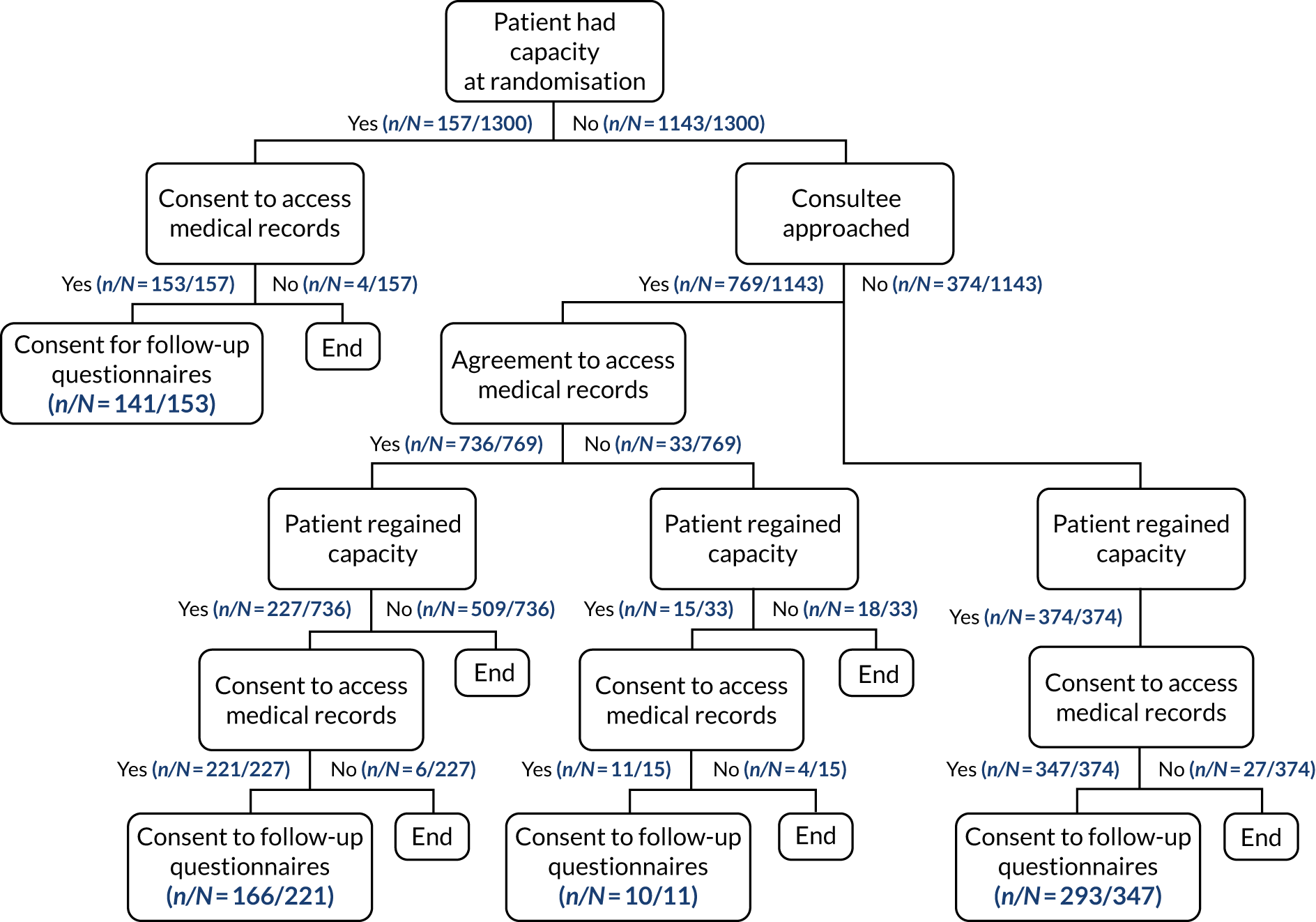
Baseline characteristics
The randomised groups were well matched at baseline (Table 7). In both groups, patients had a mean age of 75 years and over half were male (permissive hypotension, 57.2%; usual care, 55.9%). Just under half (46%) of patients in both groups had known chronic hypertension and around one-third were admitted to critical care from the emergency department (or not in hospital). The proportion of patients requiring assistance with daily activities of living showed some differences, with 417 (34.4%) patients in the permissive hypotension group compared with 380 (30.9%) patients in the usual-care group. Patients were randomised at a median of 3.1 hours after commencing vasopressors.
| Characteristic | Permissive hypotension (N = 1283a) | Usual care (N = 1300a) |
|---|---|---|
| Age (years), mean (SD) | 75.3 (6.6) | 75.2 (6.9) |
| Sex, n (%) | ||
| Male | 696 (57.2) | 692 (55.9) |
| Female | 520 (42.8) | 547 (44.1) |
| Comorbidity, n/N (%) | ||
| Chronic hypertension | 590/1283 (46.0) | 597/1299 (46.0) |
| Atherosclerotic disease | 187/1283 (14.6) | 189/1299 (14.5) |
| Chronic heart failure | 143/1283 (11.1) | 143/1298 (11.0) |
| Chronic renal replacement therapy at ICU admission | 16/1204 (1.3) | 18/1224 (1.5) |
| Dependency prior to acute hospital admission, n (%) | ||
| Able to live without assistance in daily activities | 794 (65.6) | 850 (69.1) |
| Minor/major assistance with daily activities | 409 (33.8) | 375 (30.5) |
| Total assistance with all daily activities | 8 (0.7) | 5 (0.4) |
| Location prior to admission to critical care and urgency of surgery, n (%) | ||
| Emergency department/not in hospital | 432 (35.4) | 420 (33.9) |
| Theatre: elective/scheduled surgery | 53 (4.3) | 60 (4.8) |
| Theatre: emergency/urgent surgery | 259 (21.2) | 264 (21.3) |
| Other critical care unit | 14 (1.1) | 22 (1.8) |
| Ward or intermediate care area | 461 (37.8) | 473 (38.2) |
| APACHE II score, mean (SD) | 20.9 (6.5) | 20.6 (6.1) |
| ICNARC physiology score, mean (SD) | 23.9 (8.8) | 23.5 (8.8) |
| ICNARCH–2015 predicted risk of death, median (IQR) | 0.33 (0.15–0.60) | 0.32 (0.14–0.61) |
| Sepsis-3, n (%) | ||
| No sepsis | 263 (21.6) | 275 (22.2) |
| Sepsis (not in shock) | 364 (29.9) | 369 (29.8) |
| Septic shock | 589 (48.4) | 595 (48.0) |
| MAP at randomisation (mmHg), mean (SD) | 69.9 (10.1) | 71.1 (11.5) |
| Vasopressor infusions received at time of randomisation,b n (%) | ||
| Nonec | 15 (1.2) | 25 (2.0) |
| dNoradrenaline equivalent < 0.1 µg/kg/minute | 153 (12.1) | 155 (12.1) |
| Noradrenaline equivalent ≥ 0.1 µg/kg/minute | 676 (53.4) | 677 (52.9) |
| Metaraminol | 406 (32.1) | 409 (32.0) |
| Other/combination | 43 (3.4) | 34 (2.7) |
| Duration of vasopressor infusion prior to randomisation (minutes), median (IQR) | 186 (102–277) | 186 (104–284) |
Clinical management
Vasopressors
Following randomisation and during the first episode of vasopressors, patients in the permissive hypotension group had a lower exposure to vasopressors than those in the usual-care group. Mean duration of vasopressors in the permissive hypotension group was 46.0 (SD 52.4) hours, compared with 55.9 (SD 60.8) hours in the usual-care group (difference −9.9 hours, 95% CI −14.3 to −5.5 hours). Time to discontinuation of vasopressors is shown in Figure 9. The mean total of vasopressors (noradrenaline equivalent) in the permissive hypotension group was 31.5 mg compared with 44.3 mg for the usual-care group (difference −12.8 mg, 95% CI −18.0 mg to −7.6 mg). For patients receiving metaraminol, the mean difference in metaraminol total dose was –4.1 mg (95% CI –8.3 mg to –0.0 mg). Both groups had a similar number of episodes of vasopressors, the majority with a single episode [permissive hypotension, n = 1094 (86.8%); usual care, n = 1100 (86.3%)]. Randomisation did not influence vasopressor choice. Noradrenaline was most commonly administered (received by around 80% in each group). Just over 30% of patients in each group received metaraminol and just under 10% received vasopressin (Table 8).
FIGURE 9.
Time to discontinuation of vasopressors. Reports the time (hours) to discontinuation of vasopressors from randomisation. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
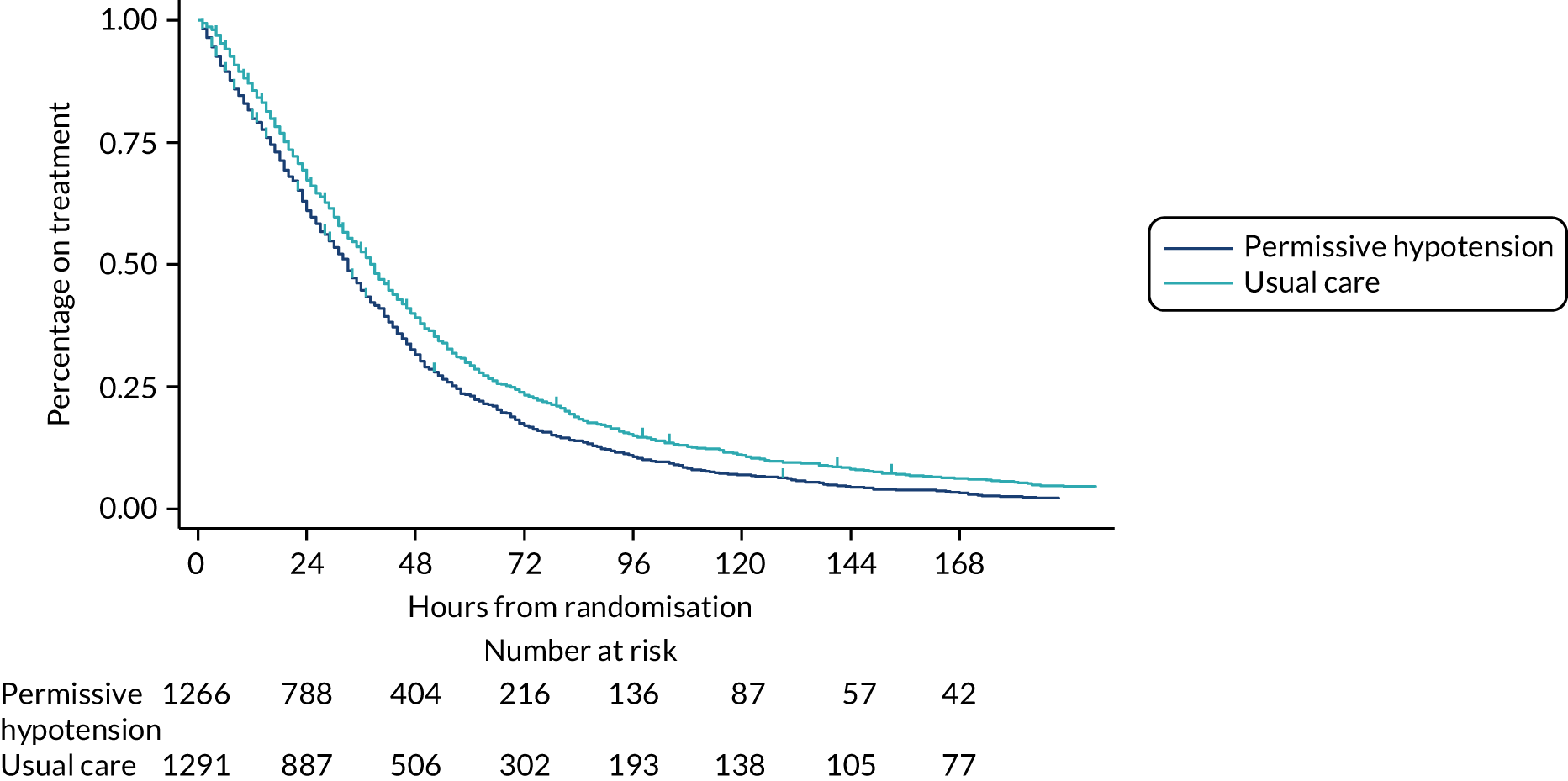
| Exposure to vasopressors | Permissive hypotension (N = 1261a) | Usual care (N = 1276a) | Difference in means (95% CI) |
|---|---|---|---|
| Total duration of vasopressors (hours) | |||
| Median (IQR) | 33.0 (15.0–56.0) | 38.0 (19.0–67.0) | |
| Mean (SD) | 46.0 (52.4) | 55.9 (60.8) | –9.9 (–14.3 to –5.5) |
| Total dose of vasopressors (mg) | |||
| Noradrenaline equivalents | n = 1008 | n = 1021 | |
| Median (IQR)b | 17.7 (5.8–47.2) | 26.4 (8.9–65.6) | |
| Mean (SD)c | 31.5 (57.4) | 44.3 (76.4) | –12.8 (–18.0 to –7.6) |
| Metaraminol | n = 395 | n = 420 | |
| Median (IQR)b | 22.0 (9.3–60.0) | 35.0 (12.7–79.8) | |
| Mean (SD)c | 15.7 (51.2) | 19.8 (53.2) | –4.1 (–8.3 to –0.0) |
| Terlipressin | n = 10 | n = 14 | |
| Median (IQR)b | 2.5 (1.0–10.8) | 3.3 (1.0–6.0) | |
| Mean (SD)c | 0.1 (1.0) | 0.1 (0.7) | 0.0 (0.0 to 0.1) |
| Mean dose rate of vasopressorsb | |||
| Noradrenaline equivalents (µg/kg/minute) | n = 1008 | n = 1021 | |
| Median (IQR) | 0.12 (0.06–0.23) | 0.15 (0.08–0.26) | |
| Mean (SD) | 0.21 (0.45) | 0.22 (0.24) | –0.02 (–0.05 to 0.02) |
| Metaraminol (mg/hour) | n = 385 | n = 408 | |
| Median (IQR) | 2.35 (1.44–4.25) | 2.83 (1.95–4.88) | |
| Mean (SD) | 3.41 (2.99) | 3.81 (3.04) | –0.40 (–0.80 to 0.01) |
| Highest dose rate of vasopressorsb | |||
| Noradrenaline equivalents (µg/kg/minute) | n = 1008 | n = 1021 | |
| Median (IQR) | 0.26 (0.13–0.57) | 0.32 (0.16–0.63) | |
| Mean (SD) | 0.44 (0.74) | 0.47 (0.49) | –0.03 (–0.09 to 0.02) |
| Metaraminol (mg/hour) | n = 385 | n = 408 | |
| Median (IQR) | 4.00 (3.00–6.50) | 5.00 (3.50–7.00) | |
| Mean (SD) | 5.39 (3.96) | 5.73 (3.60) | –0.34 (–0.88 to 0.20) |
| Total number of episodes of vasopressor treatment at critical care discharge, n (%) | |||
| One | 1094 (86.8) | 1100 (86.3) | |
| Two | 113 (9.0) | 115 (9.0) | |
| Three | 34 (2.7) | 30 (2.4) | |
| Four | 12 (1.0) | 14 (1.1) | |
| Five | 2 (0.2) | 7 (0.5) | |
| Six | 4 (0.3) | 3 (0.2) | |
| Total number of calendar days on vasopressors at critical care discharge, n (%) | |||
| One or two | 628 (49.8) | 544 (42.7) | |
| Three or four | 381 (30.2) | 414 (32.5) | |
| Five or six | 111 (8.8) | 138 (10.8) | |
| Seven or more | 139 (11.0) | 173 (13.6) | |
| Vasopressors received, n (%) | |||
| Noradrenaline | 992 (78.7) | 997 (78.1) | |
| Adrenaline | 40 (3.2) | 42 (3.3) | |
| Dopamine | 1 (0.1) | 2 (0.2) | |
| Phenylephrine | 32 (2.5) | 33 (2.6) | |
| Vasopressin | 123 (9.8) | 126 (9.9) | |
| Metaraminol | 395 (31.3) | 418 (32.8) | |
| Terlipressin | 10 (0.8) | 14 (1.1) | |
The difference in vasopressor exposure was due to permissive hypotension leading to a change in clinical management of vasopressors compared with usual care. This was observed immediately post randomisation, with permissive hypotension leading to lower vasopressor dose rates and MAPs by the end of the first 24 hours post randomisation (Figure 10), and carried on throughout the first episode of vasopressors, with clinical teams actively reducing vasopressor doses (Figure 11).
FIGURE 10.
Dose/rate of noradrenaline equivalents and MAP change post randomisation. Values reported are the mean noradrenaline dose/rate and mean MAPs for the first 24 hours post randomisation, by hour. Patients are included while receiving a noradrenaline-equivalent vasopressor on the given hour. Points are labelled by hours post randomisation, with the last available pre-randomisation value (baseline) labelled as hour 0. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
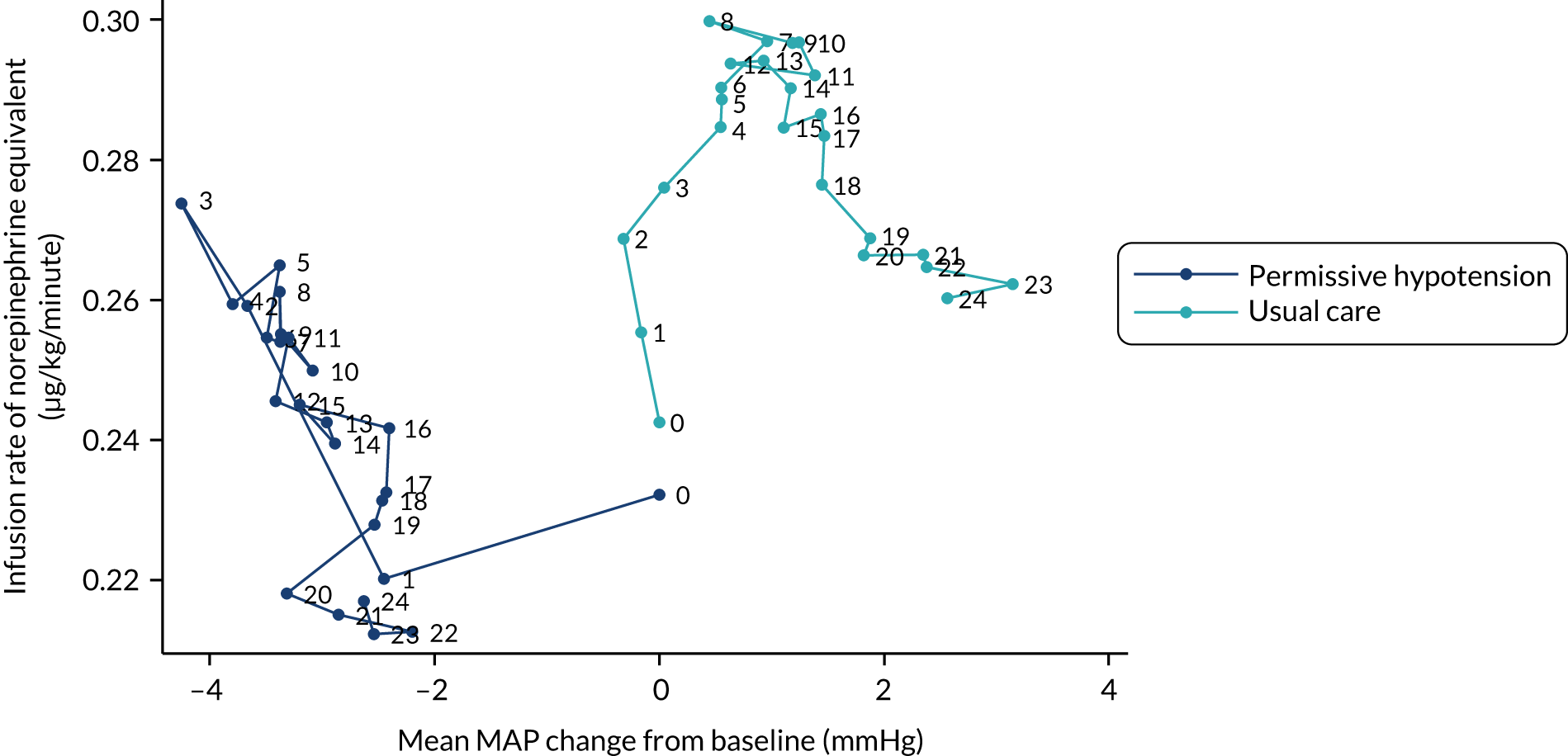
FIGURE 11.
Treatment hours: MAP and vasopressor dose reductions. (a) Mean hours of treatment; and (b) percentage of all treatment hours by randomised group. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
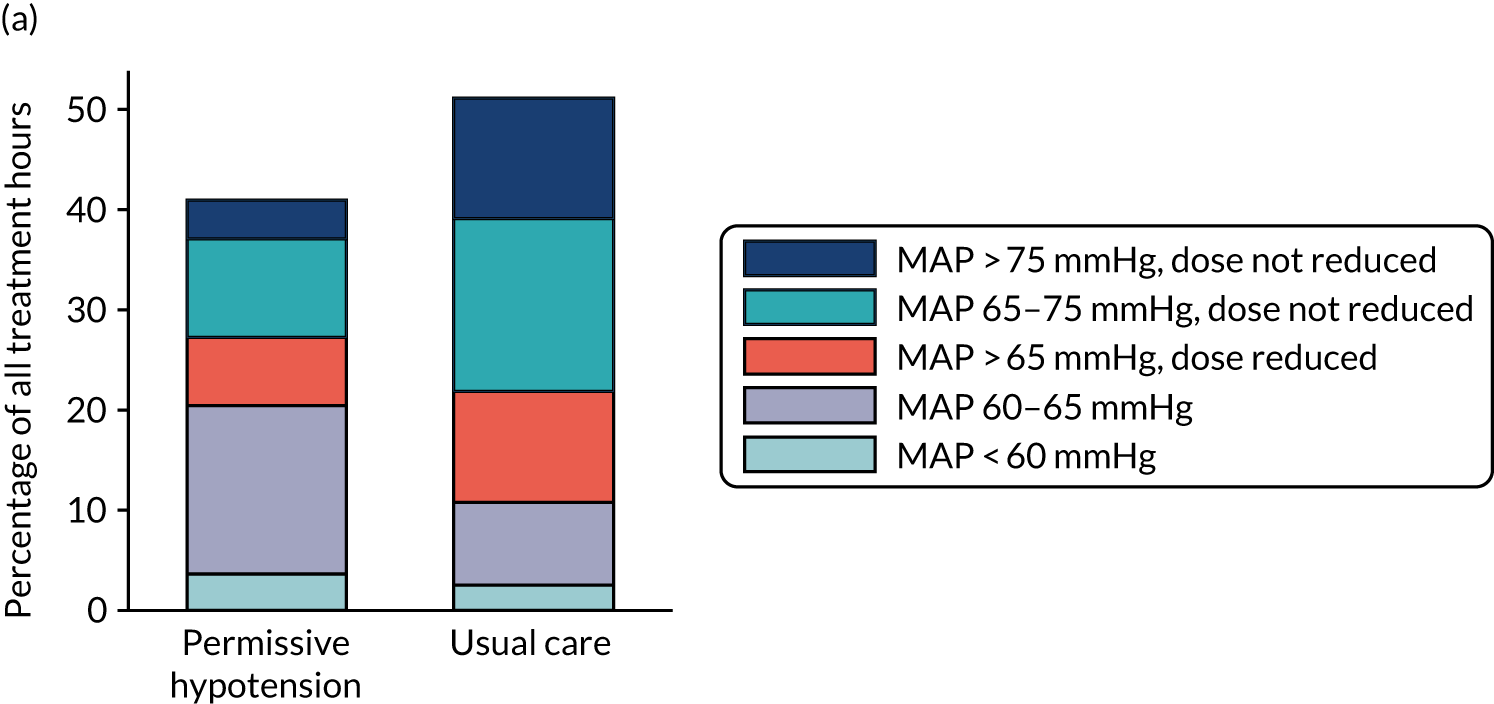
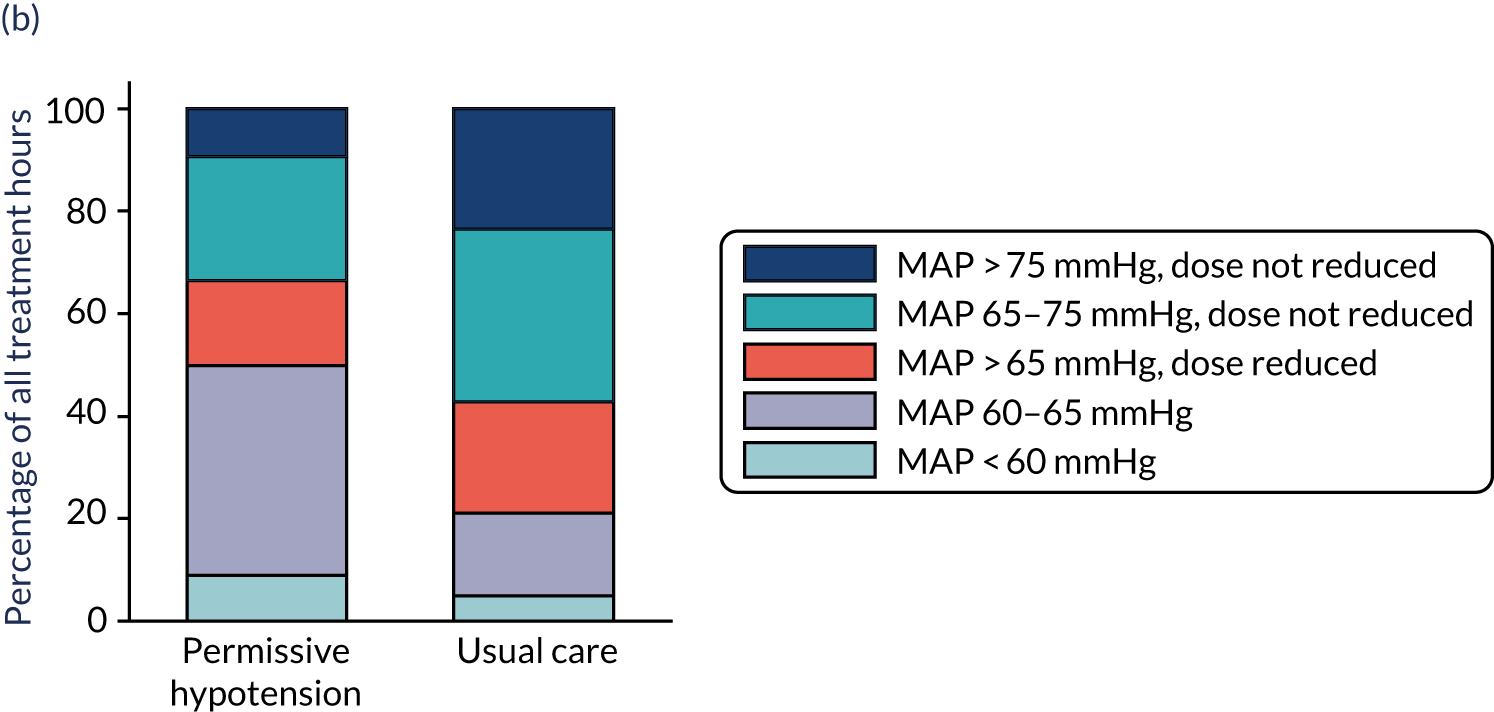
Following randomisation and during the first episode of vasopressors, mean and peak MAP values were lower in the permissive hypotension group than in the usual-care group (Figure 12 and Tables 9–11). For permissive hypotension, patients had a mean MAP of 67.6 mmHg (SD 5.2 mmHg) while receiving vasopressors compared with 72.9 mmHg (SD 5.8 mmHg) for usual care. Even though a lower MAP was targeted, this did not lead to a clinically significant increase in duration with a MAP of < 60 mmHg (see Tables 10 and 11).
FIGURE 12.
Mean arterial pressure values days 1–7 post randomisation. These box and whisker plots summarise the median of the (a) mean MAPs; and (b) peak (highest) MAPs for values recorded hourly during patient’s first episode of vasopressors for days 1–7 post randomisation. Numbers at the foot of each box represent the denominators for the numbers for that box (i.e. the number of patients receiving vasopressors on the given day). Median and quartiles (boxes) are shown with upper and lower adjacent values of median ± 1.5 × IQR (whiskers). Observations outside the adjacent values are not shown. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
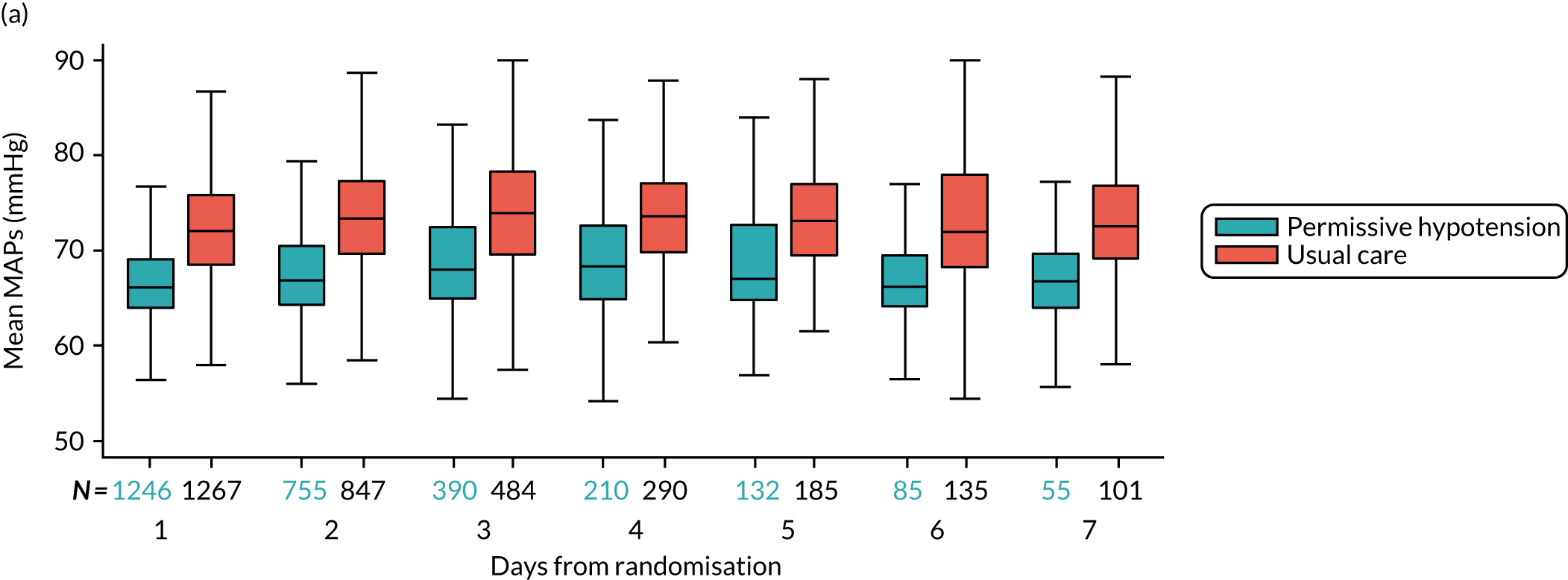
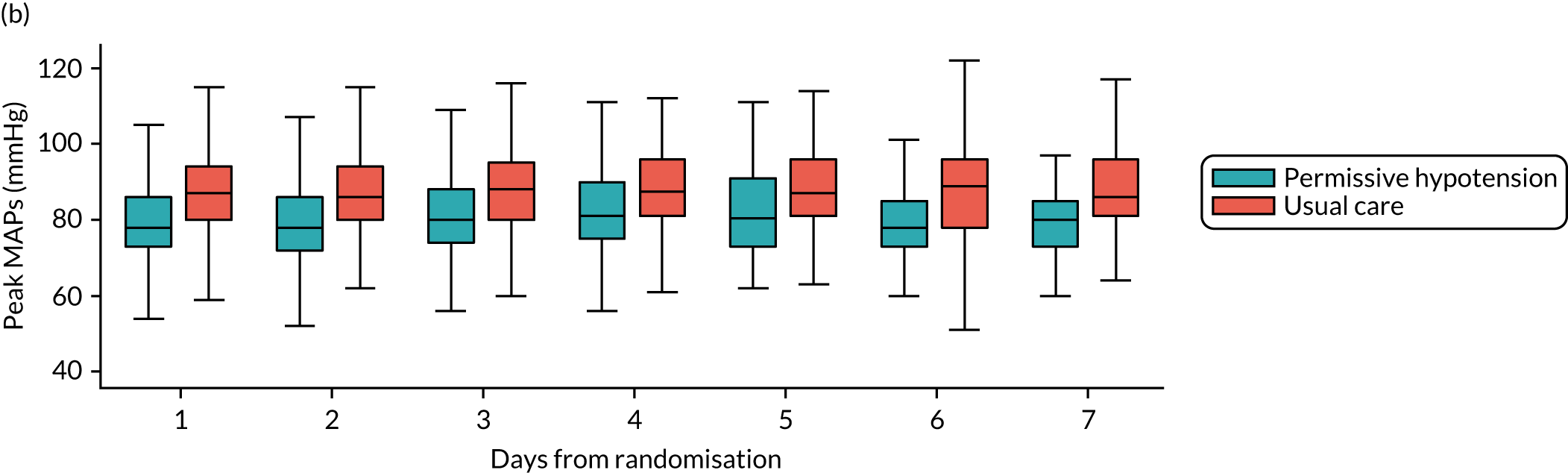
| Mean and peak MAPs while receiving vasopressors | Permissive hypotension (n = 1247a) | Usual care (n = 1267a) | Difference in means (95% CI) |
|---|---|---|---|
| Mean MAP while receiving vasopressors (mmHg) | |||
| Median (IQR) | 66.7 (64.5–69.8) | 72.6 (69.4–76.5) | |
| Mean (SD) | 67.6 (5.2) | 72.9 (5.8) | –5.3 (–5.8 to –4.9) |
| Peak MAP while receiving vasopressors (mmHg) | |||
| Median (IQR) | 83.0 (75.0–92.0) | 92.0 (85.0–100.0) | |
| Mean (SD) | 84.9 (14.9) | 93.2 (13.8) | –8.4 (–9.5 to –7.2) |
| Time (hours) on vasopressors | Permissive hypotension (n = 1261a) | Usual care (n = 1276a) |
|---|---|---|
| Time (hours) on vasopressors with recorded MAP of < 60 mmHg | ||
| Mean (SD) | 3.6 (6.5) | 2.5 (5.3) |
| Time (hours) on vasopressors with recorded MAP of between 60 and 65 mmHg | ||
| Mean (SD) | 16.8 (21.0) | 8.2 (12.4) |
| Time (hours) on vasopressors with recorded MAP between 65 and 70 mmHg | ||
| Mean (SD) | 9.3 (12.2) | 11.4 (13.9) |
| Time (hours) on vasopressors with recorded MAP of > 70 mmHg | ||
| Mean (SD) | 11.2 (18.3) | 28.9 (34.5) |
| Percentage of hours on vasopressors | Permissive hypotension (n = 1261a) | Usual care (n = 1276a) |
|---|---|---|
| Time on vasopressors with recorded MAP of < 60 mmHg (percentage of all treatment hours) | ||
| Mean (SD) | 8.8 (16.0) | 4.9 (10.4) |
| Time on vasopressors with recorded MAP of between 60 and 65 mmHg (percentage of all treatment hours) | ||
| Mean (SD) | 41.1 (51.5) | 16.1 (24.3) |
| Time on vasopressors with recorded MAP of between 65 and 70 mmHg (percentage of all treatment hours) | ||
| Mean (SD) | 22.9 (29.8) | 22.3 (27.1) |
| Time on vasopressors with recorded MAP of > 70 mmHg (percentage of all treatment hours) | ||
| Mean (SD) | 27.3 (44.7) | 56.6 (67.6) |
Adherence to permissive hypotension
Periods of non-adherence to permissive hypotension were observed in 153 (11.9%) patients, equating to 6% of the total time patients in the permissive hypotension group received vasopressors (Table 12). The main reason for not reducing or discontinuing vasopressors during a 3-hour period of a MAP of > 65 mmHg was staff/logistical issues (n = 102). This included lack of trial awareness (n = 54), focus on other clinical priorities (n = 42) and no reason documented (n = 6). In 51 of the patients with periods of non-adherence, patient concerns were the documented reason. These concerns included renal (n = 36), cardiac (n = 4), history of chronic hypertension (n = 2), gastrointestinal (n = 2) and other (n = 7).
| Failure to discontinue vasopressors or reduce the dose/rate once MAP was > 65 mmHg | Permissive hypotension (n = 1283a) |
|---|---|
| Number of potential periods of non-adherence | 456 |
| Number of periods determined not to be non-adherence | 303 |
| Number (%) of patients experiencing at least one period of non-adherence | 153 (11.9) |
| Total (%) hours of non-adherence | 3519 (6.0) |
Co-interventions
There was no clinically important difference in fluid balance, urine output or use of pure inotropes during the first episode of vasopressors. A slightly higher number of patients in the usual-care group (n = 432, 33.9%) received corticosteroids than in the permissive hypotension group (n = 398, 31.6%) (Table 13 and Figure 13).
| Co-intervention | Permissive hypotension | Usual care |
|---|---|---|
| Fluid balance at end of first episode of vasopressors (ml) | ||
| n | 1247 | 1268 |
| Median (IQR) | 3041 (1307–5744) | 2904 (1071–5789) |
| Mean (SD) | 3993 (4238) | 3976 (5202) |
| Mean urine output during first episode of vasopressors (ml/kg/hour) | ||
| n | 1246 | 1266 |
| Median (IQR) | 0.5 (0.2–0.8) | 0.6 (0.2–0.9) |
| Mean (SD) | 0.6 (0.5) | 0.6 (0.6) |
| Co-intervention received during first episode of vasopressors | ||
| n | 1261 | 1276 |
| Corticosteroids, n (%) | 398 (31.6) | 432 (33.9) |
| Inotropes, n (%) | 268 (21.3) | 256 (20.1) |
FIGURE 13.
Fluid balance and urine output days 1–7 post randomisation. These box and whisker plots summarise the median (a) daily fluid balance; and (b) daily urine output for values recorded during patient’s first episode of vasopressors for days 1–7 post randomisation. Numbers at the foot of each box represent the denominators for the numbers for that box (i.e. the number of patients receiving vasopressors on the given day). Median and quartiles (boxes) are shown with upper and lower adjacent values of median ± 1.5 × IQR (whiskers). Observations outside the adjacent values are not shown. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
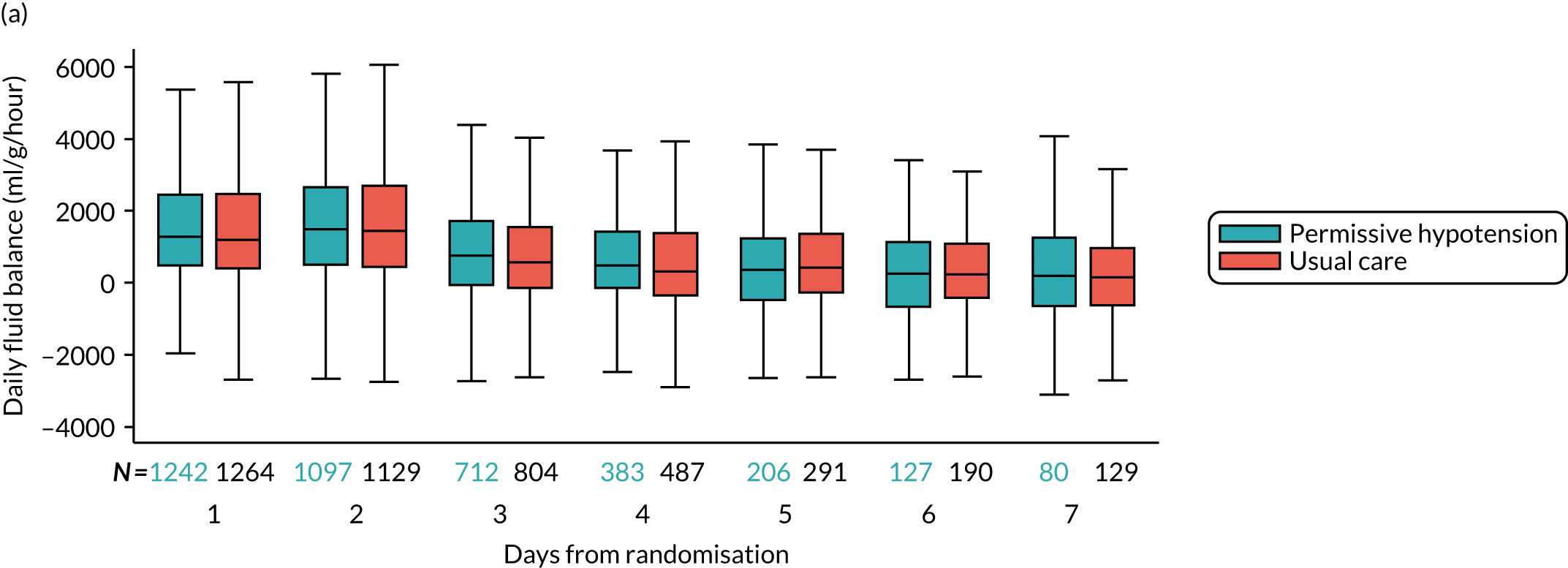
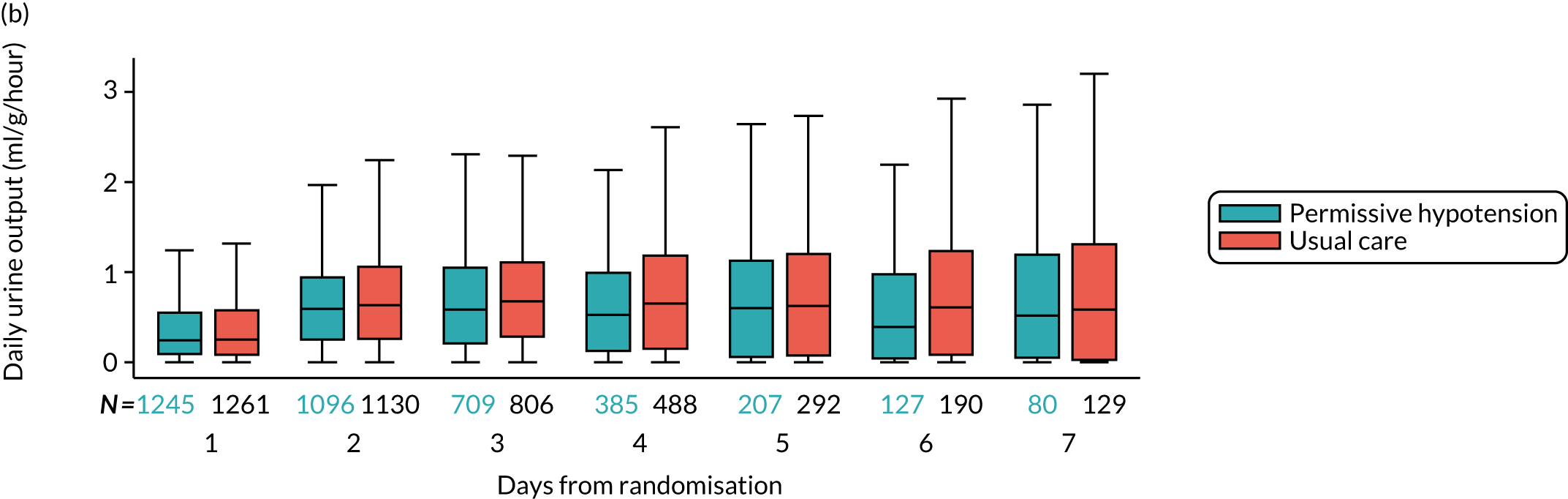
Clinical effectiveness
Primary outcome
At 90 days following randomisation, 500 (41.0%) patients randomised to permissive hypotension had died compared with 544 (43.8%) patients randomised to usual care, corresponding to an absolute risk reduction of –2.85 (95% CI –6.75 to 1.05; p = 0.154) (Table 14). When adjusted for prespecified baseline variables, the odds ratio for 90-day mortality was 0.82 (95% CI 0.68 to 0.98) compared with an unadjusted odds ratio of 0.89 (95% CI 0.76 to 1.04) (see Table 14).
| Outcome | Permissive hypotension | Usual care | Effect estimate (95% CI) | p-value |
|---|---|---|---|---|
| Primary outcome | ||||
| 90-day mortality, n/N (%) | 500/1221 (41.0) | 544/1242 (43.8) | –2.85 (–6.75 to 1.05)a | 0.154 |
| 0.93 (0.85 to 1.03)b | ||||
| 0.89 (0.76 to 1.04)c | ||||
| 0.82 (0.68 to 0.98)d | ||||
| Secondary outcomes | ||||
| Mortality at discharge from the critical care unit, n/N (%) | 362/1212 (29.9) | 380/1237 (30.7) | –0.85 (–4.49 to 2.79)a | |
| 0.97 (0.86 to 1.10)b | ||||
| 0.96 (0.81 to 1.14)c | ||||
| 0.90 (0.73 to 1.10)d | ||||
| Mortality at discharge from acute hospital, n/N (%) | 484/1232 (39.3) | 519/1250 (41.5) | –2.23 (–6.09 to 1.63)a | |
| 0.95 (0.86 to 1.04)b | ||||
| 0.91 (0.78 to 1.07)c | ||||
| 0.86 (0.71 to 1.03)d | ||||
| Advanced respiratory support | ||||
| Receipt, n/N (%) | 708/1218 (58.1) | 691/1239 (55.8) | ||
| Duration (days), median (IQR)e | 4.0 (2.0–10.0) | 4.0 (2.0–10.0) | ||
| Duration (days), mean (SD)f | 4.5 (8.3) | 4.8 (10.0) | –0.3 (–1.1 to 0.4)g | |
| –0.3 (–1.0 to 0.4)h | ||||
| Days alive and free of advanced respiratory support to day 28, mean (SD) | 15.7 (12.8) | 15.1 (13.0) | 0.6 (–0.4 to 1.7)g | |
| 0.9 (0.0 to 1.8)h | ||||
| Renal support | ||||
| Receipt, n/N (%) | 302/1218 (24.8) | 306/1239 (24.7) | ||
| Duration (days), median (IQR)e | 4.0 (2.0–7.0) | 4.0 (2.0–8.0) | ||
| Duration (days), mean (SD)f | 1.4 (3.6) | 1.5 (4.1) | –0.1 (–0.4 to 0.2)g | |
| –0.2 (–0.4 to 0.1)h | ||||
| Days alive and free of renal support to day 28, mean (SD) | 17.4 (13.2) | 16.7 (13.4) | 0.6 (–0.4 to 1.7)g | |
| 0.9 (0.0 to 1.9)h | ||||
| Duration of critical care unit stay (days), median (IQR) | ||||
| Survivors | 5.2 (2.9–10.5) | 5.4 (3.0–9.9) | ||
| Non-survivors | 3.2 (0.9–8.1) | 2.7 (0.9–8.7) | ||
| Duration of acute hospital stay (days), median (IQR) | ||||
| Survivors | 18.0 (10.0–34.0) | 18.0 (10.0–36.0) | ||
| Non-survivors | 6.0 (1.0–15.0) | 5.0 (1.0–14.5) | ||
| Cognitive decline (IQCODE score) among survivors, non-missing outcomes only, mean (SD) | ||||
| At 90 days | 2.96 (0.66) | 2.97 (0.66) | –0.01 (–0.09 to 0.07)g | |
| –0.01 (–0.09 to 0.07)h | ||||
| At 1 year | 2.95 (0.72) | 2.81 (0.81) | 0.14 (–0.00 to 0.28)g | |
| 0.12 (–0.01 to 0.25)h | ||||
| Cognitive decline (IQCODE score) among survivors, non-missing outcomes, median (IQR) | ||||
| At 90 days | 3.00 (3.00–3.13) | 3.00 (3.00–3.19) | ||
| At 1 year | 3.00 (3.00–3.19) | 3.00 (2.81–3.13) | ||
| Cognitive decline (IQCODE score) among survivors, missing outcomes imputed, mean (SD) | ||||
| At 90 days | 2.97 (0.72) | 2.98 (0.76) | –0.01 (–0.09 to 0.07)g | |
| –0.01 (–0.09 to 0.07)h | ||||
| At 1 year | 2.93 (0.81) | 2.80 (0.96) | 0.13 (–0.00 to 0.25)g | |
| 0.12 (–0.00 to 0.25)h | ||||
Secondary outcomes
All secondary outcomes were similar between the groups (see Table 14). Mortality at critical care unit discharge was 29.9% in the permissive hypotension group compared with 30.7% in the usual-care group. This increased by acute hospital discharge to 39.3% and 41.5%, respectively. There was no significant difference in duration of survival (unadjusted hazard ratio 0.96, 95% CI 0.86 to 1.07) (Figure 14).
FIGURE 14.
Kaplan–Meier survival curves. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
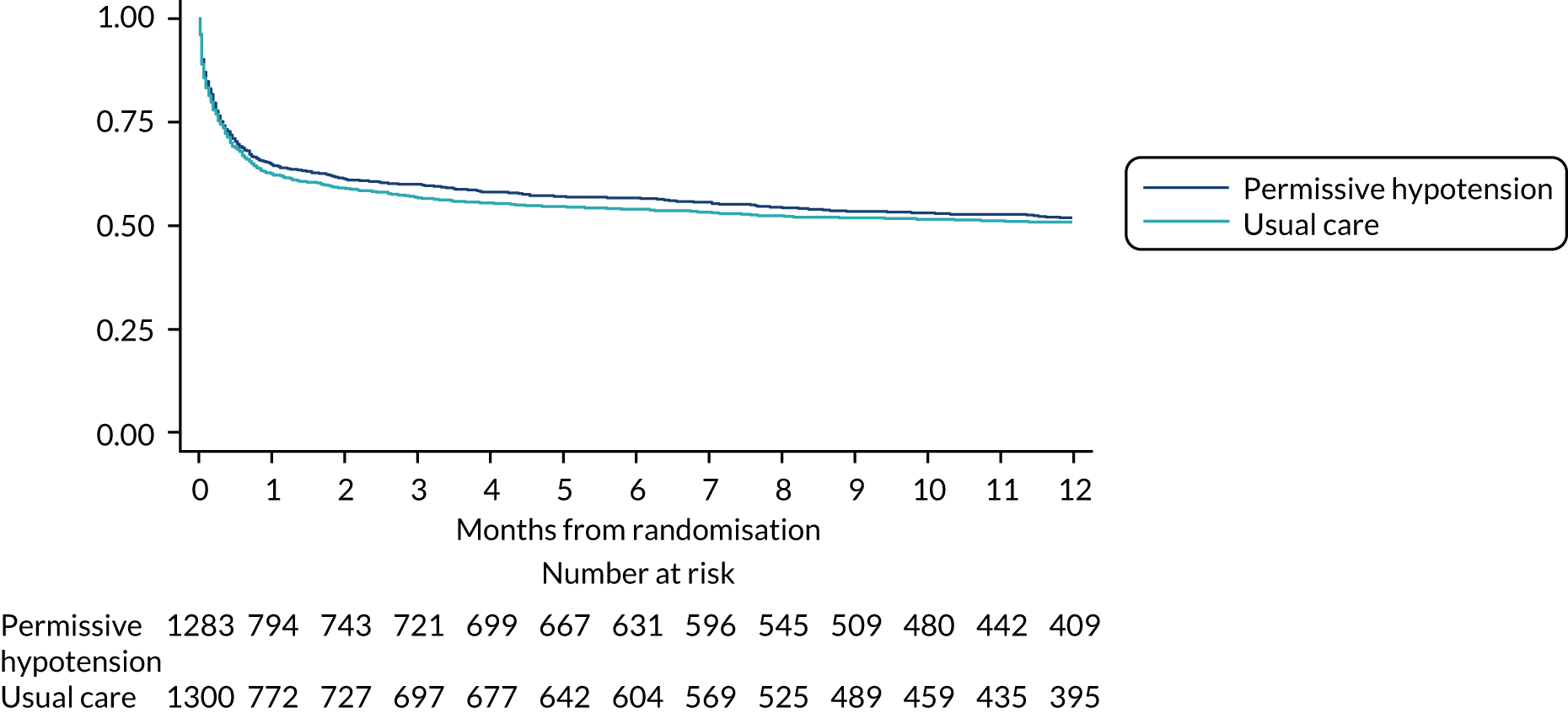
Receipt and duration of advanced respiratory and renal support were similar between the groups. In the permissive hypotension group, 24.8% of patients received renal support for a median of 4.0 (IQR 2.0–7.0) days, compared with 24.7% of patients for a median duration of 4.0 (IQR 2.0–8.0) days in the usual-care group. Among patients in the permissive hypotension group who survived to critical care discharge, median length of stay was 5.2 (IQR 2.9–10.5) days, compared with 5.4 (IQR 3.0–9.9) days in the usual-care group. With respect to acute hospital length of stay, survivors in both groups spent a median of 18 days in hospital. Cognitive function at 90 days and 1 year was similar between the groups (see Table 14).
All randomised patients are included when calculating survival, excluding 15 patients who did not consent to the trial and who refused permission for use of personal data (permissive hypotension, n = 8; usual care, n = 7). Other surviving patients were censored at last known date alive (where trial consent was obtained) or at date of withdrawal/refusal of consent (where trial consent was not obtained).
Serious adverse events
A similar number of patients in each group experienced one or more SAEs [79 (6.2%) patients in the permissive hypotension group and 75 (5.8%) patients in the usual-care group; p = 0.68] (Table 15). Of the prespecified expected adverse events, in both groups, the most commonly reported SAE was severe acute renal failure (defined in accordance with the Kidney Disease: Improving Global Outcomes66 stage 3 criteria), which was reported numerically more frequently in the permissive hypotension group. The numbers of reported myocardial injury and mesenteric ischaemia SAEs were small, although numerically more frequent in the usual-care group. Numbers of other SAEs that were not prespecified in the protocol were low, with cardiac arrests reported as a SAE in a similar proportion of patients in each group (i.e. < 1% of patients).
| SAE | Permissive hypotension (N = 1283a), n (%)b | Usual care (N = 1300a), n (%)b |
|---|---|---|
| Any SAE | 79 (6.2) | 75 (5.8) |
| Specified SAEs | ||
| Supraventricular cardiac arrhythmia | 12 (0.9) | 13 (1.0) |
| Ventricular cardiac arrhythmia | 12 (0.9) | 5 (0.4) |
| Myocardial injury | 8 (0.6) | 12 (0.9) |
| Extremity necrosis | 4 (0.3) | 3 (0.2) |
| Mesenteric ischaemia | 8 (0.6) | 12 (0.9) |
| Severe acute renal failure | 41 (3.2) | 33 (2.5) |
| Other SAEs | ||
| Cardiac arrest | 11 (0.9) | 10 (0.8) |
| Stroke | 3 (0.2) | 5 (0.4) |
| Acute ischaemic liver failure | 1 (0.1) | 0 (0.0) |
| Major haemorrhage | 1 (0.1) | 0 (0.0) |
| Massive bleed (abdominal) | 1 (0.1) | 0 (0.0) |
| Multiple kidney infarctions | 0 (0.0) | 1 (0.1) |
| Pneumobilia | 0 (0.0) | 1 (0.1) |
| Pneumothorax | 1 (0.1) | 0 (0.0) |
Subgroup analyses
For the majority of subgroups (i.e. age, chronic heart failure, atherosclerotic disease, predicted risk of death, sepsis status and vasopressor dose), there was no evidence of heterogeneity of treatment effect (Table 16 and Figure 15). Within the chronic hypertension subgroup, a mortality rate of 38.2% in the permissive hypotension group and 44.3% in usual-care group for patients with chronic hypertension (adjusted odds ratio 0.67, 95% CI 0.51 to 0.88), compared with 43.3% and 43.4%, respectively, in patients without chronic hypertension (adjusted odds ratio 0.97, 95% CI 0.76 to 1.24), was observed (test of interaction p = 0.047, not adjusted for multiple testing) (see Table 16).
| Subgroup | 90-day mortality, n/N (%) | Adjusted odds ratio (95% CI) | p-valuea | |
|---|---|---|---|---|
| Permissive hypotension | Usual care | |||
| Age (quintiles) (years) | 0.107b | |||
| 65–69c | 108/289 (37.4) | 124/304 (40.8) | 0.87 (0.60 to 1.27) | |
| 69–72 | 62/194 (32.0) | 70/224 (31.3) | 1.13 (0.71 to 1.81) | |
| 72–77 | 127/304 (41.8) | 121/274 (44.2) | 0.76 (0.52 to 1.11) | |
| 77–82 | 115/243 (47.3) | 111/219 (50.7) | 0.72 (0.48 to 1.10) | |
| > 82 | 88/191 (46.1) | 118/221 (53.4) | 0.66 (0.43 to 1.01) | |
| Chronic hypertension | 0.047 | |||
| No | 286/661 (43.3) | 291/671 (43.4) | 0.97 (0.76 to 1.24) | |
| Yes | 214/560 (38.2) | 253/571 (44.3) | 0.67 (0.51 to 0.88) | |
| Chronic heart failure | 0.811 | |||
| No | 431/1085 (39.7) | 467/1104 (42.3) | 0.82 (0.68 to 1.00) | |
| Yes | 69/136 (50.7) | 77/137 (56.2) | 0.77 (0.45 to 1.31) | |
| Atherosclerotic disease | 0.376 | |||
| No | 424/1047 (40.5) | 458/1062 (43.1) | 0.79 (0.65 to 0.96) | |
| Yes | 76/174 (43.7) | 86/180 (47.8) | 1.00 (0.62 to 1.60) | |
| Predicted risk of death (quintiles) | 0.691d | |||
| < 0.11 | 33/238 (13.9) | 34/252 (13.5) | 0.99 (0.59 to 1.68) | |
| 0.11–0.24 | 55/240 (22.9) | 63/250 (25.2) | 0.86 (0.56 to 1.31) | |
| 0.24–0.42 | 79/234 (33.8) | 114/257 (44.4) | 0.63 (0.43 to 0.92) | |
| 0.42–0.68 | 135/259 (52.1) | 124/231 (53.7) | 0.99 (0.69 to 1.43) | |
| > 0.68 | 195/242 (80.6) | 209/248 (84.3) | 0.75 (0.47 to 1.21) | |
| Sepsis-3 category | 0.062 | |||
| No sepsis | 124/263 (47.1) | 117/275 (42.5) | 1.15 (0.77 to 1.71) | |
| Sepsis (not in shock) | 112/364 (30.8) | 138/368 (37.5) | 0.62 (0.44 to 0.86) | |
| Septic shock | 262/589 (44.5) | 289/595 (48.6) | 0.83 (0.64 to 1.08) | |
| Vasopressor infusions received at randomisation | 0.363 | |||
| None | 7/15 (46.7) | 9/22 (40.9) | 1.61 (0.35 to 7.54) | |
| Noradrenaline < 0.1 µg/kg/minute | 44/142 (31.0) | 57/148 (38.5) | 0.63 (0.36 to 1.09) | |
| Noradrenaline ≥ 0.1 µg/kg/minute | 308/648 (47.5) | 324/653 (49.6) | 0.88 (0.69 to 1.13) | |
| Metaraminol | 131/385 (34.0) | 139/387 (35.9) | 0.80 (0.57 to 1.11) | |
| Other/combination | 5/15 (33.3) | 8/13 (61.5) | 0.20 (0.03 to 1.25) | |
FIGURE 15.
Subgroup analyses of the primary outcome. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association. All rights reserved.
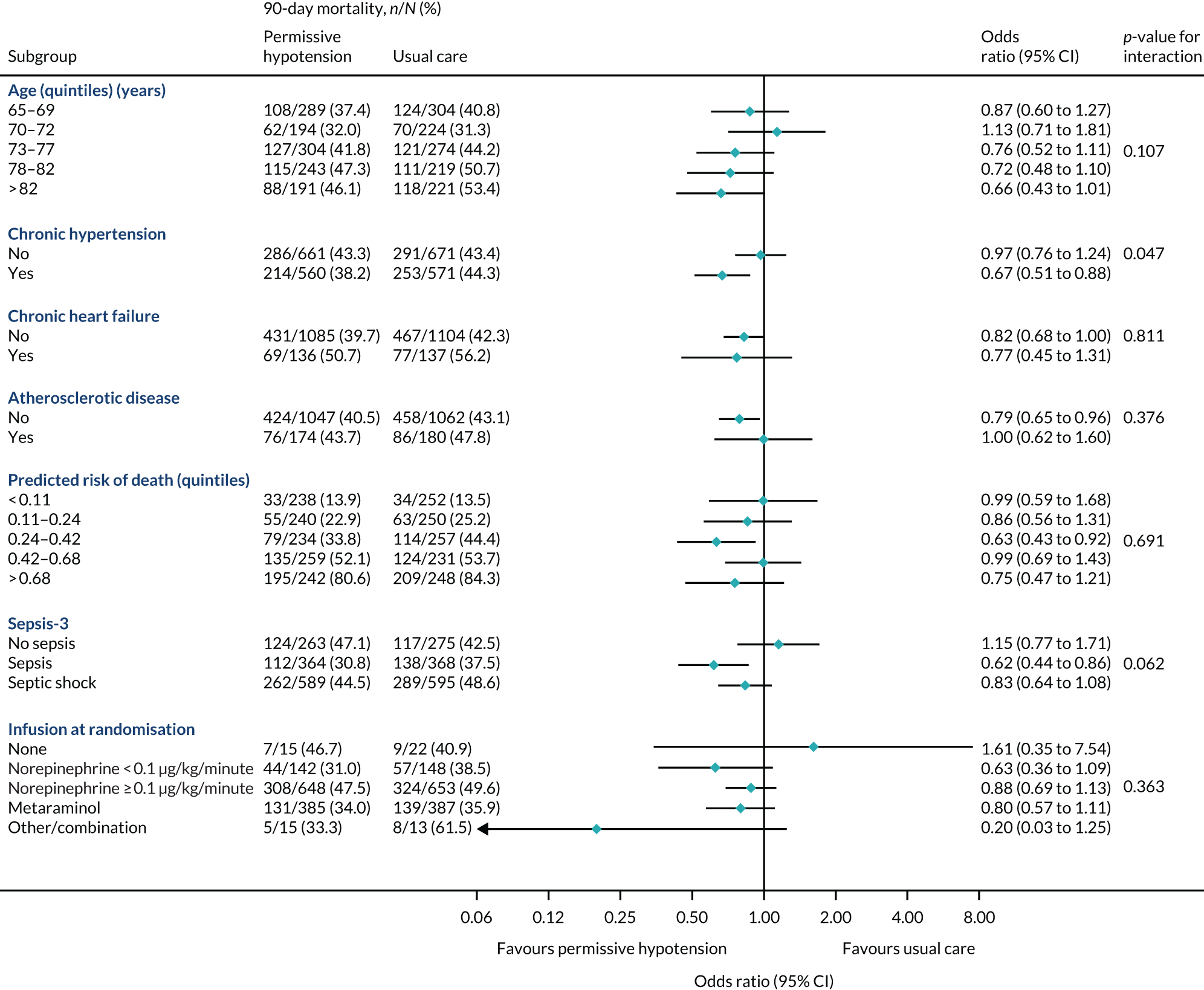
Secondary analyses of the primary outcome
Secondary and sensitivity analyses of the primary outcome, through adherence-adjusted analysis, best- and worst-case sensitivity analyses and including only patients eligible from version 2.0 of the protocol, did not alter the interpretation of the primary results (Table 17).
| Outcome | Permissive hypotension, n/N (%) | Usual care, n/N (%) | Effect estimate (95% CI) |
|---|---|---|---|
| 90-day mortality, non-missing data only | 500/1221 (41.0) | 544/1242 (43.8) | –3.24% (–7.68% to 1.20%)a |
| 90-day mortality, all missing intervention patients assumed alive, all missing control patients assumed dead (best-case scenario) | 500/1283 (39.0) | 602/1300 (46.3) | –7.34% (–11.14% to –3.53%)b |
| 0.84 (0.77 to 0.92)c | |||
| 0.74 (0.63 to 0.87)d | |||
| 90-day mortality, all missing intervention patients assumed dead, all missing control patients assumed alive (worst-case scenario) | 562/1283 (43.8) | 544/1300 (41.8) | 1.96% (–1.86% to 5.77%)b |
| 1.05 (0.96 to 1.14)c | |||
| 1.08 (0.93 to 1.27)d | |||
| 90-day mortality, patients eligible under protocol version 2.0 only (started vasopressors 1–6 hours prior to randomisation, with noradrenaline at least 0.1 µg/kg/minute) | 408/961 (42.5) | 435/965 (45.1) | –2.62% (–7.05% to 1.81%)b |
| 0.94 (0.85 to 1.04)c | |||
| 0.90 (0.75 to 1.08)d | |||
| 0.81 (0.66 to 1.00)e |
Analysis by chronic hypertension status
In a prespecified analysis by chronic hypertension status, the mortality secondary outcomes were similar to those observed in the chronic hypertension subgroup analysis of the primary outcome. For patients with chronic hypertension, critical care unit and acute mortality was 26.6% and 37.2%, respectively, in the permissive hypotension group, compared with 29.5% and 42.8%, respectively, in the usual-care group. Other secondary outcomes were similar by chronic hypertension status (Table 18).
| Outcome | Permissive hypotension | Usual care | Effect estimate (95% CI) |
|---|---|---|---|
| Mortality at discharge from the critical care unit, n/N (%) | |||
| No chronic hypertension | 214/655 (32.7) | 213/670 (31.8) | 1.05 (0.76 to 1.34)a |
| Chronic hypertension | 148/557 (26.6) | 167/567 (29.5) | 0.74 (0.51 to 0.97)a |
| Mortality at discharge from acute hospital, n/N (%) | |||
| No chronic hypertension | 274/667 (41.1) | 274/678 (40.4) | 1.03 (0.77 to 1.29)a |
| Chronic hypertension | 210/565 (37.2) | 245/572 (42.8) | 0.69 (0.50 to 0.88)a |
| Advanced respiratory support: no chronic hypertension | |||
| Receipt, n/N (%) | 373/660 (56.5) | 372/671 (55.4) | |
| Duration (days), median (IQR)b | 3.0 (2.0–9.0) | 4.0 (2.0–10.0) | |
| Duration (days), mean (SD)c | 4.4 (8.8) | 4.7 (9.1) | –0.1 (–1.0 to 0.9)d |
| Days alive and free of advanced respiratory support to day 28, mean (SD) | 15.2 (13.1) | 15.2 (13.0) | 0.1 (–1.1 to 1.2)d |
| Advanced respiratory support: chronic hypertension | |||
| Receipt, n/N (%) | 335/558 (60.0) | 319/568 (56.2) | |
| Duration (days), median (IQR)b | 4.0 (2.0–10.0) | 4.0 (2.0–11.0) | |
| Duration (days), mean (SD)c | 4.6 (7.6) | 4.9 (11.0) | –0.6 (–1.6 to 0.4)d |
| Days alive and free of advanced respiratory support to day 28, mean (SD) | 16.3 (12.6) | 14.9 (12.9) | 1.9 (0.6 to 3.2)d |
| Renal support: no chronic hypertension | |||
| Receipt, n/N (%) | 144/660 (21.8) | 166/671 (24.7) | |
| Duration (days), median (IQR)b | 4.0 (2.0–7.0) | 4.0 (2.0–8.0) | |
| Duration (days), mean (SD)c | 1.2 (3.4) | 1.5 (4.1) | –0.3 (–0.7 to 0.1)d |
| Days alive and free of renal support to day 28, mean (SD) | 16.9 (13.4) | 16.9 (13.4) | 0.1 (–1.1 to 1.4)d |
| Renal support: chronic hypertension | |||
| Receipt, n/N (%) | 158/558 (28.3) | 140/568 (24.6) | |
| Duration (days), median (IQR)b | 4.0 (2.0–8.0) | 4.0 (2.0–7.0) | |
| Duration (days), mean (SD)c | 1.7 (3.9) | 1.6 (4.2) | 0.0 (–0.4 to 0.5)d |
| Days alive and free of renal support to day 28, mean (SD) | 18.0 (12.9) | 16.6 (13.3) | 1.9 (0.5 to 3.2)d |
| Duration of ICU stay (days): no chronic hypertension, median (IQR) | |||
| Survivors | 4.8 (2.6–9.3) | 5.2 (2.9–9.2) | |
| Non-survivors | 3.8 (1.2–8.4) | 3.8 (1.2–8.9) | |
| Duration of ICU stay (days): chronic hypertension, median (IQR) | |||
| Survivors | 5.1 (3.0–10.6) | 5.4 (2.8–9.9) | |
| Non-survivors | 4.4 (1.6–10.8) | 4.2 (1.4–9.9) | |
| Duration of acute hospital stay (days): no chronic hypertension, median (IQR) | |||
| Survivors | 16.5 (9.0–33.0) | 19.0 (10.0–36.0) | |
| Non-survivors | 5.0 (1.0–13.0) | 5.0 (1.0–12.0) | |
| Duration of acute hospital stay (days): chronic hypertension, median (IQR) | |||
| Survivors | 18.0 (11.0–36.0) | 17.0 (10.0–37.5) | |
| Non-survivors | 7.0 (2.0–18.0) | 6.0 (1.0–18.0) | |
Cost-effectiveness
Primary economic outcome at 90 days
Resource use and costs at 90 days
Table 19 shows the resource use up to 90 days post randomisation. Mean total dose of noradrenaline-equivalent vasopressors was 30.3 mg in the permissive hypotension group and 43.2 mg in the usual-care group, and of metaraminol was 15.5 mg in the permissive hypotension group and 19.7 mg in the usual-care group. For the index hospital episode, the mean critical care length of stay was lower in the permissive hypotension group than in the usual-care group, but mean length of stay in general medical wards was similar. Mean length of stay in critical care and general medical wards from readmissions was low and similar between groups. The mean total length of stay up to 90 days in the permissive hypotension and usual-care groups were 20.8 days and 21.0 days, respectively.
| Resource use | Permissive hypotension (N = 1218), mean (SD) | Usual care (N = 1238), mean (SD) |
|---|---|---|
| Intervention | ||
| Noradrenaline dose infused (mg) | 30.27 (54.89) | 43.17 (74.18) |
| Dopamine dose infused (mg) | 0.44 (15.24) | 1.78 (44.58) |
| Phenylephrine dose infused (mg) | 0.29 (3.03) | 0.66 (7.00) |
| Terlipressin dose infused (mg) | 0.06 (0.98) | 0.05 (0.68) |
| Adrenaline dose infused (mg) | 0.48 (4.25) | 0.38 (3.65) |
| Vasopressin (unit) | 5.36 (30.37) | 5.35 (24.49) |
| Metaraminol dose infused (mg) | 15.50 (51.49) | 19.66 (53.38) |
| Index admission | ||
| Days in critical care | 8.29 (10.78) | 8.57 (11.94) |
| General medical bed-days | 12.32 (17.59) | 12.31 (17.24) |
| Readmissions | ||
| Readmissions | 15 (1.23) | 18 (1.45) |
| Days in critical care | 0.06 (0.74) | 0.05 (0.58) |
| General medical bed-days | 0.11 (1.34) | 0.12 (1.45) |
| Total length of stay up to 90 days | 20.79 (22.02) | 21.04 (22.46) |
Table 20 summarises resource use reported from responses to the health services questionnaire. The average number of inpatient days reported from admissions other than those to critical care was higher in the permissive hypotension group than in the usual-care group. Patients in the usual-care group had higher average numbers of outpatient visits and contacts with a GP, nurses, health visitors, occupational therapists and physiotherapists than patients in the permissive hypotension group. All other community care contacts up to 90 days were low and similar between the groups.
| Resource usea | Permissive hypotension (n = 657), mean (SD) | Usual care (n = 643), mean (SD) |
|---|---|---|
| Inpatient days (general medical) | 6.25 (14.95) | 5.47 (13.28) |
| Outpatient visits | 2.15 (3.08) | 2.22 (3.17) |
| GP contacts | 1.68 (2.66) | 1.78 (2.55) |
| Nurse contacts | 2.54 (4.83) | 2.68 (5.44) |
| Health visitor contacts | 0.94 (2.75) | 1.37 (5.05) |
| Occupational therapist contacts | 0.63 (2.92) | 1.00 (4.41) |
| Speech therapist contacts | 0.07 (0.51) | 0.08 (0.53) |
| Physiotherapist contacts | 0.60 (1.50) | 0.97 (2.77) |
| Psychiatrist contacts | 0.02 (0.20) | 0.04 (0.44) |
| Psychiatric nurse contacts | 0.07 (0.58) | 0.06 (0.50) |
| Psychologist contacts | 0.03 (0.27) | 0.02 (0.18) |
| Counsellor contacts | 0.05 (0.46) | 0.02 (0.41) |
Table 21 reports total costs per patient up to 90 days. Intervention (vasopressor) costs per patient were lower for the permissive hypotension group (£83) than for the usual-care group (£104). Index hospital stay accounts for a major share of total costs for both randomised groups. The mean total cost per patient was slightly lower in the permissive hypotension group (£19,034) than in the usual-care group (£19,413).
| Cost | Permissive hypotension (n = 1218) (£) | Usual care (n = 1238) (£) |
|---|---|---|
| Intervention costs | 83 (177) | 104 (186) |
| Hospital costs: index admission | ||
| Critical care | 13,277 (18,909) | 13,823 (21,251) |
| General medical ward | 4153 (5927) | 4147 (5810) |
| Hospital costs: readmissiona | ||
| Critical care | 92 (1186) | 67 (895) |
| General medical | 38 (450) | 41 (488) |
| Outpatient and community costsa,b | 1392 (3728) | 1231 (3512) |
| Total costs up to 90 daysa,b,c | 19,034 (21,433) | 19,413 (23,478) |
Health-related quality of life at 90 days
The health status profiles reported from EQ-5D-5L responses at 90 days are summarised by randomised group in Table 22. At 90 days, the proportion of patients who reported ‘no problems’ for each dimension of the EQ-5D-5L (except anxiety/depression) in the permissive hypotension group was higher than for the usual-care group.
| EQ-5D-5L componenta | Permissive hypotension (N = 657), n (%) | Usual care (N = 643), n (%) |
|---|---|---|
| Mobility | ||
| No problems | 141 (21.46) | 121 (18.82) |
| Slight problems | 113 (17.20) | 110 (17.11) |
| Moderate problems | 136 (20.70) | 150 (23.33) |
| Severe problems | 83 (12.63) | 60 (9.33) |
| Extreme problems | 42 (6.39) | 34 (5.29) |
| Self-care | ||
| No problems | 279 (42.47) | 261 (40.59) |
| Slight problems | 85 (12.94) | 90 (14.00) |
| Moderate problems | 84 (12.79) | 78 (12.13) |
| Severe problems | 28 (4.26) | 18 (2.80) |
| Extreme problems | 37 (5.63) | 26 (4.04) |
| Usual activities | ||
| No problems | 127 (19.33) | 111 (17.26) |
| Slight problems | 125 (19.03) | 117 (18.20) |
| Moderate problems | 117 (17.81) | 129 (20.06) |
| Severe problems | 78 (11.87) | 56 (8.71) |
| Extreme problems | 67 (10.20) | 58 (9.02) |
| Pain/discomfort | ||
| No problems | 159 (24.20) | 141 (21.93) |
| Slight problems | 152 (23.14) | 139 (21.62) |
| Moderate problems | 139 (21.16) | 140 (21.77) |
| Severe problems | 53 (8.07) | 48 (7.47) |
| Extreme problems | 9 (1.37) | 8 (1.24) |
| Anxiety/depression | ||
| No problems | 260 (39.57) | 243 (37.79) |
| Slight problems | 148 (22.53) | 136 (21.15) |
| Moderate problems | 81 (12.33) | 70 (10.89) |
| Severe problems | 17 (2.59) | 19 (2.95) |
| Extreme problems | 6 (0.91) | 9 (1.40) |
Cost-effectiveness at 90 days (primary outcome)
At 90 days, the average cost and mean EQ-5D-5L index scores were similar between groups (Table 23). After adjustment for baseline characteristics, the incremental life-years and QALYs were higher in the permissive hypotension group, with the majority of points (95%) falling on those quadrants on the cost-effectiveness plane, indicating that permissive hypotension had higher mean QALYs (Figure 16). Therefore, the INMB for permissive hypotension compared with usual care was positive, but with a wide CI. At £20,000 per QALY, the INMB was £378 (95% CI −£1347 to £2103). The probability that permissive hypotension is cost-effective is < 70% at the £20,000–30,000 per QALY threshold (Figure 17). The cost-effectiveness results were similar across prespecified subgroups (see Figure 18).
| Outcome | Permissive hypotension (n = 1283), mean (SD) | Usual care (n = 1300), mean (SD) | Effect estimate (95% CI) |
|---|---|---|---|
| Costs (£) | 19,034 (21,433) | 19,413 (23,478) | –311 (–2042 to 1420) |
| EQ-5D-5L (survivors) | 0.677 (0.274) | 0.683 (0.272) | –0.0003 (–0.031 to 0.031) |
| Life-years | 0.160 (0.108) | 0.155 (0.109) | 0.008 (0.000 to 0.015) |
| QALYs | 0.050 (0.049) | 0.048 (0.049) | 0.003 (–0.0004 to 0.007) |
| INMB (£)a | 378 (–1347 to 2103)a |
FIGURE 16.
Uncertainty in the mean costs (£) and QALY differences and their distribution for intervention vs. usual care (within 90 days post randomisation).
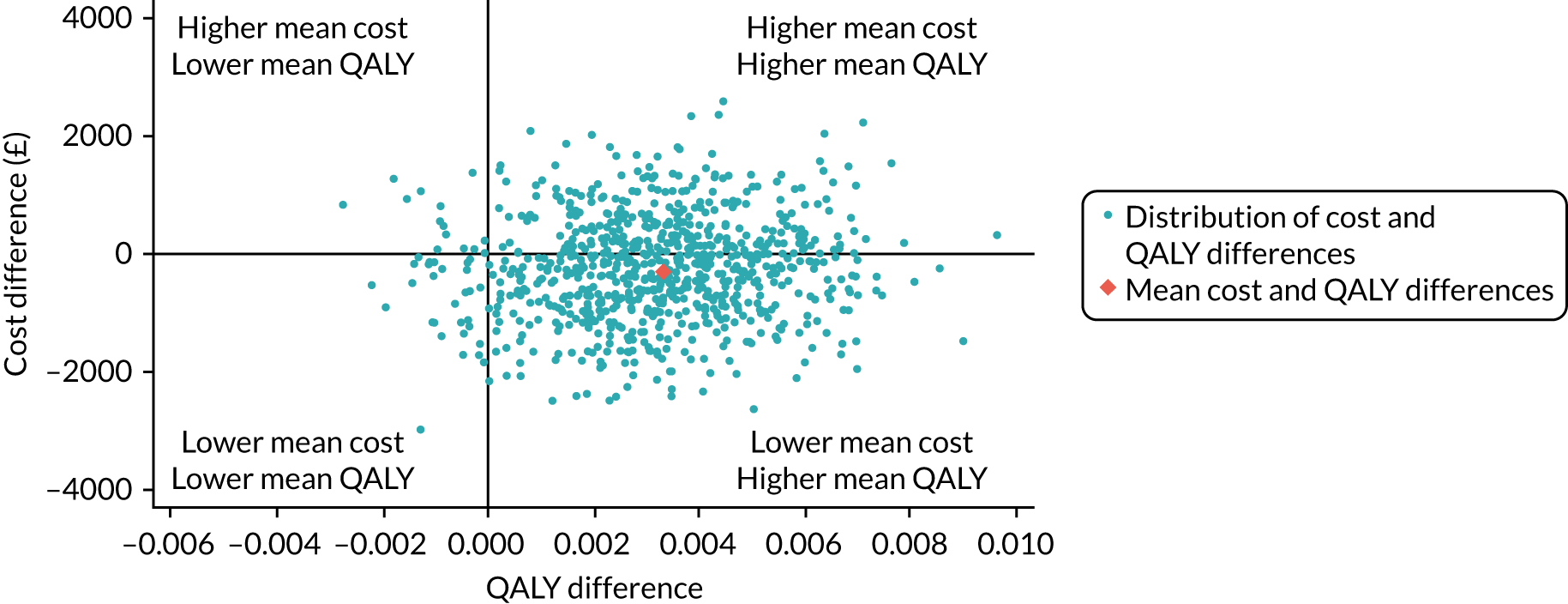
FIGURE 17.
Cost-effectiveness acceptability curve at 90 days. Reports the probability that permissive hypotension is cost-effective at alternative levels of willingness to pay for a QALY gain.
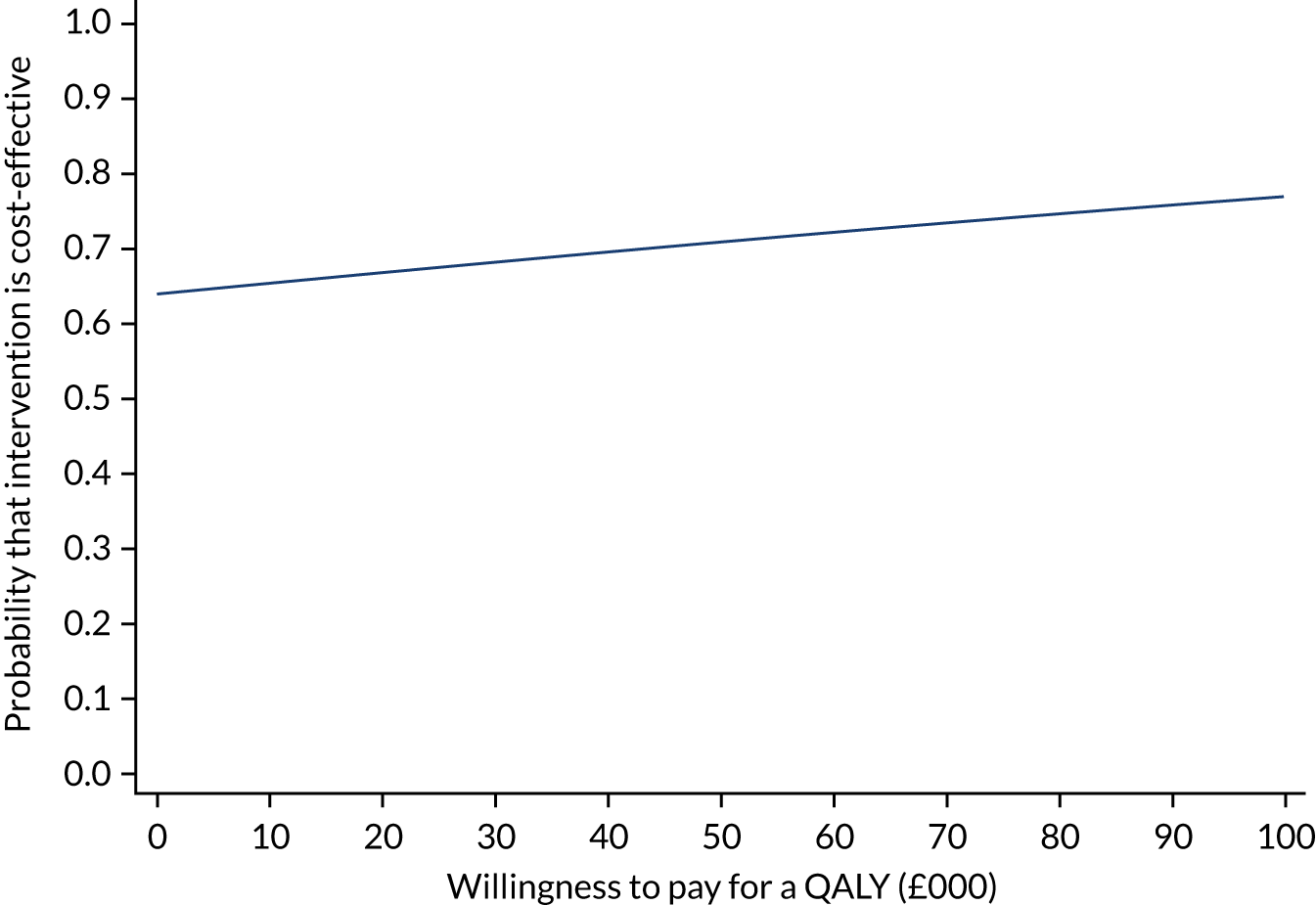
FIGURE 18.
Subgroup analysis for the cost-effectiveness primary outcome at 90 days. Vertical line indicates no difference in net monetary benefits between comparator groups.
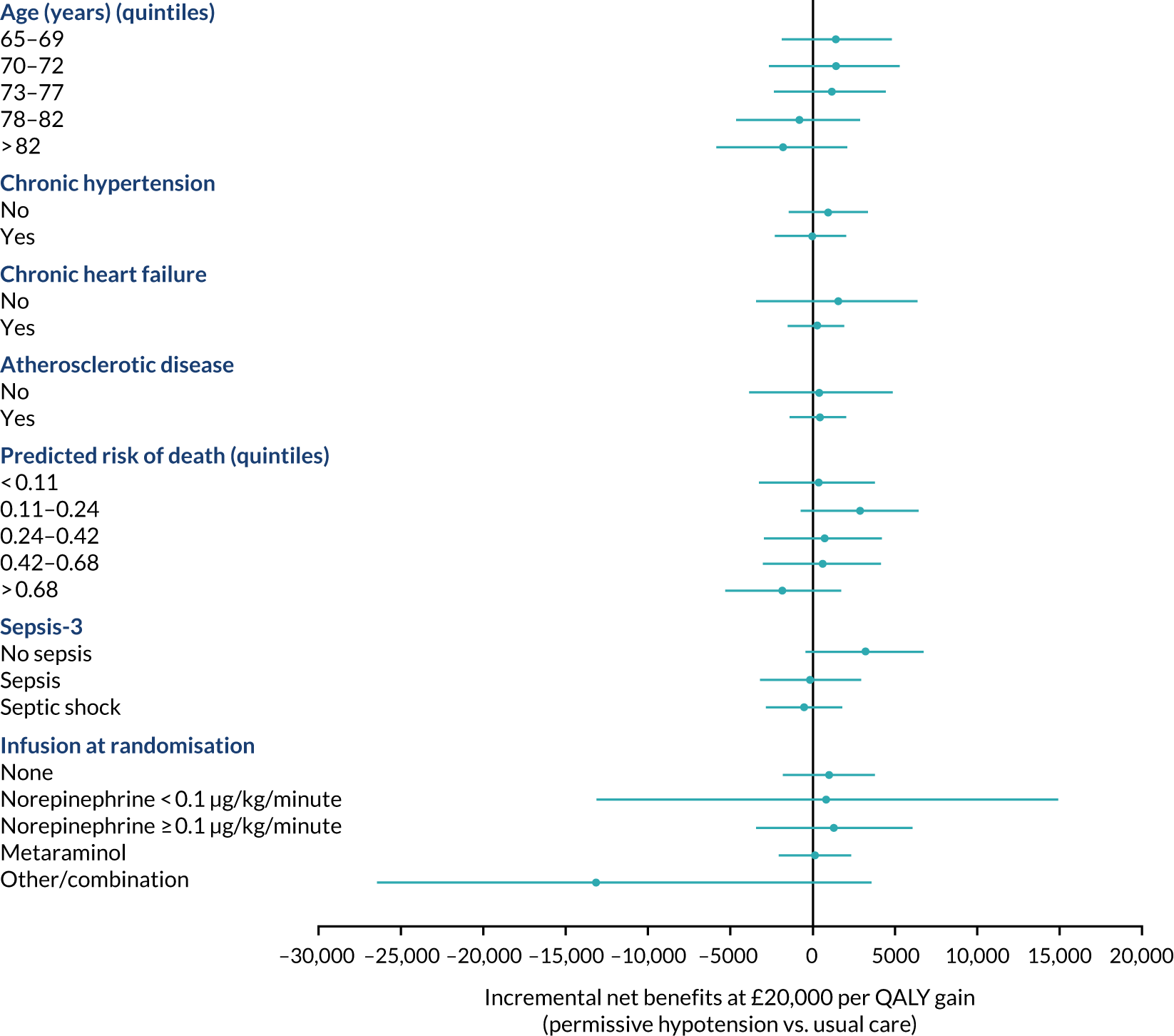
Figure 19 reports the mean (with 95% CI) of the incremental net benefit (at £20,000 per QALY) according to alternative assumptions, compared with the base case. These sensitivity analyses showed that the results were robust to alternative scenarios.
FIGURE 19.
Sensitivity analysis for the CEAs at 90 days. Vertical line indicates no difference in net monetary benefits between comparator groups. HSQ, health services questionnaire.
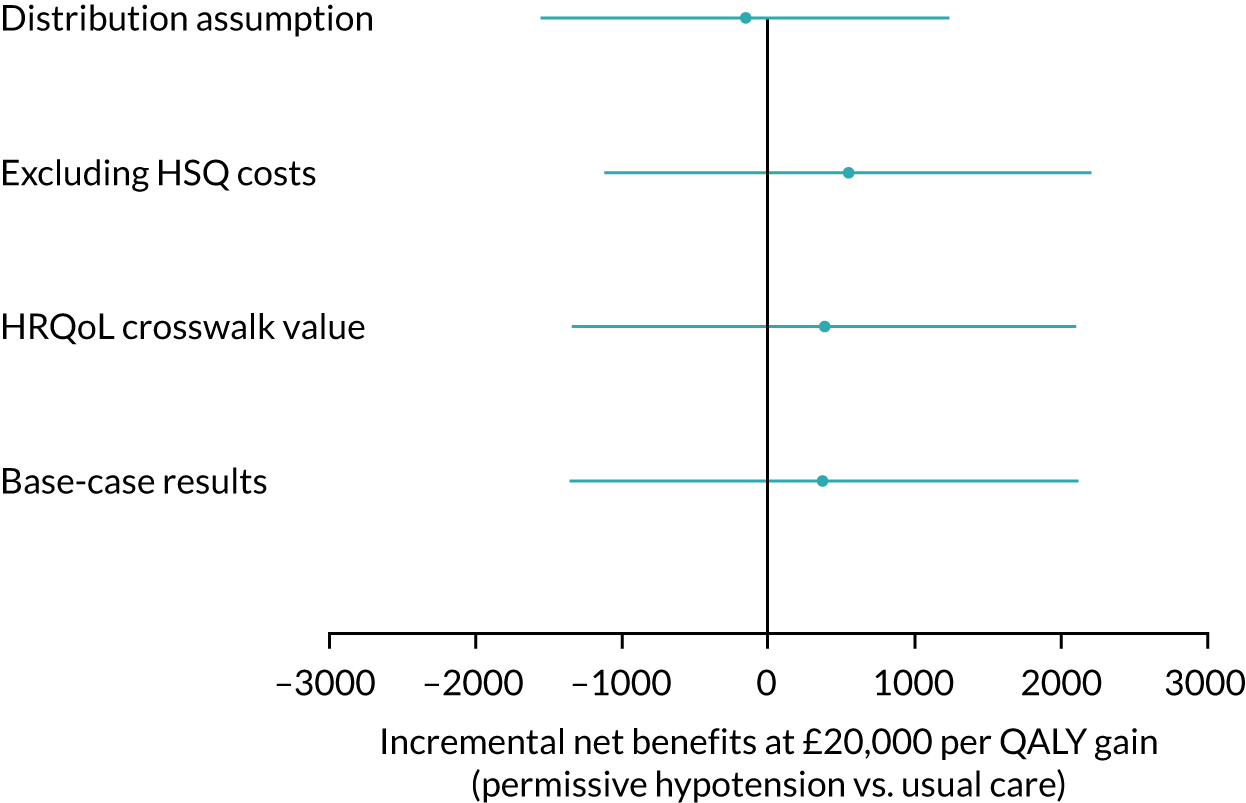
Expert elicitation
Sixty-three individuals were sent a link to the elicitation questionnaire. Thirty-five experts submitted completed questionnaires, of which 32 were classified as usable, 24 high confidence and six very high confidence. Of the usable experts, 50% were medical doctors and 31% were clinical or research nurses, 59% had been in their current role for > 7 years and 91% had work that involved following up patients in person after intensive care.
Table 24 summarises the elicitation responses across all usable experts. Overall, for patients receiving usual care, the elicited average HRQoL scores were lower for patients who did not return their questionnaire than the average from the observed data. For patients with missing HRQoL scores, the elicited values were very slightly lower for those allocated to permissive hypotension than for those allocated to usual care. There is a wide diversity in the elicited scores across experts, as indicated by the SDs.
| Most likely patient EQ-5D-5La average scores (i.e. mode) | Mean (SD) |
|---|---|
| Permissive hypotension group: did not return an EQ-5D-5L | 68 (11) |
| Usual-care group: did not return an EQ-5D-5L | 70 (11) |
| Differences in most likely patient EQ-5D-5L average scores | |
| Usual-care group: did not return an EQ-5D-5L minus did return a EQ-5D-5L | –7 (11) |
| Did not return an EQ-5D-5L: permissive hypotension minus usual care | –1 (10) |
The results of the sensitivity analysis compared with the primary analysis and complete-case analysis are summarised in Figures 20 and 21 for the HRQoL and the INMB, respectively, valuing QALY at £20,000 per QALY. These figures show (1) the posterior probability that the outcome favours permissive hypotension and (2) the posterior distribution of the 90-day HRQoL difference (permissive hypotension – usual care)/INMB. The full posterior distribution is shown as a density strip,67 where the darkness at a point is proportional to the probability density. The results from the HRQoL sensitivity analysis are broadly similar to the primary analysis in terms of point estimates, but with greater uncertainty about these. The extreme individual priors provide greater differences: the probability that the mean HRQoL score is higher for permissive hypotension is 99% and 37% for the ‘most enthusiastic’ expert and the ‘most sceptical’ expert, respectively. For INMB, there is little difference between the results of any of the analyses.
FIGURE 20.
Incremental net benefit estimates at 90 days post randomisation from sensitivity analyses exploring the effects of MNAR assumptions vs. the primary and complete-case analyses. Note that for the complete-case analysis, the posterior mean is almost zero and therefore beneath the blue line. Each shaded rectangular strip shows the full posterior distribution. The darkness at a point is proportional to the probability density, such that the strip is darkest at the maximum density and fades into the background at the minimum density. The posterior mean and 95% credible interval are marked. For INMB, positive differences favour permissive hypotension.
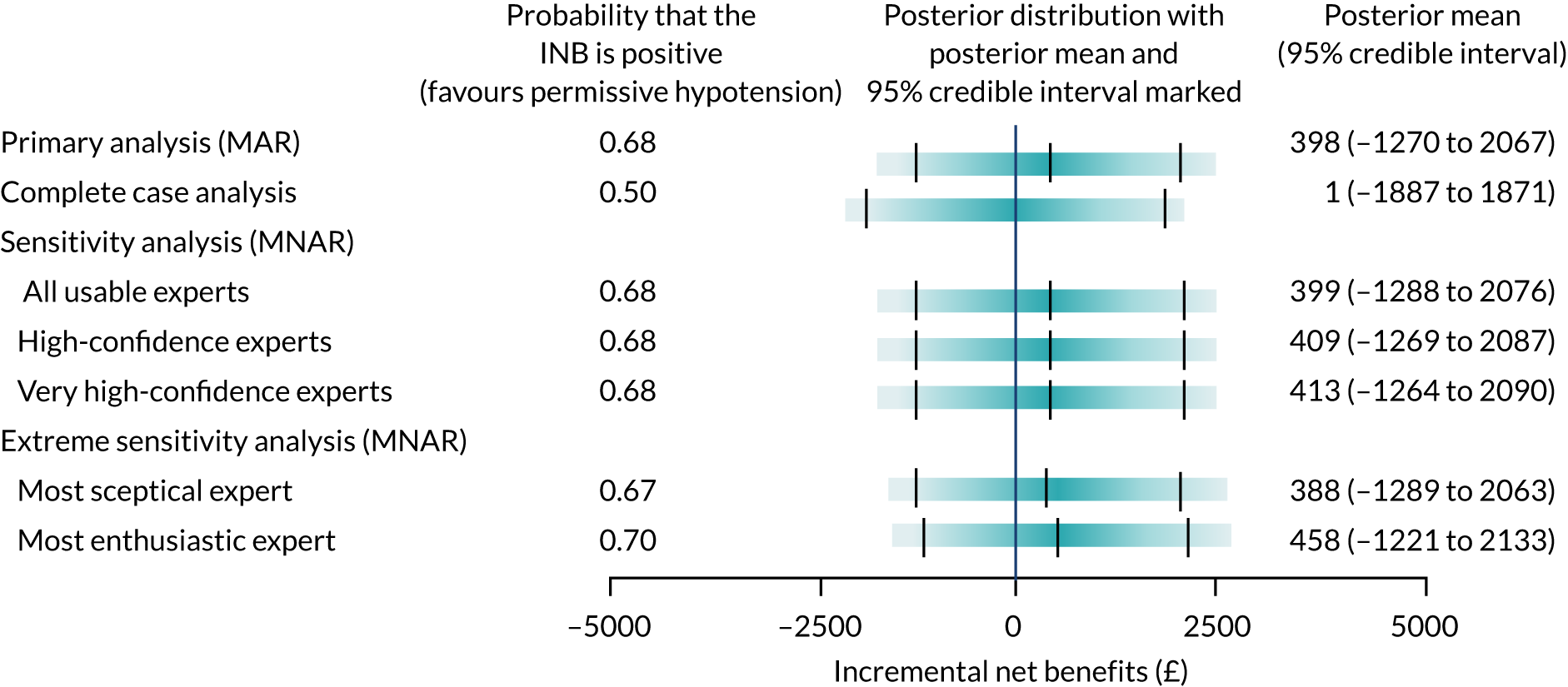
Cost-effectiveness at 1 year
Resource use and costs up to 1 year
Table 25 presents resource use up to 1 year. The permissive hypotension group had a higher mean number of days in critical care and general medical wards than the usual-care group between 90 days and 1 year post randomisation. The average hospital length of stay prior to 1 year post randomisation was 22.0 days in the permissive hypotension group and 21.1 days in the usual-care group.
| Resource use | Permissive hypotension (n = 994), mean (SD) | Usual care (n = 1005), mean (SD) |
|---|---|---|
| Total length of stay up to 90 days | 19.81 (21.88) | 19.30 (21.60) |
| Hospital length of stay from 90 days to 1 year: index admission | ||
| Days in critical care | 0.15 (2.15) | 0.03 (0.87) |
| General medical bed-days | 1.59 (15.53) | 1.07 (7.92) |
| Hospital length of stay from 90 days to 1 year: readmissions | ||
| Days in critical care | 0.14 (1.36) | 0.14 (1.72) |
| General medical bed-days | 0.33 (3.32) | 0.53 (7.08) |
| Total length of stay up to 1 year | 22.02 (32.06) | 21.07 (27.96) |
Resource use between 90 days and 1 year is summarised in Table 26. The usual-care group had a large number of inpatient days (general medical), outpatient visits, contacts with GPs and nurses, and visits to occupational therapists and physiotherapists. The permissive hypotension group had a larger number of health visitor contacts. Other contacts related to community care were small and with no difference between the groups.
| Resource usea | Permissive hypotension (n = 309), mean (SD) | Usual care (n = 303), mean (SD) |
|---|---|---|
| Inpatient days (general medical) | 5.94 (18.50) | 7.14 (16.53) |
| Outpatient visits | 4.19 (4.49) | 4.50 (4.83) |
| GP contacts | 3.35 (4.12) | 3.40 (4.15) |
| Nurse contacts | 4.36 (6.75) | 4.50 (6.38) |
| Health visitor contacts | 0.68 (3.42) | 0.48 (1.96) |
| Occupational therapist contacts | 0.51 (2.84) | 0.88 (4.01) |
| Speech therapist contacts | 0.15 (0.82) | 0.08 (0.40) |
| Physiotherapist contacts | 0.80 (1.99) | 0.93 (2.61) |
| Psychiatrist contacts | 0.02 (0.22) | 0.14 (0.86) |
| Psychiatric nurse contacts | 0.14 (0.80) | 0.08 (0.61) |
| Psychologist contacts | 0.02 (0.22) | 0.06 (0.57) |
| Counsellor contacts | 0.07 (0.58) | 0.01 (0.08) |
Table 27 reports the total costs at 1 year across all the resource use items recorded. At 1 year, the mean total cost per patient was £20,299 for the permissive hypotension group and £19,850 for the usual-care group.
| Cost | Permissive hypotension (n = 994) (£) | Usual care (n = 1005) (£) |
|---|---|---|
| Total costs up to 90 daysa,b,c | 18,455 (21,178) | 18,003 (21,732) |
| 90 days to 1 year hospital costs: index admission | ||
| Critical care | 248 (3466) | 46 (1334) |
| General medical ward | 535 (5231) | 361 (2669) |
| 90 days to 1 year hospital costs: readmissiona | ||
| Critical care | 218 (2284) | 224 (3111) |
| General medical | 113 (1118) | 178 (2385) |
| Outpatient and community costsb,c | 730 (2131) | 1038 (3601) |
| Total costs up to 1 yeara,b,c | 20,299 (25,501) | 19,850 (24,676) |
Health-related quality of life at 1 year
The health status profiles reported from responses to the EQ-5D-5L at 1 year post randomisation are summarised by randomised group in Table 28. At 1 year, the proportion who reported ‘no problems’ for each dimension of the EQ-5D-5L (except mobility) was similar in the randomised group. A higher proportion of patients in the permissive hypotension group than in the usual-care group had reported ‘extreme problems’ for each dimension of EQ-5D-5L.
| EQ-5D-5L componenta | Permissive hypotension (N = 309), n (%) | Usual care (N = 303), n (%) |
|---|---|---|
| Mobility | ||
| No problems | 79 (25.57) | 62 (20.46) |
| Slight problems | 51 (16.50) | 63 (20.79) |
| Moderate problems | 71 (22.98) | 67 (22.11) |
| Severe problems | 36 (11.65) | 45 (14.85) |
| Extreme problems | 17 (5.50) | 7 (2.31) |
| Self-care | ||
| No problems | 154 (49.84) | 154 (50.83) |
| Slight problems | 43 (13.92) | 37 (12.21) |
| Moderate problems | 31 (10.03) | 32 (10.56) |
| Severe problems | 11 (3.56) | 16 (5.28) |
| Extreme problems | 15 (4.85) | 6 (1.98) |
| Usual activities | ||
| No problems | 79 (25.57) | 80 (26.40) |
| Slight problems | 70 (22.65) | 74 (24.42) |
| Moderate problems | 54 (17.48) | 50 (16.50) |
| Severe problems | 28 (9.06) | 27 (8.91) |
| Extreme problems | 23 (7.44) | 15 (4.95) |
| Pain/discomfort | ||
| No problems | 79 (25.57) | 74 (24.42) |
| Slight problems | 81 (26.21) | 73 (24.09) |
| Moderate problems | 67 (21.68) | 68 (22.44) |
| Severe problems | 25 (8.09) | 28 (9.24) |
| Extreme problems | 3 (0.97) | 4 (1.32) |
| Anxiety/depression | ||
| No problems | 136 (44.01) | 129 (42.57) |
| Slight problems | 68 (22.01) | 75 (24.75) |
| Moderate problems | 35 (11.33) | 38 (12.54) |
| Severe problems | 10 (3.24) | 2 (0.66) |
| Extreme problems | 6 (1.94) | 2 (0.66) |
Cost-effectiveness at 1 year
At 1 year, the average cost was higher in the permissive hypotension group and mean EQ-5D-5L index scores were similar between groups (Table 29). The incremental life-years and QALYs were higher in the permissive hypotension group, with the majority of points falling into those quadrants on the cost-effectiveness plane indicating that permissive hypotension had higher mean QALYs (Figure 22). The INMB for permissive hypotension compared with usual care was negative, but with a wide CI. At £20,000 per QALY, the INMB was –£361 (95% CI −£2537 to £1815). The probability that permissive hypotension is cost-effective is < 40% and 45% at the £20,000 and £30,000 per QALY threshold, respectively (Figure 23).
| Outcome | Permissive hypotension (n = 994) | Usual care (n = 1005) | Incremental effect, mean (95% CI) |
|---|---|---|---|
| Costs (£) | 20,299 (25,501) | 19,850 (24,676) | 699 (–1466 to 2864) |
| EQ-5D-5L (survivors) | 0.706 (0.264) | 0.716 (0.245) | –0.011 (–0.050 to 0.028) |
| Life-years | 0.474 (0.465) | 0.446 (0.464) | 0.036 (–0.000 to 0.072) |
| QALYs | 0.263 (0.324) | 0.252 (0.322) | 0.017 (–0.009 to 0.043) |
| INMB (£)a | –361 (–2537 to 1815) |
FIGURE 21.
Health-related quality-of-life treatment effect estimates at 90 days post randomisation from sensitivity analyses exploring the effects of MNAR assumptions vs. the primary and complete-case analyses. Note that the posterior mean, for the primary analysis (MAR), is almost zero and below the blue line. The entire posterior distribution is indicated by each rectangular line (shaded). At each point, the shade is proportional to the probability density, meaning that a line at the maximum density is darkest and fades as density decreases. There are markings for both the posterior mean and the 95% credible interval. Positive HRQoL differences favour the permissive hypotension group. Reproduced with permission from Lamontagne et al. 59 Copyright © 2020 American Medical Association.
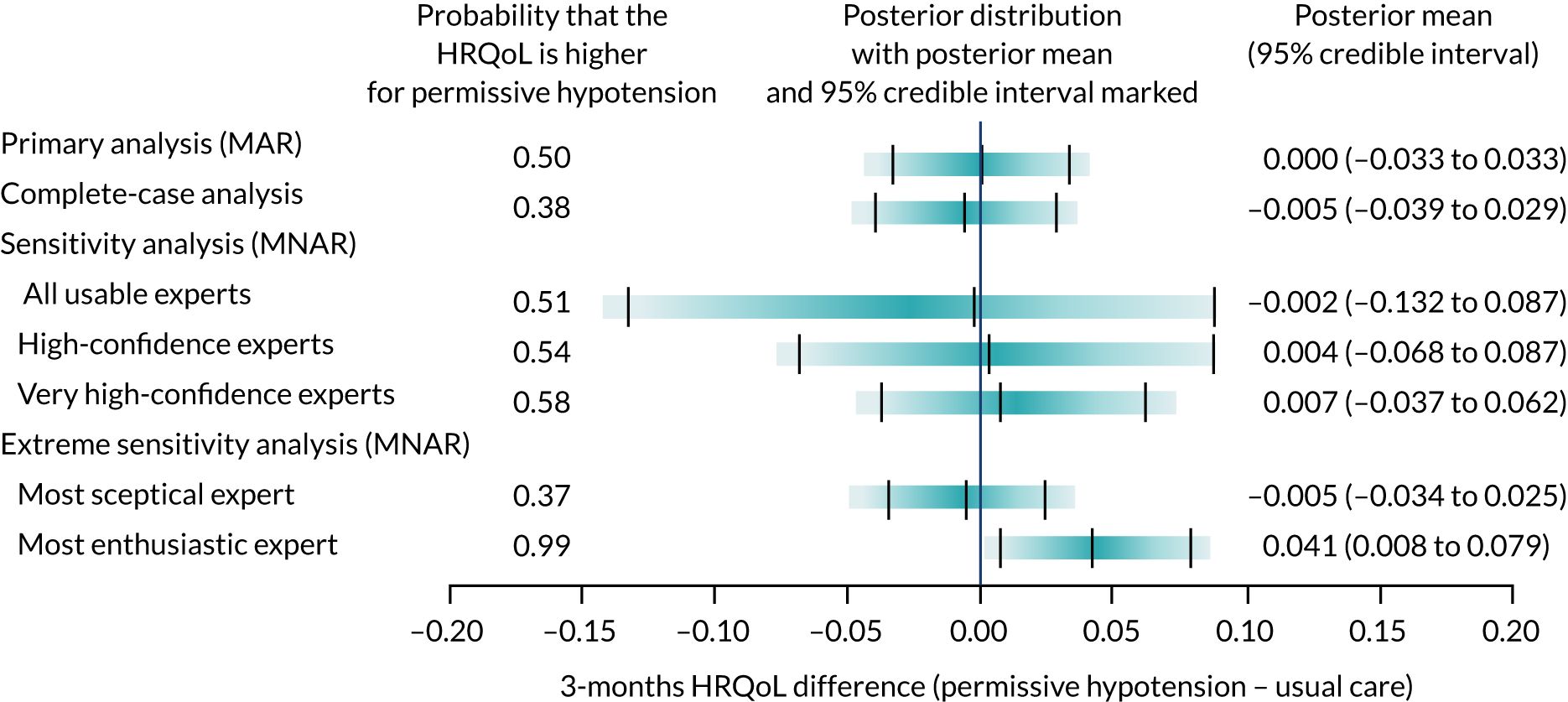
FIGURE 22.
Uncertainty in the mean costs (£) and QALY differences and their distribution for intervention vs. usual care (within 1 year post randomisation).
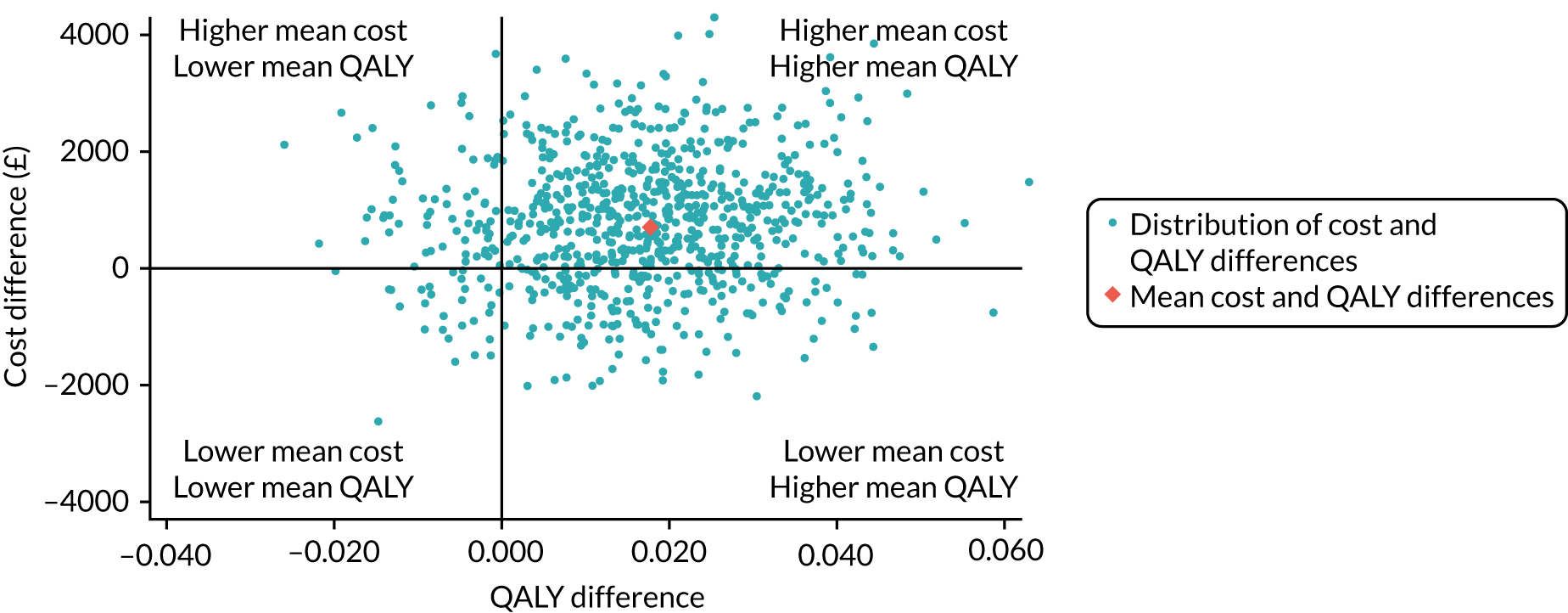
The cost-effectiveness results at 1 year were similar across prespecified subgroups (Figure 24). The results were similar for alternative scenarios considered in sensitivity analyses at 1 year (Figure 25).
FIGURE 23.
Cost-effectiveness acceptability curve at 1 year.
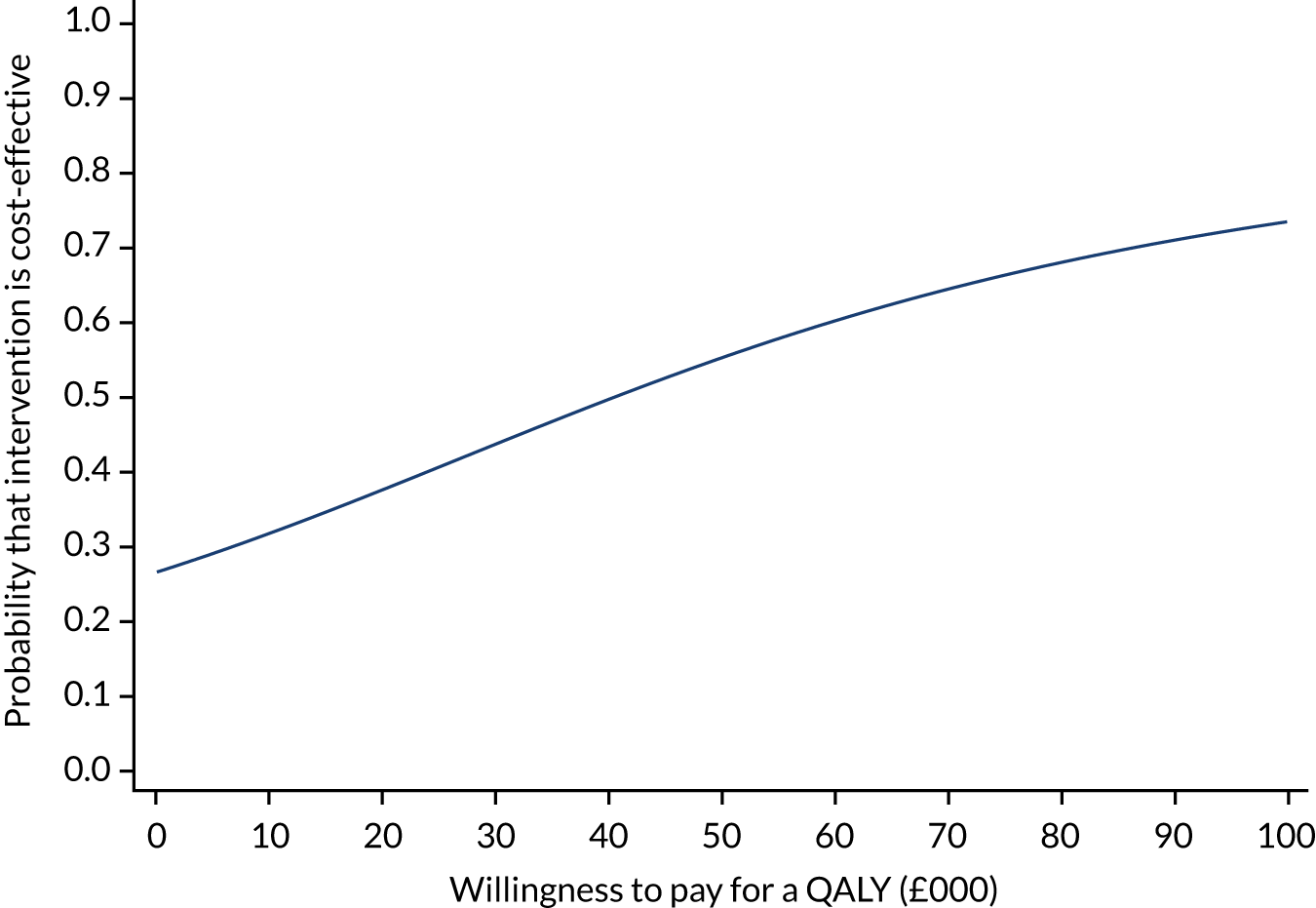
FIGURE 24.
Subgroup analysis for the cost-effectiveness outcome at 1 year. Vertical line indicates no difference in net monetary benefits between comparator groups.
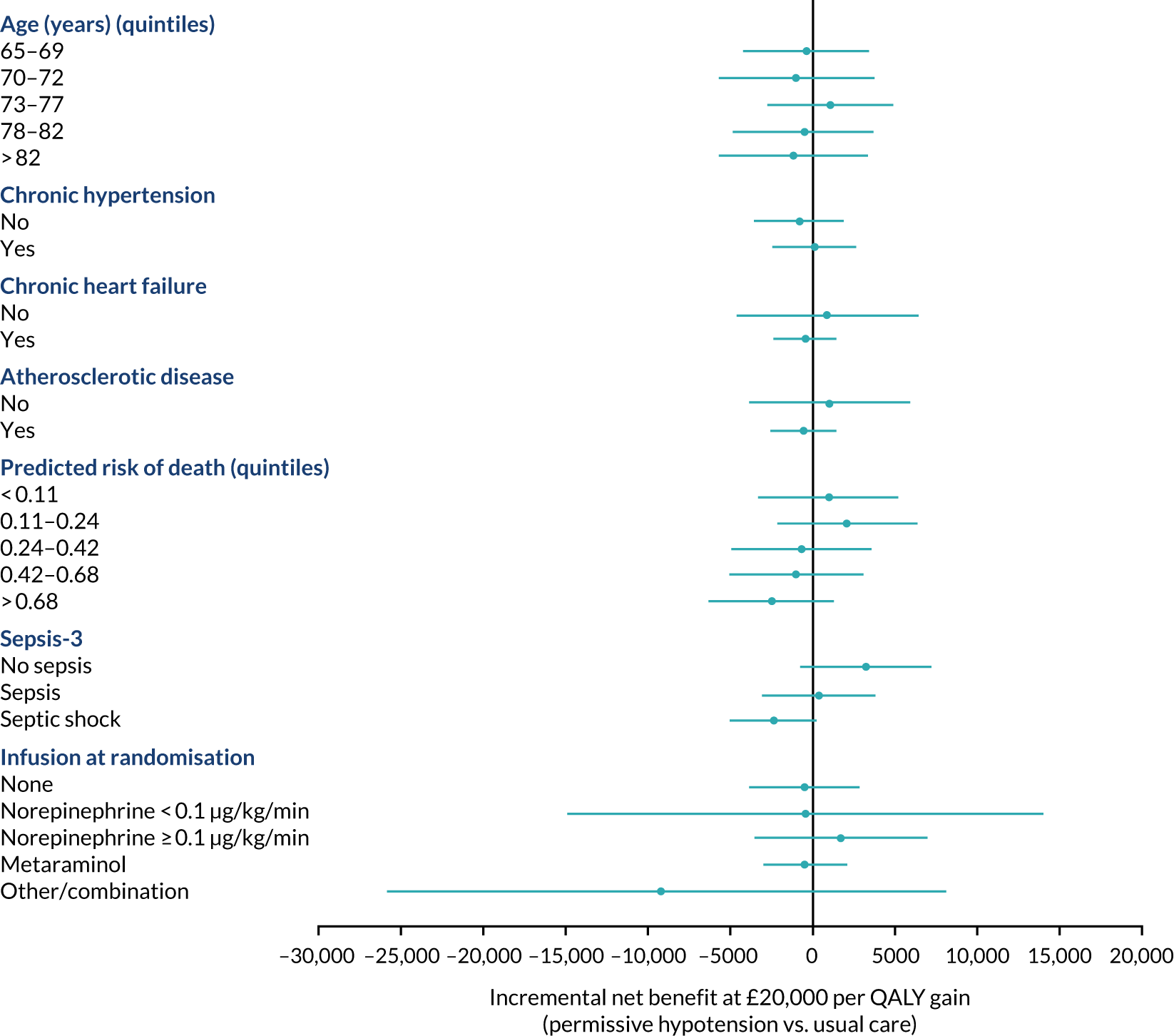
FIGURE 25.
Sensitivity analysis for the CEAs at 1 year. Reports the mean with 95% CI of the incremental net benefit (at £20,000/QALY) according to alternative assumptions. The solid vertical line indicates no difference in net monetary benefits between the treatment groups. Vertical line indicates no difference in net monetary benefits between comparator groups. HSQ, health services questionnaire.
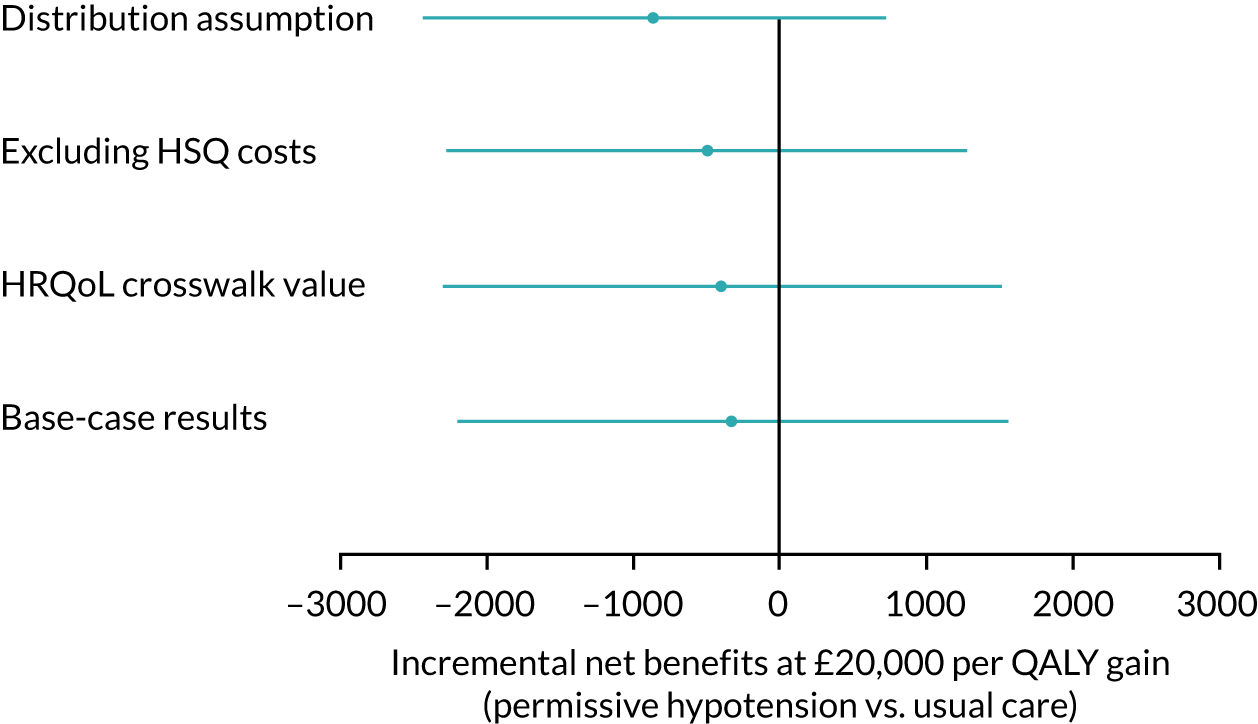
Chapter 4 Discussion and conclusions
Principal findings
In patients aged ≥ 65 years who receive vasopressors for vasodilatory hypotension, permissive hypotension did not significantly reduce mortality at 90 days. The absolute treatment effect on 90-day mortality, based on the 95% CIs, was between a 6.8-percentage reduction and a 1.1-percentage increase in mortality.
Patients in the permissive hypotension group received a lower exposure to vasopressors than those in the usual-care group, in terms of both mean duration and total dose. For mean total dose of vasopressors (noradrenaline equivalent), patients in the permissive hypotension group received 31.5 mg compared with 44.3 mg for patients in the usual-care group (difference −12.8 mg, 95% CI −18.0 mg to −7.6 mg). Adherence to permissive hypotension was good. In occurrences where a patients’ MAP was > 65 mmHg, vasopressors were not reduced or discontinued only 6% of the time. There was no clinically important difference in fluid balance, urine output or the use of pure inotropes between the groups.
Secondary outcomes and prespecified SAEs were similar in both groups. There was evidence of heterogeneity of treatment effect within the chronic hypertension subgroup, with a lower mortality rate observed in patients with chronic hypertension in the permissive hypotension than in patients with chronic hypertension in the usual-care group. There was no evidence for heterogeneity of treatment effect in other prespecified subgroups.
The economic evaluation found that permissive hypotension had similar costs and EQ-5D-5L index scores to usual care; however, given higher incremental life-years and QALYs in the permissive hypotension group, the INMB was positive at 90 days, but with high statistical uncertainty (£378, 95% CI −£1347 to £2103). The probability that permissive hypotension is cost-effective is 70% at the £20,000 per QALY threshold. The cost-effectiveness results were similar across prespecified subgroups for alternative scenarios considered in sensitivity analyses, and over 1 year. The QALY gains associated with permissive hypotension were positive, but relatively small, at both 90 days and 1 year. The incremental costs of permissive hypotension relative to usual care was lower at 90 days, but at 1 year the intervention group had higher costs. Therefore, although at 90 days the intervention was both more effective and cheaper, at 1 year it was more effective but costlier. However, it should be highlighted that these mean cost-effectiveness results at both 90 days and 1 year were surrounded by considerable statistical uncertainty. The cost-effectiveness results were sensitive to relatively small QALY gains, leading to changes in the probability of cost-effectiveness of permissive hypotension from 70% at 90 days to 40% at 1 year.
Our findings in context
Consistent with the hypothesis of the trial, the potential benefit of permissive hypotension appears that it may increase with age when patients may become frailer and more vulnerable. This is in line with the individual patient data meta-analysis11 of the SEPSISPAM (Sepsis and Mean Arterial Pressure) trial13 and the OVATION (Optimal VAsopressor TItratiON) pilot trial,12 which suggest that older patients may be at greater risk of harm from greater exposure to vasopressors. 3
It is a commonly held belief that patients with chronic hypertension benefit from higher MAPs, but our trial appeared to show a greater benefit associated with permissive hypotension in patients with chronic hypertension than in those without. However, this subgroup analysis (which was not adjusted for multiple testing) should be interpreted with caution in the light of the non-significant primary analysis. In contrast to the SEPSISPAM trial,13 in patients with chronic hypertension, we observed no clinically important increase in the use of renal replacement therapy when using a lower MAP target. Although these results suggest that it may be safe to tolerate lower blood pressure in patients with chronic hypertension, further research is required to better understand the interaction between this chronic comorbidity and vasopressors. In addition, patients who suffer from chronic hypertension are also at risk of other comorbidities, potentially rendering them more vulnerable to vasopressor-induced adverse effects.
Strengths and limitations
Strengths
The 65 trial was a pragmatic trial, set in a real-world context. It was conducted in a large, representative sample of 65 critical care units across the NHS. Using a simple intervention, designed to be implementable if adopted across the NHS, and straightforward trial procedures, the trial was designed for efficiency of delivery and generalisability of the results. This allowed central trial and local site set-up to be rapid, with > 50 sites open to recruitment within 4 months of opening. Patient recruitment followed a similar pattern, with the full sample of 2600 patients recruited in < 21 months, far exceeding the initial schedule to recruit 1440 patients over an 18-month period. This was enabled through the ability of participating sites to embed delivery of the trial into routine clinical practice, allowing randomisation to be carried out 24 hours per day, 7 days per week, which compared favourably with previous trials. The following elements contributed to this:
-
The simplicity of the telephone randomisation process, with only minimal information at the point of randomisation needed.
-
The simplicity of the intervention and control groups, with the intervention being a straightforward change to a threshold for delivery of vasopressors, which could be delivered by bedside nurses.
-
Use of ‘research without prior consent’, which meant that the minority of staff who have undergone training in taking informed consent did not need to be present prior to randomisation.
-
Data collection being nested within routinely collected data, which meant that a parsimonious data set could be defined for additional collection within the trial, focused on the key aspects of delivery of the trial interventions and safety reporting.
-
The ongoing development and experience of the UK Critical Care CRN, which has trained and funded research nurses to be able to deliver trials, such as this, efficiently and effectively.
The decision to employ a usual-care, as opposed to a higher MAP target, control group meant that we avoided the risk of potentially artificially increasing harm in the control group and ensured that the control group was representative of the current standard of care in the NHS. 68 Our design used the results of previous studies to guide enrichment of the trial population, which involved enrolling older patients who were considered to have a greater chance of benefiting from the intervention.
In contrast to the other trials in this area, the 65 trial integrated a rigorously designed and conducted economic evaluation and included assessment of important patient-centred outcomes, such as HRQoL and cognitive decline, among survivors. These important outcomes are often valued by patients above 90-day mortality. 69,70
Both the protocol22 and statistical analysis plan47 were submitted for publication during the recruitment period to ensure transparency, and all analyses were conducted following the analysis plan. Sensitivity analyses were also conducted, which comprised different approaches to handling missing data, including MAR and MNAR.
The study has measured quality of life with the EQ-5D-5L. This version of the instrument was anticipated to be sensitive to differences in health status between the treatment groups. EQ-5D-5L descriptive data at 90 days and 1 year were valued using a published EQ-5D-5L value set for England, as set out in the original proposal and statistical and health economic analysis plan. Over the time that the study was conducted, the EQ-5D-5L value set for England was subject to criticism and NICE have recommended using a crosswalk value sets (three to five level) via a ‘mapping algorithm’,63 which provides an interim means of scoring EQ-5D-5L, while a new five-level value set is developed. As both the currently available five-level value set (considered in our base case) and the crosswalk approach (considered in our sensitivity analysis) are subject to concerns, we considered that using both approaches was appropriate. In the 65 trial, there was no difference in patients described health status according to the EuroQol-5 Dimensions descriptive system and therefore the results were insensitive to the choice of approach to health state valuation.
Limitations
Owing to the nature of the intervention, it was not blinded. However, the use of outcomes collected through routine sources that are not subject to observer bias, including the objective primary outcome, would have minimised any potential bias. When compared with the sample size assumptions, there were two small differences:
-
A higher mortality rate was observed in the usual-care group. This may have been due to not being able to take into account the full eligibility criteria in routinely collected data. For example, clinical audit data would not have the granularity of data or be able to capture a subjective decision, such as whether or not the clinical team expected vasopressors to continue for an additional 6 hours or more. Therefore, a proportion of the less sick patients, who had lower vasopressor requirements, were probably excluded from the trial, but included within the sample size assumptions.
-
The rate of non-consent was slightly higher than anticipated. This was the first trial of this size carried out in this population using a ‘research without prior consent’ model in the UK. These results will be important in informing future trial design and planning in the future.
When describing exposure to vasopressors, it was not possible to convert doses of metaraminol into a noradrenaline-equivalent dose. To our knowledge, no formula for such dose conversion exists, potentially because of there being no relationship between doses of metaraminol and noradrenaline. 71
As a pragmatic trial that uses an efficient design and parsimonious data set, no mechanistic data were collected, which meant that the attributable mortality could not be adjudicated. In addition, we did not carry out an integrated process evaluation, which could have provided additional contextual factors around clinician and team behaviour.
Implications for health care/practice
The CI for the absolute risk difference, as well as the adjusted analysis, indicates that minimising exposure to vasopressors in older patients with vasodilatory hypotension is unlikely to be harmful and may be beneficial for patients. This may be because critical care patients are particularly susceptible to iatrogenic complications and ‘less may be more’. 72,73 Usual care, which allowed expert clinicians to individualise vasopressors in view of an array of parameters (e.g. patient characteristics and markers of tissue perfusion), did not outperform use of a single parameter, the MAP, to minimise exposure to vasopressors.
In addition, the longer-term patient-centred outcomes, such as quality of life and cognitive decline, supported the notion that permissive hypotension is safe. The fact that potential differences in survival were observed largely only after critical care unit discharge suggests that the risks posed by greater exposure to vasopressors therapy may not be immediate, which may explain why they are under-reported. Ongoing mechanistic studies may shed further light on this.
Although adherence to permissive hypotension was good, there were areas for improvements that our trial should cast light on. There were two main reasons for not reducing or discontinuing vasopressors when the MAP was above the target range. First, a lack of trial awareness (n = 54) meant that some staff (often not full-time critical care unit staff) did not realise that they were supposed to be actively titrating vasopressors to a lower MAP target. Second, there was a focus on other clinical priorities (n = 42), which led to vasopressors being left at the same dose rate while the clinical staff completed other tasks. This suggests that, even though less may be more beneficial, a more active involvement of clinical staff to reduce exposure to vasopressors is required. Our trial also addresses the concerns over the perceived need for higher MAP targets for patients with chronic hypertension and with low urine output. Use of these surrogate outcomes to guide vasopressors does not necessarily lead to longer-term improvements in patient-centred outcomes.
There were positive implications shown for the research infrastructure within the NHS. As reported above, there was overwhelming interest in the trial, with > 100 sites expressing interest in participating. Within the 65 participating sites, even though there was a short time frame to recruit patients (up to 6 hours from becoming eligible), patients were randomised early and recruitment rates exceeded the pre-trial estimates. This was because of the ability of sites to embed the trial procedures within their routine clinical practice, providing a template for future studies in this complex setting to use more efficient and embedded procedures.
Summaries of key research recommendations
Recommendation 1: individual patient data meta-analysis
Our trial was the next step in the evaluation of blood pressure targets in critical care. The opportunity to pool the data from the previous two RCTs (SEPSISPAM13 and OVATION12) with our data will give additional power for evaluating this group of patients and assess intervention effects in potentially important subgroups. As this will include data across the UK, France and Canada, it will enhance generalisability and improve knowledge on this important area for critical care worldwide.
Recommendation 2: evaluate heterogeneity of treatment effect
In conventional subgroup analyses alongside RCTs, a number of one-at-a-time subgroups and thresholds defining each subgroup are prespecified. Significance testing of subgroup interactions is the main approach for the evaluation of subgroup effects. This may fail to fully capture variation in complex baseline risk factors and their consequent moderation of treatment effects. RCTs are also generally underpowered to detect subgroup effects. In this context, machine learning methods that have been developed to identify subgroups with different treatment effects, allowing the estimation of individual average treatment effects, without relying on any a priori modelling assumptions, would be useful as an alternative approach to detect and estimate true subgroup effects.
Recommendation 3: further research on the conduct, including use of research without prior consent model
Further research should study the efficient conduct of our trial, which used simple, straightforward procedures and a research without prior consent model, to further understand the implications of these procedures with the hope of producing guidance for future studies in the critical care setting. One of the key aspects is that our trial is one of the largest studies in the adult critical care setting in the UK to employ a research without prior consent model. It is therefore vital to use the extensive data collected on the consent process to look into patient and family member preferences and agreement, and the mechanism of consent in the UK, for example timing of consent.
Acknowledgements
We thank the NIHR Health Technology Assessment programme for funding this trial. We are very grateful to all of the patients who took part in the 65 trial and their family members. We also thank Joseph Collins, Sian Martin, Abby Koelewyn, Laura Drikite, Akshay Patel and Sara Magnusson for their support in delivering the trial.
We acknowledged that there have been many other individuals who made a contribution within the participating sites. It is impossible to thank everyone personally; however, we would like to thank all of the 65 trial investigators (see Appendix 1).
Chief investigator and co-investigators
Mr Paul R Mouncey (chief investigator), Professor François Lamontagne (lead clinical investigator), Mrs Julie Camsooksai, Professor Anthony C Gordon, Professor Richard D Grieve, Professor David A Harrison, Ms Doreen Henry, Professor Kathryn M Rowan, Dr M Zia Sadique, Mr Chris Whitman and Professor J Duncan Young.
Trial Management Group
Mr Paul R Mouncey (chief investigator), Professor François Lamontagne (lead clinical investigator), Mr Alvin Richards-Belle, Mrs Julie Camsooksai, Mr Robert Darnell, Professor Anthony C Gordon, Professor Richard D Grieve, Professor David A Harrison, Ms Doreen Henry, Professor Kathryn M Rowan, Dr M Zia Sadique, Ms Michelle Saull, Ms Karen Thomas, Mr Chris Whitman and Professor J Duncan Young. (Previous members: Mr Nicholas Hudson and Mr Akshay Patel.)
Trial Steering Committee
Professor Tim Walsh (chairperson, independent), Dr Ben Creagh-Brown (independent), Dr Tom Lawton (independent), Mrs Theresa Melody (independent), Professor Natalie Pattison (independent), Mrs Donna Reid (independent), Professor François Lamontagne (lead clinical investigator, non-independent) and Mr Paul R Mouncey (chief investigator, non-independent).
Independent Data Monitoring and Ethics Committee
Professor John Norrie (chairperson), Professor Andreas Laupacis and Professor Danny McAuley.
Contribution of authors
Paul R Mouncey (https://orcid.org/0000-0002-8510-8517) (Head of Research) conceived the trial, led the grant application and design of the trial, contributed to acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Alvin Richards-Belle (https://orcid.org/0000-0001-8577-9380) (Trial Manager) led management of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Karen Thomas (https://orcid.org/0000-0001-7548-4466) (Senior Statistician) led the clinical effectiveness analysis, contributed to the interpretation of the data, and drafted and critically reviewed the manuscript.
David A Harrison (https://orcid.org/0000-0002-9002-9098) (Head Statistician and Honorary Professor in Medical Statistics) conceived the trial, contributed to the design of the trial, the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
M Zia Sadique (https://orcid.org/0000-0001-5814-0258) (Assistant Professor in Health Economics) conceived the trial, contributed to the design of the trial, led the health economic analysis, contributed to the interpretation of the data, and drafted and critically reviewed the manuscript.
Richard D Grieve (https://orcid.org/0000-0001-8899-1301) (Professor of Health Economics Methodology) conceived the trial, contributed to the design of the trial, led the health economic analysis, contributed to the interpretation of the data, and drafted and critically reviewed the manuscript.
Julie Camsooksai (https://orcid.org/0000-0002-1139-2252) (Senior Research Nurse in Critical Care) contributed to the design of the trial, contributed to the acquisition and interpretation of the data, and critically reviewed the manuscript.
Robert Darnell (https://orcid.org/0000-0003-4490-1962) (Research Assistant) supported management of the trial, contributed to the acquisition and interpretation of the data, and critically reviewed the manuscript.
Anthony C Gordon (https://orcid.org/0000-0002-0419-547X) (Chairperson in Anaesthesia and Critical Care and Consultant in Intensive Care Medicine) contributed to the design of the trial, contributed to the acquisition and interpretation of the data, and critically reviewed the manuscript.
Doreen Henry (https://orcid.org/0000-0003-2216-5589) (patient representative) contributed to the design of the trial, contributed to the interpretation of the data and critically reviewed the manuscript.
Nicholas Hudson (https://orcid.org/0000-0002-0127-421X) (Data Manager) contributed to the acquisition, analysis and interpretation of the data, and critically reviewed the manuscript.
Alexina J Mason (https://orcid.org/0000-0001-7319-4545) (Assistant Professor of Medical Statistics) led the elicitation exercise design and analysis, and drafted and critically reviewed the manuscript.
Michelle Saull (https://orcid.org/0000-0003-0558-0692) (Data Manager) contributed to the acquisition, analysis and interpretation of the data, and critically reviewed the manuscript.
Chris Whitman (https://orcid.org/0000-0002-1140-1147) (patient representative) contributed to the design of the trial, contributed to the interpretation of the data and critically reviewed the manuscript.
J Duncan Young (https://orcid.org/0000-0002-6838-4835) (Professor of Intensive Care Medicine) contributed to the design of the trial, contributed to the interpretation of the data and critically reviewed the manuscript.
François Lamontagne (https://orcid.org/0000-0002-0360-3427) (Professor and Intensivist) conceived the trial, led the grant application and design of the trial, contributed to acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Kathryn M Rowan (https://orcid.org/0000-0001-8217-5602) (Director of Scientific and Strategic Development/CTU Director and Honorary Professor) conceived the trial, led the grant application and design of the trial, contributed to acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Publications
Richards-Belle A, Mouncey PR, Grieve RD, Harrison DA, Sadique MZ, Henry D, et al. Evaluating the clinical and cost-effectiveness of permissive hypotension in critically ill patients aged 65 years or over with vasodilatory hypotension: protocol for the 65 randomised clinical trial. J Intensive Care Soc 2019;2019:1751143719870088.
Thomas K, Patel A, Sadique MZ, Grieve RD, Mason AJ, Moler S, et al. Evaluating the clinical and cost-effectiveness of permissive hypotension in critically ill patients aged 65 years or over with vasodilatory hypotension: statistical and health economic analysis plan for the 65 trial. J Intensive Care Soc 2019;2019:1751143719860387.
Lamontagne F, Richards-Belle A, Thomas K, Harrison DA, Sadique MZ, Grieve RD, et al. Effect of reduced exposure to vasopressors on 90-day mortality in older critically ill patients with vasodilatory hypotension: a randomized clinical trial. JAMA 2020;323:938–49.
Data-sharing statement
The authors retain exclusive use of the data until the publication of major outputs. Once data are fully analysed and published, data can be obtained from the corresponding author, following review and approval.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HS&DR programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HS&DR programme or the Department of Health and Social Care.
References
- Varpula M, Tallgren M, Saukkonen K, Voipio-Pulkki LM, Pettilä V. Hemodynamic variables related to outcome in septic shock. Intensive Care Med 2005;31:1066-71. https://doi.org/10.1007/s00134-005-2688-z.
- Maheshwari K, Nathanson BH, Munson SH, Khangulov V, Stevens M, Badani H, et al. The relationship between ICU hypotension and in-hospital mortality and morbidity in septic patients. Intensive Care Med 2018;44:857-67. https://doi.org/10.1007/s00134-018-5218-5.
- Lamontagne F, Marshall JC, Adhikari NKJ. Permissive hypotension during shock resuscitation: equipoise in all patients?. Intensive Care Med 2018;44:87-90. https://doi.org/10.1007/s00134-017-4849-2.
- Lemasle L, Blet A, Geven C, Cherifa M, Deniau B, Hollinger A, et al. Bioactive adrenomedullin, organ support therapies, and survival in the critically ill: results from the French and European Outcome Registry in ICU study. Crit Care Med 2020;48:49-55. https://doi.org/10.1097/CCM.0000000000004044.
- Vail EA, Gershengorn HB, Hua M, Walkey AJ, Wunsch H. Epidemiology of vasopressin use for adults with septic shock. Ann Am Thorac Soc 2016;13:1760-7. https://doi.org/10.1513/AnnalsATS.201604-259OC.
- Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 2013;39:165-228. https://doi.org/10.1007/s00134-012-2769-8.
- Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 2017;43:304-77. https://doi.org/10.1007/s00134-017-4683-6.
- Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med 2017;45:486-552. https://doi.org/10.1097/CCM.0000000000002255.
- Lamontagne F, Cook DJ, Meade MO, Seely A, Day AG, Charbonney E, et al. Vasopressor use for severe hypotension – a multicentre prospective observational study. PLOS One 2017;12. https://doi.org/10.1371/journal.pone.0167840.
- St-Arnaud C, Ethier JF, Hamielec C, Bersten A, Guyatt G, Meade M, et al. Prescribed targets for titration of vasopressors in septic shock: a retrospective cohort study. CMAJ Open 2013;1:E127-33. https://doi.org/10.9778/cmajo.20130006.
- Lamontagne F, Day AG, Meade MO, Cook DJ, Guyatt GH, Hylands M, et al. Pooled analysis of higher versus lower blood pressure targets for vasopressor therapy septic and vasodilatory shock. Intensive Care Med 2018;44:12-21. https://doi.org/10.1007/s00134-017-5016-5.
- Lamontagne F, Meade MO, Hébert PC, Asfar P, Lauzier F, Seely AJE, et al. Higher versus lower blood pressure targets for vasopressor therapy in shock: a multicentre pilot randomized controlled trial. Intensive Care Med 2016;42:542-50. https://doi.org/10.1007/s00134-016-4237-3.
- Asfar P, Meziani F, Hamel JF, Grelon F, Megarbane B, Anguel N, et al. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014;370:1583-93. https://doi.org/10.1056/NEJMoa1312173.
- Rochwerg B, Hylands M, Møller MH, Asfar P, Cohen D, Khadaroo RG, et al. CCCS-SSAI WikiRecs clinical practice guideline: vasopressor blood pressure targets in critically ill adults with hypotension and vasopressor use in early traumatic shock. Intensive Care Med 2017;43:1062-4. https://doi.org/10.1007/s00134-016-4539-5.
- Girardis M, Busani S, Damiani E, Donati A, Rinaldi L, Marudi A, et al. Effect of conservative vs. conventional oxygen therapy on mortality among patients in an intensive care unit: the Oxygen-ICU Randomized Clinical Trial. JAMA 2016;316:1583-9. https://doi.org/10.1001/jama.2016.11993.
- Arabi YM, Aldawood AS, Al-Dorzi HM, Tamim HM, Haddad SH, Jones G, et al. Permissive underfeeding or standard enteral feeding in high- and low-nutritional-risk critically ill adults. Post hoc analysis of the PermiT trial. Am J Respir Crit Care Med 2017;195:652-62. https://doi.org/10.1164/rccm.201605-1012OC.
- Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000;342:1301-8. https://doi.org/10.1056/NEJM200005043421801.
- Hébert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409-17. https://doi.org/10.1056/NEJM199902113400601.
- Bickell WH, Wall MJ, Pepe PE, Martin RR, Ginger VF, Allen MK, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med 1994;331:1105-9. https://doi.org/10.1056/NEJM199410273311701.
- Maitland K, Kiguli S, Opoka RO, Engoru C, Olupot-Olupot P, Akech SO, et al. Mortality after fluid bolus in African children with severe infection. N Engl J Med 2011;364:2483-95. https://doi.org/10.1056/NEJMoa1101549.
- Harrison DA, Brady AR, Rowan K. Case mix, outcome and length of stay for admissions to adult, general critical care units in England, Wales and Northern Ireland: the Intensive Care National Audit & Research Centre Case Mix Programme Database. Crit Care 2004;8:R99-111. https://doi.org/10.1186/cc2834.
- Richards-Belle A, Mouncey PR, Grieve RD, Harrison DA, Sadique MZ, Henry D, et al. Evaluating the clinical and cost-effectiveness of permissive hypotension in critically ill patients aged 65 years or over with vasodilatory hypotension: protocol for the 65 randomised clinical trial. J Intensive Care Soc 2019. https://doi.org/10.1177/1751143719870088.
- Health Research Authority . UK Policy Framework for Health and Social Care Research 2017.
- Guideline for Good Clinical Practice n.d.
- Great Britain . Mental Capacity Act 2005 2005.
- Cook D, Lauzier F, Rocha MG, Sayles MJ, Finfer S. Serious adverse events in academic critical care research. CMAJ 2008;178:1181-4. https://doi.org/10.1503/cmaj.071366.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Jorm AF. A short form of the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): development and cross-validation. Psychol Med 1994;24:145-53. https://doi.org/10.1017/s003329170002691x.
- NHS Information Standard Board . Critical Care Minimum Data Set 2012.
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal 2013.
- Sanders GD, Neumann PJ, Basu A, Brock DW, Feeny D, Krahn M, et al. Recommendations for conduct, methodological practices, and reporting of cost-effectiveness analyses: second panel on cost-effectiveness in health and medicine. JAMA 2016;316:1093-103. https://doi.org/10.1001/jama.2016.12195.
- Devlin NJ, Shah KK, Feng Y, Mulhern B, van Hout B. Valuing health-related quality of life: an EQ-5D-5L value set for England. Health Econ 2018;27:7-22. https://doi.org/10.1002/hec.3564.
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96. https://doi.org/10.1002/hec.944.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2014.
- Department of Health and Social Care (DHSC) . NHS Reference Costs 2017–2018 2018.
- Curtis LA, Burns A. Unit Costs of Health and Social Care. Canterbury: PSSRU, University of Kent; 2018.
- Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine – reporting of subgroup analyses in clinical trials. N Engl J Med 2007;357:2189-94. https://doi.org/10.1056/NEJMsr077003.
- Sun X, Ioannidis JP, Agoritsas T, Alba AC, Guyatt G. How to use a subgroup analysis: users’ guide to the medical literature. JAMA 2014;311:405-11. https://doi.org/10.1001/jama.2013.285063.
- Schulz KF, Altman DG, Moher D. CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. https://doi.org/10.1136/bmj.c332.
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med 1985;13:818-29. https://doi.org/10.1097/00003246-198510000-00009.
- Harrison DA, Parry GJ, Carpenter JR, Short A, Rowan K. A new risk prediction model for critical care: the Intensive Care National Audit & Research Centre (ICNARC) model. Crit Care Med 2007;35:1091-8. https://doi.org/10.1097/01.CCM.0000259468.24532.44.
- Ferrando-Vivas P, Jones A, Rowan KM, Harrison DA. Development and validation of the new ICNARC model for prediction of acute hospital mortality in adult critical care. J Crit Care 2017;38:335-9. https://doi.org/10.1016/j.jcrc.2016.11.031.
- Shankar-Hari M, Harrison DA, Rubenfeld GD, Rowan K. Epidemiology of sepsis and septic shock in critical care units: comparison between sepsis-2 and sepsis-3 populations using a national critical care database. Br J Anaesth 2017;119:626-36. https://doi.org/10.1093/bja/aex234.
- Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016;315:775-87. https://doi.org/10.1001/jama.2016.0289.
- Khanna A, English SW, Wang XS, Ham K, Tumlin J, Szerlip H, et al. Angiotensin II for the treatment of vasodilatory shock. N Engl J Med 2017;377:419-30. https://doi.org/10.1056/NEJMoa1704154.
- Brown SM, Lanspa MJ, Jones JP, Kuttler KG, Li Y, Carlson R, et al. Survival after shock requiring high-dose vasopressor therapy. Chest 2013;143:664-71. https://doi.org/10.1378/chest.12-1106.
- Thomas K, Patel A, Sadique MZ, Grieve RD, Mason AJ, Moler S, et al. Evaluating the clinical and cost-effectiveness of permissive hypotension in critically ill patients aged 65 years or over with vasodilatory hypotension: statistical and health economic analysis plan for the 65 trial. J Intensive Care Soc 2019. https://doi.org/10.1177/1751143719860387.
- Iwashyna TJ, Burke JF, Sussman JB, Prescott HC, Hayward RA, Angus DC. Implications of heterogeneity of treatment effect for reporting and analysis of randomized trials in critical care. Am J Respir Crit Care Med 2015;192:1045-51. https://doi.org/10.1164/rccm.201411-2125CP.
- Dodd S, White IR, Williamson P. A framework for the design, conduct and interpretation of randomised controlled trials in the presence of treatment changes. Trials 2017;18. https://doi.org/10.1186/s13063-017-2240-9.
- Barber JA, Thompson SG. Analysis of cost data in randomized trials: an application of the non-parametric bootstrap. Stat Med 2000;19:3219-36. https://doi.org/10.1002/1097-0258(20001215)19:23<3219::AID-SIM623>3.0.CO;2-P.
- Rubin D. Multiple Imputation for Nonresponse in Surveys. New York, NY: John Wiley & Sons; 1987.
- Mason AJ, Grieve RD, Richards-Belle A, Mouncey PR, Harrison DA, Carpenter JR. A framework for extending trial design to facilitate missing data sensitivity analyses. BMC Med Res Methodol 2020;20. https://doi.org/10.1186/s12874-020-00930-2.
- Molenberghs G, Fitzmaurice G, Kenward MG, Tsiatas A, Verbeke G. Handbook of Missing Data Methodology. Boca Raton, FL: Chapman & Hall/CRC; 2015.
- Mason AJ, Gomes M, Grieve R, Ulug P, Powell JT, Carpenter J. Development of a practical approach to expert elicitation for randomised controlled trials with missing health outcomes: application to the IMPROVE trial. Clin Trials 2017;14:357-67. https://doi.org/10.1177/1740774517711442.
- Plummer M. JAGS: A Program for Analysis of Bayesian Graphical Models Using Gibbs Sampling 2003.
- Wade DM, Mouncey PR, Richards-Belle A, Wulff J, Harrison DA, Sadique MZ, et al. Effect of a nurse-led preventive psychological intervention on symptoms of posttraumatic stress disorder among critically ill patients: a randomized clinical trial. JAMA 2019;321:665-75. https://doi.org/10.1001/jama.2019.0073.
- Brooks SP, Gelman A. General methods for monitoring convergence of iterative simulations. J Comput Graph Stat 1998;7:434-55. https://doi.org/10.1080/10618600.1998.10474787.
- Ramsey SD, Willke RJ, Glick H, Reed SD, Augustovski F, Jonsson B, et al. Cost-effectiveness analysis alongside clinical trials II – an ISPOR Good Research Practices Task Force report. Value Health 2015;18:161-72. https://doi.org/10.1016/j.jval.2015.02.001.
- Lamontagne F, Richards-Belle A, Thomas K, Harrison DA, Sadique MZ, Grieve RD, et al. Effect of reduced exposure to vasopressors on 90-day mortality in older critically ill patients with vasodilatory hypotension: a randomized clinical trial. JAMA 2020;323:938-49. https://doi.org/10.1001/jama.2020.0930.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. https://doi.org/10.1002/sim.4067.
- Claxton K. Exploring uncertainty in cost-effectiveness analysis. PharmacoEconomics 2008;26:781-98. https://doi.org/10.2165/00019053-200826090-00008.
- Hernandez Alava M, Wailoo A, Grimm S, Pudney S, Gomes M, Sadique Z, et al. EQ-5D-5L versus EQ-5D-3L: the impact on cost effectiveness in the United Kingdom. Value Health 2018;21:49-56. https://doi.org/10.1016/j.jval.2017.09.004.
- van Hout B, Janssen MF, Feng YS, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15. https://doi.org/10.1016/j.jval.2012.02.008.
- Thompson SG, Nixon RM. How sensitive are cost-effectiveness analyses to choice of parametric distributions?. Med Decis Making 2005;25:416-23. https://doi.org/10.1177/0272989x05276862.
- Willan AR, Briggs AH, Hoch JS. Regression methods for covariate adjustment and subgroup analysis for non-censored cost-effectiveness data. Health Econ 2004;13:461-75. https://doi.org/10.1002/hec.843.
- Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 2012;120:c179-84. https://doi.org/10.1159/000339789.
- Jackson CH. Displaying uncertainty with shading. Am Stat 2008;62:340-7. https://doi.org/10.1198/000313008X370843.
- Kavanagh BP, Nurok M. Standardized Intensive Care. Protocol misalignment and impact misattribution. Am J Respir Crit Care Med 2016;193:17-22. https://doi.org/10.1164/rccm.201502-0314CP.
- Needham DM, Sepulveda KA, Dinglas VD, Chessare CM, Friedman LA, Bingham CO, et al. Core outcome measures for clinical research in acute respiratory failure survivors. An International Modified Delphi Consensus Study. Am J Respir Crit Care Med 2017;196:1122-30. https://doi.org/10.1164/rccm.201702-0372OC.
- Lamontagne F, Cohen D, Herridge M. Understanding patient-centredness: contrasting expert versus patient perspectives on vasopressor therapy for shock. Intensive Care Med 2017;43:1052-4. https://doi.org/10.1007/s00134-016-4518-x.
- Natalini G, Schivalocchi V, Rosano A, Taranto M, Pletti C, Bernardini A. Norepinephrine and metaraminol in septic shock: a comparison of the hemodynamic effects. Intensive Care Med 2005;31:634-7. https://doi.org/10.1007/s00134-005-2607-3.
- Auriemma CL, Van den Berghe G, Halpern SD. Less is more in critical care is supported by evidence-based medicine. Intensive Care Med 2019;45:1806-9. https://doi.org/10.1007/s00134-019-05771-2.
- Venkatesh B, Khanna AK, Cohen J. Less is more: catecholamine-sparing strategies in septic shock. Intensive Care Med 2019;45:1810-12. https://doi.org/10.1007/s00134-019-05770-3.
Appendix 1 The 65 trial investigators
Lead investigators
Paul R Mouncey, Intensive Care National Audit & Research Centre, London, UK
Alvin Richards-Belle, Intensive Care National Audit & Research Centre, London, UK
Karen Thomas, Intensive Care National Audit & Research Centre, London, UK
David A Harrison, Intensive Care National Audit & Research Centre, London, UK
M Zia Sadique, London School of Hygiene & Tropical Medicine, London, UK
Richard D Grieve, London School of Hygiene & Tropical Medicine, London, UK
Julie Camsooksai, Poole Hospital NHS Foundation Trust, Poole, UK
Robert Darnell, Intensive Care National Audit & Research Centre, London, UK
Anthony C Gordon, Imperial College London, London, UK, and Imperial College Healthcare NHS Trust, St Mary’s Hospital, London, UK
Doreen Henry (patient representative), UK
Nicholas Hudson, Intensive Care National Audit & Research Centre, London, UK
Alexina J Mason, Imperial College London, London, UK
Michelle Saull, Intensive Care National Audit & Research Centre, London, UK
Chris Whitman (patient representative), UK
J Duncan Young, University of Oxford, Oxford, UK
François Lamontagne, Université de Sherbrooke, Sherbrooke, QC, Canada
Kathryn M Rowan, Intensive Care National Audit & Research Centre, London, UK
Site investigators
Petra Polgarova, Addenbrookes Hospital, Cambridge, UK
Peter Featherstone, Addenbrookes Hospital, Cambridge, UK
Sofia Teixeira, Addenbrookes Hospital, Cambridge, UK
Colette Jones-Criddle, Aintree University Hospital, Liverpool, UK
Ben Morton, Aintree University Hospital, Liverpool, UK
Ian Turner-Bone, Aintree University Hospital, Liverpool, UK
Laura Wilding, Aintree University Hospital, Liverpool, UK
Gail Quigley, Altnagelvin Area Hospital, Derry, UK
Noel Hemmings, Altnagelvin Area Hospital, Derry, UK
Adrian Donnelly, Altnagelvin Area Hospital, Derry, UK
Aidan Campbell, Altnagelvin Area Hospital, Derry, UK
Sinéad O’Kane, Altnagelvin Area Hospital, Derry, UK
Emma McKay, Antrim Area Hospital, Antrim, UK
Paul Johnston, Antrim Area Hospital, Antrim, UK
Orla O’Neill, Antrim Area Hospital, Antrim, UK
Emma Totten, Antrim Area Hospital, Antrim, UK
Nadine Weeks, Arrowe Park Hospital, Upton, UK
Paul Jeanrenaud, Arrowe Park Hospital, Upton, UK
Cathy Jones, Arrowe Park Hospital, Upton, UK
Reni Jacob, Arrowe Park Hospital, Upton, UK
Ron Mathew Jacob, Arrowe Park Hospital, Upton, UK
Maria Alpuerto, Basingstoke and North Hampshire Hospital, Basingstoke, UK
Antony Ashton, Basingstoke and North Hampshire Hospital, Basingstoke, UK
Denise Griffin, Basingstoke and North Hampshire Hospital, Basingstoke, UK
McDonald Mupudzi, Basingstoke and North Hampshire Hospital, Basingstoke, UK
Jason Cuppitt, Blackpool Victoria Hospital, Blackpool, UK
Emma Stoddard, Blackpool Victoria Hospital, Blackpool, UK
Gemma Brown, Blackpool Victoria Hospital, Blackpool, UK
Jazmine McCooey, Blackpool Victoria Hospital, Blackpool, UK
Lisa Grimmer, Bristol Royal Infirmary, Bristol, UK
Jeremy Bewley, Bristol Royal Infirmary, Bristol, UK
Katie Sweet, Bristol Royal Infirmary, Bristol, UK
Chloe Searles, Bristol Royal Infirmary, Bristol, UK
Rebecca Keskeys, Broomfield Hospital, Chelmsford, UK
Jayachandran Radhakrishnan, Broomfield Hospital, Chelmsford, UK
Fiona McNeela, Broomfield Hospital, Chelmsford, UK
Sue Smolen, Broomfield Hospital, Chelmsford, UK
Laura Curran, Charing Cross Hospital, London, UK
David Antcliffe, Charing Cross Hospital, London, UK
Roceld Rojo, Charing Cross Hospital, London, UK
Kim Zantua, Charing Cross Hospital, London, UK
Helen Robertson, Countess of Chester Hospital, Chester, UK
Lyndsay Cheater, Countess of Chester Hospital, Chester, UK
Maria Faulkner, Countess of Chester Hospital, Chester, UK
Laura Parry, Countess of Chester Hospital, Chester, UK
Phillipa Wakefield, Darent Valley Hospital, Dartford, UK
Zakaulla Belagodu, Darent Valley Hospital, Dartford, UK
Danielle Vosper, Darent Valley Hospital, Dartford, UK
Carmel Stuart, Darent Valley Hospital, Dartford, UK
Binu Ravindran, Darent Valley Hospital, Dartford, UK
Amanda Cowton, Darlington Memorial Hospital, Darlington, UK
James Limb, Darlington Memorial Hospital, Darlington, UK
Julie O’Brien, Darlington Memorial Hospital, Darlington, UK
Rosalyn Squires, Derriford Hospital, Plymouth, UK
Sam Waddy, Derriford Hospital, Plymouth, UK
Esme Elloway, Derriford Hospital, Plymouth, UK
Helen McMillan, Derriford Hospital, Plymouth, UK
Sarah Williams, Dorset County Hospital, Dorchester, UK
Andrew Ball, Dorset County Hospital, Dorchester, UK
Patricia Williams, Dorset County Hospital, Dorchester, UK
Sharon Hiscox, Dorset County Hospital, Dorchester, UK
Sarah Horton, Dorset County Hospital, Dorchester, UK
Ulla Chappell, Glangwili General Hospital, Carmarthen, UK
Igor Otahal, Glangwili General Hospital, Carmarthen, UK
Peter Havalda, Glangwili General Hospital, Carmarthen, UK
Samantha Coetzee, Glangwili General Hospital, Carmarthen, UK
Kelly Matthews, Gloucestershire Royal Hospital, Gloucester, UK
Andrew Foo, Gloucestershire Royal Hospital, Gloucester, UK
Izzy King, Gloucestershire Royal Hospital, Gloucester, UK
Kirsty Manns, Gloucestershire Royal Hospital, Gloucester, UK
Sonia Sousa Arias, Hammersmith Hospital, London, UK
Stephen Brett, Hammersmith Hospital, London, UK
Leilani Cabreros, Hammersmith Hospital, London, UK
Rhoda Rosal, Hammersmith Hospital, London, UK
Stephanie Bell, Ipswich Hospital, Ipswich, UK
Kate Turner, Ipswich Hospital, Ipswich, UK
Vanessa Rivers, Ipswich Hospital, Ipswich, UK
Susan Brixey, Ipswich Hospital, Ipswich, UK
Lindsay Garcia, James Cook University Hospital, Middlesbrough, UK
Judith Wright, James Cook University Hospital, Middlesbrough, UK
Keith Hugill, James Cook University Hospital, Middlesbrough, UK
Susan Mortimer, James Cook University Hospital, Middlesbrough, UK
Nicola Cree, James Cook University Hospital, Middlesbrough, UK
Fiona Bartley, King’s College Hospital, London, UK
Philip Hopkins, King’s College Hospital, London, UK
Su Jeffreys, King’s College Hospital, London, UK
Harriet Noble, King’s College Hospital, London, UK
Clare Finney, King’s College Hospital, London, UK
Louise Houslip, Leicester Royal Infirmary, Leicester, UK
Neil Flint, Leicester Royal Infirmary, Leicester, UK
Dawn Hales, Leicester Royal Infirmary, Leicester, UK
Prematie Andreou, Leicester Royal Infirmary, Leicester, UK
Iain McLaren, Leicester Royal Infirmary, Leicester, UK
Carina Cruz, Lister Hospital, Stevenage, UK
Sunil Jamadarkhana, Lister Hospital, Stevenage, UK
Naomi Brice, Lister Hospital, Stevenage, UK
Katie Goodyer, Lister Hospital, Stevenage, UK
Richard Clark, Manchester Royal Infirmary, Manchester, UK
Jonathan Bannard-Smith, Manchester Royal Infirmary, Manchester, UK
Emma Connaughton, Manchester Royal Infirmary, Manchester, UK
Abigail Williams, Manchester Royal Infirmary, Manchester, UK
Amanda Cameron, Medway Maritime Hospital, Gillingham, UK
Rahuldeb Sarkar, Medway Maritime Hospital, Gillingham, UK
Vongayi Ogbeide, Medway Maritime Hospital, Gillingham, UK
Mary Everett, Medway Maritime Hospital, Gillingham, UK
Ceri Battle, Morriston Hospital, Swansea, UK
Milercy Oliveros, Morriston Hospital, Swansea, UK
Tracy Owen, Morriston Hospital, Swansea, UK
Sharon Storton, Morriston Hospital, Swansea, UK
Patricia Doble, Musgrove Park Hospital, Taunton, UK
Richard Innes, Musgrove Park Hospital, Taunton, UK
Joanne Hutter, Musgrove Park Hospital, Taunton, UK
Stephen Harris, Musgrove Park Hospital, Taunton, UK
Georgina Randell, Norfolk and Norwich University Hospital, Norwich, UK
Steve Hutchinson, Norfolk and Norwich University Hospital, Norwich, UK
Deirdre Fottrell-Gould, Norfolk and Norwich University Hospital, Norwich, UK
Lisa Hudig, Norfolk and Norwich University Hospital, Norwich, UK
Tracey Shanley, North Devon District Hospital, Barnstaple, UK
Guy Rousseau, North Devon District Hospital, Barnstaple, UK
Max Coupe King, North Devon District Hospital, Barnstaple, UK
Nicolas Stafford, North Devon District Hospital, Barnstaple, UK
Joy Grewcock, Northampton General Hospital, Northampton, UK
Jonathan Wilkinson, Northampton General Hospital, Northampton, UK
Kathryn Hall, Northampton General Hospital, Northampton, UK
Lorraine Campey, Northampton General Hospital, Northampton, UK
Joanne Pons, Northern General Hospital, Sheffield, UK
Gary Mills, Northern General Hospital, Sheffield, UK
Sarah Bird, Northern General Hospital, Sheffield, UK
Joshua Cooper, Northern General Hospital, Sheffield, UK
Alan Pope, Peterborough City Hospital, Peterborough, UK
Matthew Davies, Peterborough City Hospital, Peterborough, UK
Coralie Carle, Peterborough City Hospital, Peterborough, UK
Nicola Butterworth-Cowin, Peterborough City Hospital, Peterborough, UK
Loran Davies, Pinderfields Hospital, Wakefield, UK
Alastair Rose, Pinderfields Hospital, Wakefield, UK
Sarah Buckley, Pinderfields Hospital, Wakefield, UK
Lucy Brooks, Pinderfields Hospital, Wakefield, UK
Sarah Smith, Pinderfields Hospital, Wakefield, UK
Julie Camsooksai, Poole Hospital, Poole, UK
Henrik Reschreiter, Poole Hospital, Poole, UK
Sarah Patch, Poole Hospital, Poole, UK
Sarah Jenkins, Poole Hospital, Poole, UK
Olivia Rowe, Princess Royal University Hospital, Farnborough, UK
Tom Williams, Princess Royal University Hospital, Farnborough, UK
Emma Clarey, Princess Royal University Hospital, Farnborough, UK
Jane Wilson, Princess Royal University Hospital, Farnborough, UK
Jenny Ritzema, Queen Elizabeth Hospital, Gateshead, UK
Vanessa Linnett, Queen Elizabeth Hospital, Gateshead, UK
Amanda Sanderson, Queen Elizabeth Hospital, Gateshead, UK
Steve Rose, Queen Alexandra Hospital, Portsmouth, UK
David Pogson, Queen Alexandra Hospital, Portsmouth, UK
Zoe Daly, Queen Alexandra Hospital, Portsmouth, UK
Aimi Collins, Queen Alexandra Hospital, Portsmouth, UK
Amy Collins, Queen Elizabeth Hospital, London, UK
Ashraf Roshdy, Queen Elizabeth Hospital, London, UK
Ahmed Zaki, Queen Elizabeth Hospital, London, UK
Estefania Treus, Queen Elizabeth Hospital, London, UK
Yvonna Marasigan, Queen Elizabeth Hospital, London, UK
Lucy Ryan, Queens Medical Centre, Nottingham, UK
Daniel Harvey, Queens Medical Centre, Nottingham, UK
Megan Meredith, Queens Medical Centre, Nottingham, UK
Louise Hughes, Queens Medical Centre, Nottingham, UK
Nicola Jacques, Royal Berkshire Hospital, Reading, UK
Andrew Walden, Royal Berkshire Hospital, Reading, UK
Parminder Bhuie, Royal Berkshire Hospital, Reading, UK
Aoife Dowling, Royal Berkshire Hospital, Reading, UK
Sarah Bean, Royal Cornwall Hospital, Truro, UK
Jonathan Paddle, Royal Cornwall Hospital, Truro, UK
Karen Burt, Royal Cornwall Hospital, Truro, UK
Caroline Aherne, Royal Blackburn Hospital, Blackburn, UK
Justin Roberts, Royal Blackburn Hospital, Blackburn, UK
Rebecca Crosby, Royal Blackburn Hospital, Blackburn, UK
Carole Boulanger, Royal Devon and Exeter Hospital, Exeter, UK
Charly Gibson, Royal Devon and Exeter Hospital, Exeter, UK
Sinead Kelly, Royal Devon and Exeter Hospital, Exeter, UK
Ceri Lynch, Royal Glamorgan Hospital, Ynysmaerdy, UK
Bethan Gibson, Royal Glamorgan Hospital, Ynysmaerdy, UK
Lisa Roche, Royal Glamorgan Hospital, Ynysmaerdy, UK
Keri Turner, Royal Glamorgan Hospital, Ynysmaerdy, UK
Kelly Thomas, Royal Glamorgan Hospital, Ynysmaerdy, UK
Gemma Hodkinson, Royal Gwent Hospital, Newport, UK
Tamas Szakmany, Royal Gwent Hospital, Newport, UK
Una Gunter, Royal Gwent Hospital, Newport, UK
Samantha Hendry, Royal Liverpool University Hospital, Liverpool, UK
Ingeborg Welters, Royal Liverpool University Hospital, Liverpool, UK
Karen Williams, Royal Liverpool University Hospital, Liverpool, UK
Victoria Waugh, Royal Liverpool University Hospital, Liverpool, UK
Ian Angus, Royal Oldham Hospital, Oldham, UK
Redmond Tully, Royal Oldham Hospital, Oldham, UK
Karen Hallett, Royal Oldham Hospital, Oldham, UK
Susan Dermody, Royal Oldham Hospital, Oldham, UK
Mark Verlander, Royal Preston Hospital, Preston, UK
Shondipon Laha, Royal Preston Hospital, Preston, UK
Alexandra Williams, Royal Preston Hospital, Preston, UK
Donna Doyle, Royal Preston Hospital, Preston, UK
David Cartlidge, Royal Stoke Hospital, Stoke-on-Trent, UK
Moses Chikungwa, Royal Stoke Hospital, Stoke-on-Trent, UK
Minnie Gellamucho, Royal Stoke Hospital, Stoke-on-Trent, UK
Ruth Salt, Royal Stoke Hospital, Stoke-on-Trent, UK
Patricia Piercy, Royal Victoria Infirmary, Newcastle upon Tyne, UK
Ian Clement, Royal Victoria Infirmary, Newcastle upon Tyne, UK
Leigh Dunn, Royal Victoria Infirmary, Newcastle upon Tyne, UK
Carmen Bradshaw, Royal Victoria Infirmary, Newcastle upon Tyne, UK
Abigail Harrison, Royal Victoria Infirmary, Newcastle upon Tyne, UK
Davinder Kaur, Russells Hall Hospital, Dudley, UK
Mike Reay, Russells Hall Hospital, Dudley, UK
Vikram Anumakonda, Russells Hall Hospital, Dudley, UK
Rachel Collins, Russells Hall Hospital, Dudley, UK
Angela Watts, Russells Hall Hospital, Dudley, UK
Julie Matthews, Russells Hall Hospital, Dudley, UK
Alexandra Larkin, Salford Royal Hospital, Salford, UK
Paul Ferris, Salford Royal Hospital, Salford, UK
Kathryn Cawley, Salford Royal Hospital, Salford, UK
Joy Dearden, Salford Royal Hospital, Salford, UK
Beverley Faulkner, Southmead Hospital, Bristol, UK
Matt Thomas, Southmead Hospital, Bristol, UK
Kati Hayes, Southmead Hospital, Bristol, UK
Ruth Worner, Southmead Hospital, Bristol, UK
Dorota Banach, St Mary’s Hospital, London, UK
Anthony Gordon, St Mary’s Hospital, London, UK
John Adams, St Mary’s Hospital, London, UK
Maie Templeton, St Mary’s Hospital, London, UK
Aneta Bociek, St Thomas’ Hospital, London, UK
Marlies Ostermann, St Thomas’ Hospital, London, UK
Simon Sparkes, St Thomas’ Hospital, London, UK
Ruth Wan, St Thomas’ Hospital, London, UK
Andrea Kelly, St Thomas’ Hospital, London, UK
Joanne Holman, Torbay Hospital, Torquay, UK
Thomas Clark, Torbay Hospital, Torquay, UK
Alison Cornwell, Torbay Hospital, Torquay, UK
Ilona Cassar, Tunbridge Wells Hospital, Royal Tunbridge Wells, UK
David Golden, Tunbridge Wells Hospital, Royal Tunbridge Wells, UK
Joanne Jones, Tunbridge Wells Hospital, Royal Tunbridge Wells, UK
Miriam Davey, Tunbridge Wells Hospital, Royal Tunbridge Wells, UK
Thomas Billyard, University Hospital Coventry, Coventry, UK
Geraldine Ward, University Hospital Coventry, Coventry, UK
Laura Wild, University Hospital Coventry, Coventry, UK
Pamela Bremmer, University Hospital Coventry, Coventry, UK
Christopher Bassford, University Hospital Coventry, Coventry, UK
Rosaleeta Reece-Anthony, University Hospital Lewisham, London, UK
Waqas Khaliq, University Hospital Lewisham, London, UK
Jayson Clarke, University Hospital Lewisham, London, UK
Babita Gurung, University Hospital Lewisham, London, UK
Michele Clark, University Hospital of North Tees, Stockton-on-Tees, UK
Farooq Brohi, University Hospital of North Tees, Stockton-on-Tees, UK
Tracey Oldfield, University Hospital of North Tees, Stockton-on-Tees, UK
Sophie Mason, Warwick Hospital, Warwick, UK
Ben Attwood, Warwick Hospital, Warwick, UK
Camilla Stagg, Warwick Hospital, Warwick, UK
Penny Parsons, Warwick Hospital, Warwick, UK
Carl Boswell, William Harvey Hospital, Willesborough, UK
Neil Anthony Richardson, William Harvey Hospital, Willesborough, UK
Tracy Hazelton, William Harvey Hospital, Willesborough, UK
Natasha Schumacher, William Harvey Hospital, Willesborough, UK
Nicholas Dalmon, William Harvey Hospital, Willesborough, UK
Jenny Lord, Worthing Hospital, Worthing, UK
David Helm, Worthing Hospital, Worthing, UK
Charalice Ramiro, Worthing Hospital, Worthing, UK
Jordi Margalef, Worthing Hospital, Worthing, UK
Liliana Silva, Yeovil District Hospital, Yeovil, UK
Agnieszka Kubisz-Pudelko, Yeovil District Hospital, Yeovil, UK
Alison Lewis, Yeovil District Hospital, Yeovil, UK
Johnyta Panakal, Yeovil District Hospital, Yeovil, UK
Danielle Wilcox, York Hospital, York, UK
Jonathan Redman, York Hospital, York, UK
Joseph Carter, York Hospital, York, UK
Kate Howard, York Hospital, York, UK
List of abbreviations
- APACHE II
- Acute Physiology and Chronic Health Evaluation II
- CEA
- cost-effectiveness analysis
- CI
- confidence interval
- CMP
- Case Mix Programme
- CRF
- case report form
- CRN
- Clinical Research Network
- CTU
- Clinical Trials Unit
- DMEC
- Data Monitoring and Ethics Committee
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- GP
- general practitioner
- HRG
- Healthcare Resource Group
- HRQoL
- health-related quality of life
- ICNARC
- Intensive Care National Audit & Research Centre
- ICU
- intensive care unit
- INMB
- incremental net monetary benefit
- IQCODE
- Informant Questionnaire on Cognitive Decline in the Elderly
- IQR
- interquartile range
- ISF
- investigator site file
- MAP
- mean arterial pressure
- MAR
- missing at random
- MNAR
- missing not at random
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- OVATION
- Optimal VAsopressor TItratiON
- PI
- principal investigator
- PIS
- patient information sheet
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- RCT
- randomised clinical trial
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SD
- standard deviation
- SEPSISPAM
- Sepsis and Mean Arterial Pressure
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/hta25140).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.