

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 14/190/01. The contractual start date was in October 2014. The draft report began editorial review in February 2020 and was accepted for publication in July 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2021 Roberts et al. This work was produced by Roberts et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2021 Roberts et al.
Chapter 1 Introduction
Each year, worldwide, there are over 60 million new cases of traumatic brain injury (TBI). 1 Low- and middle-income countries (LMIC) bear a disproportionate burden of morbidity and mortality due to TBI compared with high-income countries. In LMIC, TBI is most commonly the result of road traffic accidents, whereas in high-income countries the mechanism of injury for TBI appears to be shifting from road traffic accidents to falls by the elderly. 1 With rapid urbanisation in LMIC and the resulting increase in motorisation, cases of TBI are expected to rise. 2
The impact of TBI can be devastating for individuals and their families. Survivors of TBI may experience long-term physical, emotional and cognitive dysfunction. This, in turn, has considerable financial consequences through health and social costs and wider economic impacts due to reduced productivity.
Traumatic brain injury mechanism
Traumatic brain injury is an acute injury to the brain from an external mechanical force that temporarily or permanently impairs brain function. TBI is often classified as mild, moderate or severe according to the patient’s level of consciousness. This is assessed clinically using the Glasgow Coma Scale (GCS). 3
Bleeding within the skull (known as intracranial haemorrhage) is common after TBI and is associated with increased mortality and morbidity. 4 Although bleeding can start from the moment of impact, it often continues for several hours after injury. 5,6 In the CRASH-1 (Corticosteroid Randomisation After Significant Head Injury) trial,7 which included 10,008 TBI patients, 73% of patients with moderate or severe TBI had intracranial haemorrhage on computerised tomography (CT) scan. Bleeding progressed in 84% of these patients with confirmed intracranial haemorrhage and moderate or severe TBI.
Management of traumatic brain injury
The skull is a rigid compartment containing three components: brain, blood and cerebrospinal fluid. An increase in one of these components, such as blood, from an intracranial haemorrhage, will need to be compensated by a decrease in one or more of the other components. 8 Initially, this increase in volume can be accommodated; however, once these compensatory mechanisms become exhausted, intracranial pressure will rise. 8 This may result in the brain tissue shifting and becoming displaced (known as brain herniation), which if left untreated can lead to respiratory depression and ultimately death.
Management of TBI is concerned with reducing intracranial pressures and can be broadly classified as either surgical or medical. Surgical interventions include draining cerebrospinal fluid and decompressive craniectomy. 9 This involves removing a portion of the skull to relieve intracranial pressure. Medical options include therapeutic hypothermia, sedation and analgesia, hyperosmolar therapy and hyperventilation. 9 Many of the current TBI management options require skilled medical professionals and specialist health-care facilities.
An inexpensive, simple and widely practicable treatment that improves outcomes in patients with TBI could save many thousands of lives and reduce the burden of disability.
Tranexamic acid and traumatic brain injury
Tranexamic acid (TXA) is an antifibrinolytic drug that inhibits the enzymatic breakdown of fibrin blood clots. It is possible that early administration of TXA in patients with TBI might prevent or reduce intracranial haemorrhage expansion and thus avert brain herniation and death.
Approximately one-third of patients with TBI have laboratory evidence of abnormal coagulation at hospital admission. 10 These patients have an increased risk of intracranial haemorrhage and higher mortality. Increased clot breakdown (fibrinolysis), as indicated by elevated levels of fibrinogen degradation products, is often seen in patients with TBI and predicts intracranial haemorrhage expansion. 11
In addition, it has been shown that progressive tissue damage and oedema develop in regions surrounding intracranial bleeding lesions, and are associated with worse outcomes. 12 Tissue plasminogen activator (tPA) has been shown to be an important factor in this process of perilesional oedema. 13–15 By blocking the conversion from plasminogen to plasmin, TXA counteracts the effect of tPA and, therefore, it is possible that TXA might also be beneficial in traumatic intracerebral haemorrhage by decreasing perilesional oedema through a specific neuroprotective effect.
Existing research on tranexamic acid
Tranexamic acid is commonly given to surgical patients to reduce bleeding and the need for blood transfusion. A systematic review of randomised trials of TXA in elective surgical patients shows that TXA reduces the number of patients receiving a blood transfusion by about one-third, reduces the volume of blood transfused by about 1 unit and halves the need for further surgery to control bleeding. 16 These differences are all highly statistically significant. Furthermore, there is no evidence of any increased risk of vascular occlusive events with TXA. 16
More recently, the CRASH-2 (Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage-2) trial17,18 showed that, in trauma patients with significant extracranial bleeding, early administration (within 3 hours of injury) of TXA reduces bleeding deaths by one-third. Subsequent analyses showed that even a short delay in treatment reduces the benefit of TXA administration. 19 Based on these results, TXA was included in guidelines for the pre-hospital care of trauma patients, although patients with isolated TBI were specifically excluded.
Two studies have evaluated the effect of TXA in TBI. The CRASH-2 Intracranial Bleeding Study20 was a nested randomised trial conducted in 270 trauma patients who had evidence of TBI on a pre-randomisation CT scan. A second scan was conducted 24–48 hours after randomisation. There was a reduction in intracranial haemorrhage growth [risk ratio (RR) 0.80, 95% confidence interval (CI) 0.59 to 1.09], fewer ischaemic lesions and lower all-cause mortality (RR 0.60, 95% CI 0.32 to 1.11) in TXA-allocated patients, but these results were not statistically significant. 20 A second randomised trial conducted in 240 patients with isolated TBI also found reductions in haemorrhage growth (RR 0.56, 95% CI 0.32 to 0.97) and mortality (RR 0.67, 95% CI 0.34 to 1.32) with TXA, but this trial did not collect data on ischaemic lesions. 21
Rationale for trial
Meta-analysis of the two trials shows a significant reduction in haemorrhage growth (RR 0.72, 95% CI 0.55 to 0.94) and mortality (RR 0.63, 95% CI 0.40 to 0.99) with TXA. However, the studies provided no evidence about the effect of TXA on disability or adverse events. The CRASH-3 (Clinical Randomisation of an Antifibrinolytic in Significant Head Injury-3) trial aimed to quantify the effects of TXA on head injury death, disability and adverse events in patients with TBI. 19 We also wanted to assess the cost-effectiveness of treating TBI patients with TXA.
Chapter 2 Methods
The trial protocol,22 statistical analysis plan23 and results24 have been previously published and parts of these published articles are reproduced throughout this report. The protocol was published in Trials (reproduced from Dewan et al. 22). This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The text below includes minor additions and formatting changes to the original text. The statistical analysis plan was published in Wellcome Open Research (© 2018 Roberts et al. 23 This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text). The trial results were published in The Lancet [copyright © 2019 the CRASH-3 trial collaborators. 24 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text].
The protocol and statistical analysis plan can be found at www.journalslibrary.nihr.ac.uk/programmes/hta/1419001/#/documentation (accessed November 2020).
Trial design
CRASH-3 is an international, multicentre, randomised, placebo-controlled trial of the effects of TXA on death and disability in patients with TBI. The trial protocol was peer reviewed and published in BioMed Central Trials journal as an open access article in 2012 (see the trial protocol). 22
CRASH-3 is the third international, multicentre, randomised, placebo-controlled trial in trauma patients conducted by the London School of Hygiene & Tropical Medicine (LSHTM) trial co-ordinating centre. CRASH-1 investigated corticosteroid use in head injury and recruited 10,000 patients with TBI from across the world. 7 CRASH-217 examined the effects of early administration of a short course of TXA in trauma patients. The trial recruited 20,211 patients from 274 hospitals in 40 countries.
Through these many years of collaboration, LSHTM has developed good working relationships with a large number of trauma doctors and an excellent global network of collaborating trauma hospitals.
CRASH-3 was undertaken in 175 hospitals in 29 countries. Suitable collaborating hospitals and investigators were assessed in terms of the trauma service that they provide and their ability to conduct the trial. Before the trial could begin at any site, the local principal investigator must have agreed to adhere to good clinical practice guidelines and all relevant national regulations. In addition, all relevant regulatory and ethics approvals were in place before the trial started at a site. See Appendix 1 for a list of the trial collaborators by country.
There is a wide spectrum of treatments for TBI. As the trial was conducted worldwide, each participating site was instructed to follow its own clinical guidelines for the treatment of TBI patients. No clinically indicated treatment was required to be withheld for the trial. TXA or placebo was provided as an additional treatment to the usual management of TBI.
Approvals
The Medical Research and Ethics Committee and Health Research Authority reviewed the protocol and supporting documents for the CRASH-3 trial and provided a favourable ethics opinion on 19 July 2012 (Research Ethics Committee reference 12/EE/0274). One substantial amendment to the protocol was submitted to limit the time window for eligibility from within 8 hours of injury to within 3 hours of injury. Favourable opinion was received on 6 September 2016. Two non-substantial amendments were submitted to extend the recruitment period, and were categorised on 1 August 2017 and 2 February 2018. The Medicines and Healthcare products Regulatory Agency authorised the CRASH-3 trial on 8 August 2012 (reference 17072/0007/001-0001). A favourable ethics opinion was received from the Observational/Interventions Research Ethics Committee at LSHTM on 17 November 2011 (reference 6060).
Participants (inclusion and exclusion)
Adults with TBI who were within 3 hours of injury and had a GCS score of ≤ 12 or any intracranial bleeding noted on their CT scan and no significant extracranial bleeding (i.e. not in need of immediate blood transfusion) were eligible. The time window for eligibility was originally within 8 hours of injury; however, in 2016, the protocol was changed to limit the time window for eligibility from within 8 hours to within 3 hours of injury. This change was made blind to the trial data, in response to external evidence suggesting that delayed treatment is unlikely to be effective.
The fundamental eligibility criterion was the responsible clinician’s ‘uncertainty’ about whether or not to use TXA in a particular patient with TBI. This is based on the uncertainty principle, which is a well-established approach for assessing trial eligibility. 25 A patient can be enrolled if, and only if, the responsible clinician is substantially uncertain as to which of the trial treatments is most appropriate for that particular patient. A patient should not be enrolled if the responsible clinician or the patient (or his/her representative) is, for any medical or non-medical reasons, reasonably certain that one of the treatments that might be allocated is inappropriate for that particular individual (in comparison with either no treatment or some other treatment that could be offered to the patient in or outside the trial). Using the uncertainty principle allowed the process of this trial to be closer to what is appropriate in normal medical practice. The pragmatic design allowed us to find out how effective the treatment actually is in routine everyday practice.
Consent
Owing to the nature of their injury, most TBI patients are unable to provide prior informed consent to participate in a clinical trial. As acknowledged in the Declaration of Helsinki,26 patients who are incapable of giving consent are an exception to the general rule of informed consent in clinical trials. In the CRASH-3 trial, consent was usually sought from the patient’s relative or a legal representative. If no such representative was available, the study proceeded with the agreement of two clinicians (one independent of the trial). If and when the patient regained capacity, they were told about the trial and written consent was sought to continue participation. If the patient or their representative declined consent, participation stopped. If patients were included in the trial but did not regain capacity, consent was sought from a relative or legal representative. We adhered to the requirements of the local and national ethics committees. See Appendix 2 for an overview of the consent procedure.
Randomisation and blinding
Sites were advised to randomise patients who were eligible for inclusion as soon as possible. The entry form was used to assess eligibility and collect baseline information. Following confirmation of eligibility, patients were randomly allocated to receive TXA or matching placebo (0.9% sodium chloride) by intravenous (i.v.) infusion. An independent statistician from Sealed Envelope Ltd (London, UK) prepared the randomisation codes and gave them to the drug packers so that treatment packs could be prepared. After baseline information was collected on the entry form, the lowest numbered treatment pack remaining was taken from a box of eight treatment packs. If the treatment ampoules were intact, the patient was considered randomised. Entry form data were entered into a secure online database by the trial investigators. Both participants and study staff (site investigators and trial co-ordinating centre staff) were masked to allocation. An emergency unblinding service was available for use in those rare situations when the clinician believed that clinical management depended on knowledge of whether the patient received TXA or placebo.
The TXA (Cyklokapron® injection) was manufactured by Pfizer Ltd (Sandwich, UK). The Torbay and South Devon NHS Foundation Trust prepared the 0.9% sodium chloride placebo. Ampoules and packaging were identical in appearance. The blinding was done by Bilcare GCS (Europe) Ltd (Crickhowell, UK). This entailed removal of the manufacturer’s label and replacement with the trial label and treatment pack number. Pack label texts were identical for TXA and placebo. We checked the coding of the blinded ampoules by randomly testing each batch of treatments and doing high-performance liquid chromatography to determine the contents.
Trial intervention
Patients were randomly allocated to receive a loading dose of 1 g of TXA infused over 10 minutes, started immediately after randomisation, followed by an i.v. infusion of 1 g over 8 hours, or matching placebo. Every patient was assigned a treatment pack with a unique number, which contained four ampoules of either 500 mg of TXA or placebo, one 100-ml bag of 0.9% sodium chloride (to use with the loading dose), a syringe and needle, stickers with the trial details and randomisation number (for attaching to the infusion bags, forms and medical records), and instructions. We separately provided information for patients and representatives, consent forms and data collection forms. The stickers, instructions, leaflets and forms were in local languages.
Dose selection
Tranexamic acid has been used to reduce bleeding in elective surgery for many years. A systematic review of randomised trials of TXA in surgery shows that dose regimens of TXA vary widely. 16 Loading doses range from 2.5 mg/kg to 100 mg/kg and maintenance doses range from 0.25 mg/kg/hour to 4 mg/kg/hour delivered over periods of 1 to 12 hours. Studies examining the impact of different doses of TXA on bleeding and transfusion requirements showed no significant difference between a high dose and a low dose. 16,27 In emergency situations, the administration of a fixed dose is more practicable because weighing patients in such situations is difficult. In the CRASH-3 trial, a fixed dose of 1-g loading dose of TXA, followed by a 1-g maintenance dose over 8 hours was selected. This fixed dose is within the dose range that has been shown to inhibit fibrinolysis and provide haemostatic benefit. It should be efficacious for heavier patients (> 100 kg) but also safe for lighter patients (< 50 kg), as the estimated dose/kg that the latter group would receive has been used in other trials without adverse effects. Furthermore, this fixed dose was used for 20,211 patients enrolled in the CRASH-2 trial and was found to be both effective and safe. 17 The same fixed dose was also used in two studies of TXA in TBI patients, again with no evidence of adverse effects. 20,21
Sites
We recruited patients with TBI from 175 hospitals in 29 countries. We enrolled the first patient on 20 July 2012 and the last patient on 31 January 2019. We stopped recruiting when the trial treatment expired. See Appendix 3, Table 17, for the total number of randomisations by geographical region.
Data collection
Baseline data
The trial entry form was used to collect baseline information including age, sex, time since injury, systolic blood pressure (SBP), GCS score, pupil reaction and, if relevant, the location of intracranial haemorrhage.
Outcome data
An outcome form was required to be completed 28 days after randomisation, or at death or hospital discharge if either event had already occurred. Once randomised, outcome data were collected even if the trial treatment was interrupted or not actually given. Short-term disability was assessed on the outcome form using the Disability Rating Scale (DRS). This scale measures the level of disability in six diagnostic categories of (1) eye opening, (2) best verbal response, (3) best motor response, (4) self-care ability for feeding, grooming and toileting, (5) level of cognitive functioning and (6) employability, and it can be used across the span of recovery. The maximum score a patient can obtain is 29, which represents an extreme vegetative state. A person without disability would score zero. 28 Specific patient-orientated outcomes were also assessed. These measures were identified from the literature and then considered and agreed by patient representatives from RoadPeace (London, UK), the UK national charity for those killed or injured in road crashes.
Monitoring
As the trial was assessed as low risk (TXA is widely used and the trial was considered to have a low risk of bias), central trial monitoring and central statistical monitoring were used in conjunction with investigator training, meetings and written guidance. Trial investigators and their institutions provided direct access to the source data for trial-related monitoring, audits and regulatory inspections. We planned to monitor about 10% of patient records on site; however, after changing the primary outcome, we expanded our monitoring plan to include patients enrolled within 3 hours of injury who subsequently died. We monitored 2436 (19%) patient records on site or remotely (using video call or telephone). This included 1161 (67%) of the patients who died from head injury (the primary outcome). The team of monitors worked alongside local trial teams to verify data from the source data, including pre-hospital ambulance cards, admission registers, emergency department notes, CT scans, surgery notes, blood transfusion registers, death registers and death certificates.
Outcome measures
Primary outcome
The primary outcome was head injury death in hospital within 28 days of injury in patients randomised within 3 hours of injury. The primary end point was originally 8 hours but, in 2016, the protocol was changed to patients treated within 3 hours of injury. Cause of death was assessed by the responsible clinician.
Secondary outcome
Secondary outcomes were early head injury death (within 24 and 48 hours after injury), all-cause and cause-specific mortality, disability, vascular occlusive events [myocardial infarction (MI), stroke, deep-vein thrombosis (DVT), pulmonary embolism (PE)], seizures, complications, neurosurgery, days in intensive care unit and adverse events within 28 days of randomisation. A diagnosis of DVT or PE was recorded only if there was a positive result on imaging (e.g. ultrasound) or at post-mortem examination.
Adverse events
Tranexamic acid has a well-documented safety profile. Although the summary of product characteristics29 suggests that rare cases of thromboembolic events might be associated with TXA administration, there is no evidence that the TXA treatment regimen used in this trial is associated with an increased risk of vascular occlusive events. Nevertheless, data on vascular occlusive events and seizures were collected as secondary outcomes and presented to the independent Data Monitoring Committee for unblinded review.
Change to the protocol
In September 2016, in response to evidence external to the trial indicating that TXA is unlikely to be effective when initiated beyond 3 hours of injury, the Trial Steering Committee (TSC) amended the protocol to limit recruitment to within 3 hours of injury. 18,30,31 Consequently, the primary end point was changed to ‘head injury death in hospital within 28 days of injury for patients treated within 3 hours of injury’.
To ensure that the trial would be large enough to reliably confirm or refute an early (< 3 hours) treatment benefit, the sample size was increased from 10,000 to 13,000 patients with the aim of enrolling 10,000 patients within 3 hours of injury.
The changes were made without reference to the unblinded trial data. The Data Monitoring Committee was not consulted about the change. The change was therefore not driven by the unblinded trial data seen by the Data Monitoring Committee, but instead driven by accumulating evidence external to the trial. The trial was conducted in accordance with International Conference on Harmonisation-Good Clinical Practice Guidelines. 32
Rationale for protocol change
During the CRASH-3 trial, new research emerged suggesting that TXA is likely to be most effective in the first few hours after injury and less effective when given later. 18 Trauma triggers the early release of tPA, the enzyme that converts plasminogen to the fibrinolytic enzyme plasmin, resulting in increased clot breakdown and bleeding. 33,34 tPA levels peak about 30 minutes after injury and plasmin peaks at 1 hour. 34
By inhibiting early fibrinolysis, TXA prevents coagulopathic bleeding;35 however, the effects appear to be short lived. Around 2 hours after injury, plasminogen activator inhibitor (PAI-1) levels increase, reaching a peak at 3 hours. 34 Plasminogen activator inhibitor inhibits fibrinolysis, resulting in ‘fibrinolytic shutdown’. 36 This might explain why the benefits of TXA in polytrauma patients appear to be limited to the first 3 hours. 18 As recent research shows that the coagulopathy after TBI is similar to that in poly-trauma, a similar time-dependent effect might be expected after TBI. 37,38 If the pathophysiological mechanisms affected by TXA are most relevant in the early hours after injury, the effect of TXA in this early period is the outcome of greatest importance. Nevertheless, intracranial bleeding can continue for up to 24 hours after injury and, therefore, examination of the effects of TXA within and beyond 3 hours remains an important scientific objective that will be addressed in preplanned subgroup analyses.
Sample size
Prior to implementing the amendment on limiting recruitment to within 3 hours of injury, 3535 participants had been recruited. It was originally estimated that a trial with about 10,000 patients would have 90% power (two-sided alpha of 1%) to detect a 15% relative reduction (20% to 17%) in mortality. We increased the sample size to 13,000 to get enough patients (about 10,000 as per the original sample size calculation) within 3 hours of injury to confirm or refute an early benefit. With 10,000 patients, the study would also have > 90% power to detect a difference in mean DRS score of 1.0 [assuming a standard deviation (SD) of DRS score of 9.0]. Experience from the CRASH-1 and CRASH-2 trials suggests that the anticipated rates of loss to follow-up (< 1%) would not have an important impact on study power. 17,39
Statistical methods and analysis plan
The statistical analysis plan was published before unblinding (see www.journalslibrary.nihr.ac.uk/programmes/hta/1419001/#/documentation; accessed November 2020). 19 The plan gave our reasons for limiting recruitment to within 3 hours of injury and stated that outcomes for patients treated after 3 hours of injury would be presented separately. All analyses were on an ‘intention-to-treat’ basis. For each binary outcome, we calculated RRs and 95% CIs. We conducted a complete-case analysis with no imputation for missing data. The safety of participants was overseen by an independent Data Monitoring Committee, which reviewed four unblinded interim analyses.
Subgroup analyses
In order to test the hypothesis that TXA is most effective when given soon after injury, a subgroup analysis was conducted of the effect of TXA according to the time interval between injury and TXA treatment (≤ 1 hour, > 1 to ≤ 3 hours, > 3 hours). We prespecified that this analysis would include patients treated within and beyond 3 hours of injury. As TBI severity, SBP and age could confound the impact of time to treatment on treatment effectiveness, we planned to control for these variables in a multivariable model. We expected that any beneficial effect of TXA would vary by time to treatment, with earlier treatment being most effective. We examined this hypothesis in a subgroup analysis of the effect of TXA according to the estimated time interval between injury and treatment (≤ 1 hour, > 1 to ≤ 3 hours, > 3 hours).
The effects of TXA on the primary outcome were also stratified by severity of head injury and age. Severity of head injury was assessed using the baseline GCS score, mild to moderate (GCS score of 9–15) or severe (GCS score of 3–8), and by pupil reactivity. In addition, we assessed the impact of severity in a regression analysis that included continuous terms for GCS and its square.
Traumatic brain injury patients who have a GCS score of 3 and bilateral unreactive pupils have a very poor prognosis, with a mortality risk of about 75%. The inclusion in the CRASH-3 trial of such severely injured patients, who may have little potential to benefit from the trial treatment, would bias the treatment effect towards the null. We therefore prespecified a sensitivity analysis that excluded patients with a GCS score of 3 and bilateral unreactive pupils.
As fibrinolytic activation after TBI may increase with age, we examined the effect of TXA on head injury death stratified by age: younger (≤ 30 years), middle (31–60 years) and older (> 60 years). For subgroup analyses, we report p-values for the test for heterogeneity.
Economic evaluation methods
An economic model was developed to analyse the cost-effectiveness of TXA treatment versus no treatment for patients with TBI. The analysis was performed in line with National Institute for Health and Care Excellence (NICE) guidance for economic evaluations, comparing the incremental costs and outcomes associated with providing TXA, over a lifetime time horizon, from the perspective of the UK NHS. 40 Full details of the methods and results are provided in Chapter 5.
Patient and public involvement
The CRASH-3 trial included patient and public involvement (PPI) to achieve the following objectives, namely to:
-
gain a lay perspective on PPI involvement in the design and management of emergency care clinical trials
-
identify an appropriate consent procedure for entering critically ill trauma patients into emergency clinical trials, which could be used for CRASH-3
-
ensure that we collect outcomes that are of primary concern to patients and their families after TBI
-
ensure that patient-facing documents for the trial were appropriate and clear
-
provide a lay perspective on the management of the trial and interpretation of the results
-
assist in developing and implementing the results dissemination strategy, and to help with presenting the trial results in a public-friendly format.
We included PPI groups to input to different stages of the trial. This included people who are at high risk of TBI, charitable organisations that support victims of trauma (RoadPeace) and people who have suffered TBI (Headway, Nottingham, UK).
Prior to working with our group, we carried out formative research to help guide PPI activities.
Formative research
Method
A qualitative study was conducted to elicit views on how best to involve patients and the public in the design, conduct and reporting of clinical trials involving people in emergency situations, gathering perspectives on which areas of the research programme they believed public contribution would be most appropriate. Approaches to designing a consent process to enter patients into emergency clinical trials were also explored.
Three focus group discussions were conducted, one with young people involved in an amateur boxing club, the other with a group of older men belonging to a social club and the third with a group of older women who were involved in a continuing education project and crafts-based activities. In total, 19 people took part (12 men and 7 women).
The sessions included a PowerPoint® (Microsoft Corporation, Redmond, WA, USA) presentation detailing why clinical trials are conducted in emergency medicine, how they are conducted and the key principles, including issues of consent, randomisation and the use of placebos. This was followed by three exercises using group work and discussion techniques.
Two key areas of inquiry emerged from these discussions: public involvement in the design and management of clinical trials and decisions about entering patients into clinical trials in an emergency.
Involvement in clinical trial design and management
Participants were highly supportive of clinical medical research, seeing it as essential for the progress of medical science. They also had a sense that the public should be consulted in principle. However, they struggled to identify how they might usefully contribute to the design and management of clinical trials in practice, seeing this as the province of highly skilled and qualified experts. Although there were individuals who could envisage a role for themselves with appropriate information and preparation, it was important to acknowledge that others felt that they had neither the inclination nor the aptitude to become involved, trusting in the expertise and competence of clinical researchers. Participants did have strong opinions in one area: that decision-making about the outcomes of clinical research must take account of quality-of-life issues and not be confined to treatment efficacy or safety, which they saw patients and the public as being well placed to comment on.
Consent process for involving patients in clinical trials in an emergency
Initially, a minority opposed entering patients into trials without their consent but these views tended be modified as participants considered the comments of others about the incapacitation of patients, the time-critical nature of emergency medicine and the necessity of clinical trials for medical progress.
Overall, among all groups, there was a very high regard for the medical profession and a strong faith in the skills and competence of medics, as well as the belief that clinicians would always act in the best interest of the patient. This was reflected in a sense that clinicians should be allowed to exercise their clinical judgement without undue burden to seek consent from next of kin when patients could not consent for themselves. However, moderating this perspective for some was a belief in the principle that, where practicable, next of kin should be consulted. Others argued that this might place a heavy burden of responsibility on families, and that the clinician’s greater expertise may in fact render better decisions.
Interestingly, when the participants were asked what they would want for themselves, all the participants expressed a desire for the clinician (or their family) to enter them into the trial.
Patient and public involvement group
The PPI group was responsible for providing input on the development of quality-of-life outcome measures to be used in the trial. They provided feedback from individuals with TBI and their caregivers on items of primary concern to patients after TBI
The PPI group reviewed drafts of the patient representative and patient information sheets, and consent forms.
A member of the PPI group from RoadPeace provided a lay perspective on the management of the trial as part of the TSC. RoadPeace is the national charity for road crash victims in the UK. Road traffic collisions are responsible for the majority of cases of TBI globally. RoadPeace supports survivors and their families and works to prevent serious injury and deaths from road crashes.
RoadPeace provided input in the CRASH-3 dissemination strategy (see Chapter 7). RoadPeace was involved in interpreting the data as part of the writing committee responsible for the main result publication. Both RoadPeace and Headway (National Head Injuries Association) provided help in dissemination of the results. A film to report the main trial results was led by a member of Headway (https://crash3.lshtm.ac.uk/blog/crash-3-trial-results/; accessed 23 February 2020).
Outcome of patient and public involvement
Patient and public involvement contributed to the success of the trial. The consent process that was developed with PPI groups was used in all countries that took part in the trial. The main structure and content of the brief information sheet, participant/legal representative information sheets and consent forms were utilised globally. They were accepted by all ethics committees and regulatory agencies with only local modifications needed.
The outcome measure developed with the PPI group included the following domains, which were considered to be important to TBI patients and their families: (a) walking, (b) washing/dressing, (c) pain/discomfort, (d) anxiety/depression, (e) agitation/aggression and (f) fatigue. A three-point scale response for each domain was used (none, moderate, extreme).
Role of funding source
The run-in phase (the first 500 patients) was funded by the JP Moulton Charitable Trust. The main phase was funded jointly by the National Institute for Health Research Health Technology Assessment (HTA) (project number 14/190/01) and Joint Global Health Trials [Medical Research Council (MRC), Department for International Development, Wellcome Trust] (project number MRM0092111). Dr Paul Atkinson, Saint John Regional Hospital, Canada, received a CA$10,000 grant from the New Brunswick Trauma Program to support the trial in Canada. The funders of the study had no role in study design, data collection, data analysis, data interpretation or writing the report. The corresponding author/writing committee had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Chapter 3 Baseline results
The first patient was randomised on 20 July 2012 and the last patient on 31 January 2019. Recruitment ended when the trial treatment expired.
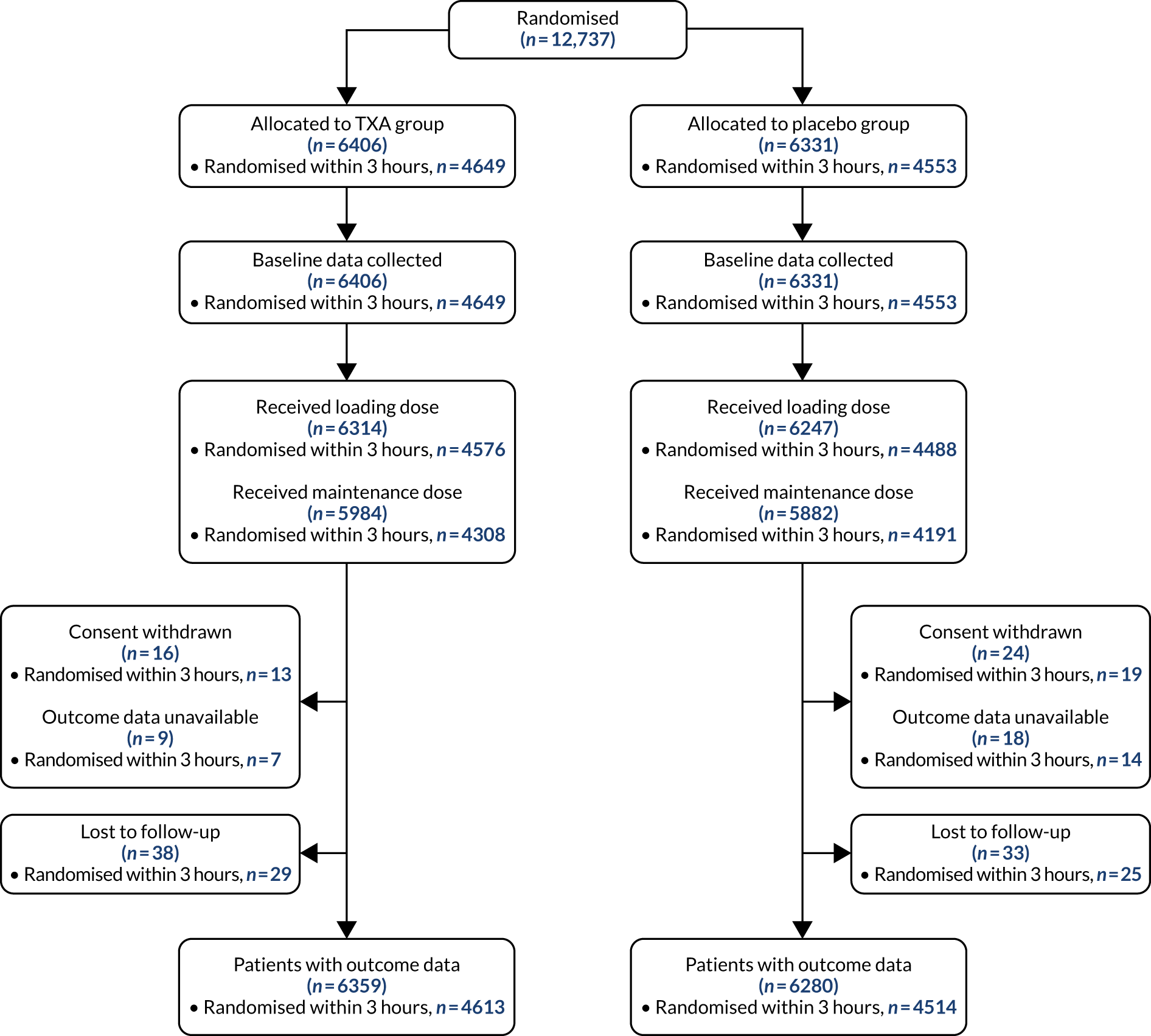
Figure 1 shows the trial profile. A total of 12,737 patients were randomly allocated to receive TXA (6406 patients) or matching placebo (6331 patients). A total of 9202 patients were enrolled within 3 hours of injury. Forty patients withdrew consent after randomisation, but 13 of them agreed to outcome data collection or had outcome data collected as part of adverse event reporting. We did not obtain primary outcome data for 75 patients (0.8%). There were 98 protocol violations. Sixty-six patients did not meet the inclusion criteria (32 patients had a GCS score of > 12 and no bleeding on CT scan, 11 had significant extracranial bleeding, eight had a time since injury of > 8 hours, six were aged < 16 years, three had non-traumatic bleeding, five had a combination of the above reasons, and one patient received TXA before randomisation). Thirty-two patients were recruited during a lapse in ethics approval in country. These patients were recruited in accordance with the approved procedure and approval was reissued after the lapse. Thirteen patients were unblinded. Baseline characteristics were similar between treatment groups for patients treated within 3 hours of injury (Table 1) and for those treated after 3 hours (Table 2).
FIGURE 1.
Trial profile.
| TXA (N = 4649), n | % | Placebo (N = 4553), n | % | |
|---|---|---|---|---|
| Sex | ||||
| Male | 3742 | 80 | 3660 | 80 |
| Female | 906 | 19 | 893 | 20 |
| Unknown | 1 | < 1 | 0 | 0 |
| Age (years) | ||||
| Mean (SD) | 41.7 | 19.0 | 41.9 | 19.0 |
| < 25 | 1042 | 22 | 996 | 22 |
| 25–44 | 1716 | 37 | 1672 | 37 |
| 45–64 | 1169 | 25 | 1184 | 26 |
| ≥ 65 | 722 | 16 | 701 | 15 |
| Time since injury (hours) | ||||
| Mean (SD) | 1.9 | 0.7 | 1.9 | 0.7 |
| ≤ 1 | 877 | 19 | 869 | 19 |
| > 1–2 | 2003 | 43 | 1889 | 41 |
| > 2–3 | 1769 | 38 | 1795 | 39 |
| SBP (mmHg) | ||||
| < 90 | 89 | 2 | 85 | 2 |
| 90–119 | 1508 | 32 | 1490 | 33 |
| 120–139 | 1461 | 31 | 1504 | 33 |
| ≥ 140 | 1576 | 34 | 1466 | 32 |
| Unknown | 15 | < 1 | 8 | < 1 |
| GCS scorea | ||||
| 3 | 495 | 11 | 506 | 11 |
| 4 | 213 | 5 | 213 | 5 |
| 5 | 163 | 4 | 172 | 4 |
| 6 | 221 | 5 | 232 | 5 |
| 7 | 311 | 7 | 294 | 6 |
| 8 | 354 | 8 | 315 | 7 |
| 9 | 335 | 7 | 292 | 6 |
| 10 | 371 | 8 | 364 | 8 |
| 11 | 375 | 8 | 390 | 9 |
| 12 | 476 | 10 | 478 | 10 |
| 13 | 297 | 6 | 312 | 7 |
| 14 | 526 | 11 | 458 | 10 |
| 15 | 484 | 10 | 492 | 11 |
| Unknown | 28 | 1 | 35 | 1 |
| Pupil reaction | ||||
| None react | 425 | 9 | 440 | 10 |
| One reacts | 374 | 8 | 353 | 8 |
| Both react | 3706 | 80 | 3636 | 80 |
| Unable to assess/unknown | 144 | 3 | 124 | 3 |
| All | > 3 hours | |||||||
|---|---|---|---|---|---|---|---|---|
| TXA (N = 6406) | Placebo (N = 6331) | TXA (N = 1757) | Placebo (N = 1778) | |||||
| n | % | n | % | n | % | n | % | |
| Sex | ||||||||
| Male | 5104 | 80 | 5013 | 79 | 1362 | 78 | 1353 | 76 |
| Female | 1301 | 20 | 1318 | 21 | 395 | 22 | 425 | 24 |
| Unknown | 1 | < 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Age (years) | ||||||||
| Mean (SD) | 43 | 19.8 | 43.1 | 19.7 | 46.4 | 21.3 | 46.2 | 21.1 |
| < 25 | 1362 | 21 | 1326 | 21 | 320 | 18 | 330 | 19 |
| 25–44 | 2285 | 36 | 2264 | 36 | 569 | 32 | 592 | 33 |
| 45–64 | 1625 | 25 | 1613 | 25 | 456 | 26 | 429 | 24 |
| ≥ 65 | 1134 | 18 | 1128 | 18 | 412 | 23 | 427 | 24 |
| Time since injury (hours) | ||||||||
| Mean (SD) | 2.9 | 3.2 | 2.9 | 2.3 | 5.5 | 5.2 | 5.4 | 2.9 |
| ≤ 1 | 877 | 14 | 869 | 14 | – | – | – | – |
| 1–3 | 3772 | 59 | 3684 | 58 | – | – | – | – |
| 3–8 | 1737 | 27 | 1760 | 28 | 1737 | 99 | 1760 | 99 |
| > 8 | 20 | < 1 | 18 | < 1 | 20 | 1 | 18 | 1 |
| SBP (mmHg) | ||||||||
| < 90 | 108 | 2 | 109 | 2 | 19 | 1 | 24 | 1 |
| 90–119 | 2001 | 31 | 1988 | 31 | 493 | 28 | 498 | 28 |
| 120–139 | 2107 | 33 | 2120 | 33 | 646 | 37 | 616 | 35 |
| ≥ 140 | 2167 | 34 | 2097 | 33 | 591 | 34 | 631 | 35 |
| Unknown | 23 | < 1 | 17 | < 1 | 8 | < 1 | 9 | 1 |
| GCS scorea | ||||||||
| 3 | 630 | 10 | 642 | 10 | 135 | 3 | 136 | 3 |
| 4 | 261 | 4 | 275 | 4 | 48 | 1 | 62 | 1 |
| 5 | 211 | 3 | 242 | 4 | 48 | 1 | 70 | 2 |
| 6 | 304 | 5 | 308 | 5 | 83 | 2 | 76 | 2 |
| 7 | 413 | 6 | 400 | 6 | 102 | 2 | 106 | 2 |
| 8 | 465 | 7 | 406 | 6 | 111 | 2 | 91 | 2 |
| 9 | 416 | 6 | 382 | 6 | 81 | 2 | 90 | 2 |
| 10 | 463 | 7 | 444 | 7 | 92 | 2 | 80 | 2 |
| 11 | 465 | 7 | 502 | 8 | 90 | 2 | 112 | 2 |
| 12 | 600 | 9 | 601 | 9 | 124 | 3 | 123 | 3 |
| 13 | 460 | 7 | 453 | 7 | 163 | 4 | 141 | 3 |
| 14 | 790 | 12 | 754 | 12 | 264 | 6 | 296 | 7 |
| 15 | 899 | 14 | 886 | 14 | 415 | 9 | 394 | 9 |
| Unknown | 29 | < 1 | 36 | 1 | 1 | < 1 | 1 | < 1 |
| Pupil reaction | ||||||||
| None react | 536 | 8 | 575 | 9 | 111 | 6 | 135 | 8 |
| One reacts | 511 | 8 | 482 | 8 | 137 | 8 | 129 | 7 |
| Both react | 5174 | 81 | 5113 | 81 | 1468 | 84 | 1477 | 83 |
| Unable to assess/unknown | 185 | 3 | 161 | 3 | 41 | 2 | 37 | |
Chapter 4 Outcome and results
Outcome data were available for 12,639 randomised patients (6359 patients allocated to the TXA group and 6280 patients to the placebo group). For patients randomised within 3 hours of injury, outcome data were available for 9127 patients (4613 patients allocated to the TXA group and 4514 patients to the placebo group). A total of 12,561 (98.6%) patients were known to have completed the loading dose, and 11,866 (93.2%) patients completed the 8-hour maintenance dose.
Primary outcome
Figure 2 shows the number of deaths and cause of death by days since injury in all patients randomised. There were 2560 deaths in total and the median time to death was 59 hours after injury (interquartile range 20–151 hours). Among patients treated within 3 hours of injury, there were 1878 deaths overall. Appendix 4, Figure 12, shows the cumulative incidence of head injury death in patients randomised within 3 hours of injury.
FIGURE 2.
Mortality by days since injury among all participants randomised.
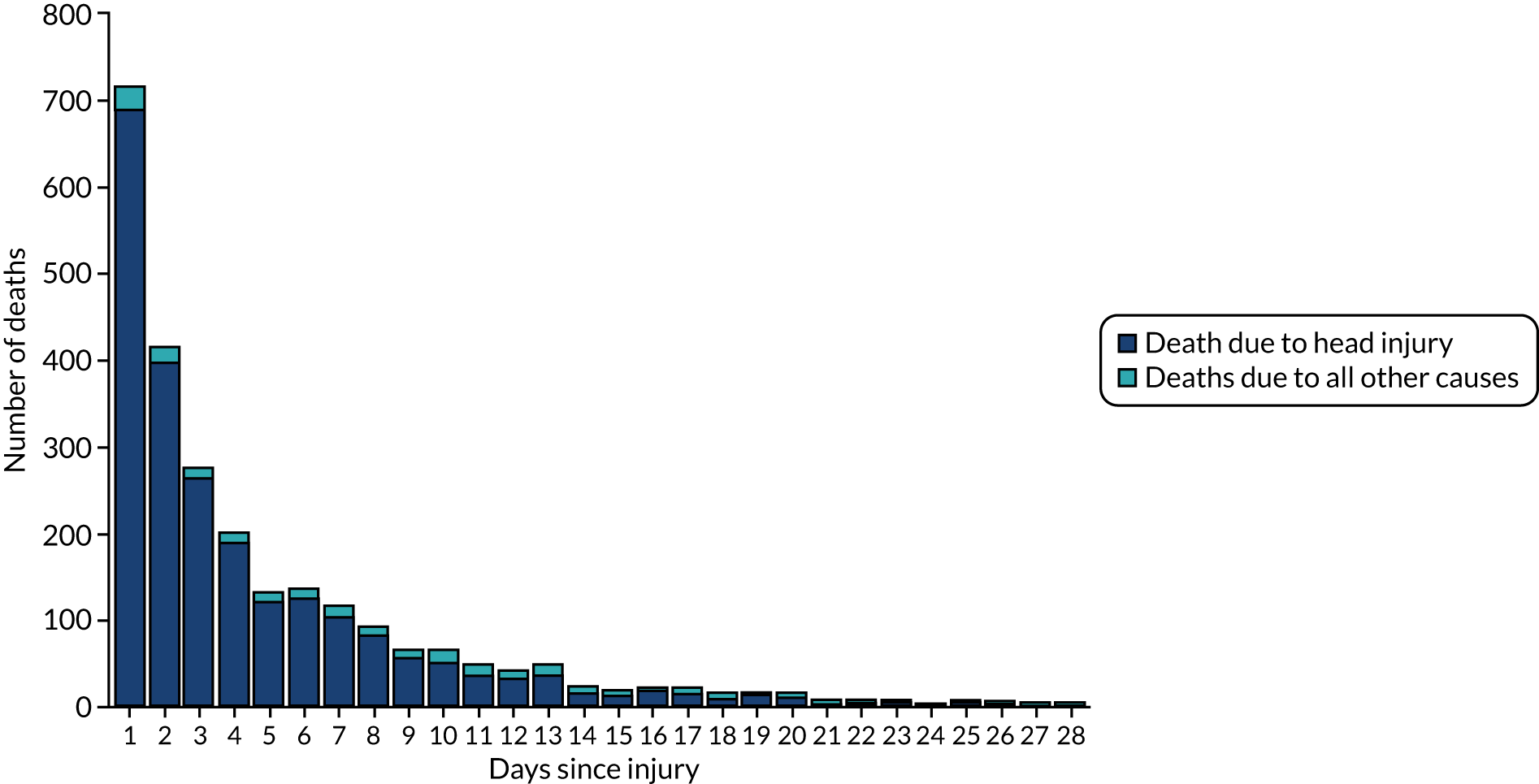
Table 3 shows the effect of TXA on head injury death in the 9127 patients randomised within 3 hours of injury with outcome data. Among patients treated within 3 hours of injury, the risk of head injury death was 18.5% in the TXA group versus 19.8% in the placebo group (855 vs. 892 events; RR = 0.94, 95% CI 0.86 to 1.02). In the prespecified sensitivity analysis that excluded patients with a GCS score of 3 or bilateral unreactive pupils at baseline, the results were 12.5% in the TXA group versus 14.0% in the placebo group (485 vs. 525 events; RR = 0.89, 95% CI 0.80 to 1.00).
| Head injury death | TXA | Placebo | RR (95% CI) | ||||
|---|---|---|---|---|---|---|---|
| n | N | % | n | N | % | ||
| All | 855 | 4613 | 18.5 | 892 | 4514 | 19.8 | 0.94 (0.86 to 1.02) |
| Excluding GCS score of 3, both unreactivea | 485 | 3880 | 12.5 | 525 | 3757 | 14.0 | 0.89 (0.80 to 1.00) |
Subgroup analysis
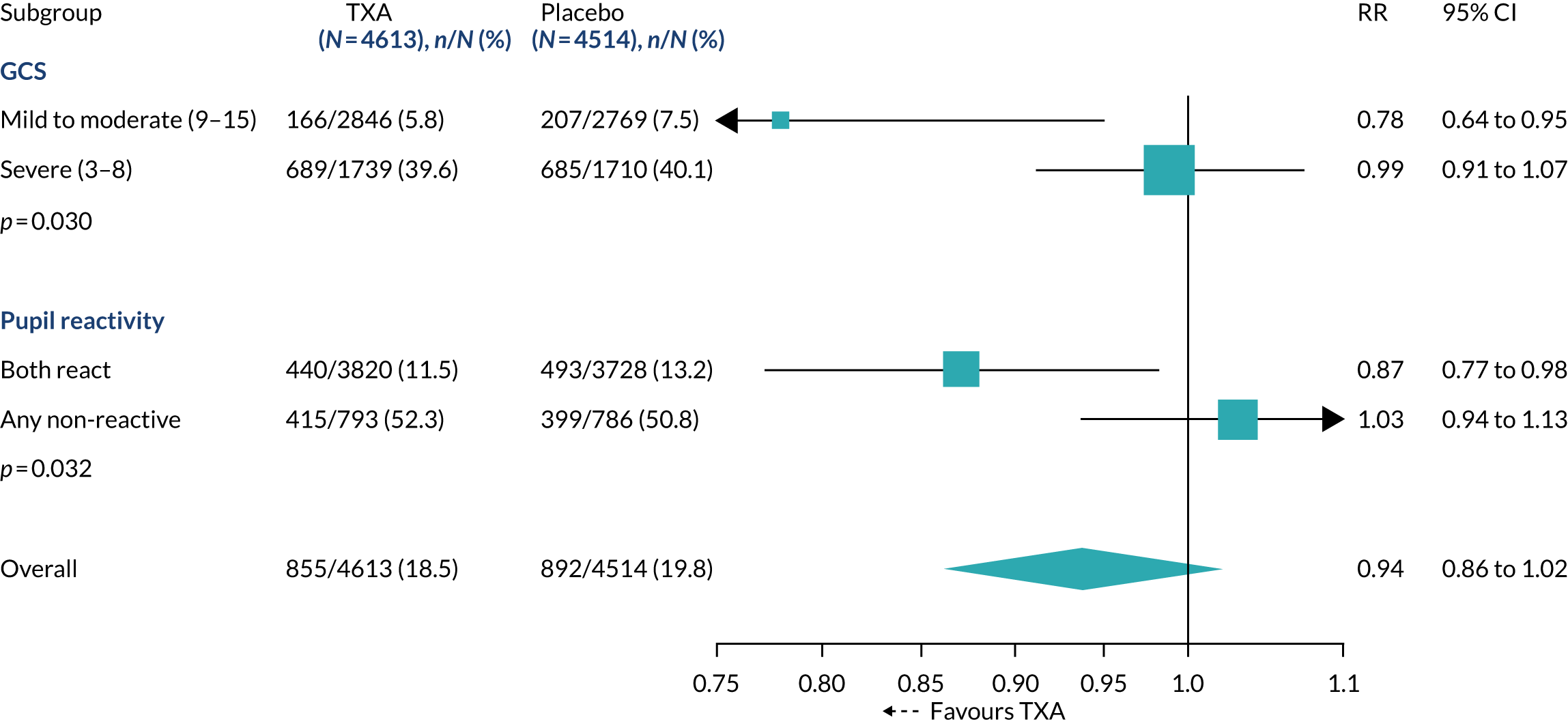
The effect of TXA on head injury death stratified by baseline GCS and pupillary reactions was examined (Figure 3). There was a reduction in the risk of head injury death with TXA in mild to moderate head injury (RR 0.78, 95% CI 0.64 to 0.95), but in severe head injury (RR 0.99, 95% CI 0.91 to 1.07) there was no clear evidence of a reduction (p-value for heterogeneity = 0.030). When we examined the impact of baseline GCS score in a regression analysis, there was evidence (p = 0.007) that TXA is more effective in less severely injured patients. Among patients with reactive pupils, head injury deaths were reduced with TXA (RR 0.87, 95% CI 0.77 to 0.98).
FIGURE 3.
Effect of TXA on head injury death stratified by baseline severity in participants randomised within 3 hours of injury.
We examined the effect of TXA on head injury death stratified by time to treatment and recorded no evidence of heterogeneity (p = 0.96). The RR of head injury death with TXA was 0.96 (95% CI 0.79 to 1.17) in patients randomised ≤ 1 hour after injury, 0.93 (95% CI 0.85 to 1.02) in those randomised > 1 to ≤ 3 hours after injury and 0.94 (95% CI 0.81 to 1.09) in those randomised > 3 hours after injury. However, as anticipated in the statistical analysis plan, patients treated soon after injury often have more severe head injury and so the impact of time to treatment could be confounded by severity.
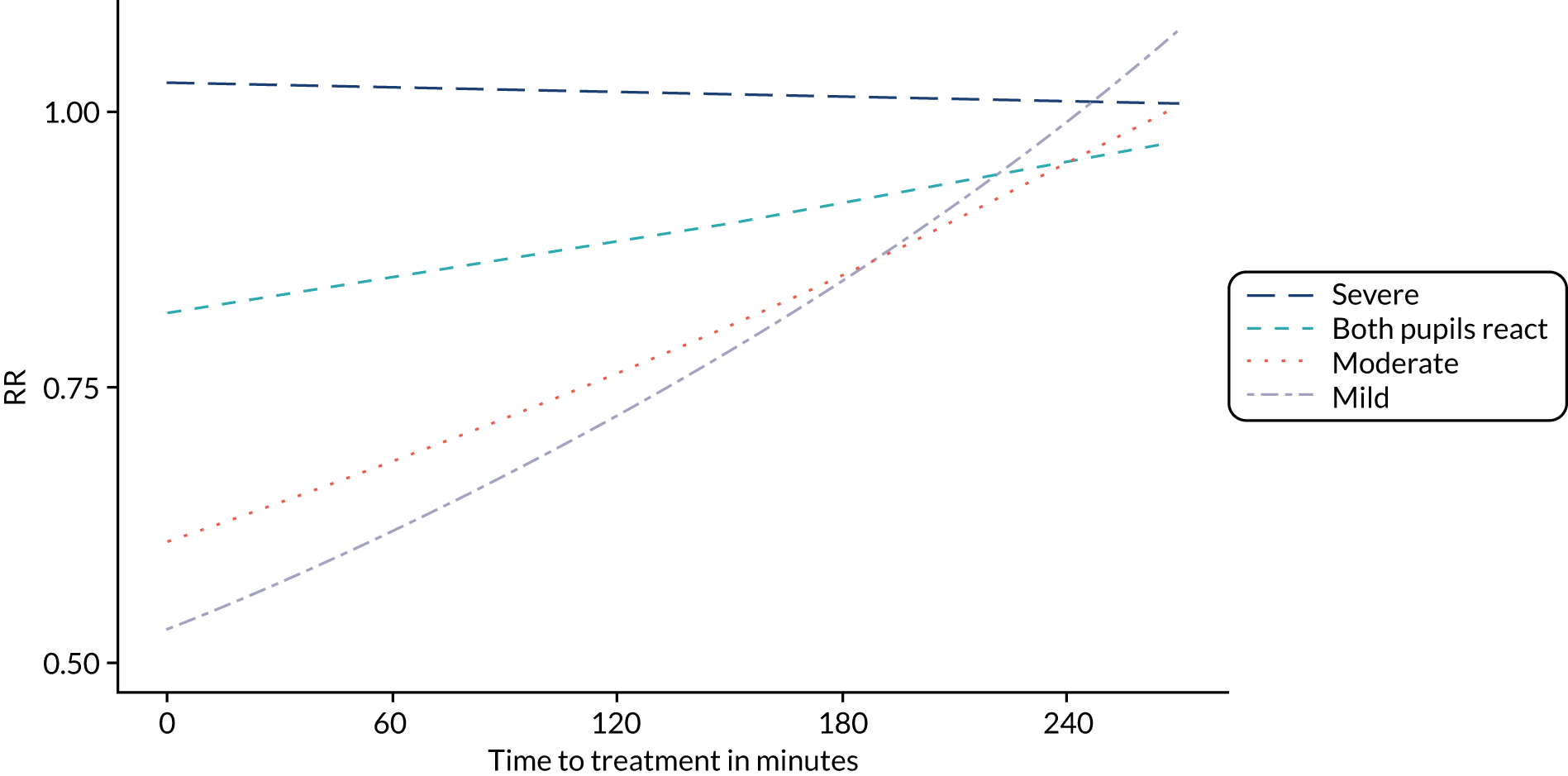
Figure 4 shows the impact of time to treatment on the effect of TXA in patients with a mild or moderate head injury and in those with severe head injury after adjusting for GCS score, SBP and age in a multivariable model including all participants. Early treatment was more effective in patients with mild or moderate head injury (p = 0.005), but there was no obvious impact of time to treatment in severe head injury (p = 0.73). The effectiveness of TXA by time to treatment stratified by severity is further demonstrated in Figure 5. We recorded no evidence of heterogeneity in the effect of TXA by patient age (p = 0.45).
FIGURE 4.
Effect of TXA on head injury death by severity and time to treatment in all participants with (a) mild and moderate head injury; and (b) severe head injury. Dotted lines represent 95% confidence limits. Mild/moderate, n = 8107; severe, n = 2703.
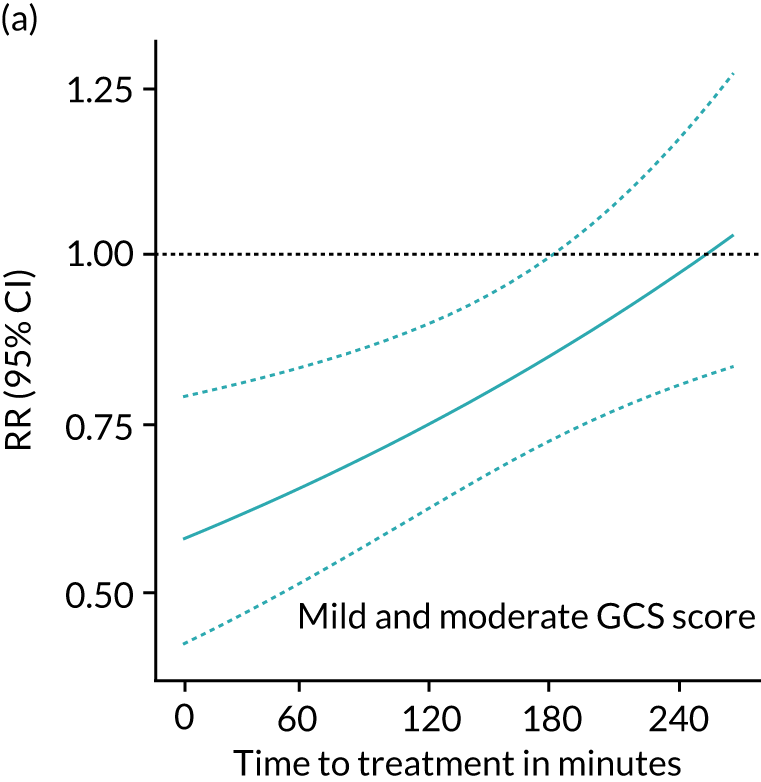
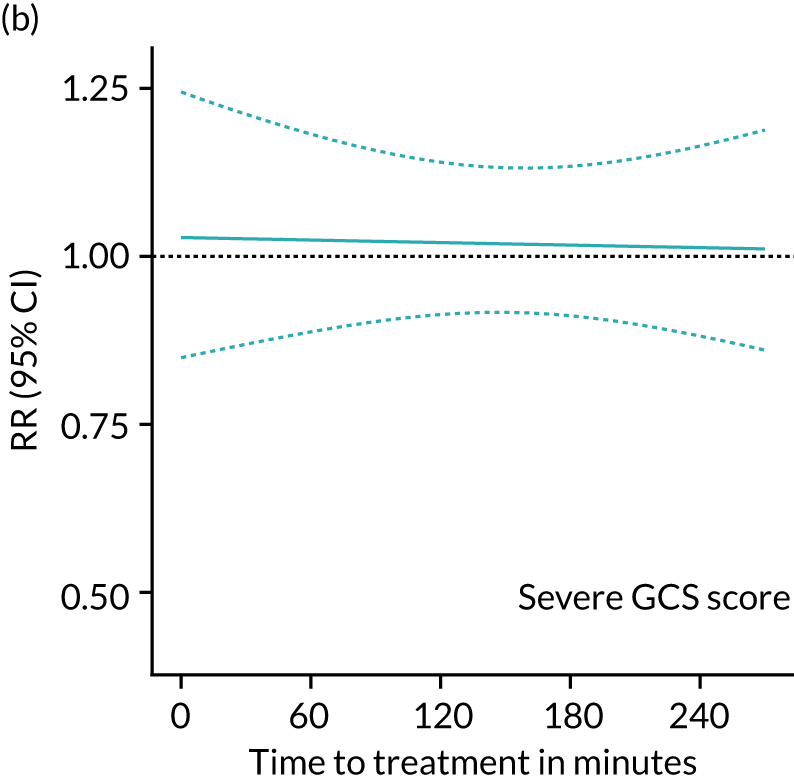
FIGURE 5.
Effectiveness of TXA on head injury death vs. time to treatment stratified by severity in all patients. Severe, n = 2703; both pupils react, n = 2204; moderate, n = 3897; mild, n = 4275.
We examined the effect of TXA on head injury death stratified by World Bank country income level (high income vs. low and middle income). This analysis was not prespecified. Although the reduction in the risk of head injury death with TXA was higher in high-income countries (RR 0.76, 95% CI 0.55 to 1.04) than in LMIC (RR 0.92, 95% CI 0.81 to 1.04), there was no statistical evidence of heterogeneity by country income level (p = 0.258). As early head injury deaths are more likely than late head injury deaths to result from intracranial haemorrhage, we examined the effect of TXA on head injury deaths within 24 and 48 hours of injury. The RRs of head injury death were 0.81 (95% CI 0.69 to 0.95) and 0.89 (95% CI 0.79 to 1.02) within 24 and 48 hours, respectively. When patients with a GCS score of 3 and those with bilateral unreactive pupils at baseline were excluded, the corresponding values were 0.72 (95% CI 0.56 to 0.92) and 0.84 (95% CI 0.69 to 1.01).
The models are adjusted for GCS score, age and SBP. In patients with a mild and moderate GCS score (9–15) there were 537 head injury deaths. In patients with a severe GCS score (3–8) there were 918 head injury deaths, excluding those with a GCS score of 3 and those with unreactive pupils.
Secondary outcomes
In patients randomised within 3 hours of injury, the RRs for non-head injury deaths and for all-cause mortality were 1.31 (95% CI 0.93 to 1.85; 75 vs. 56 events) and 0.96 (95% CI 0.89 to 1.04; 930 vs. 948 events), respectively. The results for non-head injury deaths broken down by cause and all-cause mortality in all patients randomised are presented in Table 4.
| Cause of death | TXA group, N = 6359 | Placebo group, N = 6280 | RR (95% CI) | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Bleeding | 9 | 0.1 | 7 | 0.1 | 1.27 (0.47 to 3.41) |
| PE | 9 | 0.1 | 7 | 0.1 | 1.27 (0.47 to 3.41) |
| Stroke | 10 | 0.2 | 4 | 0.1 | 2.47 (0.77 to 7.87) |
| MI | 9 | 0.1 | 3 | 0.0 | 2.96 (0.80 to 10.94) |
| Multiorgan failure | 27 | 0.4 | 24 | 0.4 | 1.11 (0.64 to 1.92) |
| Aspiration/pneumonia | 30 | 0.5 | 34 | 0.5 | 0.87 (0.53 to 1.42) |
| Sepsis | 9 | 0.1 | 6 | 0.1 | 1.48 (0.53 to 4.16) |
| Cervical spine injury | 3 | 0.0 | 4 | 0.1 | 0.74 (0.17 to 3.31) |
| Other | 16 | 0.3 | 11 | 0.2 | 1.44 (0.67 to 3.09) |
| Any cause | 1262 | 0.2 | 1298 | 0.2 | 0.96 (0.90 to 1.03) |
We assessed the effect of TXA on disability in survivors by comparing the mean DRS score (lower score means less disabled) between the TXA and placebo groups. The scores were similar between groups for patients treated within 3 hours of injury (mean = 4.99, SD = 7.6, for TXA group, vs. mean = 5.03, SD = 7.6, for placebo group) and for those treated after 3 hours of injury (mean = 4.52, SD = 7.0 for TXA group, vs. mean = 5.00, SD = 7.4 for placebo group). We also examined the effect of TXA on disability (Table 5) using an outcome measure designed by patient representatives by estimating the RR of being in the most extreme category for six areas of functioning: (1) walking, (2) washing, (3) pain and discomfort, (4) anxiety or depression, (5) agitation or aggression and (6) fatigue. The prevalence of disability among survivors was similar in the TXA and placebo groups.
| < 3 hours | ≥ 3 hours | All | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TXA (N = 4613) | Placebo (N = 4514) | RR (95% CI) | TXA (N = 1746) | Placebo (N = 1766) | RR (95% CI) | TXA (N = 6359) | Placebo (N = 6280) | RR (95% CI) | |||||||
| n | % | n | % | n | % | n | % | n | % | n | % | ||||
| Patient-derived disability measuresa | |||||||||||||||
| Confined to bed | 579 | 12.6 | 549 | 12.2 | 1.03 (0.93 to 1.15) | 190 | 10.9 | 222 | 12.6 | 0.87 (0.72 to 1.04) | 769 | 12.1 | 771 | 12.3 | 0.99 (0.90 to 1.08) |
| Unable to wash or dress | 580 | 12.6 | 583 | 12.9 | 0.97 (0.87 to 1.08) | 195 | 11.2 | 228 | 12.9 | 0.87 (0.72 to 1.04) | 775 | 12.2 | 811 | 12.9 | 0.94 (0.86 to 1.03) |
| Extreme pain or discomfort | 38 | 0.8 | 29 | 0.6 | 1.28 (0.79 to 2.08) | 10 | 0.6 | 10 | 0.6 | 1.01 (0.42 to 2.42) | 48 | 0.8 | 39 | 0.6 | 1.22 (0.80 to 1.85) |
| Extreme anxiety or depression | 43 | 0.9 | 41 | 0.9 | 1.03 (0.67 to 1.57) | 19 | 1.1 | 20 | 1.1 | 0.96 (0.51 to 1.79) | 62 | 1.0 | 61 | 1.0 | 1.00 (0.71 to 1.43) |
| Extreme agitation or aggression | 53 | 1.1 | 53 | 1.2 | 0.98 (0.67 to 1.43) | 14 | 0.8 | 27 | 1.5 | 0.52 (0.28 to 1.00) | 67 | 1.1 | 80 | 1.3 | 0.83 (0.60 to 1.14) |
| Extreme fatigue | 100 | 2.2 | 101 | 2.2 | 0.97 (0.74 to 1.27) | 40 | 2.3 | 43 | 2.4 | 0.94 (0.61 to 1.44) | 140 | 2.2 | 144 | 2.3 | 0.96 (0.76 to 1.21) |
| Complicationsb | |||||||||||||||
| All vascular occlusive events | 69 | 1.5 | 60 | 1.3 | 1.13 (0.80 to 1.59) | 32 | 1.8 | 42 | 2.4 | 0.77 (0.49 to 1.21) | 101 | 1.6 | 102 | 1.6 | 0.98 (0.74 to 1.28) |
| PE | 18 | 0.4 | 18 | 0.4 | 0.98 (0.51 to 1.88) | 6 | 0.3 | 14 | 0.8 | 0.43 (0.17 to 1.13) | 24 | 0.4 | 32 | 0.5 | 0.74 (0.44 to 1.26) |
| DVT | 15 | 0.3 | 12 | 0.3 | 1.22 (0.57 to 2.61) | 4 | 0.2 | 4 | 0.2 | 1.01 (0.25 to 4.04) | 19 | 0.3 | 16 | 0.3 | 1.17 (0.60 to 2.28) |
| Stroke | 29 | 0.6 | 23 | 0.5 | 1.23 (0.71 to 2.13) | 17 | 1.0 | 19 | 1.1 | 0.90 (0.47 to 1.74) | 46 | 0.7 | 42 | 0.7 | 1.08 (0.71 to 1.64) |
| MI | 9 | 0.2 | 12 | 0.3 | 0.73 (0.31 to 1.74) | 9 | 0.5 | 8 | 0.5 | 1.14 (0.44 to 2.94) | 18 | 0.3 | 20 | 0.3 | 0.89 (0.47 to 1.68) |
| Renal failure | 73 | 1.6 | 56 | 1.2 | 1.28 (0.90 to 1.80) | 27 | 1.5 | 28 | 1.6 | 0.98 (0.58 to 1.65) | 100 | 1.6 | 84 | 1.3 | 1.18 (0.88 to 1.57) |
| Sepsis | 297 | 6.4 | 279 | 6.2 | 1.04 (0.89 to 1.22) | 114 | 6.5 | 133 | 7.5 | 0.87 (0.68 to 1.10) | 411 | 6.5 | 412 | 6.6 | 0.99 (0.86 to 1.12) |
| Seizure | 130 | 2.8 | 105 | 2.3 | 1.21 (0.94 to 1.56) | 76 | 4.4 | 81 | 4.6 | 0.95 (0.70 to 1.29) | 206 | 3.2 | 186 | 3.0 | 1.09 (0.90 to 1.33) |
| Gastrointestinal bleeding | 16 | 0.3 | 22 | 0.5 | 0.71 (0.37 to 1.35) | 8 | 0.5 | 13 | 0.7 | 0.62 (0.26 to 1.50) | 24 | 0.4 | 35 | 0.6 | 0.68 (0.40 to 1.14) |
Adverse events
The risk of vascular occlusive events and other complications was similar in the TXA and placebo groups (see Table 5). There was no evidence that TXA increased fatal or non-fatal stroke (RR = 1.08, 95% CI 0.71 to 1.64). The risk of seizures was similar between groups (RR = 1.09, 95% CI 0.90 to 1.33). The numbers of other adverse events were similar between groups (see Appendix 5, Table 18).
Unblinding
Clinicians requested unblinding of the treatment allocation for 13 patients after randomisation for the following reasons: TXA became indicated after randomisation, n = 7; clinical management depended on knowing the treatment allocation, n = 3; patient requested unblinding, n = 1; required for suspected unexpected serious adverse reaction reporting, n = 1; unblinded in error, n = 1.
Forty patients received TXA in addition to the trial treatment after randomisation. In 36 cases, this was because the clinician believed that it was clinically indicated, and in four cases it was given in error instead of the trial drug.
Chapter 5 Economic evaluation results
Model analysis and model population characteristics
The cost-effectiveness analysis has been published in BMJ Global Health. 41 Parts of this chapter have been reproduced from Williams et al. 41 in accordance with © Williams et al. 41 [or their employer(s)] 2020. [Re-use permitted under CC BY. Published by BMJ. https://creativecommons.org/licenses/by/4.0/. This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.]
The economic analysis assessed the cost-effectiveness of treating TBI patients with TXA and without TXA, as per the trial treatment arms. The following health economic section has been reported to meet the criteria of the CHEERS (Consolidated Health Economic Evaluation Reporting Standards) checklist. 42
As stated above, the trial included patients treated within 3 hours of their injury with either a GCS score of ≤ 12 or any intracranial bleeding on their CT scan, and without extracranial bleeding. The trial found that TXA reduced head injury deaths among those with TBI, with a RR of 0.94 (95% CI 0.86 to 1.02). However, there was evidence that people with mild or moderate TBI (baseline GCS score of 9–15) had a greater benefit from TXA treatment, in terms of reduction in head injury death (RR 0.78, 95% CI 0.64 to 0.95), than those with a severe head injury (GCS score of 3–8; RR 0.99, 95% CI 0.91 to 1.07). For this reason, the mild and moderate population was used as the base-case population, excluding those with severe head injury. In addition to considering the cost-effectiveness of TXA based on baseline GCS score, we also evaluated the cost-effectiveness of TXA for an alternative subgroup of patients: those with head injury of any severity with both pupils reactive (RR 0.87, 95% CI 0.77 to 0.98), based on the clinical results presented in Figure 3. We excluded those with either pupil unreactive (RR 1.03, 95% CI 0.94 to 1.13), as there was no evidence of a reduction in head injury deaths for this subgroup.
The model was analysed over a lifetime time horizon with costs presented in Great British pounds, and outcomes presented as life-years (LYs) and quality-adjusted life-years (QALYs). The analysis was performed from a UK NHS and personal social services perspective. The model estimates the incremental cost-effectiveness ratio (ICER) by dividing the incremental costs of TXA by the incremental health outcomes associated with TXA treatment, to give a cost per LY or QALY gained. We used the lower bound of the £20,000 to £30,000 per QALY cost-effectiveness threshold stated by NICE to estimate the cost-effectiveness of TXA. 40 If the ICER falls below the cost-effectiveness threshold, then that intervention can be considered cost-effective. Both costs and outcomes were discounted at a rate of 3.5%, in accordance with NICE guidelines,40 to capture the higher value of current costs and outcomes compared with those occurring in the future. The mean age of individuals entering the model was derived directly from the CRASH-3 trial (41.7 years for patients with mild and moderate injury and 41.6 years for those with both pupils reactive). Deterministic sensitivity analyses were performed, in which alternative discount rates (0% and 6%) were evaluated. The cost-effectiveness model was developed in Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA), with the analysis of trial data performed in Stata® 16 (StataCorp LP, College Station, TX, USA).
Model structure
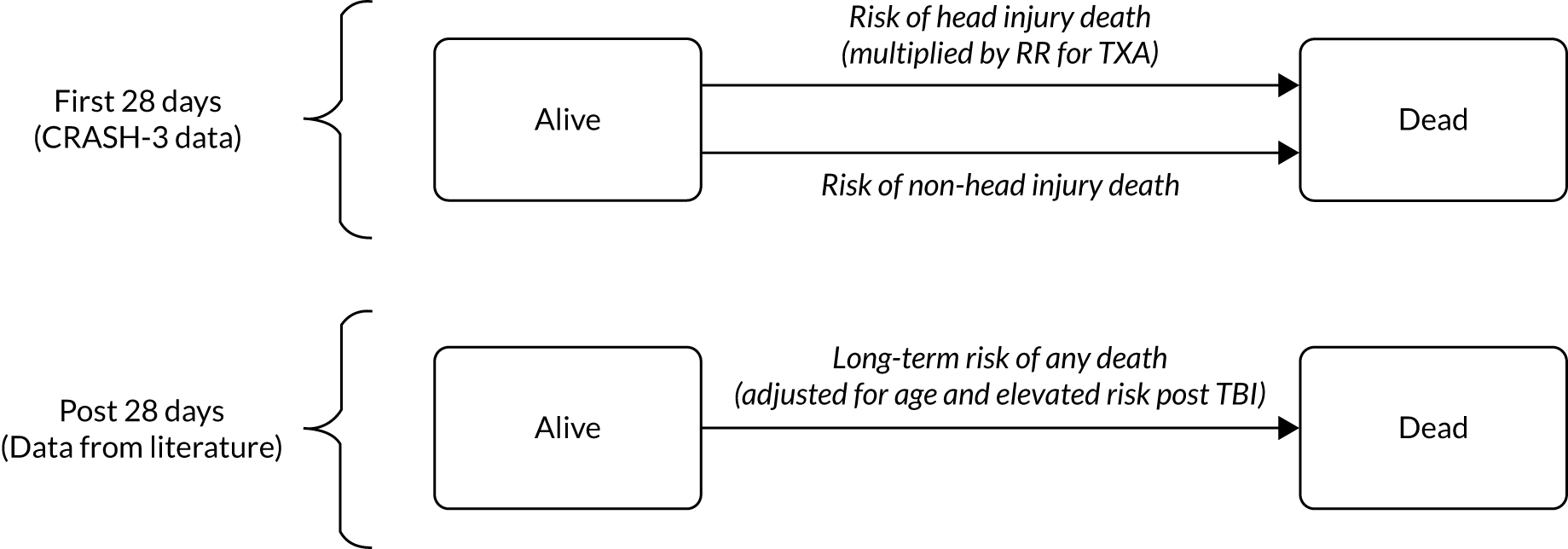
A Markov model captured the long-term outcomes associated with head injury, and is shown in Figure 6. It consists of two health states, alive and dead, and includes the risk of death during the first 28 days of the trial from both head injuries and non-head injuries along with estimates of longer-term mortality. The model uses a daily cycle length for the first year, to allow the events during the trial period to be accurately modelled, followed by an annual cycle length thereafter.
FIGURE 6.
Model structure.
Clinical outcomes
The 28-day risk of head injury and non-head injury death for the placebo group were derived from the CRASH-3 trial, with the risk in high-income countries used to estimate the risk in the UK. A RR of head injury death was applied for patients receiving TXA, as derived directly from the CRASH-3 trial. The risk of non-head injury death was equal for placebo and TXA groups in the model, based on the CRASH-3 trial. The risk of head injury and non-head injury death within the 28-day follow-up period, and the head injury rate ratio associated with TXA, are presented for the mild and moderate CRASH-3 population in Table 6 and for patients with both pupils reactive in Table 7.
| Parameter | Value | Distribution | Source |
|---|---|---|---|
| TXA rate ratio treatment effect | |||
| Head injury | 0.78 | Log-normal(µ = –0.248, σ = 0.1) | CRASH-324 |
| Non-head injury | 1 | N/A | CRASH-324 |
| 28-day risk of death | |||
| Head injury death | 0.061 | Beta(α = 42, β = 643) | CRASH-324 |
| Non-head injury death | 0.018 | Beta(α = 12, β = 673) | CRASH-324 |
| Long-term standardised mortality ratios | |||
| First year, post injury | 4.00 | Normal(95% CI 3.27 to 4.90) | McMillan et al.43 |
| Beyond first year, post injury | 2.26 | Normal(95% CI 1.84 to 2.77) | McMillan et al.43 |
| Parameter | Value | Distribution | Source |
|---|---|---|---|
| TXA rate ratio treatment effect | |||
| Head injury | 0.87 | Log-normal(µ = –0.138, σ = 0.06) | CRASH-324 |
| Non-head injury | 1 | N/A | CRASH-324 |
| 28-day risk of death | |||
| Head injury death | 0.105 | Beta(α = 42, β = 643) | CRASH-324 |
| Non-head injury death | 0.019 | Beta(α = 12, β = 673) | CRASH-324 |
| Long-term standardised mortality ratios | |||
| First year, post injury | 4.00 | Normal(95% CI 3.27 to 4.90) | McMillan et al.43 |
| Beyond first year, post injury | 2.26 | Normal(95% CI 1.84 to 2.77) | McMillan et al.43 |
Following the 28-day trial follow-up period, the risk of death was assumed equal for people treated with and people treated without TXA. Standardised mortality ratios (SMRs) were used to account for the higher risk of death post TBI compared with the general population. SMRs were derived from a Scottish study that included a variety of head injury severities. It estimated a SMR of 4 for the first year following injury, and 2.26 thereafter, compared with a group of matched community controls. 43 These SMRs were applied relative to age-based, UK general population mortality estimates, and were assumed to be the same for those with mild or moderate TBI and those with both pupils reactive. 44 It was assumed that the additional long-term risk of death continued throughout the duration of the model; however, a sensitivity analysis that excluded this long-term risk of death was performed, to assess the impact of this parameter.
Health status, utility and quality-adjusted life-years
In the CRASH-3 trial, there was little difference between the DRS scores for each treatment arm reported for those with mild or moderate TBI [TXA 3.12 (SD 5.6) vs. placebo 2.91 (SD 5.1), with lower scores representing better outcomes] and those with both pupils reactive [TXA 4.38 (SD 7) vs. placebo 4.33 (SD 6.9)].
To capture the quality of life for patients post TBI, utility values for the ‘alive’ health state were derived from a systematic review and EuroQol-5 Dimensions utility mapping study, which identified five studies reporting utility values stratified by the severity of TBI outcomes. 45 This mapping studies then estimated utility by Glasgow Outcome Scale (GOS) outcomes, using a UK value set. 45 The utilities associated with each GOS outcome is shown in Table 8. 45 In our analysis, we estimated the overall utility by estimating the corresponding GOS outcome for each patient by using the DRS outcomes reported in the CRASH-3 trial. The mapping of each DRS outcome to the GOS outcome is presented in Appendix 6, Table 19. Once this mapping was performed, a weighted average of GOS outcomes was used to estimate the average utility for each population. The average utility was 0.74 for the mild and moderate population and 0.70 for those with both pupils reactive (Table 9). It was assumed that individuals who died within the 28-day study period had a utility of 0 between their injury and death.
| GOS outcomea | DRS scores | Mild/moderate population (n) | Both pupils react population (n) | Utility value | Distribution |
|---|---|---|---|---|---|
| Good recovery | 0–1 | 3094 | 3478 | 0.894 | Beta(α = 50, β = 5.9) |
| Moderate disability | 2–6 | 1288 | 1545 | 0.675 | Beta(α = 30.5, β = 14.7) |
| Severe disability | 7–21 | 677 | 1084 | 0.382 | Beta(α = 10.9, β = 17.7) |
| Vegetative state | 22–29 | 124 | 359 | –0.178 | Beta(α = 16.1, β = –106.3) |
| Population | Utility value |
|---|---|
| Mild/moderate TBI | 0.74 |
| Both pupils react | 0.70 |
Owing to the uncertainty around the utility estimates used in the base-case analysis, three sensitivity analyses were performed to consider the impact of alternative utility values on the cost-effectiveness of TXA. First, a sensitivity analysis was performed in which the DRS scores for those receiving TXA and placebo were modelled independently, and independently mapped to utility scores, which resulted in a marginally lower utility among those receiving TXA. This resulted in utility values of 0.74 for TXA and 0.75 for placebo in the mild and moderate group, and 0.69 for TXA and 0.7 for placebo for patients with both pupils reactive. Second, a sensitivity analysis considered an alternative method to estimate GOS outcomes among CRASH-3 patients, using a previous study reporting the correlation between GCS score at injury and GOS outcomes (see Appendix 6, Table 20, for additional details). 45,46 This allowed for the distribution of GCS scores for patients in the CRASH-3 trial to be used to estimate the distribution of GOS outcomes, to which utility values could be applied. This produced higher estimated utilities of 0.79 for the mild and moderate population and 0.76 for patients with both pupils reactive (see Appendix 6, Table 21). Last, a sensitivity analysis considered the impact of a lower utility value, of 0.63, for both treatment groups, and for both model populations (patients with mild and moderate injury and patients with both pupils reactive), to assess the impact of a lower utility estimate on cost-effectiveness. This was an average estimate derived from a Swiss study of trauma patients reporting utility values in mild (0.7) and moderate (0.56) TBI patients, with GCS score of 9–15, and with an abbreviated injury score of 0–2, representing mild or no TBI. 47
Age-based utility estimates for the UK general population were used to account for the decline in utility with age (Table 10). 48 The post-TBI utility estimates (see Table 9) were derived from a cohort with a median age of 50 years. Therefore, a utility decrement model population for those reaching the age of ≥ 55 years.
| Age (years) | Utility | Utility decrement |
|---|---|---|
| 35–44 | 0.91 | 0 |
| 45–54 | 0.85 | 0 |
| 55–64 | 0.8 | 0.05 |
| 65–74 | 0.78 | 0.07 |
| ≥ 75 | 0.73 | 0.12 |
For example, the average utility for patients with mild or moderate TBI after discharge would be 0.75 until they reach 55 years, when the utility would decrease to 0.70 (0.75 minus 0.05). The utility would then decrease to 0.68 at 65 years (0.75 minus 0.07). The utility estimates were not inflated between 42 (average starting age in the model) and 44 years, to remain conservative.
Costs
Treatment costs
The model captured the costs of TXA treatment, including the cost of TXA, needle and syringe, and nurse administration time, which were applied to the TXA intervention only (see Table 14). The total cost of TXA was derived from the British National Formulary49 (£6 per person), as were the costs of the infusion bags (£3.25 for a 100-ml and 500-ml bag). The costs of needles and syringes were derived from a NICE costing template for the UK. 50
The nurse time required to administer TXA was assumed to be 21 minutes (as per the CRASH-218 economic analysis), and the hourly cost of a nurse was derived from UK social service costs, based on the hourly cost of a band 5, hospital-based NHS nurse. 51,52 This gave a total cost of £22.25 for treatment, equipment and treatment administration.
Hospital costs
There was little difference in hospital length of stay for those treated with and those treated without TXA in high-income countries [TXA 14 days (SD 9.8 days), placebo 13.3 days (SD 9.3 days), overall 13.7 days (SD 9.6 days)], and, therefore, this was assumed to be the same for both arms. A sensitivity analysis was performed to assess this assumption, in which the trial data for hospital length of stay were modelled specifically for each treatment arm. Inpatient hospital costs were derived from UK NHS reference costs,53 using the cost associated with head injury admissions. A weighted average of all head injury admission costs was calculated, based on the severity of the head injury (case mix adjusted). As the length of stay was assumed to be the same for those treated with TXA and those treated without TXA, hospital costs did not affect the incremental cost-effectiveness, except in the sensitivity analysis to assess this assumption.
Monitoring costs
Patients were assumed to incur additional health-care resources post discharge. These long-term monitoring costs include the increased use of health services, such as outpatient clinic visits and more frequent visits to GPs. It also includes rehabilitation and physiotherapy, and community care, such as formal carers. These costs were assumed to differ between the first year post injury and after 12 months. First-year monitoring costs were derived from a UK costing study,54 for those with good recovery, moderate disability and severe disability. These costs were mapped from DRS scores (using the same method described above to map from DRS to GOS outcomes) to estimate the average annual monitoring costs (Table 11). These costs have also been used in a previous HTA analysis. 55
| GOS status | Estimated equivalent DRS scores | Cost, first year (£) | Distribution | Cost, after first year (£) | Distribution |
|---|---|---|---|---|---|
| Good recovery | 0–1 | £290 | Gamma(k = 25, θ = 9.6) | £26 | Gamma(k = 25, θ = 0.96) |
| Moderate disability | 2–6 | £20,745 | Gamma(k = 25, θ = 686) | £1710 | Gamma(k = 25, θ = 64) |
| Severe disability | 7–21 | £40,983 | Gamma(k = 25, θ = 1356) | £13,363 | Gamma(k = 25, θ = 500) |
| Vegetative state | 22–29 | £40,983a | Gamma(k = 25, θ = 1356) | £13,363a | Gamma(k = 25, θ = 500) |
The average first-year monitoring cost was estimated to be £11,662 for those with mild or moderate head injury and £14,259 for those with both pupils reactive. 54 Long-term monitoring costs (applied after the first year post injury) were estimated by expert opinion in a previous HTA. 55 The average cost was £2505 per year for patients with mild or moderate TBI and £3405 for patients with both pupils reactive, and was assumed to be incurred until the patient died. We explored the impact of excluding monitoring costs beyond the first year post injury and applying monitoring costs until 5 years post injury in sensitivity analyses, owing to the uncertainty in these estimates.
The average monitoring costs for the UK were estimated by combining the annual cost by GOS status (see Table 11) with the proportion of patients across each GOS outcome (see Appendix 6, Table 19). A weighted average was used to provide the average annual monitoring cost for each population, as displayed in Table 12.
| Population | Cost, 0–12 months (£) | Cost, > 12 months (£) |
|---|---|---|
| Patients with mild/moderate TBI | £11,662 | £2505 |
| Patients with both pupils reactive | £14,259 | £3405 |
All costs for the mild and moderate TBI population are shown in Table 13, and for the both pupils reactive population in Table 14. All costs were inflated to 2018 prices using a UK hospitals and community service index. 52
| Parameter | Cost (£) | Distribution | Source |
|---|---|---|---|
| TXA (full dose) | £6.00 | N/A | British National Formulary 56 |
| Sodium chloride | £3.25 | N/A | British National Formulary 57 |
| Needle and syringe | £0.05 | N/A | NICE50 |
| Hospital cost | £4751 | N/A | CRASH-324/Department of Health and Social Care53 |
| Monitoring costs (first year post injury) | £11662 | By component (see Table 11) | Lecky et al.,55 Beecham et al.54 |
| Monitoring costs (after first year post injury) | £2505 | By component (see Table 11) | Lecky et al.55 |
| Parameter | Cost (£) | Distribution | Source |
|---|---|---|---|
| TXA (full dose) | £6.00 | N/A | British National Formulary 56 |
| Sodium chloride | £3.25 | N/A | British National Formulary 57 |
| Needle and syringe | £0.05 | N/A | NICE50 |
| Hospital cost | £5158 | N/A | CRASH-324/Department of Health and Social Care53 |
| Monitoring costs (first year post injury) | £14,259 | By component (see Table 11) | Lecky et al.,55 Beecham et al.54 |
| Monitoring costs (after first year post injury) | £3405 | By component (see Table 11) | Lecky et al.55 |
Sensitivity analyses
The main analysis was performed using probabilistic sensitivity analyses to simultaneously capture the uncertainty in model parameters. Distributions were assigned to each probabilistic parameter, with each sampled simultaneously across 1000 Monte Carlo simulations. One-way deterministic sensitivity analyses were also performed to assess the sensitivity of specific parameters on the cost-effectiveness estimates, and are presented relative to the base case as a tornado diagram.
Primary analysis of base-case incremental costs, quality-adjusted life-years and incremental cost-effectiveness ratio: mild and moderate traumatic brain injury patients
The costs, LYs and QALYs associated with TXA treatment and without TXA treatment are presented in Table 15. In the base-case analysis, TXA is highly cost-effective in the UK for those with mild and those with moderate TBI, at £4288 per QALY gained. When considering LYs only, the ICER was £3078 per LY gained.
| Treatment group | Costs (£) | LYs | QALYs | ICER (per LY) | ICER (per QALY) |
|---|---|---|---|---|---|
| Placebo | £55,108 | 16.87 | 12.10 | ||
| TXA | £55,867 | 17.12 | 12.28 | £3078 | £4288 |
The cost of purchasing and administering TXA represented a very small proportion of the incremental costs (3%), with long-term monitoring costs contributing to most of the incremental costs for the TXA group (97%). These higher costs are due to a higher proportion of patients surviving when given TXA, as monitoring costs per person were the same in both treatment groups.
The long-term survival projections of the model for patients with mild or moderate TBI receiving TXA or placebo are presented in Appendix 6, Figures 13 and 14.
Sensitivity analyses of base-case population: mild and moderate traumatic brain injury
Probabilistic sensivity analysis
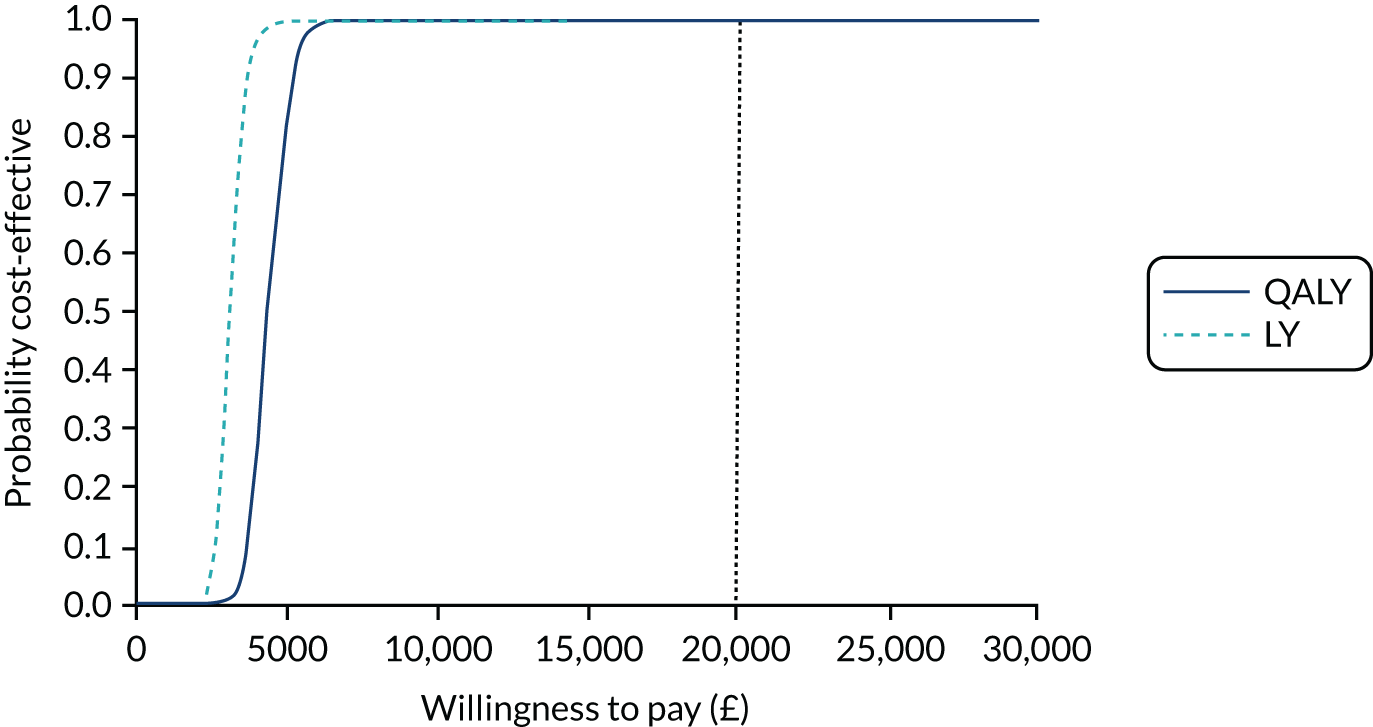
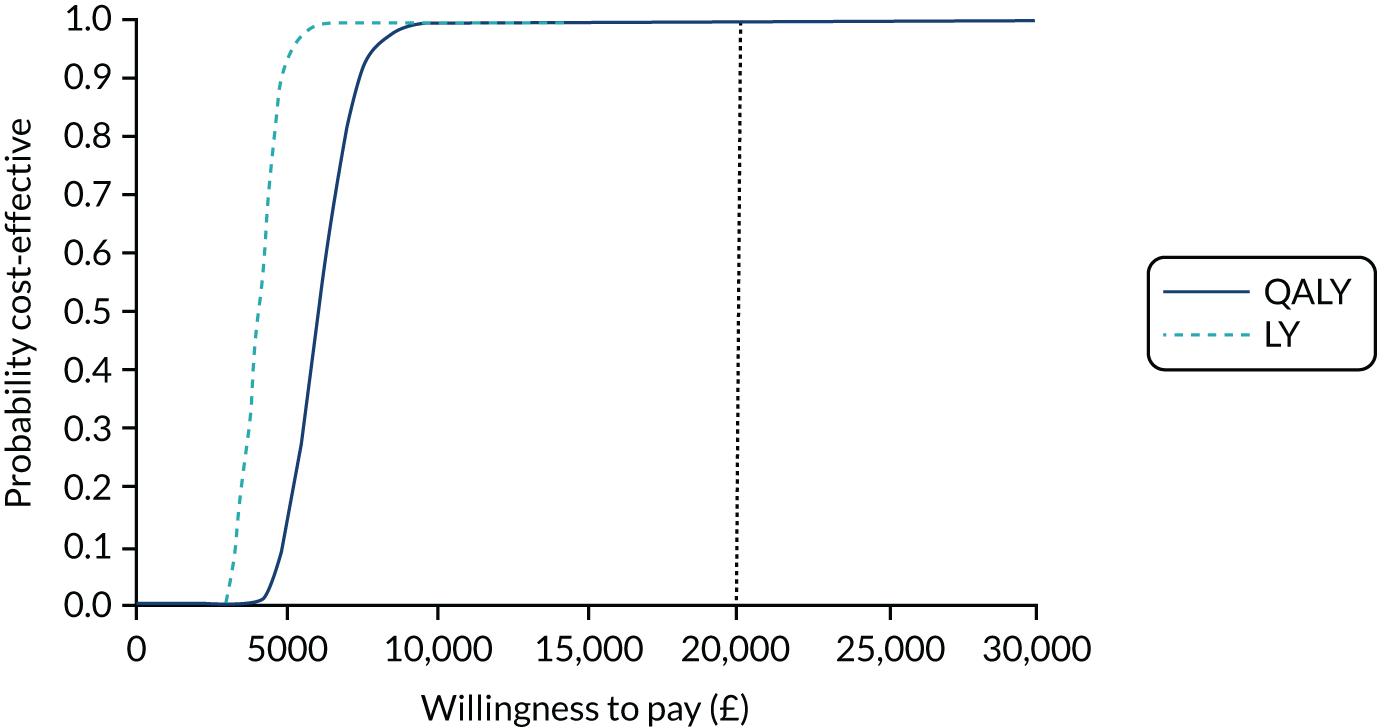
Tranexamic acid was highly likely to be cost-effective in the probabilistic sensitivity analysis (PSA), with a 99% probability of being cost-effective at the NICE £20,000 per QALY willingness-to-pay threshold (Figure 7).
FIGURE 7.
Cost-effectiveness acceptability curve for TXA for patients with mild or moderate TBI.
Deterministic sensitivity analysis
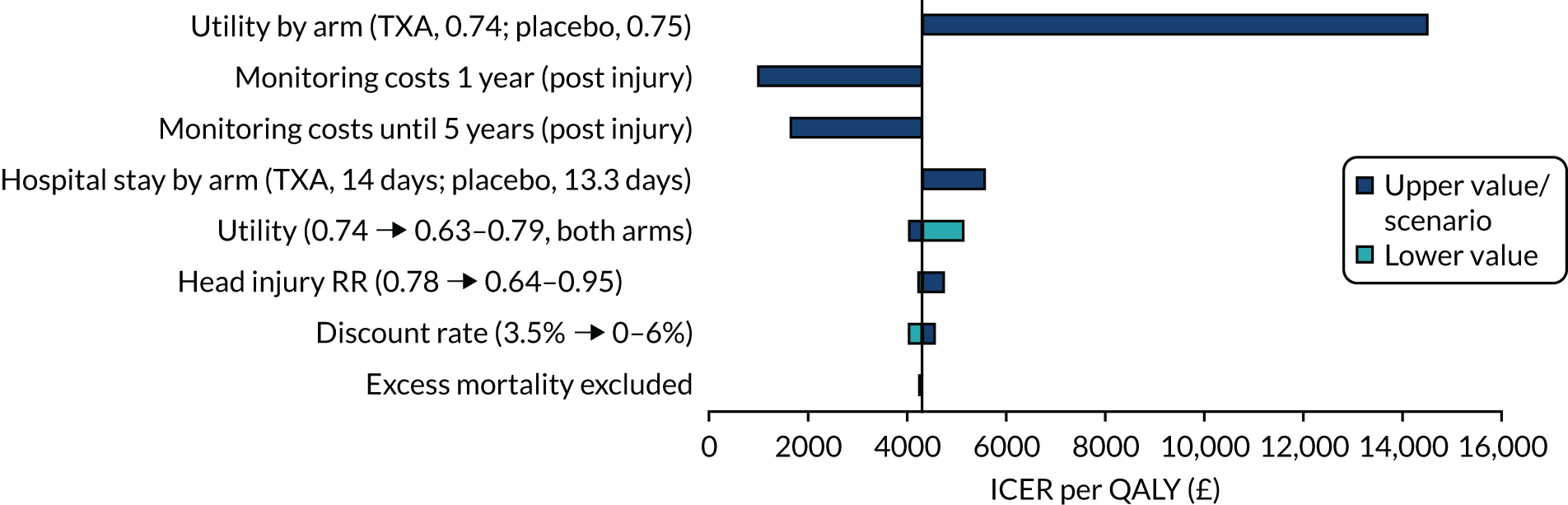
A number of sensitivity analyses were performed, but none increased the ICER above the cost-effectiveness threshold, meaning that TXA remained cost-effective in all deterministic sensitivity analyses (Figure 8). Assuming a lower utility among those receiving TXA than among those receiving placebo increased the ICER the most, to £14,465 per QALY. Restricting monitoring costs to only the first year or first 5 years post injury reduces the ICER to £979 and £1646 per QALY, respectively. When considering a longer length of hospital stay for those receiving TXA than for those receiving placebo, the ICER increased to £5567, whereas assuming a lower utility (0.63 for both arms) increased the ICER to £5112 per QALY. TXA remained cost-effective when the RR increased to 0.95, with the ICER increasing to £4721 per QALY. The discount rate, and excluding excess mortality after the trial period, had little impact on the ICER.
FIGURE 8.
Tornado diagram showing deterministic sensitivity analyses and the impact on the ICER per QALY gained, for those with mild or moderate TBI.
Analyses for patients with both pupils reactive: incremental costs, quality-adjusted life-years and incremental cost-effectiveness ratio
Deterministic results
When considering individuals with both pupils reactive, treatment remained highly cost-effective with an ICER of £6097 per QALY in the UK. When considering LYs only, the ICER was £4066 per LY gained (Table 16).
| Treatment group | Costs (£) | LYs | QALYs | ICER (per LY) | ICER (per QALY) |
|---|---|---|---|---|---|
| Placebo | £68,894 | 16.04 | 10.69 | ||
| TXA | £69,901 | 16.29 | 10.86 | £4066 | £6097 |
Probabilistic sensitivity analysis
At the UK cost-effectiveness threshold of £20,000 per QALY, TXA is 99% likely to be cost-effective in the PSA (Figure 9).
FIGURE 9.
Cost-effectiveness acceptability curve for TXA treatment for patients with both pupils reactive.
Deterministic sensitivity analysis
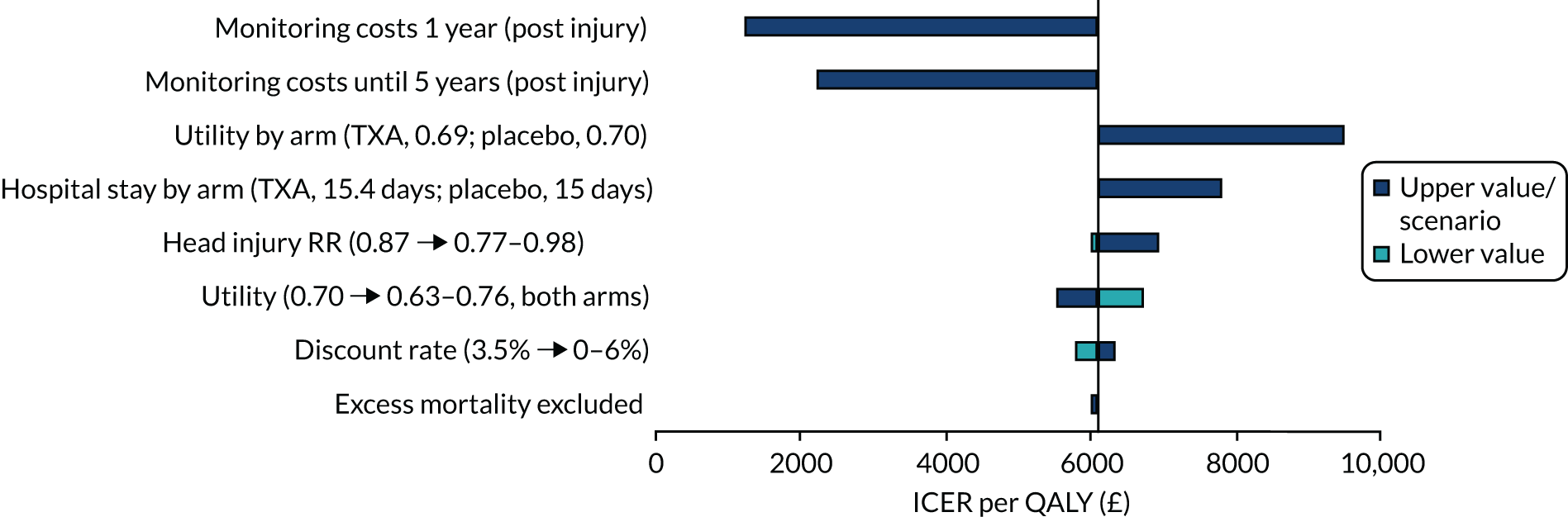
The deterministic results for patients with both pupils reactive show that, for all sensitivity analyses in the UK, TXA remained highly cost-effective (Figure 10). Similarly to the results for the mild and moderate TBI population, a reduction in monitoring costs being applied for either 1 year only or 5 years reduced the ICER to £1257 and £2233 per QALY, respectively. Assuming a lower utility among those receiving TXA than among those receiving placebo increased the ICER to £9512 per QALY. When assuming a longer length of hospital stay for TXA based on the CRASH-3 trial, the ICER increased to £7804. At a head injury treatment effect rate ratio of 0.98 (representing the upper 95% CI), TXA remained cost-effective, with the ICER increasing to £6949 per QALY. The ICER also increased when considering a lower utility (0.63), to £6753 per QALY. The discount rate, and excluding excess mortality after the trial period, had little impact on the ICER.
FIGURE 10.
Tornado diagram showing deterministic sensitivity analyses and the impact on the ICER per QALY gained for patients with both pupils reactive.
Chapter 6 Discussion
This trial provides evidence that administration of TXA to TBI patients within 3 hours of injury reduces head injury deaths, with no evidence of adverse effects or complications. There was a substantial reduction in head injury deaths with TXA in patients with mild or moderate head injuries, but no apparent reduction in those with severe head injuries. There was no increase in disability among survivors.
The effect of TXA on head injury death appears to depend on the time interval between injury and the initiation of the trial treatment, and on the severity of the TBI. Early treatment of patients with mild (GCS score of 13–15 and intracranial bleeding on baseline CT scan) and moderate head injury seems to confer the greatest mortality benefit. This is consistent with the hypothesis that TXA improves outcomes by reducing intracranial bleeding. As haemorrhage expansion occurs in the hours immediately after injury, treatment delay would reduce the potential for TXA to prevent intracranial bleeding. 5,6 Patients with severe head injury may have less to gain from TXA treatment because they already have extensive intracranial haemorrhage prior to treatment, or other potentially life-threatening intracranial pathologies that are not affected by TXA. We anticipated in our statistical analysis plan that the effect of TXA would be greatest for head injury deaths occurring in the first few days after injury than for later head injury deaths, because early head injury deaths are more likely as a result of bleeding. Our data support this hypothesis, showing a substantial reduction in head injury deaths within 24 hours of injury (RR 0.72, 95% CI 0.56 to 0.92). Similar results were obtained in the CRASH-2 trial35 of TXA in traumatic extracranial bleeding, in which the effect of TXA on death from bleeding was greatest on the day of the injury (RR 0.72, 95% CI 0.60 to 0.86). However, thereafter, the benefit of TXA for head injury patients is slightly attenuated, probably as patients succumbed to non-bleeding-related pathophysiological mechanisms. This may explain why the effect of early TXA treatment on head injury death is slightly smaller than the effect of TXA on death due to bleeding seen in the CRASH-2 trial. 35
Tranexamic acid did not appear to increase disability among survivors, and there was no evidence of any increased risk of adverse events. In particular, the risk of DVT, PE, stroke and MI was similar in the TXA and placebo groups. This is consistent with the results of the CRASH-2 trial35 in traumatic extracranial bleeding, which also recorded no increased risk of vascular occlusive events with TXA. Unlike the CRASH-2 trial,35 there was no evidence that administration beyond 3 hours of injury increased the risk of head injury death or any other adverse events. Indeed, given the absence of any adverse effects in this trial, the implications of wrongly concluding that TXA is ineffective are likely to be far more consequential than wrongly concluding that TXA is effective.
The CRASH-3 trial provides evidence that TXA is safe in TBI patients and that treatment within 3 hours of injury reduces head injury deaths.
Strengths and limitations
Our trial had several strengths but also some limitations. The method of randomisation ensured that participating clinicians had no foreknowledge of the treatment allocation, and the use of placebo control ensured that outcome assessment was blind to the intervention. Although the eligibility criteria required the recruiting doctor to be uncertain as to the appropriateness of TXA treatment, because TXA is not a recommended treatment for isolated TBI, almost all TBIs meeting the inclusion criteria were recruited. Baseline prognostic factors were well balanced and, because almost all randomly assigned patients were followed up, there is little potential for bias. The analysis was by intention to treat (176 patients did not receive any of the trial treatment). The primary outcome was head injury death as assessed by the responsible clinician. Although some misclassification of cause of death is inevitable, the assessment was blinded to the trial treatment. All-cause mortality combines causes of death that might be affected by TXA (e.g. head injury death due to intracranial bleeding) with causes that we do not expect to be affected by TXA (e.g. sepsis) and, therefore, would be biased towards the null. Although the CRASH-3 trial is one of the largest trials of TBI, the CIs were wide and compatible with a substantial reduction in head injury death and little or no benefit. On the other hand, when set in the context of all the available randomised trials of TXA in TBI, the possibility of no mortality benefit appears remote. 21,22,58 When assessing outcome measures in clinical trials, provided that there are few false positives (high specificity), estimates of the RR are unbiased even when sensitivity is imperfect. 59 For this reason a diagnosis of DVT or PE was recorded only if there was a positive result on imaging (e.g. ultrasound) or at post-mortem examination. As a result, although the trial may have underestimated the risk of DVT or PE, the RR estimates for this outcome should be unbiased.
We anticipated that TBI patients with a GCS score of 3 and those with bilateral unreactive pupils prior to treatment would have little potential to benefit from TXA and that their inclusion in the analysis would bias the treatment effect towards the null. Most patients with bilateral unreactive pupils already have extensive intracranial haemorrhage and brain herniation and so it is unlikely that TXA could improve the outcome in these cases. We therefore prespecified a sensitivity analysis that excluded these patients. However, patients with unilateral unreactive pupils were not excluded, and because many of these patients have brain herniation their inclusion might also have diluted the treatment effect. Indeed, when patients with a GCS score of 3 and those with unilateral or bilateral unreactive pupils prior to treatment are excluded in a post hoc analysis, the treatment effect is noticeably larger (RR 0.85, 95% CI 0.74 to 0.96).
Cost-effectiveness
Although the cost of TXA treatment is low, providing treatment will still incur additional costs to the health service and, therefore, questions arise regarding whether or not this cost represents an efficient use of resources, based on the benefit associated with treatment. Our analysis shows that TXA is highly cost-effective in the UK for those with complicated mild and moderate TBI, and is also highly cost-effective for patients with both pupils reactive. These results were robust across sensitivity analyses, as probabilistic analyses showed that the intervention is 99% likely to be cost-effective in both model populations at the NICE willingness-to-pay threshold of £20,000 per QALY. Furthermore, all deterministic sensitivity analyses produced ICERs below the lower limit of the NICE cost-effectiveness threshold of £20,000.
The NICE guidelines included TXA for the pre-hospital care of patients with trauma, following the results of the CRASH-2 trial. 60 Our analysis suggests that TXA should also be recommended for patients with complicated mild and moderate TBI and for patients with both pupils reactive, when treatment can be provided within 3 hours of injury, as treatment is highly cost-effective.
The cost-effectiveness analysis has some limitations. One limitation is that the trial followed patients for only 28 days post injury, leading to uncertainty about patient outcomes beyond this time. Furthermore, evidence on long-term outcomes post TBI in the literature is limited. To capture the long-term additional risk of death for these patients, we assumed that, after the trial period, the risk of death remained elevated compared with the risk of death in the general population (four times higher for the first year and two times higher thereafter). However, these estimates were derived from a case–control study performed in Scotland, and there is uncertainty as to whether or not the additional risk of death reported in this study is likely to be reflective of the patients in this trial. Sensitivity analyses were performed to consider the uncertainty in future outcomes, first, using higher discount rates (giving lower weighting to future events), and, second, performing a scenario excluding this additional mortality. Both had little impact on the estimated ICER, with TXA remaining cost-effective in both analyses, suggesting that this uncertainty is unlikely to affect the overall cost-effectiveness of the treatment.
In addition, the CRASH-3 trial did not collect direct utility estimates, meaning that they were estimated from the DRS outcomes at 28 days (or at time of discharge). Although just over half of all mild and moderate TBI patients had no disability at discharge, there was uncertainty in this estimation process of overall utility, as well as uncertainty regarding the long-term disability of patients compared with their status at discharge or 28 days, when some patients’ utility would be expected to improve over time. To address this, sensitivity analyses with lower utility values, and a lower utility value for those receiving TXA, were performed. Neither sensitivity analysis influenced the decision on cost-effectiveness.
Last, our analysis was performed from a health service perspective, and therefore did not capture the potential long-term costs that could be associated with caregiver burden or out-of-pocket medical payments that might be associated with those living with disability. However, it should be noted that the disability scores for survivors are similar between groups, and therefore any additional societal burden associated with TXA treatment would result from a higher proportion of patients surviving only, as the outcomes among survivors were comparable.
Despite the limitations stated above, we have used robust trial results and supporting evidence from the literature to show that TXA treatment is highly likely to be cost-effective for the treatment of patients with complicated mild and moderate TBI and for patients with both pupils reactive, when provided within 3 hours of injury.
Findings in context
Evidence before this study
Evidence from the CRASH-218 trial that administration of TXA within 3 hours of injury reduces death in patients with traumatic extracranial bleeding raised the possibility that it might reduce death from traumatic intracranial bleeding. Intracranial bleeding is common after TBI, and increases head injury death and disability. Prior to the CRASH-3 trial, we made a systematic search for all randomised trials of TXA in acute traumatic injury. We searched PubMed, Science Citation Index, National Research Register, Zetoc, System for Information on Grey Literature in Europe (SIGLE), Global Health, Latin American and Caribbean Health Sciences Literature (LILACS), Current Controlled Trials, the Cochrane Injuries Group Specialised Register, CENTRAL, MEDLINE and EMBASE to July 2010. Details of our search were published previously. 61 We found two small randomised trials of TXA in TBI with a total of 510 patients. Meta-analysis of the two trials showed a statistically significant reduction (RR 0.63, 95% CI 0.40 to 0.99) in death with TXA. However, given the small size of the trials, we considered this evidence to be hypothesis generating requiring confirmation in larger randomised trials.
Added value of this study
The CRASH-3 trial included 9202 TBI patients who were within 3 hours of injury with either a GCS score of ≤ 12 or any intracranial bleeding on CT scan and no major extracranial bleeding. The risk of head injury death was lower with TXA, particularly when patients who had a GCS score of 3 and those with bilateral unreactive pupils at baseline were excluded as prespecified in the statistical analysis plan (RR 0.89, 95% CI 0.80 to 1.00). There was no evidence of any increase in disability among survivors. The risk of vascular occlusive events was similar in both groups.
Implications of all the available evidence
An updated search for randomised trials of the early administration of TXA in patients with TBI identified one randomised trial in addition to the CRASH-3 trial. This was a randomised trial of pre-hospital TXA in 967 patients with TBI, which was funded by the US National Institutes of Health and sponsored by the University of Washington. The dose of TXA was the same as in the CRASH-3 trial and it also excluded patients with a GCS score of 3 and those with unreactive pupils at baseline. When the two trials are pooled (Figure 11), there is a reduction in head injury death with TXA (RR 0.89, 95% CI 0.80 to 0.99) and no evidence of an increased risk in vascular occlusive events (RR 0.89, 95% CI 0.71 to 1.13) or seizures (RR 1.08, 95% CI 0.89 to 1.31). When the results of all available randomised trials are combined there is a reduction in head injury death with TXA (RR 0.88, 95% CI 0.79 to 0.97). Early administration of TXA should be considered in patients with TBI.
FIGURE 11.
Summary of (a) previous evidence and (b) current evidence on the effect of TXA on head injury death.
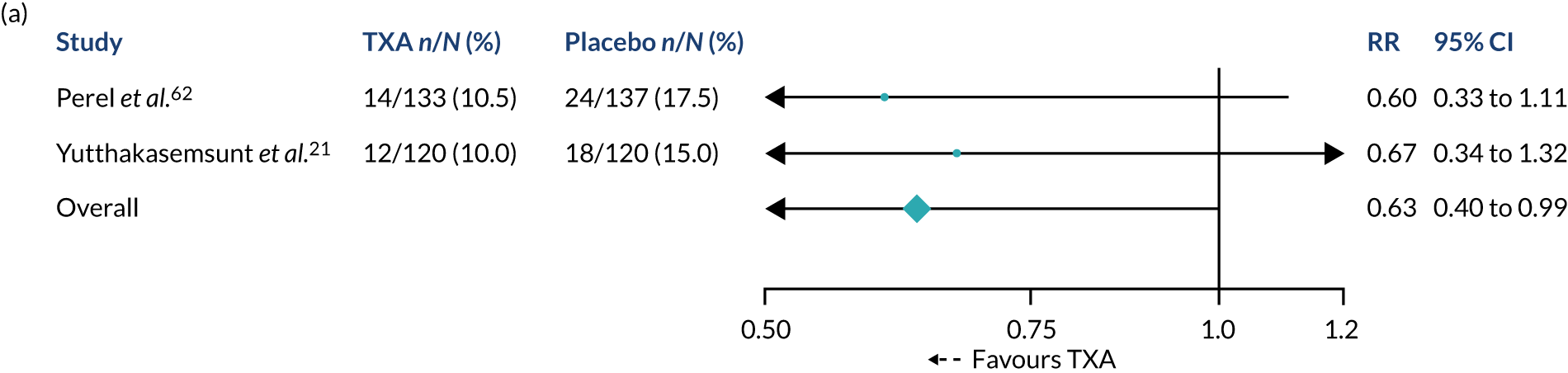
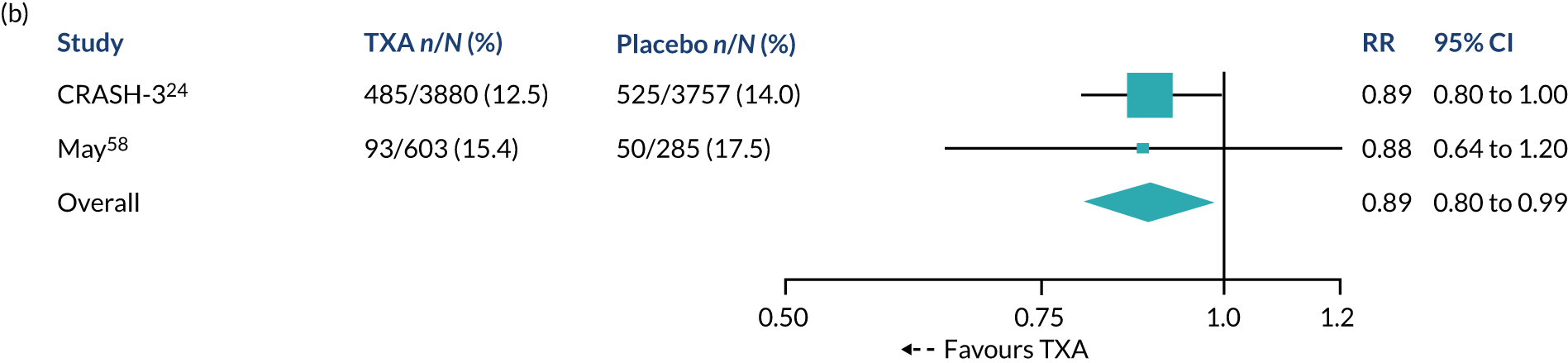
Implications for practice in the NHS
Based on the CRASH-218 trial results, TXA was included in guidelines for the pre-hospital care of trauma patients. Box 1 shows the TXA trauma guideline from the Joint Royal Colleges Ambulance Liaison Committee (JRCALC). 63
Treatment of known or suspected severe traumatic internal or external haemorrhage as soon as clinically possible on arrival at the scene and within 3 hours of bleeding starting in adults and children who are considered to be at risk of significant haemorrhage. This may be demonstrated by one or more of:
-
SBP of < 90 mmHg or absent radial pulse or heart rate of > 110 b.p.m. believed to be due to bleeding in adults. In children this may be demonstrated by changes in the normal physiological parameters for age (see Joint Royal Colleges Ambulance Liaison Committee page for age).
-
Any patient where haemostatic gauze, arterial tourniquet(s), chest dressing(s) or pressure dressing(s) have been applied.
-
Patient who has suffered a traumatic cardiac arrest.
Contraindications
-
Known previous anaphylactic reaction to TXA.
-
Bleeding started > 3 hours ago.
-
Obvious resolution of haemorrhage.
-
Isolated head injury.
-
Critical interventions required [must be given only after critical interventions have been performed (i.e. airway managed, control or splinting of major haemorrhage, etc.), and if administration does not delay transfer, noting that it may be administered en route].
b.p.m., beats per minute.
As can be seen, patients with isolated TBI are specifically excluded. The CRASH-3 trial provides evidence that TXA is safe in TBI patients and that treatment within 3 hours of injury reduces head injury deaths. 24 In the light of this evidence, the exclusion of patients with isolated TBI from TXA treatment guidelines seems unnecessary.
The effect of TXA on head injury-related death appears to depend on the time interval between injury and the initiation of the trial treatment and on the severity of the TBI. Early treatment of patients with mild (GCS score of 13–15 and intracranial bleeding on baseline CT scan) and moderate head injury seemed to confer the greatest mortality benefit. This finding is consistent with the hypothesis that TXA improves outcomes by reducing intracranial bleeding. Haemorrhage expansion occurs in the hours immediately after injury and, therefore, treatment delay would reduce the potential for TXA to prevent intracranial bleeding. Patients with mild or moderate head injury have the most to gain from TXA treatment because, if intracranial haemorrhage can be prevented, these patients are less likely to die from other life-threatening intracranial pathologies such as generalised brain swelling, which may not be affected by TXA.
However, the need to rapidly treat the large number of patients who attend emergency departments with mild or moderate TBI presents challenges for implementation in the NHS. Each year, about 1.4 million people attend emergency departments in England and Wales with a recent head injury. 64 Around 95% of these patients present with a normal or minimally impaired consciousness level (GCS score of < 12) and are classified as having mild TBI. 64 It is unlikely that all patients attending hospital with mild TBI would be treated with TXA because the inclusion criteria of the CRASH-3 trial included only those patients with mild TBI with evidence of intracranial bleeding on their CT scan. Although patients with intracranial bleeding on their CT scan represent only about 5–10% of patients with mild TBI, this is still a large number of patients. The indications for TXA treatment in mild TBI are clearly a matter for discussion between clinicians and policy-makers and will need to take into account considerations of practicality and cost-effectiveness.
Implications for research in the NHS
The CRASH-217 and CRASH-324 clinical trials have shown that i.v. administration of TXA significantly reduces mortality in trauma patients; however, patients must be treated urgently. Many deaths occur on the day of the injury and treatment delay reduces the survival benefit from TXA. Immediate TXA treatment improves survival but the treatment benefit decreases by about 10% for every 15 minutes of treatment delay until 3 hours, after which there is no benefit. 19 To reduce delay, TXA is increasingly given by paramedics at the scene of injury. Trauma audit data for England and Wales (2016) show that when TXA is given by paramedics the median time to treatment is 50 minutes, compared with 110 minutes when TXA is given in hospital. 65
One of the main obstacles to further reducing treatment delay is the need for an i.v. injection. Securing i.v. access at the injury scene can be difficult, particularly for trapped patients. Moreover, on-scene i.v. cannulation increases pre-hospital times, thus delaying definitive surgical control of bleeding. If TXA could be given by intramuscular (i.m.) injection, this might reduce the time to TXA treatment and pre-hospital times. It would also facilitate the more rapid treatment of the large number of patients with mild TBI seen in UK emergency departments. If, for example, mild TBI patients could be rapidly triaged to identify those who would benefit from TXA treatment, nursing staff could administer an i.m. injection of TXA while the patient was waiting to see an emergency physician. Although absorption of TXA from muscle tissue would involve some delay, the available pharmacokinetic data suggest that an immediate i.m. injection might achieve therapeutic TXA levels faster than a delayed i.v. injection. As TXA has a wide therapeutic index, i.m. TXA injection can be followed by an i.v. injection.
The British military also has a strong interest in i.m. TXA use in trauma and is in the early stages of developing an i.m. TXA auto-injector for combat use. 66 An easy-to-use TXA auto-injector would allow soldiers to administer i.m. TXA to themselves or their colleagues as soon as possible after wounding to maximise survival. Such a device could also have implications for civilian trauma, particularly mass casualty events (the UK incidence is three or four events per year67), as it would facilitate rapid treatment of a large number of trauma patients. An easy-to-use auto-injector would also raise the possibility of use by non-medical first aiders.
Studies of i.m. TXA in healthy volunteers show that therapeutic levels (plasma TXA > 10 mg/l) are reached within 30 minutes of i.m. injection of 500 mg of TXA. 68 Administration of 1000 mg (the dose used in trauma) would achieve therapeutic levels even sooner. 69 If absorption was as rapid in trauma patients, this would strongly suggest the i.m. route as an alternative to i.v. use. The main uncertainty is the impact of bleeding on muscle absorption of TXA. Acute blood loss leads to compensatory cardiovascular responses that maintain blood flow to the vital organs at the expense of the peripheral tissues. 70 Skin and skeletal muscle are major targets for these responses, with significant reductions in muscle blood flow. This could reduce the rate of absorption of TXA from muscle. Studies of i.m. atropine in animal shock models show that hypovolaemia significantly reduces absorption, although the reductions are modest (10 minutes). 71 In most cases, on-scene i.m. injection would be given before shock onset, as only the most severely bleeding patients have early shock and shock is rare in patients with isolated TBI. Furthermore, because low-risk patients greatly outnumber high-risk patients, early treatment of low-risk patients prevents more deaths. To resolve this uncertainty, studies of the pharmacokinetics of i.m. TXA in a spectrum of trauma patients to assess the time taken to reach therapeutic levels would be a research priority.
To determine whether i.m. TXA has the potential to improve the care of trauma patients, we need to understand the pharmacokinetics of TXA following i.m. use. If we find that i.m. TXA is well absorbed, with therapeutic TXA levels achieved in a timely manner, i.m. TXA would provide a rapid alternative to i.v. use when immediate i.v. administration is not possible. This would expand the treatment options available to UK paramedics at the scene of a crash and facilitate the development of a TXA auto-injector for use on the battlefield and during mass casualty events. It would also facilitate early treatment of the large numbers of patients with mild or moderate TBI seen in UK emergency departments. Because TXA safely reduces mortality after trauma, this research would provide immediate benefits to patients.
Chapter 7 Dissemination
A dissemination plan and a detailed communication strategy were written to guide the dissemination of the study. These documents expressed the goals of dissemination, identified target audiences and credible messengers, developed key messages and set out the activities that we planned to undertake. See Appendix 7 for the dissemination plan.
Audiences
Stakeholder mapping was used to assess the power, influence and interest of each stakeholder. Neurosurgeons involved in the trial, emergency medicine consultants and paramedics working in high-income countries emerged as the priority target audiences for the first stage of dissemination activity. This process helped to hone the communication strategy and ensured that resources were allocated efficiently to maximise impact.
Messengers
Informal interviews were conducted with medical practitioners in the UK to understand where they typically access information on medical research and what sources they view as respected and credible. Interview respondents described the difficulty of keeping up to date with the large volume of new research being published. Interviewees explained that they increasingly relied on informal, online sources of medical information rather than on journal articles. One online source highlighted was FOAMed (Free Open Access Medical education), a collection of open access medical education resources. Contributors to these informal education resources summarise and appraise important journal articles and present the information in a variety of formats including blogs, podcasts and videos.
Mediums
Publications and conferences
The trial results were published on an open access basis in The Lancet. 24 The results were also presented at two large international conferences72,73 on the same date to coincide with the publication of the journal article.
Media
A list of the key online, print and broadcast media outlets based in the UK and the USA was compiled. Selected journalists were given 5 days’ notice of the press release. The press release was issued to other media outlets 3 days in advance of the results paper. The press were provided with a media pack comprising a quote sheet, statistics on the impact of TBI in the UK and globally, and an animation explaining the trial and study results. Two short films were also produced: one featuring interviews with the trial chief investigator, a trial neurosurgeon and a trial participant, and the other focusing solely on the experiences of the trial participant. High-resolution stills from the films were provided.
Furthermore, in order to encourage content production, journalists were offered access to a large London trauma hospital for filming, interviews with one of the study participants and the trial investigators, and B-roll footage.
Social media
A social media pack, containing suggested tweets, newsletter copy and Facebook (Facebook, Inc., Menlo Park, CA, USA) and Instagram (Facebook, Inc., Menlo Park, CA, USA) posts, was shared with trial sites and charity collaborators. The study funders, which have a large social media following and influence, were given advance notice of the results to enable them to plan their social media activity and to sign off on the branding. FOAMed medical influencers also had advance notice of the results and access to the trial investigators for questions and interviews.
Out-takes
Press coverage
Online
Between 1 September 2019 and 24 October 2019, CRASH-3 had more than 500 mentions across online global news sources. The majority of mentions were from UK-based media sites, with just over 10% from US media sources.
Most media sites cross-posted the CRASH-3 team press release or used the resources that we provided. The British Broadcasting Corporation (BBC) conducted its own interviews, including one with a study participant.
Although there was a lot of mainstream digital media coverage of CRASH-3, including in The Guardian, the Independent and the Daily Mail, a number of the articles with broader reach were in science-specific publications such as Medical News Today, WebMD and BBC Health.
Unexpectedly, a number of regional papers also featured the story. Although they have a lower reach individually, cumulatively this resulted in broad coverage.
Broadcast media
There were 86 broadcast pieces featuring CRASH-3 between 1 September and 24 October 2019. The majority were covered by the BBC as well as various radio stations in the USA, the Pakistani news channel City 42 Pakistan and the Kurdish news channel Rudaw.
Social media
Between 1 October 2019 and 31 October 2019, there were over 2000 mentions of CRASH-3 on Twitter (Twitter, Inc., San Francisco, CA, USA). Over one-quarter of the mentions originated from UK sources (28%), followed by the USA (12%) and Canada (10%).
Twitter activity from The Lancet and the Department for International Development achieved the highest reach. The Wellcome Trust, the National Institute for Health Research and the MRC, which funded the study, also featured in the top 10 posters by reach. FOAMed channels were particularly successful in achieving a large and targeted reach. The Resus Room podcast, which is a FOAMed site, had > 16,000 downloads of their podcast on CRASH-3.
What worked well
Advance notice of results
Certain media contacts, including the BBC, the study funders and FOAMed medical influencers, were given advance notice of the trial results. This allowed them to plan media and social media content and share the results on our behalf:
CRASH 3 has been our most popular episode ever (approximately 10% greater than previous, which is a jump from what we expect) . . . From experience, advertising the paper prior to publication in the way that was done makes a large impact on its reach and it is something that we plan to continue with future papers that we will be covering.
Reproduced with permission from Simon Laing, The Resus Room, 2021, personal communication
Know your audience
Interviews with medical professionals during the development of the communication strategies identified FOAMed as a credible information source for our key audiences. Owing to their large social media following, FOAMed contributors proved to be our most successful targeted route for dissemination. As well as access to the results paper prior to the publication of the article, medical influencers were also given complete creative and intellectual freedom on the interpretation of the results.
Flexible content
A variety of resources were developed, including short videos, photographs, infographics and animations, allowing content to be shared through different channels. Permission was also given to adapt the resources, making it much easier for busy communications teams to work with and share our material. UK Aid Direct (© 2019 UK Aid Direct) made a branded version of one of the videos that we produced to tie in with an ongoing campaign that it was running. In addition, by offering a filming site and interviews with a study participant and trial investigators, we supported others to develop their own content.
What we learned
Choose content carefully
An animation explaining the trial results was created to support the dissemination of the study; however, the animation was costly to produce and generated little engagement. The infographics, on the other hand, were inexpensive to make and were shared widely. Owing to the long lead time required for producing an animation, it was commissioned prior to knowledge of the trial results. Consequently, the messaging was not as strong as we would have liked. It is possible that a high-tech video explaining the mechanism of action of TXA would have been a more successful angle.
Consider patient case studies for dissemination as part of trial design
The BBC interviewed one of the study participants as part of its coverage of the trial. This provided the human interest element to the story and was well received. Studies should consider cultivating a case study portfolio of trial participants who would be interested in speaking about their experiences. This could be achieved by giving study participants the option, in the participant information sheet, to opt in to communications from the trial team.
Make the most of collaborators
Most hospitals have communications teams that are keen to share information relating to research in which they have been involved. Support from communications teams to share trial results on social media can be helpful to target messages to relevant local and regional hospital staff and decision-makers.
Debate can be good
There was a lack of consensus among FOAMed contributors on the conclusions that were drawn from the results of the trial. This, in turn, generated further discussion, debate and social media activity. The CRASH-3 team did not directly engage in these debates, but several of our trial collaborators who are active on social media responded to the comments and questions tweeted. Individuals who are respected in their field, active on Twitter and defenders of your work, can become your social media champions.
Think outside the box
Although it is important to utilise existing tried-and-tested dissemination approaches, this field benefits from a willingness to innovate, take risks and try new things. Gamifcation is an area of growing interest in medical education. One idea we are exploring is designing a mobile app-based (application-based) game aimed at medical practitioners to explain the mechanism of action of TXA.
Chapter 8 Reflections and concluding remarks
The CRASH-3 trial found that a low-cost, widely available drug reduces death after TBI by up to 20%, depending on the severity of injury. 24 The CRASH-3 trial is the largest clinical trial in TBI and the first to identify a safe and effective neuroprotective drug. If widely implemented, TXA could prevent over 100,000 deaths each year worldwide. The CRASH-3 trial builds on the success of the CRASH-2 trial,17 which showed that TXA reduces deaths in traumatic extracranial bleeding. Based on the CRASH-2 trial results, TXA was included on the WHO Model List of Essential Medicines74 and incorporated into trauma treatment guidelines worldwide. The CRASH-2 trial was considered by RAND Europe as providing an excellent return on the research investement. 75
Obtaining funding support for the CRASH-3 trial was not straightforward, with applications to the MRC and HTA initially rejected. Commissioners’ concerns, which in the authors’ opinion were unfounded, included the potential for bias in a study with many hospitals; the challenge of obtaining participant consent; value for money of initial proposals; and complexity of managing international trials. The authors believe that proper randomisation, placebo control, complete follow-up and objective outcomes (e.g. death) avoid bias and that unconscious patients with life-threatening emergencies are an exception to the general rule of patient informed consent. As regards value for money, the authors believe that providing reliable and definitive answers in a large adequately powered trial provides much better value for money than by conducting many smaller trials over a longer period. Fortunately, the successful international pilot phase involving over 1000 patients, funded by the JP Moulton Charitable Trust, demonstrated the feasibility of the CRASH-3 approach. The CRASH-218 and CRASH-3 trials show that early treatment with TXA safely reduces mortality in low-, middle- and high-income countries. There is no evidence that the effects of TXA vary by a country’s income level. NHS patients were the first to benefit from the results of these global trials. Even sooner than this, the British Army incorporated TXA into combat care treatment protocols, resulting in a demonstrable reduction in combat deaths. 76 The authors believe that it is more efficient to conduct adequately powered international trials that provide reliable answers for patients everywhere than to conduct smaller or less efficient trials within the borders of the UK.
Although recruitment was rapid, we extended the trial for scientific reasons. New research had suggested that the recruitment window of 8 hours was too long and that it should be shortened to 3 hours. The protocol was amended accordingly. This substantially reduced recruitment; however, UK research nurses and international collaborators worked hard to ensure that few eligible patients were missed and that patients were randomised and treated urgently. Although UK research nurses were critical to the success of the trial, many hospitals have no research nurse cover at night and weekends, periods when trauma is most common, and this resulted in reduced recruitment.
Trials have become more expensive. The CRASH-218 trial (20,210 patients) cost approximately £2M; however, 10 years later, the CRASH-3 trial (12,737 patients) cost approximately twice this amount. One reason for this increase in cost is burgeoning clinical trial bureaucracy. To conduct a multinational trial, approval must be obtained from the competent authority and National Ethics Committee of each participating country. In the European Union (EU), the Clinical Trials Directive was introduced to simplify and harmonise the administrative processes around clinical trials. 77 The Directive stated that each Member State should:
-
require a standard list of documents for review of a trial
-
accept the English language in their communications with applicants and for documentation that is not aimed at the public or the trial participant
-
provide an opinion on the trial within a maximum of 60 days.
Hospitals from 15 EU countries expressed an interest in taking part in the CRASH-3 trial. Seven of these countries required documents that are not part of the EU Directive standard list. Six countries required the full application form in their local language (i.e. Croatia, France, Greece, Italy, Lithuania and Portugal) and seven countries required the Investigational Medicinal Product labelling in their local language (i.e. Belgium, the Czechia, Greece, Hungary, Latvia, Lithuania and Portugal). Of the seven countries where competent authority approval was obtained, Belgium and the UK were the only two that complied with the 60-day timeline. For the other five countries (i.e. Ireland, Italy, Romania, Slovenia and Spain), the review time ranged between 64 and 291 days.
Of the six countries in which national ethics committee review approval was obtained, only two complied with the 60-day review timeline (i.e. Spain and the UK). In the other countries (i.e. Ireland, Italy, Romania and Slovenia), the review times ranged between 114 and 293 days. The cost of ethical review varied widely, with the Czechia charging £1162 for up to 10 sites and £116 for each additional site. There was no charge in the UK.
The sponsor global insurance policy (a worldwide policy excluding the USA) did not meet with the requirements of some EU countries, including Belgium, Germany and Italy. As there was no budget for additional insurance, we could not run the trial in Germany. In Italy, the investigators institutional ethics committee (Comitato Etico della Provincia di Brescia) found and paid for insurance. In Belgium, the investigators’ institutional ethics committee (Ethisch Comite UZ Gent) suggested that we delegate the responsibility for insurance to it. These responses were commendable and show the commitment to patient-relevant research.
With the implementation of the EU Clinical Trials Directive, it was anticipated that the approvals process throughout the EU would be streamlined and standardised. However, the additional costs for translation and review, and delays in obtaining approvals added to the costs of the trial.
We are grateful to the UK taxpayer for the opportunity to conduct this trial. We believe that the results will improve the care of patients with TBI in the UK and worldwide and we sincerely hope that, like the CRASH-218 trial, the CRASH-3 trial will be seen as providing a good return on the research investment. The trial team will continue to work with policy-makers to ensure that patients benefit.
Acknowledgements
We would like to acknowledge and thank the significant contribution made by our oversight committees in guiding the trial.
Trial Steering Committee
Peter Sandercock (Chairperson), Henry Benjamin Hartzenberg, Manjul Joshipura (2011–16), Amy Aeron-Thomas (Patient Representative), Ian Roberts, Pablo Perel and Haleema Shakur-Still.
Data Monitoring Committee
Michael J Clarke (Chairperson), Samuel C Ohaegbulam, Anthony Rodgers and Tony Brady (Independent Statistician).
We would especially like to thank the patients and their families who had to make the decision in a critical emergency to take part in this trial.
Contributions of authors
Ian Roberts (https://orcid.org/0000-0003-1596-6054) (Professor of Epidemiology and Public Heath) was involved in the design, conduct, analysis and reporting phases of the study.
Haleema Shakur-Still (https://orcid.org/0000-0002-6511-109X) (Professor of Global Health Clinical Trials) was involved in the design, conduct, analysis and reporting phases of the study.
Amy Aeron-Thomas (https://orcid.org/0000-0002-2379-5833) (Advocacy and Justice Manager, RoadPeace) was involved in the design, conduct, analysis and reporting phases of the study.
Danielle Beaumont (https://orcid.org/0000-0002-2530-9608) (Senior Trial Manager/Research Fellow) was involved in the conduct and reporting phases of the study.
Antonio Belli (https://orcid.org/0000-0002-3211-9933) (Professor of Trauma Neurosurgery) was involved in the design, conduct, analysis and reporting phases of the study.
Amy Brenner (https://orcid.org/0000-0003-2017-5994) (Research Fellow in Epidemiology) was involved in the analysis and reporting phases of the study.
Madeleine Cargill (https://orcid.org/0000-0001-9050-877X) (Data Assistant) was involved in the conduct phase of the study.
Rizwana Chaudhri (https://orcid.org/0000-0002-5428-3988) (Dean and Professor of Obstetrics and Gynaecology) was involved in the conduct and reporting phases of the study.
Nicolas Douglas (https://orcid.org/0000-0002-2921-4198) (Research Fellow) was involved in the design phase of the study.
Lauren Frimley (https://orcid.org/0000-0002-2478-348X) (Trial Manager/Research Assistant) was involved in the conduct and reporting phases of the study.
Catherine Gilliam (https://orcid.org/0000-0003-4010-7139) (Trial Administrator) was involved in the conduct phase of the study.
Amber Geer (https://orcid.org/0000-0001-5126-1436) (Assistant Data Manager) was involved in the conduct phase of the study.
Zahra Jamal (https://orcid.org/0000-0002-3817-6795) (Trial Assistant) was involved in drafting this report.
Rashid Jooma (https://orcid.org/0000-0001-6627-3245) (Professor of Neurosurgery) was involved in the conduct and reporting phases of the study.
Raoul Mansukhani (https://orcid.org/0000-0002-9456-5859) (Research Fellow in Medical Statistics) was involved in the analysis and reporting phases of the study.
Alec Miners (https://orcid.org/0000-0003-1850-1463) (Associate Professor in Health Economics) was involved in the analysis and reporting phases of the study.
Jason Pott (https://orcid.org/0000-0003-2068-0560) (Lead Research Nurse for Emergency Medicine) was involved in the conduct and reporting phases of the study.
Danielle Prowse (https://orcid.org/0000-0002-7470-4823) (Assistant Data Manager) was involved in the conduct phase of the study.
Temitayo Shokunbi (https://orcid.org/0000-0002-7819-6862) [Professor of Anatomy (joint appointment with surgery) and Consultant Neurological Surgeon] was involved in the conduct and reporting phases of the study.
Jack Williams (https://orcid.org/0000-0002-1331-387X) (Research Fellow in Health Economics) was involved in the analysis and reporting phases of the study.
All authors made substantial contributions to conception and design of the study, or the acquisition, analysis and interpretation of data. All authors were involved in the drafting of the manuscript or revising it critically, and all authors approved the final version to be published.
Publications
The CRASH-3 trial collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet 2019;394:1713–23.
Brenner A, Belli A, Chaudhri R, Coats T, Frimley L, Jamaluddin SF, et al. Understanding the neuroprotective effect of tranexamic acid: an exploratory analysis of the CRASH-3 randomised trial. Crit Care 2020;24:560.
Mansukhani R, Frimley L, Shakur-Still H, Sharples L, Roberts I. Accuracy of time to treatment estimates in the CRASH-3 clinical trial: impact on the trial results. Trials 2020;21:681.
Williams J, Roberts I, Shakur-Still H, Lecky FE, Chaudhri R, Miners A. Cost-effectiveness analysis of tranexamic acid for the treatment of traumatic brain injury, based on the results of the CRASH-3 randomised trial: a decision modelling approach. BMJ Global Health 2020;5:e002716.
Data-sharing statement
After publication of the planned primary and secondary analyses, the totally anonymised trial data will be made available via our data-sharing portal, the Free Bank of Injury and emergency Research Data (freeBIRD) website (http://freebird.lshtm.ac.uk). Please contact the corresponding author for more information.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg 2018;1:1-18. https://doi.org/10.3171/2017.10.JNS17352.
- Peden N, Scurfield R, Sleet D, Mohan D, Hyder AA, Jarawan E, et al. World Report on Road Traffic Injury Prevention 2004.
- Haydel M. Assessment of Traumatic Brain Injury, Acute. BMJ Best Practice; 2018.
- Perel P, Roberts I, Bouamra O, Woodford M, Mooney J, Lecky F. Intracranial bleeding in patients with traumatic brain injury: a prognostic study. BMC Emerg Med 2009;9. https://doi.org/10.1186/1471-227X-9-15.
- Oertel M, Kelly DF, McArthur D, Boscardin WJ, Glenn TC, Lee JH, et al. Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J Neurosurg 2002;96:109-16. https://doi.org/10.3171/jns.2002.96.1.0109.
- Narayan RK, Maas AI, Servadei F, Skolnick BE, Tillinger MN, Marshall LF. Traumatic Intracerebral Hemorrhage Study Group . Progression of traumatic intracerebral hemorrhage: a prospective observational study. J Neurotrauma 2008;25:629-39. https://doi.org/10.1089/neu.2007.0385.
- Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 2005;365:1957-9. https://doi.org/10.1016/S0140-6736(05)66552-X.
- Rodríguez-Boto G, Rivero-Garvía M, Gutiérrez-González R, Márquez-Rivas J. Basic concepts about brain pathophysiology and intracranial pressure monitoring. Neurologia 2015;30:16-22. https://doi.org/10.1016/j.nrl.2012.09.002.
- Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 2017;80:6-15. https://doi.org/10.1227/NEU.0000000000001432.
- Harhangi BS, Kompanje EJ, Leebeek FW, Maas AI. Coagulation disorders after traumatic brain injury. Acta Neurochir (Wien) 2008;150:165-75. https://doi.org/10.1007/s00701-007-1475-8.
- Bayir A, Kalkan E, Koçak S, Ak A, Cander B, Bodur S. Fibrinolytic markers and neurologic outcome in traumatic brain injury. Neurol India 2006;54:363-5. https://doi.org/10.4103/0028-3886.28106.
- Gebel JM, Jauch EC, Brott TG, Khoury J, Sauerbeck L, Salisbury S, et al. Relative edema volume is a predictor of outcome in patients with hyperacute spontaneous intracerebral hemorrhage. Stroke 2002;33:2636-41. https://doi.org/10.1161/01.str.0000035283.34109.ea.
- Figueroa BE, Keep RF, Betz AL, Hoff JT. Plasminogen activators potentiate thrombin-induced brain injury. Stroke 1998;29:1202-7. https://doi.org/10.1161/01.str.29.6.1202.
- Thiex R, Küker W, Müller HD, Rohde I, Schröder JM, Gilsbach JM, et al. The long-term effect of recombinant tissue-plasminogen-activator (rt-PA) on edema formation in a large-animal model of intracerebral hemorrhage. Neurol Res 2003;25:254-62. https://doi.org/10.1179/016164103101201463.
- Thiex R, Mayfrank L, Rohde V, Gilsbach JM, Tsirka SA. The role of endogenous versus exogenous tPA on edema formation in murine ICH. Exp Neurol 2004;189:25-32. https://doi.org/10.1016/j.expneurol.2004.05.021.
- Henry DA, Carless PA, Moxey AJ, O’Connell D, Stokes BJ, Fergusson DA, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev 2011;1. https://doi.org/10.1002/14651858.CD001886.pub3.
- Shakur H, Roberts I, Bautista R, Caballero J, Coats T, Dewan Y, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23-32. https://doi.org/10.1016/S0140-6736(10)60835-5.
- Roberts I, Shakur H, Afolabi A, Brohi K, Coats T, Dewan Y, et al. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011;377. https://doi.org/10.1016/S0140-6736(11)60278-X.
- Gayet-Ageron A, Prieto-Merino D, Ker K, Shakur H, Ageron FX, Roberts I. Antifibrinolytic Trials Collaboration. Effect of treatment delay on the effectiveness and safety of antifibrinolytics in acute severe haemorrhage: a meta-analysis of individual patient-level data from 40138 bleeding patients. Lancet 2018;391:125-32. https://doi.org/10.1016/S0140-6736(17)32455-8.
- Crash-2 Collaborators (Intracranial Bleeding Study) . Effect of tranexamic acid in traumatic brain injury: a nested randomised, placebo controlled trial (CRASH-2 Intracranial Bleeding Study). BMJ 2011;343. https://doi.org/10.1136/bmj.d3795.
- Yutthakasemsunt S, Kittiwatanagul W, Piyavechvirat P, Thinkamrop B, Phuenpathom N, Lumbiganon P. Tranexamic acid for patients with traumatic brain injury: a randomized, double-blinded, placebo-controlled trial. BMC Emerg Med 2013;13. https://doi.org/10.1186/1471-227X-13-20.
- Dewan Y, Komolafe EO, Mejía-Mantilla JH, Perel P, Roberts I, Shakur H. CRASH-3 Collaborators. CRASH-3 – tranexamic acid for the treatment of significant traumatic brain injury: study protocol for an international randomized, double-blind, placebo-controlled trial. Trials 2012;13. https://doi.org/10.1186/1745-6215-13-87.
- Roberts I, Belli A, Brenner A, Chaudhri R, Fawole B, Harris T. Tranexamic acid for significant traumatic brain injury (The CRASH-3 trial): Statistical analysis plan for an international, randomised, double-blind, placebo-controlled trial. Wellcome Open Res 2018;3. https://doi.org/10.12688/wellcomeopenres.14700.2.
- The CRASH-3 trial collaborators . Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet 2019;394:1713-23. https://doi.org/10.1016/S0140-6736(19)32233-0.
- Weijer C, Shapiro SH, Cranley Glass K. For and against: clinical equipoise and not the uncertainty principle is the moral underpinning of the randomised controlled trial. BMJ 2000;321:756-8. https://doi.org/10.1136/bmj.321.7263.756.
- World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013;310:2191-4. https://doi.org/10.1001/jama.2013.281053.
- Horrow JC, Van Riper DF, Strong MD, Grunewald KE, Parmet JL. The dose–response relationship of tranexamic acid. Anesthesiology 1995;82:383-92. https://doi.org/10.1097/00000542-199502000-00009.
- Rappaport M, Hall KM, Hopkins K, Belleza T, Cope DN. Disability rating scale for severe head trauma: coma to community. Arch Phys Med Rehabil 1982;63:118-23. https://doi.org/10.1037/t29015-000.
- Electronic Medicines Compendium . Tranexamic Acid 100 mg Ml Solution for Injection 2018. www.medicines.org.uk/emc/product/1220/smpc#gref (accessed 28 November 2019).
- Hijazi N, Abu Fanne R, Abramovitch R, Yarovoi S, Higazi M, Abdeen S, et al. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood 2015;125:2558-67. https://doi.org/10.1182/blood-2014-08-588442.
- Medcalf RL. The traumatic side of fibrinolysis. Blood 2015;125:2457-8. https://doi.org/10.1182/blood-2015-02-629808.
- Barton BL. International conference on harmonization-good clinical practices update. Drug Inf J 1998;32:1143-7. https://doi.org/10.1177/00928615980320043410.
- Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandleret JG, et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J Trauma Acute Care Surg 2016;80:16-23. https://doi.org/10.1097/TA.0000000000000885.
- Wu X, Darlington DN, Cap AP. Procoagulant and fibrinolytic activity after polytrauma in rat. Am J Physiol Regul Integr Comp Physiol 2016;310:R323-9. https://doi.org/10.1152/ajpregu.00401.2015.
- Roberts I, Prieto-Merino D, Manno D. Mechanism of action of tranexamic acid in bleeding trauma patients: an exploratory analysis of data from the CRASH-2 trial. Crit Care 2014;18. https://doi.org/10.1186/s13054-014-0685-8.
- Moore EE, Moore HB, Gonzalez E, Chapman MP, Hansen KC, Sauaia A, et al. Postinjury fibrinolysis shutdown: Rationale for selective tranexamic acid. J Trauma Acute Care Surg 2015;78:65-9. https://doi.org/10.1097/TA.0000000000000634.
- Epstein DS, Mitra B, O’Reilly G, Rosenfeld JV, Cameron PA. Acute traumatic coagulopathy in the setting of isolated traumatic brain injury: a systematic review and meta-analysis. Injury 2014;45:819-24. https://doi.org/10.1016/j.injury.2014.01.011.
- Abdelmalik PA, Boorman DW, Tracy J, Jallo J, Rincon F. Acute traumatic coagulopathy accompanying isolated traumatic brain injury is associated with worse long-term functional and cognitive outcomes. Neurocrit Care 2016;24:361-70. https://doi.org/10.1007/s12028-015-0191-0.
- Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet 2004;364:1321-8. https://doi.org/10.1016/S0140-6736(04)17188-2.
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal 2013 (PMG9) 2013.
- Williams J, Roberts I, Shakur-Still H, Lecky FE, Chaudhri R, Miners A. Cost-effectiveness analysis of tranexamic acid for the treatment of traumatic brain injury, based on the results of the CRASH-3 randomised trial: a decision modelling approach. BMJ Global Health 2020;5. https://doi.org/10.1136/bmjgh-2020-002716.
- Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. BMJ 2013;346. https://doi.org/10.1046/j.1365-2958.2001.02411.x.
- McMillan TM, Teasdale GM, Weir CJ, Stewart E. Death after head injury: the 13 year outcome of a case control study. J Neurol Neurosurg Psychiatry 2011;82:931-5. https://doi.org/10.1136/jnnp.2010.222232.
- Office for National Statistics . National Life Tables: UK. 2016–2018 2019. www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/datasets/nationallifetablesunitedkingdomreferencetables (accessed 30 October 2019).
- Ward Fuller G, Hernandez M, Pallot D, Lecky F, Stevenson M, Gabbe B. Health state preference weights for the Glasgow Outcome Scale following traumatic brain injury: a systematic review and mapping study. Value Health 2017;20:141-51. https://doi.org/10.1016/j.jval.2016.09.2398.
- Teasdale G, Maas A, Lecky F, Manley G, Stocchetti N, Murray G. The Glasgow Coma Scale at 40 years: standing the test of time. Lancet Neurol 2014;13:844-54. https://doi.org/10.1016/S1474-4422(14)70120-6.
- Born K, Amsler F, Gross T. Prospective evaluation of the Quality of Life after Brain Injury (QOLIBRI) score: minor differences in patients with major versus no or mild traumatic brain injury at one-year follow up. Health Qual Life Outcomes 2018;16. https://doi.org/10.1186/s12955-018-0966-z.
- Kind P, Hardman G, Macran S. UK Population Norms for EQ-5D. York: Centre for Health Economics, University of York; 1999.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2019.
- National Institute for Health and Care Excellence (NICE) . Costing Statement: Needle and Syringe Programmes 2014.
- Guerriero C, Cairns J, Perel P, Shakur H, Roberts I. CRASH 2 trial collaborators . Cost-effectiveness analysis of administering tranexamic acid to bleeding trauma patients using evidence from the CRASH-2 trial. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0018987.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2018. Canterbury: Personal Social Services Research Unit, University of Kent; 2018.
- Department of Health and Social Care (DHSC) . NHS Reference Costs 2017–18 2018.
- Beecham J, Perkins M, Snell T, Knapp M. Treatment paths and costs for young adults with acquired brain injury in the United Kingdom. Brain Inj 2009;23:30-8. https://doi.org/10.1080/02699050802590338.
- Lecky F, Russell W, Fuller G, McClelland G, Pennington E, Goodacre S, et al. The Head Injury Transportation Straight to Neurosurgery (HITS-NS) randomised trial: a feasibility study. Health Technol Assess 2016;20. https://doi.org/10.3310/hta20010.
- Joint Formulary Committee . British National Formulary n.d. https://bnf.nice.org.uk/medicinal-forms/tranexamic-acid.html; 2019 (accessed 24 September 2019).
- Joint Formulary Committee . British National Formulary n.d. https://bnf.nice.org.uk/medicinal-forms/sodium-chloride.html; 2019 (accessed 24 September 2019).
- May S. Prehospital Tranexamic Acid Use for Traumatic Brain Injury (TXA n.d. https://clinicaltrials.gov/ct2/show/NCT01990768 (accessed 19 November 2019).
- Rodgers A, MacMahon S. Systematic underestimation of treatment effects as a result of diagnostic test inaccuracy: implications for the interpretation and design of thromboprophylaxis trials. Thromb Haemost 1995;73:167-71. https://doi.org/10.1055/s-0038-1653746.
- National Institute for Health and Care Excellence (NICE) . Trauma Quality Standard (QS166) 2018.
- Mahmood A, Roberts I, Shakur H, Harris T, Belli A. Does tranexamic acid improve outcomes in traumatic brain injury?. BMJ 2016;354. https://doi.org/10.1136/bmj.i4814.
- Perel P, Al-Shahi Salman R, Kawahara T, Morris Z, Prieto-Merino D. CRASH-2 (Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage) intracranial bleeding study: the effect of tranexamic acid in traumatic brain injury – a nested randomised, placebo-controlled trial. Health Technol Assess 2012;16. https://doi.org/10.3310/hta16130.
- Joint Royal Colleges Ambulance Liaison Committee, Association of Ambulance Chief Executives . JRCALC Clinical Guidelines 2019 2019.
- National Institute for Health and Care Excellence (NICE) . Head Injury: Assessment and Early Management 2019.
- Coats TJ, Fragoso-Iñiguez M, Roberts I. Implementation of tranexamic acid for bleeding trauma patients: a longitudinal and cross-sectional study. Emerg Med J 2019;36:78-81. https://doi.org/10.1136/emermed-2018-207693.
- Wright C. Battlefield administration of tranexamic acid by combat troops: a feasibility analysis. J R Army Med Corps 2014;160:271-2. https://doi.org/10.1136/jramc-2013-000152.
- Carley S, Mackway-Jones K, Donnan S. Major incidents in Britain over the past 28 years: the case for the centralised reporting of major incidents. J Epidemiol Community Health 1998;52:392-8. https://doi.org/10.1136/jech.52.6.392.
- Puigdellívol E, Carral ME, Moreno J, Plà-Delfina JM, Jané F. Pharmacokinetics and absolute bioavailability of intramuscular tranexamic acid in man. Int J Clin Pharmacol Ther Toxicol 1985;23:298-301.
- Sano M, Hakusui H, Kojima C, Akimoto T. Absorption and excretion of tranexamic acid following intravenous, intramuscular and oral administrations in healthy volunteers. Rinsho Yakuri Japanese J Clin Pharmacol Ther 1976;7:375-82. https://doi.org/10.3999/jscpt.7.375.
- Haljamäe H. Microcirculation and hemorrhagic shock. Am J Emerg Med 1984;2:100-7. https://doi.org/10.1016/0735-6757(84)90117-7.
- Yost J, Baldwin P, Bellenger S, Bradshaw F, Causapin E, Demotica R, et al. The pharmacokinetics of intraosseous atropine in hypovolemic swine. Am J Disaster Med 2015;10:217-22. https://doi.org/10.5055/ajdm.2015.0204.
- First Release of CRASH-3 Trial Results. Conference Presentation n.d.
- Effects of Tranexamic Acid on Death, Disability, Vascular Occlusive Events and Other Morbidities in Patients With Acute Traumatic Brain Injury (CRASH-3): A Randomised, Placebo-Controlled Trial. Conference Presentation n.d.
- World Health Organization (WHO) . WHO Model List of Essential Medicines 2019.
- Guthrie S, Hafner M, Bienkowska-Gibbs T, Wooding S. Returns on research funded under the NIHR Health Technology Assessment (HTA) programme. Rand Health Q 2016;5.
- Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military application of tranexamic acid in trauma emergency resuscitation (MATTERs) study. Arch Surg 2012;147:113-19. https://doi.org/10.1001/archsurg.2011.287.
- European Council . Directive 2001 20 EC of the European Parliament and of the Council of 4 April 2001 on the Approximation of the Laws, Regulations and Administrative Provisions of the Member States Relating to the Implementation of Good Clinical Practice in the Conduct of Clinical Trials on Medicinal Products for Human Use 2001. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_20/dir_2001_20_en.pdf (accessed 20 November 2020).
- Perel P, Arango M, Clayton T, Edwards P, Komolafe E, Poccock S, et al. Predicting outcome after traumatic brain injury: practical prognostic models based on large cohort of international patients. BMJ 2008;336:425-9. https://doi.org/10.1136/bmj.39461.643438.25.
Appendix 1 CRASH-3 trial organisation
Trial Steering Committee
Peter Sandercock (Chairperson), Henry Benjamin Hartzenberg, Manjul Joshipura (2011–16), Amy Aeron-Thomas (Patient Representative), Ian Roberts, Pablo Perel and Haleema Shakur-Still.
Data Monitoring Committee
Michael J Clarke (Chairperson), Samuel C Ohaegbulam, Anthony Rodgers and Tony Brady (Independent Statistician).
Trial Co-ordinating Centre Team
Nigeria co-ordinating team: Bukola Fawole (Co-ordinating Centre Director), Olusade Adetayo (Assistant Trial Co-ordinator), Olujide Okunade (Assistant Trial Co-ordinator) and Temitayo Shokunbi (Clinical Lead).
Pakistan co-ordinating team: Rizwana Chaudhri (Co-ordinating Centre Director), Kiran Javaid (Assistant Research Co-ordinator), Rashid Jooma (Clinical Lead) and Aasia Kayani (Research Co-ordinator).
National Co-ordinators: Rizwana Chaudhri (Pakistan), Rashid Jooma (Pakistan), Sabariah Faizah Bt Jamaluddin (Malaysia), Julina Md Noor (National Co-ordinator’s Assistant, Malaysia), Tamar Gogichaishvili (Georgia), Maria de los Angeles Munoz-Sanchez (Spain), Bukola Fawole (Nigeria), Temitayo Shokunbi (Nigeria), Jorge Mejia-Mantilla (Colombia), Liliana Vallecilla (Colombia), Fatos Olldashi (Albania), Satish Krishnan (United Arab Emirates), Vincent Djientcheu (Cameroon), Jorge Loria Castellanos (Mexico), Frank Rasulo (Italy), Qadamkhear Hama (Iraq), Yakub Mulla (Zambia), Ioan Stefan Florian (Romania), Juan Tobar (El Salvador), Hussein Khamis (Egypt), Conor Deasy (Ireland), Bobby Wellsh (Papua New Guinea), Jean Williams-Johnson (Jamaica), Susilo Chandra (Indonesia) and Vincent Mutiso (Kenya).
CRASH-3 trial collaborators by country
The number of participants recruited is shown in brackets.
Pakistan (4567). Lahore General Hospital Neurosurgery Unit I (1178): Rizwan Butt, Muhammad Hammad Nasir, Salman Ahmad, Farwah Aslam, Khurram Ishaque, Faheem Usmani, Shahrukh Rizvi, Farhad Ali, Omair Sajjad and Ali Zunair. Jinnah Postgraduate Medical Centre (700): Lal Rehman, Raza Rizvi, Farrukh Javeed, Shakeel Ahmed, Asad Abbas, Ali Afzal and Ali Mikdad. Lahore General Hospital Neurosurgery Unit III (648): Asif Bashir, Anwar Chaudary, Tariq Salahuddin, Bashir Ahemed, Shahrukh Rizvi, Faheem Usmani and Amir Aziz. Jinnah Hospital Lahore (619): Naveed Ashraf, Shahzad Hussain, Usman Ahmad, Muhammad Asif, Muhammad Adil and Adeel Rauf. Lahore General Hospital Neurosurgery Unit II (607): Khalid Mahmood, Rizwan Khan, Bilal Ahmad, Umair Afzal, Hassan Raza and Quratul Ain. District Headquarters Hospital Narowal (303): Sajjad Yaqoob, Qaiser Waseem, Muffasser Nishat, Suneel Semvel and Javed Iqbal. Services Hospital Lahore (226): Samra Majeed, Sana Zulfiqar, Madeeha Iqbal, Nazia Majeed and Manzoor Ahmed. District Headquarters Rawalpindi (137): Nadeem Akhtar, Mohammad Malik, Yasir Shehzad and Muhammad Yousaf. District Headquarters Hospital Khuzdar (65): Abdul Wahid, Abdul Samad and Saifullah Shah. Lady Reading Hospital (31): Mumtaz Ali and Jehan Zeb. Shifa International Hospital (29): Abdus Salam Khan and Adeela Irfan. Liaquat National Hospital and Medical College (14): Salman Sharif. Liaquat University Hospital (7): Riaz Memon. Aga Khan University Hospital (3): Rashid Jooma.
UK (3143). Royal London Hospital (501): Ben Bloom, Tim Harris, Jason Pott, Imogen Skene, Geoffrey Bellhouse and Olivia Boulton. University Hospital Coventry (312): Caroline Leech, Geraldine Ward, Catherine Jarvis, Carly Swann and Sathananathan Ratnam. Queen Elizabeth Hospital Birmingham (302): Antonio Belli, Ronald Carrera, Kamal Yakoub, David Davies and Emma Fellows. St George’s Hospital (280): Phil Moss, Heather Jarman, Sarah Rounding, Elizabeth Johnson and Catherine Loughran. Salford Royal Hospital (176): Fiona Lecky, Kate Clayton, Angiy Michael and Angela Coumbarides. Southmead Hospital (156): Jason Kendall, Beverley Faulkner, Ruth Worner and Emma Gendall. King’s College Hospital (155): Philip Hopkins, Paul Riozzi, Hannah Cotton and Raine Astin-Chamberlain. St Mary’s Hospital, London (117): Mark Wilson, Jan Bodnar, Rachel Williams and Alberto Rigoni. Aintree University Hospital (108): Abdo Sattout, John Fletcher, Calum Edge and Nina Maryanji. Addenbrooke’s Hospital (103): Adrian Boyle, Susie Hardwick, Ellen Nichols and Catherine Hayhurst. Queen’s Medical Centre (100): Frank Coffey, Chris Gough, Philip Miller and Lucy Ryan. John Radcliffe Hospital (76): Melanie Darwent, Alexis Espinosa and Sally Beer. Royal Stoke University Hospital (71): Julie Norton, Holly Maguire and Kay Finney. Derriford Hospital (67): Anthony Kehoe, Rosalyn Squire and Alison Jeffery. Queen Alexandra Hospital (60): Christiane Vorwerk, Denise Foord and Eliot Wilkinson. Northern General Hospital (57): Avril Kuhrt, Shammi Ramlakhan and Stuart Reid. Royal Preston Hospital (41): Andy Curran and Sean McMullan. Leeds General Infirmary (39): Tajek Hassan and Stuart Nuttall. Great Western Hospital (32): Stephen Haig and Saif Al-Nahhas. Southampton General Hospital (31): Diederik Bulters and Ardalan Zolnourian. Dorset County Hospital (27): Tamsin Ribbons and Ian Mew. Gloucestershire Royal Hospital (27): Tanya de Weymarn and Victoria Hughes. Royal Liverpool Hospital (21): Jane McVicar. Queen Elizabeth University Hospital (20): Cieran McKiernan. Royal Berkshire Hospital (20): Liza Keating. Poole Hospital (17): Henrik Reschreiter. James Cook University Hospital (16): Judith Wright. Basingstoke and North Hampshire Hospital (13): Louisa Chan. Whiston Hospital (13): Himanshu Kataria. Glasgow Royal Infirmary (12): Alastair Ireland. Manchester Royal Infirmary (12): Richard Body. Royal Alexandra Hospital (12): Alasdair Corfield. Milton Keynes University Hospital (11): Shindo Francis. Hull Royal Infirmary (10): William Townend. Leicester Royal Infirmary (10): Timothy Coats. Musgrove Park Hospital (10): James Gagg. Wexham Park Hospital (10): Sarah Wilson. Royal Sussex County Hospital (8): Rowley Cottingham. Blackpool Victoria Hospital (7): Simon Tucker. Norfolk and Norwich University Hospital (7): Frank Sutherland. North Devon District Hospital (7): Louisa Mitchell. Whipps Cross University Hospital (7): Tim Harris. Whittington Hospital (7): Lucy Parker. Darlington Memorial Hospital (6): Ola Afolabi. Monklands Hospital (6): Fiona Hunter. Royal Cornwall Hospital (6): Mark Jadav. University Hospital of North Tees (6): Kayode Adeboye. Worthing Hospital (5): Mandy Grocutt. Royal Oldham Hospital (4): Gabrielle May. Royal United Hospitals Bath (4): David Watson. Arrowe Park Hospital (3): Andrea Wootten. Pinderfields General Hospital (3): Sarah Robertshaw. Birmingham Heartlands Hospital (2): Susan Dorrian. Gwynedd Hospital, Bangor (2): Rob Perry. Newham University Hospital (2): Tim Harris. University Hospital Lewisham (2): Hyun Choi. Western Infirmary (2): Claire McGroarty. Worcestershire Royal Hospital (1): Paul Shone. Yeovil District Hospital (1): David Maritz.
Malaysia (1567). Hospital Sungai Buloh (410): Sabariah Jamaluddin, Julina Noor, Norizan Rosli, Leonard Leong Sang Xian and Yong De Jun. Hospital Sultanah Bahiyah (241): Fatahul Mohamed, Cheng Hee Song, Arman Hawari, Leong Yuen Chin and Hardawani Mohd Hussein. Hospital Sultanah Nur Zahirah (205): Mohd Lotfi, Hafiq Hamid, Nujaimin Udin, Peck Lian and See Choo. Penang General Hospital (161): Kwanhathai Wong, Fathiyah Gani, Mardhiah Jusoh and Darrsini Rajakumar. Miri General Hospital (111): Chia Boon Yang, Nur Shahidah Binti Dzulkiflee, Wong Chok Ky and Muhaimin Azwan Bin Mohd Azman. Hospital Raja Permaisuri Bainun (101): Adi Bin Osman, Azma Haryaty Ahmad, Ramzuzaman Ismail and Si Qi Lai. Hospital Sultanah Aminah (94): Mohd Amin Bin Mohidin, Norwani Binti Deraman and Salliza Binti Selamat. Hospital Tuanku Fauziah (72): Ida Abidin, Nurkhairulnizam Halim and Zuraini Bakar. Hospital Tengku Ampuan Afzan (41): Zainalabidin Mohamed Ismail, Badrul Hisham and Ruhaida Kamal. Hospital Sultan Abdul Halim (36): Zainal Effendy and Mashitah Ismail. Hospital Seberang Jaya (30): Noor Azleen and Liu Yeo Seng. Universiti Sains Malaysia (26): Kamarul Aryffin Baharuddin and Regunath Kandasamy. Hospital Langkawi (13): Azlan Kamalludin. Hospital Kulim (8): Shamsul Asmee. Hospital Kemaman (7): Mohd Fadzil. Hospital Segamat (6): Ahmad Basitz. Hospital Pakar Sultanah Fatimah (5): Norhaya Abdullah.
Georgia (771). High Technology Medical Center, University Clinic (751): Tamar Gogichaishvili, Giorgi Ingorokva, Shota Ingorokva, Iamze Agdgomelashvili, Kote Mumladze, Ioseb Maisuradze and Iulia Kugusheva. Archangel St Michael Multiprofile Clinical Hospital (18): Buba Shalamberidze. City Hospital 1 (2): Gia Tomadze.
Spain (425). Hospital Regional Universitario Carlos Haya (102): Juan Fernandez-Ortega, Raimundo Seara-Valero, Guillermo Ibañez-Botella and Victoria Garcia-Martinez. Hospital Alvaro Cunqueiro VIGO (82): Melida Garcia Martul, Santiago Freita Ramos and Guillermo Lago Preciado. Hospital Universitario Virgen del Rocio (77): Claudio Garcia-Alfaro, Angeles Munoz-Sanchez and Rafael Bellido-Alba. Hospital 14 General Universitario de Ciudad Real (67): Carmen Corcobado, Ana Bueno and Alfonso Ambros. Complejo Hospitalario de Navarra (44): Juan Tihista Jimenez and Jose Roldan Ramirez. Hospital Torrecardenas (21): José Martín. Hospital de Lucus Augusti (13): Laura Inés Rodríguez. Hospital Clinico de Barcelona (9): Jaime Fontanals. Hospital Universitario Puerta del Mar de Cadiz (9): José Manuel Jiménez-Moragas. Hospital General Universitario De Albacete (1): Joaquín Paya Berbegal.
Nigeria (409). National Hospital Abuja (64): Olaomi Oluwole, Raji Mahmud and Nancy Ukwu. Lagos University Teaching Hospital (55): Femi Bankole, Abidemi Oseni and Bamidele Adebayo. University College Hospital, Ibadan (53): Adefolarin Malomo, Liadi Tiamiyu and Adefisayo Adekanmbi. Olabisi Onabanjo University Teaching Hospital (38): Lateef Thanni and Ayodeji Olubodun. Federal Medical Centre Abeokuta (36): Fidelis Ojeblenu and Michael Uwaezuoke. Obafemi Awolowo University Teaching Hospitals (31): Edward Komolafe and Oluwafemi Owagbemi. Lagos State Accident and Emergency Centre (22): Fatai Ishola. Bowen University Teaching Hospital Ogbomoso (17): Adewumi Durodola. Federal Medical Centre Lokoja (13): Ukpong Udoffa. Federal Medical Centre Bida (12): Adeniran James. Abubakar Tafawa Balewa University Teaching Hospital (11): Azeez Tella. Irrua Specialist Teaching Hospital (9): Andrew Dongo. Federal Medical Centre Umuahia (8): Uchechi Ekpemiro. Nnamdi Azikiwe University Teaching Hospital (8): Stanley Anyanwu. State Hospital, Ijaiye, Abeokuta (8): Nafiu Aigoro. University of Nigeria Teaching Hospital Enugu (7): Wilfred Mezue. Jos University Teaching Hospital (6): Danaan Shilong. University of Benin Teaching Hospital (6): Abiodun Azeez. Federal Medical Centre Ido-Ekiti (2): Olakunle Babalola. Federal Teaching Hospital, Gombe (2): Mohammed Ibrahim. University of Abuja Teaching Hospital (1): Joseph Obande.
Colombia (335). Hospital Pablo Tobon Uribe (127): Alfredo Constain Franco, Edwin Vasquez Salazar, Sebastian Betancur Londoño and Viviana Medina Cardona. Hospital Universitario San Vicente Fundacion (112): Carlos Morales, Santiago Upegui, Santiago Naranjo and July Agudelo. Fundacion Valle del Lili (96): Jorge Mejia-Mantilla, Sandra Carvajal and Yidhira Fajardo-Gaviria.
Nepal (255). Neuro Hospital (103): Yam Roka, Ushma Ghising, Narayani Roka and Manzil Shrestha. National Institute of Neurological and Allied Sciences (64): Upendra Devkota, Bivek Vaidya and Pankaj Nepal. Kathmandu Medical College Teaching Hospital (47): Amit Thapa and Bidur KC. Chitwan Medical College Teaching Hospital (24): Ajit Shrestha. Bir Hospital (11): Rajiv Jha. B & B Hospital Ltd (6): Prabin Shrestha.
Albania (214). University Hospital of Trauma (214): Fatos Olldashi, Irgen Hodaj, Erion Spaho, Asllan Selaj and Nirian Bendo.
Japan (165). Matsudo City Hospital (64): Tomohisa Shoko, Hideki Endo and Atsushi Senda. Senshu Trauma and Critical Care Centre (61): Yasushi Hagihara, Takashi Fuse and Naohisa Masunaga. Tokyo Medical and Dental University (28): Yasuhiro Otomo and Ryuichiro Egashira. Teikyo University Hospital (12): Takahiro Ohnuki.
The United Arab Emirates (126). Al Qassimi Hospital (126): Satish Krishnan, Alya Al Mazmi, Subrata Saha and Alexander Suvarov.
Myanmar (121). 1000 Bedded Nay Pyi Taw Hospital (121): Than Latt Aung, Kaung Myat Tun, Tint Khaing and Thinzar Maw.
Cameroon (116). Yaounde Central Hospital (38): Vincent Djientcheu and Orlane Ndome. Hopital General Douala (31): Mireille Moumi and André Mbida. Hopital Laquintinie de Douala (28): Joseph Fondop and N’Diaye. Yaounde General Hospital (19): Mba Sebastien.
Afghanistan (87). Nangarhar University Teaching Hospital (87): Abdul Azim, Jan Adil and Zabiullah Amiry.
Mexico (79). Hospital Regional 25 IMSS (24): Jorge Loría-Castellanos. Hospital General Jose G Parres (21): Nancy Guevara Rubio. Hospital General de Uruapan, Dr Pedro Daniel Martinez (11): Patricia Ortega Leon. Hospital General Regional No. 1 (10): Francisco Estrada. Hospital General de Zona 197 15 (8): Erandy Montes de Oca-García. Hospital General Regional Bernardo Sepulveda (3): Hafid Sanchez. Hospital General La Perla (2): Angélica Soria.
Italy (72). Azienda Ospedaliera Universitaria Senese (35): Paola Bonucci and Federico Franchi. Fondazione Poliambulanza (19): Alan Girardini. Spedali Civili Di Brescia (18): Frank Rasulo.
Iraq (55). Rozhawa Emergency Hospital (51): Qadamkhear Hama, Himdad Hameed and Muhammad Basim. Rojhelat Emergency Hospital (3): Qadamkhear Hama. Par Hospital (1): Qadamkhear Hama.
Cambodia (45). World Mate Emergency Hospital (45): Simon Stock and Eap Hourt.
Zambia (44). University Teaching Hospital Lusaka (40): Yakub Mulla and Ali Ilunga. Kitwe Central Hospital (4): Jonathan Mulenga.
Romania (35). Timisoara County Hospital (17): Horia Ples. Spitalul Sf. Pantelimon Bucharest (11): Adam Danil. Bagdasar-Arseni Emergency Clinical Hospital (5): Mircea Gorgan. Cluj County Emergency Hospital (2): Ioan Florian.
El Salvador (28). Hospital Nacional Rosales (28): Juan Tobar Fernandez.
Egypt (20). Mataria Teaching Hospital (20): Hussein Khamis.
Slovenia (15). University Medical Centre Ljubljana (15): Dusan Vlahovic.
Ireland (12). Cork University Hospital (12): Conor Deasy.
Papua New Guinea (10). Port Moresby General Hospital (10): Bobby Wellsh.
Canada (7). Saint John Regional Hospital (7): James French.
Jamaica (7). Cornwall Regional Hospital (5): Jeffrey East. University Hospital of the West Indies (2): Jean Williams-Johnson.
Indonesia (6). Rumah Sakit Sekar Kamulyan (6): Antonius Kurniawan.
Kenya (1). Kenyatta National Hospital, University of Nairobi (1): Julius Kiboi.
Appendix 2 Consent procedure overview
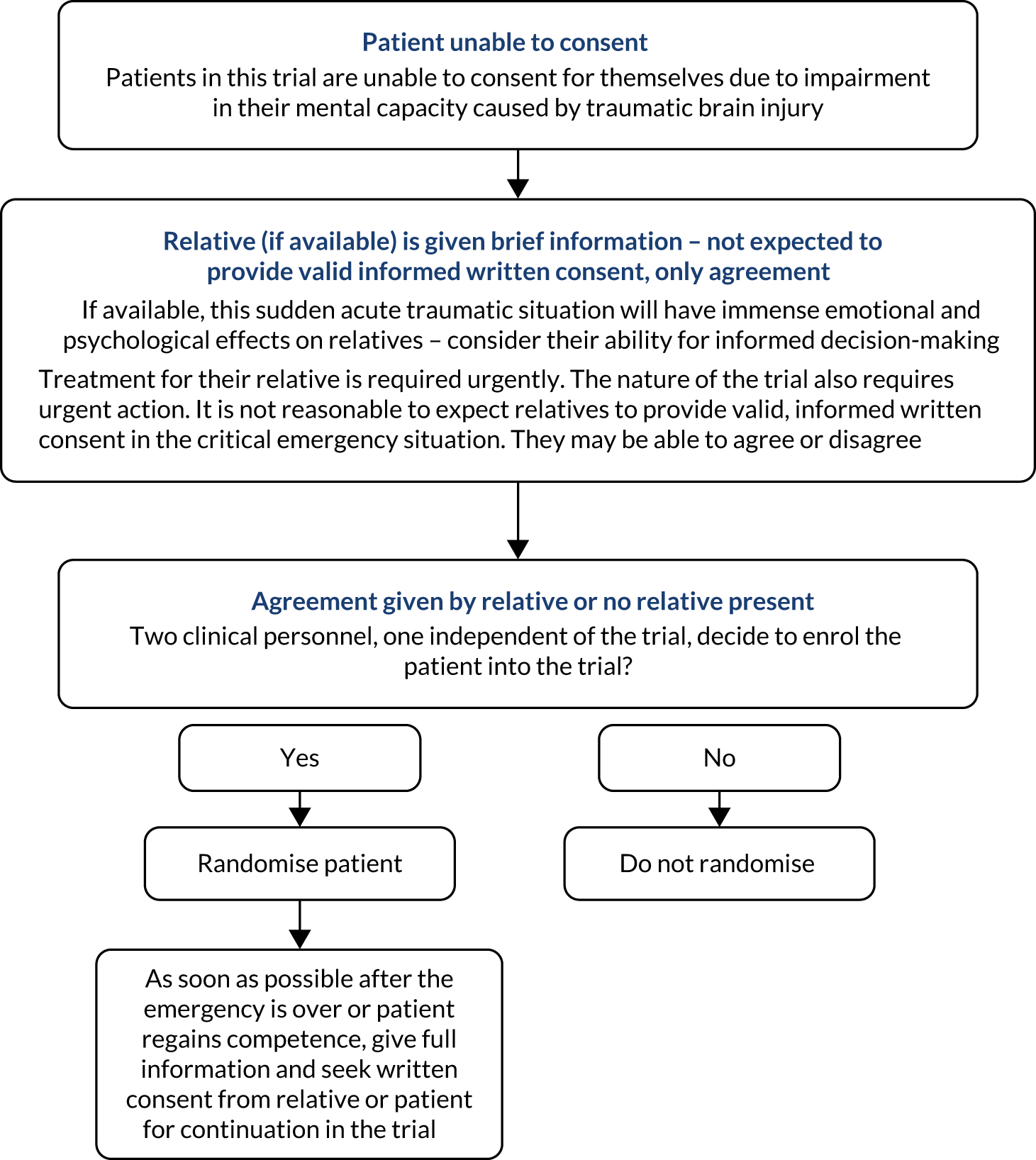
Appendix 3 Total randomisations by geographical region
| Geographical region | TXA (n) | Placebo (n) | Total (N) |
|---|---|---|---|
| Africa | 301 | 289 | 590 |
| Asia | 3905 | 3860 | 7765 |
| Europe, Australia and North America | 2009 | 1993 | 4002 |
| Caribbean, Central and South America | 186 | 184 | 370 |
| Oceania | 5 | 5 | 10 |
| Total | 6406 | 6331 | 12,737 |
Appendix 4 Cumulative incidence of head injury death by treatment group in patients randomised within 3 hours of injury
Figure 12 shows the cumulative incidence of head injury death in the TXA and placebo groups by days since randomisation in all patients randomised within 3 hours of injury. The numbers at risk at time points 0, 7, 14, 21 and 28 days after randomisation are presented in the risk table.
FIGURE 12.
Cumulative incidence plot of the prespecified primary outcome.
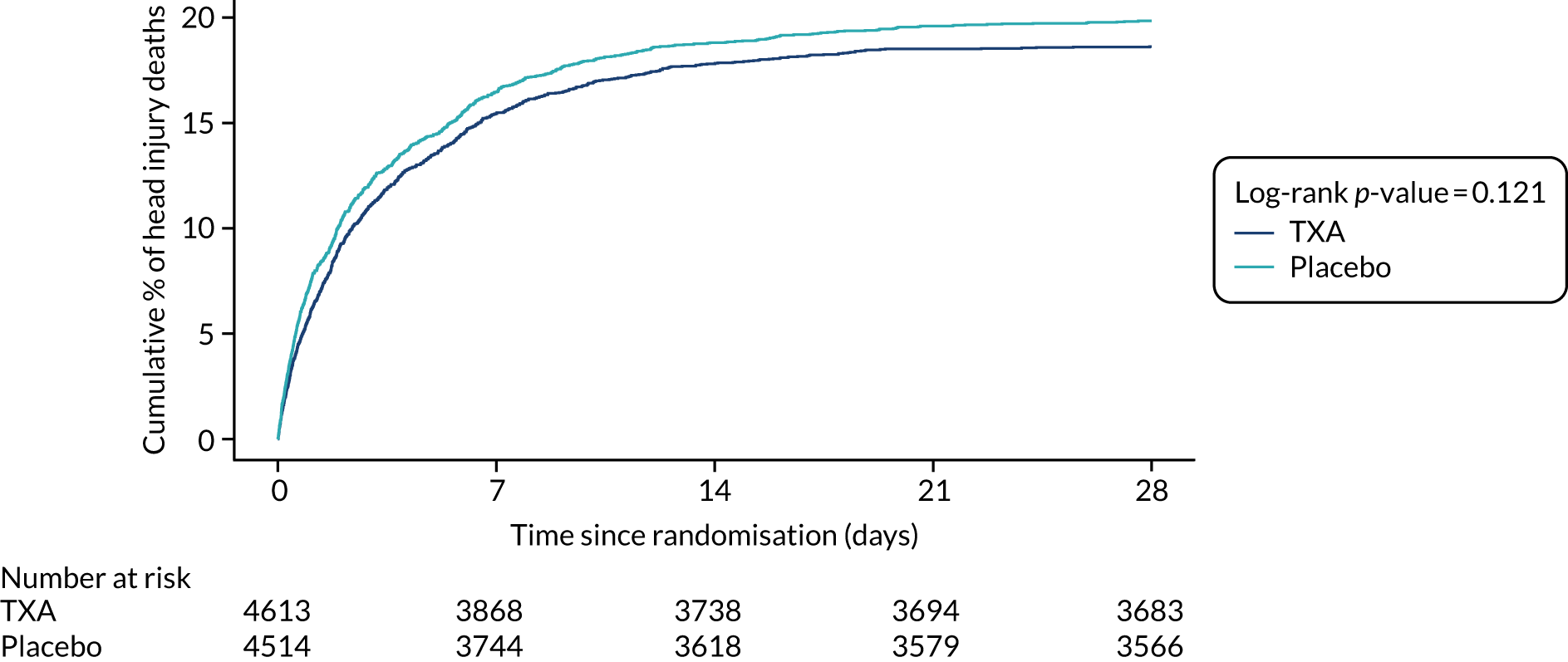
Appendix 5 Adverse events by treatment group in all patients
| Adverse event | TXA (n = 6359) | Placebo (n = 6280) | Total (n = 12,639) |
|---|---|---|---|
| Any adverse event | 198 | 168 | 366 |
| Pneumonia | 51 | 50 | 101 |
| Respiratory infection | 10 | 7 | 17 |
| Fall | 11 | 5 | 16 |
| Urinary tract infection | 9 | 5 | 14 |
| Abnormal liver function tests | 6 | 6 | 12 |
| Allergic reaction | 4 | 5 | 9 |
| Cellulitis | 4 | 4 | 8 |
| Wound infection | 4 | 3 | 7 |
| Atrial fibrillation | 5 | 1 | 6 |
| Headache | 5 | 1 | 6 |
| Pneumothorax | 4 | 2 | 6 |
| Supraventricular tachycardia | 3 | 2 | 5 |
| Cerebral haemorrhage | 1 | 3 | 4 |
| Ileus | 1 | 3 | 4 |
| Pyrexia | 2 | 2 | 4 |
| Urinary retention | 3 | 1 | 4 |
| Cardiac arrest | 3 | 0 | 3 |
| Chest pain | 3 | 0 | 3 |
| Constipation | 1 | 2 | 3 |
| Haemothorax | 1 | 2 | 3 |
| Heart block | 1 | 2 | 3 |
| Infection – MRSA | 1 | 2 | 3 |
| Intracranial venous sinus thrombosis | 1 | 2 | 3 |
| Meningitis | 1 | 2 | 3 |
| PE | 3 | 0 | 3 |
| Respiratory failure | 0 | 3 | 3 |
| Acute respiratory distress syndrome | 1 | 1 | 2 |
| Anaemia | 0 | 2 | 2 |
| Atrial flutter | 1 | 1 | 2 |
| Cerebral haematoma | 2 | 0 | 2 |
| Clostridium difficile infection | 2 | 0 | 2 |
| Diarrhoea | 2 | 0 | 2 |
| Epilepsy | 0 | 2 | 2 |
| Gangrene | 2 | 0 | 2 |
| Hypertension | 0 | 2 | 2 |
| Hypokalaemia | 1 | 1 | 2 |
| Intestinal pseudo-obstruction | 1 | 1 | 2 |
| Ischaemic stroke | 2 | 0 | 2 |
| Neutropenia | 2 | 0 | 2 |
| Pancreatitis | 1 | 1 | 2 |
| Rash | 1 | 1 | 2 |
| Respiratory arrest | 1 | 1 | 2 |
| Seizure | 1 | 1 | 2 |
| Sepsis | 1 | 1 | 2 |
| Thrombocytopenia | 2 | 0 | 2 |
| Thrombocytosis | 1 | 1 | 2 |
| TBI | 1 | 1 | 2 |
| Unintended unilateral bronchial intubation | 1 | 1 | 2 |
| Wound dehiscence | 0 | 2 | 2 |
| Abdominal compartment syndrome | 1 | 0 | 1 |
| Abdominal distension | 0 | 1 | 1 |
| Acute alcoholic intoxication | 1 | 0 | 1 |
| Agitation | 1 | 0 | 1 |
| Atelectasis | 0 | 1 | 1 |
| Bacteraemia | 0 | 1 | 1 |
| Bowel obstruction | 1 | 0 | 1 |
| Bradycardia | 0 | 1 | 1 |
| Central line infection | 1 | 0 | 1 |
| Cerebral salt-wasting syndrome | 1 | 0 | 1 |
| Cerebrospinal fluid leakage | 0 | 1 | 1 |
| Cerebrospinal infection | 1 | 0 | 1 |
| Cervical pain | 0 | 1 | 1 |
| Corneal ulcer | 1 | 0 | 1 |
| Cranial nerve palsies multiple | 0 | 1 | 1 |
| Cranial nerve paralysis | 1 | 0 | 1 |
| Depression | 1 | 0 | 1 |
| Diabetic ketoacidosis | 1 | 0 | 1 |
| Electrocardiographic signs of myocardial ischaemia | 0 | 1 | 1 |
| Eye injury | 1 | 0 | 1 |
| Facial palsy | 0 | 1 | 1 |
| Foot drop | 0 | 1 | 1 |
| Fractured zygomatic arch reduction | 0 | 1 | 1 |
| Haematoma | 1 | 0 | 1 |
| Haematuria | 0 | 1 | 1 |
| Haemophilus influenza pneumonia | 0 | 1 | 1 |
| Herpes zoster infection | 1 | 0 | 1 |
| Hip dislocation | 0 | 1 | 1 |
| Humerus fracture | 1 | 0 | 1 |
| Hydrocephalus | 0 | 1 | 1 |
| Hyperbilirubinaemia | 1 | 0 | 1 |
| Hypernatraemia | 1 | 0 | 1 |
| Hyponatraemia | 1 | 0 | 1 |
| Hypotension | 1 | 0 | 1 |
| Hypothermia | 0 | 1 | 1 |
| Jaw pain | 1 | 0 | 1 |
| Laceration of head | 1 | 0 | 1 |
| Laryngopharyngitis | 1 | 0 | 1 |
| Leg pain | 0 | 1 | 1 |
| Liver failure | 0 | 1 | 1 |
| Metabolic encephalopathy | 1 | 0 | 1 |
| Necrotising fasciitis | 0 | 1 | 1 |
| Neuroleptic malignant syndrome | 0 | 1 | 1 |
| Obstructive jaundice | 0 | 1 | 1 |
| Overdose | 0 | 1 | 1 |
| Painful urination | 0 | 1 | 1 |
| Paraesthesia | 1 | 0 | 1 |
| Pleural effusion | 0 | 1 | 1 |
| Post-procedural infection | 0 | 1 | 1 |
| Psychotic episode | 1 | 0 | 1 |
| Pulmonary haemorrhage | 1 | 0 | 1 |
| Pulmonary oedema | 0 | 1 | 1 |
| Rectal bleeding | 1 | 0 | 1 |
| Shunt infection | 1 | 0 | 1 |
| Sinus pause | 1 | 0 | 1 |
| Stroke | 1 | 0 | 1 |
| Thrombocythaemia | 0 | 1 | 1 |
| Thyroid haemorrhage | 1 | 0 | 1 |
| Toothache | 0 | 1 | 1 |
| Tracheostomy | 0 | 1 | 1 |
| Tracheostomy complication | 0 | 1 | 1 |
| Tracheostomy infection | 1 | 0 | 1 |
| Vasovagal reaction | 0 | 1 | 1 |
| Ventricular fibrillation | 0 | 1 | 1 |
| Ventricular tachycardia | 0 | 1 | 1 |
| Ventriculitis | 1 | 0 | 1 |
| Vocal cord paresis | 1 | 0 | 1 |
Appendix 6 Cost-effectiveness analysis
Disability Rating Scale outcomes
The DRS outcomes, stratified by population, are presented in Table 19. In order to estimate the utility and monitoring costs post TBI, we estimated the GOS score corresponding to each level of disability, as reported for the DRS score. We also utilised clinical feedback for this estimation process.
| DRS score | Level of disability (based on DRS score)28 | Mild/moderate (n) | Both pupils react (n) | Estimated corresponding GOS outcome |
|---|---|---|---|---|
| 0 | None | 2845 | 3172 | Good recovery |
| 1 | Mild | 249 | 306 | |
| 2–3 | Partial | 775 | 907 | Moderate disability |
| 4–6 | Moderate | 513 | 638 | |
| 7–11 | Moderately severe | 384 | 539 | Severe disability |
| 12–16 | Severe | 157 | 245 | |
| 17–21 | Extremely severe | 136 | 300 | |
| 22–24 | Vegetative state | 78 | 205 | Vegetative state |
| 25–29 | Extreme vegetative state | 46 | 154 | |
| Total | 5183 | 6466 |
Utility estimation: correlation between Glasgow Coma Scale score and Glasgow Outcome Scale from previous randomised controlled trial (scenario)
An alternative estimation process was considered, to predict the utility in each population. A previous analysis showed the distribution of GOS outcomes (good recovery, moderate disability, severe disability) stratified by GCS score. 46,78
For a sensitivity analysis, we used the GCS scores from the CRASH-3 patients to estimate a distribution of GOS scores, to which the utility values estimated by Ward Fuller et al. 45 (see Table 9) were applied.
| GCS score at injury | GOS outcome among survivors | ||
|---|---|---|---|
| Good recovery (%) | Moderate disability (%) | Severe disability (%) | |
| 3 | 28.9 | 30.8 | 40.3 |
| 4 | 20.6 | 25.8 | 53.6 |
| 5 | 22.9 | 30.6 | 46.5 |
| 6 | 33.4 | 34.0 | 32.6 |
| 7 | 44.0 | 29.9 | 26.1 |
| 8 | 45.9 | 32.7 | 21.4 |
| 9 | 56.8 | 26.0 | 17.2 |
| 10 | 57.7 | 27.1 | 15.2 |
| 11 | 65.2 | 22.7 | 12.0 |
| 12 | 68.5 | 19.7 | 11.8 |
| 13 | 75.2 | 16.2 | 8.6 |
| 14 | 74.5 | 16.6 | 9.0 |
| 15a | 74.5 | 16.6 | 9.0 |
| CRASH-3 population | GOS outcome among survivors | Estimated utilitya | ||
|---|---|---|---|---|
| Good recovery (%) | Moderate disability (%) | Severe disability (%) | ||
| Mild or moderate TBI | 59.4 | 23.2 | 17.4 | 0.79 |
| Both pupils reactive | 68.5 | 20.1 | 11.4 | 0.76 |
Long-term model survival predictions
The survival of patients by treatment group is shown for the first 3 months of the model (Figure 13) and for the duration of the model time horizon (Figure 14).
FIGURE 13.
Model predictions for survival for 3 months by treatment group. Vertical dotted line represents 28-day trial period.
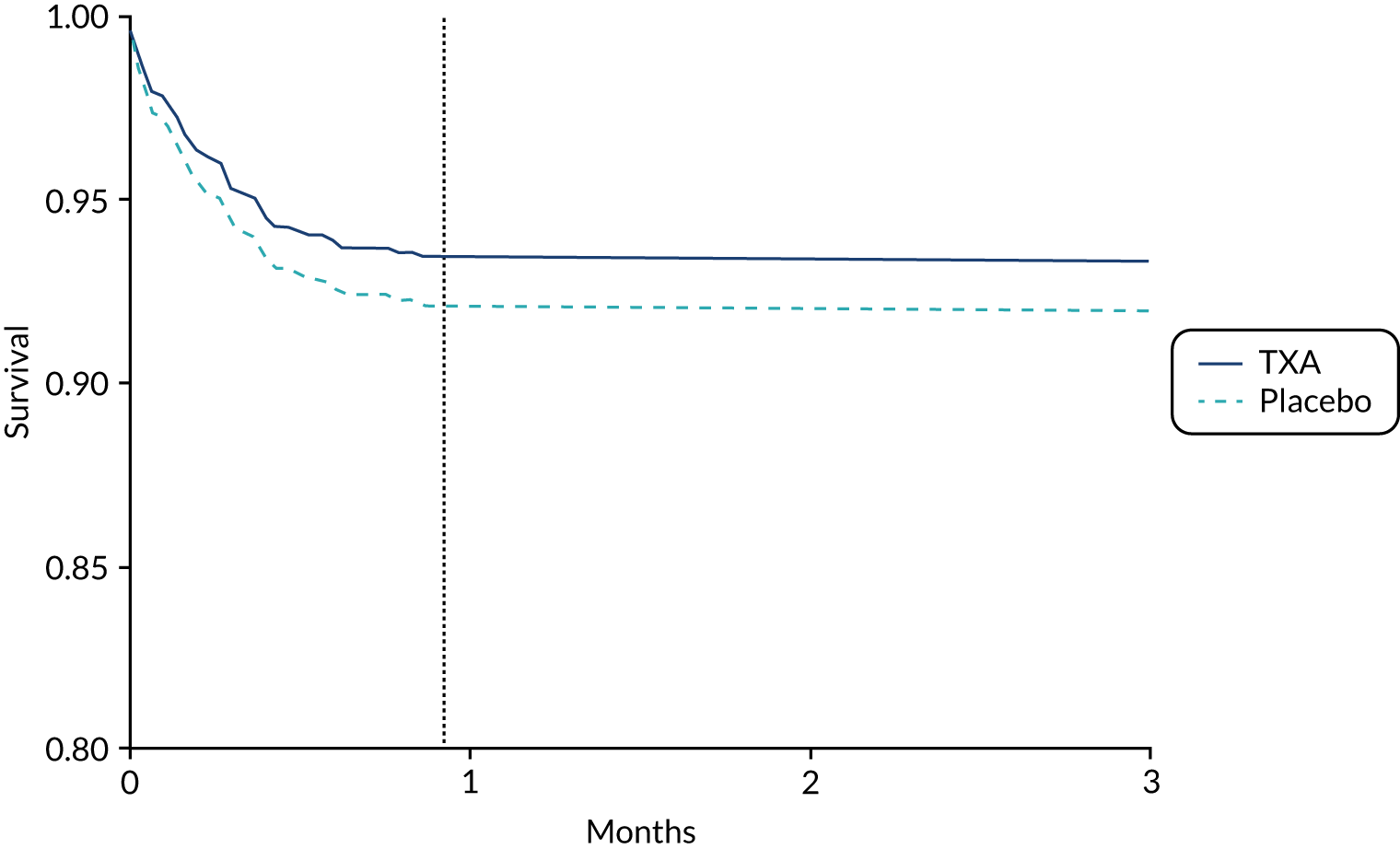
FIGURE 14.
Model predictions for survival for the duration of the analysis time horizon by treatment group.
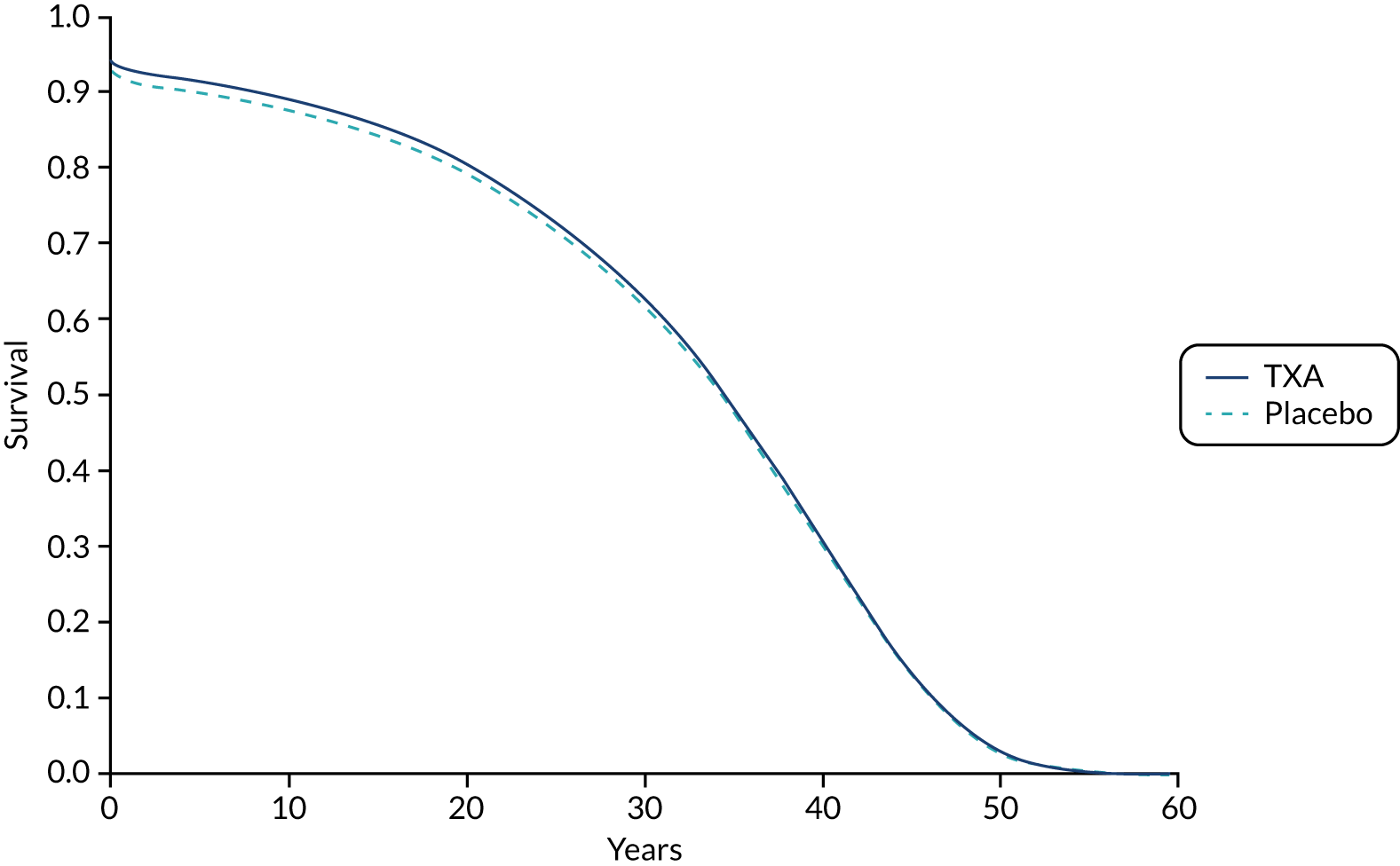
Appendix 7 Dissemination plan
List of abbreviations
- BBC
- British Broadcasting Corporation
- CI
- confidence interval
- CRASH-1
- Corticosteroid Randomisation After Significant Head Injury
- CRASH-2
- Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage-2
- CRASH-3
- Clinical Randomisation of an Antifibrinolytic in Significant Head Injury-3
- CT
- computerised tomography
- DRS
- Disability Rating Scale
- DVT
- deep-vein thrombosis
- EU
- European Union
- FOAMed
- Free Open Access Medical education
- GCS
- Glasgow Coma Scale
- GOS
- Glasgow Outcome Scale
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- i.m.
- intramuscular
- i.v.
- intravenous
- LMIC
- low- and middle-income countries
- LSHTM
- London School of Hygiene & Tropical Medicine
- LY
- life-year
- MI
- myocardial infarction
- MRC
- Medical Research Council
- NICE
- National Institute for Health and Care Excellence
- PE
- pulmonary embolism
- PPI
- patient and public involvement
- PSA
- probabilistic sensitivity analysis
- QALY
- quality-adjusted life-year
- RR
- risk ratio
- SBP
- systolic blood pressure
- SD
- standard deviation
- SMR
- standardised mortality ratio
- TBI
- traumatic brain injury
- tPA
- tissue plasminogen activator
- TSC
- Trial Steering Committee
- TXA
- tranexamic acid