

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 13/88/11. The contractual start date was in March 2016. The draft report began editorial review in October 2020 and was accepted for publication in June 2021. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2021 Barratt et al. This work was produced by Barratt et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2021 Barratt et al.
Chapter 1 Introduction
This chapter includes material that has been adapted from the trial protocol, which has been published in BMJ Open. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Background
Antibiotics are among the most frequently prescribed medicines for children worldwide. 2,3 In the UK, Italy and the Netherlands, almost 50% of children have received antibiotics by their second birthday. Annually, it is estimated that 30% of children aged 2–11 years receive antibiotics. 3
Of the possible indications in children aged < 5 years, the most common are acute respiratory tract infections, including community-acquired pneumonia (CAP). 4–6 CAP is one of the most common serious bacterial childhood infections. Although the majority of pneumonia deaths occur in low- and middle-income countries, CAP is a major cause of morbidity in Europe and North America. 5,7 In the UK, 62% of all antibiotics prescribed for community-acquired infections are for CAP. 8 In the USA, respiratory symptoms, fever or cough are responsible for one-third of all childhood medical visits, and 7–15% of these children will be diagnosed with CAP. 9,10
Emergency department (ED) attendances and hospital admissions of children with respiratory complaints have increased in recent decades, mostly in preschool children. 9,11,12 According to Hospital Episode Statistics,13 children aged 0–4 years accounted for around 2.11 million ED attendances in 2017–18. More than 11,000 children aged < 15 years were admitted to hospitals in England with a diagnosis of bacterial pneumonia in 2008, and 9000 1- to 4-year-old inpatients with non-influenza pneumonia were recorded in 2012/13. 13,14
The bacterial pathogen most commonly associated with childhood CAP is Streptococcus pneumoniae, including in countries where pneumococcal conjugate vaccine (PCV) is routinely administered. 7,15–17 In 2010, PCV13 (which covers 13 S. pneumoniae serotypes) was introduced in the UK, with almost 95% uptake in young children. 18,19 However, despite an observed impact on invasive pneumococcal disease, a decrease in CAP-related hospital admissions in young children has not been observed. 11,14,20,21
What are the current challenges in the management of childhood community-acquired pneumonia?
There is no test capable of accurately distinguishing between bacterial and viral CAP. 22 Interobserver agreement for chest radigoraphic findings is poor, casting doubt on the usefulness of chest radiographs for identifying bacterial CAP, and culturing of microbiological samples, such as sputum, has low diagnostic value and samples are often difficult to take from young children. 23–25 Diagnosis of bacterial CAP presents a challenge for treating clinicians, who rely largely on clinical criteria. 22 Children presenting with fever, raised respiratory rate, focal chest signs and other respiratory signs and symptoms (such as cough) are commonly ascribed a diagnosis of bacterial CAP,10,26–28 whereas wheezing is associated with the absence of radiographic pneumonia and failure to detect bacteria in clinical samples. 26,29 If bacterial CAP is considered the likely diagnosis, treatment with antibiotics is instituted. 10,30 This diagnostic challenge is particularly problematic in secondary care, where the proportion of children presenting with serious bacterial infections is higher than in primary practice. 31,32
A further challenge for clinicians is severity assessment. Available validated predictive scoring systems for CAP severity include the Pneumonia Severity Index and the CURB-65 (confusion, urea, respiratory rate, blood pressure, and 65 years of age or older) score, but these are not applicable to children. 33,34 Pneumonia mortality risk scores for children have been developed in low-resource settings, but do not differentiate between viral and bacterial pneumonia. 35,36 Low oxygen saturation in room air is included as one component in these risk scores, and is an important factor for differentiating between non-severe and severe pneumonia. 37–39
Finally, assessing the efficacy of childhood CAP treatment is complex. Key measures in studies assessing efficacy early in the treatment course include lack of improvement or worsening of clinical symptoms and signs, such as respiratory rate and oxygen saturation. 40 According to British Thoracic Society (BTS) guidance, such criteria should trigger clinical review of children treated with oral antibiotics for CAP,22 including where the following features are present at 48 hours: (1) persistent high fever, (2) increasing or persistently increased effort of breathing and (3) persistent or increasing oxygen requirement to maintain saturations ≥ 92%. 22 Approximately 15% of children with CAP receive further antibiotics within 28 days of starting treatment because of symptoms that concern parents. 41–44 However, only half of children show recovery from symptoms of acute respiratory illness by day 9 or 10, and 90% of children recover by 3.5 weeks after symptom onset. 45–47
What are the current management recommendations for childhood community-acquired pneumonia?
Amoxicillin is the drug of choice for the treatment of childhood CAP according to the British National Formulary for Children (BNFc) and BTS and National Institute for Health and Care Excellence guidelines, as well as several international guidelines,22,48–51 as it can effectively target and treat S. pneumoniae in the absence of high-level penicillin resistance. As a result, amoxicillin accounts for a very high proportion of overall oral antibiotic use among young children in many settings. Despite this, there is insufficient evidence to inform optimal treatment dose or duration.
What are the current dose recommendations?
Antibiotic dose selection should be driven by pharmacokinetic/pharmacodynamic considerations. The key pharmacokinetic/pharmacodynamic parameter for beta-lactams (including amoxicillin) is time spent above the minimum inhibitory concentration (T > MIC) (mainly focused here on pneumococcus). The recommended T > MIC is 40–50% of the dosing interval; however, the exact relationship between blood pharmacokinetics and concentrations of amoxicillin in the lungs is unclear. 48,52 The half-life of oral amoxicillin is about 1.0–1.5 hours and, on this basis, a three times daily regimen has been widely recommended. 53 However, there are few data to inform whether or not three times daily dosing is likely to achieve pharmacokinetic/pharmacodynamic parameters better than twice-daily dosing. The available data suggest that, in the case of total amoxicillin doses of 25–50 mg/kg/day, twice-daily dosing should be sufficient to achieve adequate T > MIC53 and a Brazilian group recently demonstrated non-inferiority of twice-daily dosing compared with thrice-daily dosing in childhood CAP. 54 Together with a likely improvement in adherence to less frequent administration, twice-daily dosing is, therefore, widely recommended. 48–50,52 Currently, the BNFc recommends amoxicillin (250 mg) thrice daily for children aged 1–5 years with CAP, resulting in highly variable dosing, between approximately 40 mg/kg/day and 80 mg/kg/day, depending on the weight of the child. 55 Therefore, alternative strategies, such as weight-banded dosing, may be more appropriate. 56 Furthermore, much higher daily doses of amoxicillin, up to 200 mg/kg/day, are recommended for the treatment of severe infections. 55
What are the current duration recommendations?
Several large randomised controlled trials (RCTs) have found shorter treatment courses in childhood CAP to be effective in low- and middle-income settings in terms of clinical cure, treatment failure and relapse rate. 57,58 However, these trials enrolled children with symptoms indicative of a viral infection not requiring antibiotics, and generalisability to the UK has, therefore, been questioned. 22 The BTS recognises that there are no robust data to inform guidance on duration of antibiotic treatment in childhood CAP. 22 The BNFc guidance relevant at the start of this trial recommended a 7-day course, whereas European and World Health Organization (WHO) guidance suggests a 3- to 5-day course. 48,55 In 2019, the National Institute for Health and Care Excellence published guidance recommending stopping amoxicillin treatment after 5 days (250 mg thrice daily) for children aged 1–4 years, unless microbiological results suggest that a longer course length is needed or the patient is not clinically stable. 51
What is the impact of antimicrobial resistance in childhood community-acquired pneumonia?
In the UK, the rates of penicillin non-susceptibility of S. pneumoniae are relatively low, at approximately 15% for respiratory samples (mainly from adults) and 4–6% for blood culture isolates. 59 Penicillin resistance [i.e. minimal inhibitory concentration (MIC) > 2 μg/ml] has not been observed in blood culture isolates and has been found in < 1% of respiratory S. pneumoniae isolates in the UK since 2010. 59 However, some worrying trends are observed in resistance of gut bacteria, and this situation will be exacerbated in a setting where antibiotics are used injudiciously. 60
The relationship between MIC (an in vitro phenomenon) and clinical outcome in CAP is complex, and data on the level of S. pneumoniae antimicrobial resistance that reduces amoxicillin effectiveness are limited. Harmonisation of European breakpoints (i.e. the MIC at which an isolate is considered susceptible, intermediate or resistant) attempts to provide a link between clinical impact and in vitro observation of resistance. 61 Clinical breakpoints are determined based on a variety of data, in addition to efficacy studies. This includes pharmacokinetic/pharmacodynamic data, which for penicillin usually take T > MIC of 40% as the key exposure measure.
Children have high rates of bacterial colonisation, which often represents an increased level of carriage of resistant organisms62,63 These may be passed on to others in the community, especially within child-care settings. 64,65 Interventions to maintain a low level of antimicrobial resistance among colonising bacteria may, therefore, have population implications.
The limited existing data on the specific impact of duration and dose of antibiotic treatment on subsequent colonisation with resistant bacteria in vivo suggest a complex and dynamic relationship. 62–73 Experimental models suggest that insufficiently high dosing could promote selection of resistant pathogens. In addition, although most of the effect on bacterial load is achieved early during antibiotic exposure, resistant isolates emerge after 4 or 5 days. 74–78 RCTs assessing the effect of antibiotic duration and dose have been called for, as they will probably provide the strongest evidence for the relationship between antibiotic exposure and colonisation with resistant bacteria. 79 One such RCT found that higher-dose shorter-duration amoxicillin therapy for childhood CAP led to less colonisation with resistant bacteria after 4 weeks, and was associated with better treatment adherence. 72 However, mathematical modelling indicates that this may come at the price of selecting isolates with higher levels of resistance, and clinical efficacy was not addressed in the trial. 72,78
Trial rationale
Despite the reduction in incidence of invasive pneumococcal disease since the introduction of the conjugate vaccine,20 CAP remains one of the most commonly identified and treated childhood infections in the UK. Although there is clear agreement that amoxicillin should be the first-line treatment, there are insufficient data to inform selection of dose and duration, and the impact that different regimens have on antimicrobial resistance is unknown.
Effectiveness and resistance-outcome data pertaining to dose and duration of amoxicillin could inform antimicrobial stewardship strategies in the large group of children with a high likelihood of bacterial CAP targeted by CAP-IT (Community-Acquired Pneumonia: a protocol for a randomIsed controlled Trial). A better understanding of the relationship between dose and duration of antibiotic treatment, and the impact on clinical outcomes and antimicrobial resistance, would make it possible to formulate improved evidence-based treatment recommendations for childhood CAP.
Objectives
The main objective of CAP-IT was to determine the following for young children with uncomplicated CAP treated after discharge from hospital if:
-
a 3-day course of amoxicillin is non-inferior to a 7-day course, determined by receipt of a clinically indicated systemic antibiotic other than trial medication for respiratory tract infection (including CAP) in the 4 weeks after randomisation up to day 28
-
lower-dose amoxicillin is non-inferior to higher-dose amoxicillin under the same conditions.
Secondary objectives were to evaluate the impact of lower-dose and shorter-duration amoxicillin on antimicrobial resistance, severity and duration of parent/guardian-reported CAP symptoms and specified clinical adverse events (AEs).
Chapter 2 Methods
Trial design
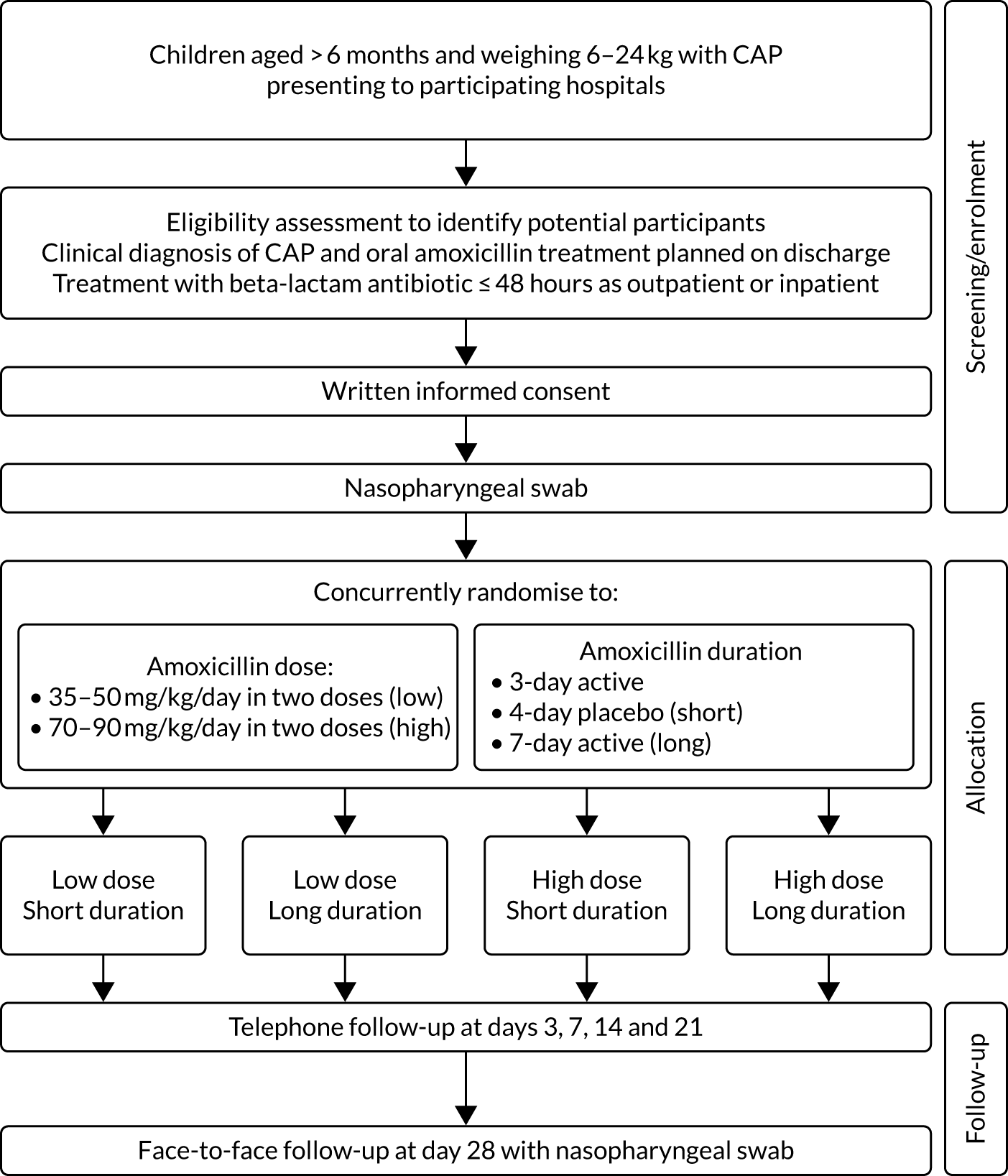
The CAP-IT study was a multicentre clinical trial with a target sample size of 800 participants in the UK and Ireland. In design, it was a randomised double-blind placebo-controlled 2 × 2 factorial non-inferiority trial of amoxicillin dose and duration in young children with CAP (Figure 1).
FIGURE 1.
The CAP-IT schema.
Trial setting
Participants were recruited from 28 UK NHS hospitals and one children’s hospital in Ireland:
-
Alder Hey Children’s Hospital NHS Foundation Trust (Liverpool, UK)
-
Barts Health NHS Trust (London, UK)
-
Birmingham Women’s and Children’s NHS Foundation Trust (Birmingham, UK)
-
Brighton and Sussex University Hospitals NHS Trust (Brighton, UK)
-
Chelsea and Westminster Hospital NHS Foundation Trust (London, UK)
-
Children’s Health Ireland (Dublin, Ireland)
-
City Hospitals Sunderland NHS Foundation Trust (Sunderland, UK)
-
Countess of Chester Hospital NHS Foundation Trust (Chester, UK)
-
County Durham and Darlington NHS Foundation Trust (Darlington, UK)
-
Guy’s and St Thomas’ NHS Foundation Trust (London, UK)
-
Hull and East Yorkshire Teaching Hospitals NHS Trust (Hull, UK)
-
Imperial College Healthcare NHS Trust (London, UK)
-
King’s College Hospital NHS Foundation Trust (London, UK)
-
The Leeds Teaching Hospitals NHS Trust (Leeds, UK)
-
Manchester University NHS Foundation Trust (Manchester, UK)
-
Nottingham University Hospitals NHS Trust (Nottingham, UK)
-
Oxford University Hospitals NHS Foundation Trust (Oxford, UK)
-
Southport and Ormskirk Hospital NHS Trust (Southport, UK)
-
Royal Hospital for Children (Glasgow, UK)
-
Sheffield Children’s NHS Foundation Trust (Sheffield, UK)
-
South Tees Hospitals NHS Foundation Trust (Middlesbrough, UK)
-
St George’s University Hospitals NHS Foundation Trust (London, UK)
-
University Hospitals Bristol and Weston NHS Foundation Trust (Bristol, UK)
-
University Hospitals of Derby and Burton NHS Foundation Trust (Derby, UK)
-
University Hospitals of Leicester NHS Trust (Leicester, UK)
-
University Hospitals Lewisham (London, UK)
-
University Hospital Southampton NHS Foundation Trust (Southampton, UK)
-
University Hospital of Wales (Cardiff, UK).
Participating sites were tertiary or secondary hospitals with paediatric emergency departments (PEDs) and inpatient facilities, and were selected in collaboration with Paediatric Emergency Research in the UK & Ireland80 on the basis of clinical and research infrastructure, experience in clinical research and likely eligible population size.
Participants
Patients presenting to participating hospitals were identified in PEDs, assessment/observation units or inpatient wards. Potential participants were screened as early as possible during the initial clinical assessment. Informed consent was sought from a parent/guardian once eligibility had been confirmed, but only after full explanation of the trial aims, methods and potential risks and benefits. Discussions regarding the trial took place between families and clinical teams when the child’s clinical condition was stable, to minimise distress. Extensive information and recruitment materials were available for recruiting sites, including printed and video materials [accessible at URL: www.capitstudy.org.uk (accessed 29 July 2021)]. CAP-IT information film was designed to assist research teams in the recruitment process and provided information to parents/guardians about the purpose of the trial, the use of placebo and trial procedures. Parents/guardians could watch the film in their own time while in hospital, and research teams reported that the film was a useful tool during the recruitment process. The film was made with input from the trial patient and public involvement (PPI) representative and featured a site principal investigator and research nurse, as well as graphics to aid explanation of trial procedures. [It can be viewed at https://vimeo.com/217849985 (accessed 29 July 2021).] Families were able to decline participation in the trial at any time without providing a reason and without incurring any penalty or affecting clinical management.
Recruitment pathways
Children were recruited through two different pathways based on whether they received any inpatient antibiotic treatment (ward group) or not (PED group). Children in either group may have had up to 48 hours of oral or parenteral beta-lactam treatment before enrolment. The PED group contained children who had not received any in-hospital antibiotic treatment (but may have had up to 48 hours of beta-lactam antibiotics in the community), whereas the ward group contained children who received any in-hospital oral or intravenous beta-lactam therapy prior to randomisation. Children in the latter group may have received beta-lactam treatment in the community first and subsequently in hospital, without interruption, for a total of < 48 hours.
Inclusion criteria
Children were eligible if they had a clinical diagnosis of uncomplicated CAP, were aged > 6 months and weighed 6–24 kg, and treatment with amoxicillin as the sole antibiotic was planned on discharge. Box 1 shows the clinical criteria required for a diagnosis of CAP in CAP-IT.
Clinical diagnosis of CAP is defined as:
-
cough (reported by parents/guardians within 96 hours before presentation)
-
temperature ≥ 38 °C measured by any method or likely fever within 48 hours before presentation
-
signs of laboured/difficult breathing or focal chest signs (i.e. one or more of nasal flaring, chest retractions, abdominal breathing, focal dullness to percussion, focal reduced breath sounds, crackles with asymmetry or lobar pneumonia on chest radiograph).
Exclusion criteria
Children were excluded if they had received ≥ 48 hours of beta-lactam antibiotics or any non-beta-lactam agents, or if they had severe underlying chronic disease with increased risk of complicated CAP (including sickle cell anaemia, immunodeficiency, chronic lung disease and cystic fibrosis), documented penicillin allergy or other contraindication to amoxicillin, complicated pneumonia (including shock, hypotension, altered mental state, ventilatory support, empyema, pneumothorax and pulmonary abscess) or bilateral wheezing without focal chest signs.
Changes to selection criteria
During the trial enrolment period, eligibility criteria were modified based on emerging data to better reflect clinical management and facilitate inclusion of all children to whom the results of the trial may be of relevance.
Age and weight criteria were amended from ‘age from 1 to 5 years (up to their 6th birthday)’ in protocol v2.0 to ‘greater than 6 months and weighing 6–24 kg’ in protocol v3.0. Children recruited to protocol v2.0 were excluded if they were receiving systemic antibiotic treatment at presentation. This was modified in protocol v3.0 for the PED group and in protocol v4.0 for the ward group, such that children were eligible if they had received ≤ 48 hours’ systemic antibiotic treatment at trial entry, as per section 2.3 of the protocol.
Children in the ward group were excluded in protocol v2.0 if they had ‘current oxygen requirement’ or ‘current age-specific tachypnoea’; however, these criteria were removed in protocol v3.0 and replaced with the inclusion criterion ‘child is considered fit for discharge at randomisation’.
The CAP diagnostic criterion relating to fever changed from ‘temperature ≥ 38 °C measured by any method OR history of fever in last 24 hours reported by parents/guardians’ in protocol v2.0 to ‘temperature ≥ 38 °C measured by any method OR likely fever in last 48 hours’ in protocol v3.0 to account for the accompanying parent/guardian not measuring temperature in the preceding 24 hours.
Interventions
The investigational medicinal product (IMP) for treatment at home was provided as a powder to be suspended on the day of randomisation. Children received oral amoxicillin suspension twice daily, commencing on the day of randomisation. All children were weighed during eligibility screening and was used to determine dose volume according to seven weight bands (Table 1).
| Weight range (kg) | Dosing intructions |
|---|---|
| ≤ 6.4 | 4.5 ml twice a day |
| 6.5–8.4 | 6 ml twice a day |
| 8.5–10.4 | 7.5 ml twice a day |
| 10.5–13.4 | 9.5 ml twice a day |
| 13.5–16.9 | 12 ml twice a day |
| 17.0–20.9 | 15 ml twice a day |
| 21.0–24.0 | 16.5 ml twice a day |
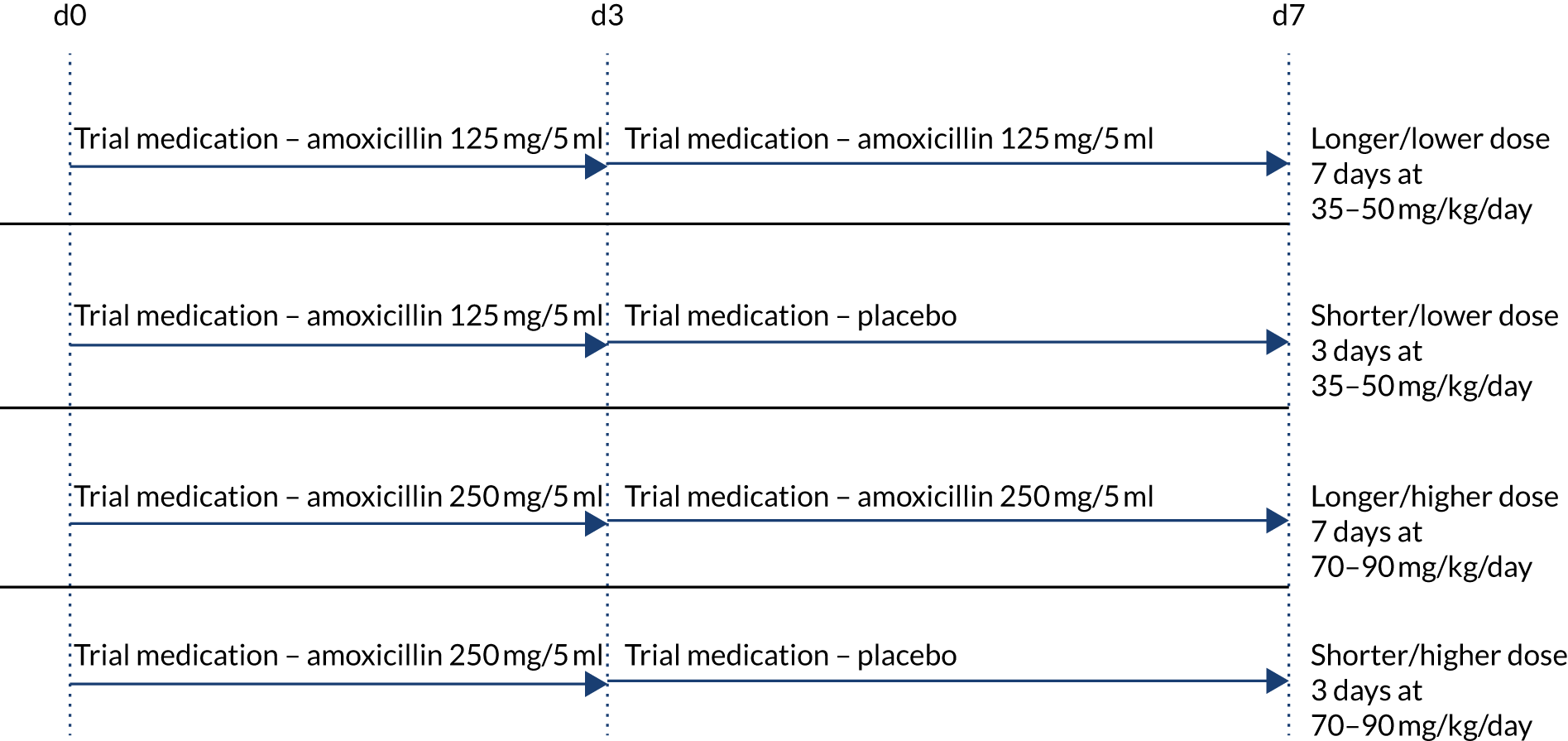
Participants were randomised to receive either a lower (35–50 mg/kg/day) or a higher (70–90 mg/kg/day) dose, concealment of which was achieved by using amoxicillin products of two different strengths (125 mg/5 ml and 250 mg/5 ml). Therefore, children in each dose arm in the same weight band were administered the same volume of suspension .
Participants were simultaneously randomised to receive either 3 or 7 days of amoxicillin treatment at home. A placebo manufactured to match the characteristics of oral amoxicillin suspension was used to blind parents/guardians and clinical staff to the duration allocation. Both active drug and placebo formed a yellow-coloured similar-tasting suspension. However, because of difficulties in exactly taste-matching the placebo suspension to amoxicillin, one brand of amoxicillin was used for the first 3 days of treatment followed by a second brand for days 4–7 when duration of treatment was 7 days. Parents were instructed to expect a taste change between bottles, but they did not know whether this was due to moving to placebo or to a new brand of amoxicillin. Allocated treatment duration to be given after discharge from hospital was fixed at 3 or 7 days independently of any antibiotics received before randomisation, with up to 48 hours of oral or parenteral beta-lactam treatment permitted before enrolment.
This resulted in four treatment arms, as shown in Figure 2.
FIGURE 2.
Treatment arms.
The hypothesis is that higher doses of amoxicillin given for a longer duration are non-inferior to lower doses of amoxicillin given for a shorter duration for the treatment of children attending hospital with CAP in terms of antibiotic retreatment.
The objective is to conduct a RCT in children attending hospital with CAP comparing higher and lower doses of amoxicillin given for 3 or 7 days.
Drug substitutions and discontinuations of trial treatment
Substitution of an alternative amoxicillin formulation or another antibiotic was permitted where tolerability issues could not be overcome by improving acceptability (e.g. by mixing the suspension with formula milk, other liquids or foods) or where a clinical need for continued treatment persisted. In situations of toxicity, for example if an allergic reaction to penicillin was suspected, substitution with an alternative class of antibiotic was permitted.
Discontinuation of trial treatment was permitted if, on clinical review, a change in the child’s condition justified discontinuation or modification of trial treatment, if use of a medication with a known major or moderate drug interaction with amoxicillin was essential for the child’s management or if the parent/guardian withdrew consent for treatment.
In situations where retreatment was deemed necessary, the choice of antibiotic was left to the treating clinician.
Trial assessments and follow-up
Participants were screened as described in Participants, and, following receipt of informed consent, randomisation was performed at the point of discharge from hospital. Following randomisation, all participants were followed up for 29 days for evaluation of the primary and secondary end points described in Outcomes. The timing and frequency of assessments are summarised in the trial schedule (Table 2) and described below.
| Assessment | Pre randomisation:a ≤ 48 hours before randomisation | Days in trial | ||||||
|---|---|---|---|---|---|---|---|---|
| Day 0 (randomisation) | Day 3 | Days 7–9 (week 1) | Days 14–16 (week 2) | Days 21–23 (week 3) | Days 28–30 (week 4) | Any acute event | ||
| Trial participation | ||||||||
| Parent/guardian information sheetb | ✗ | ✗ | ||||||
| Informed consent | ✗ | |||||||
| Drug supply dispensing | ✗ | |||||||
| Adherence questionnaire | ✗ | ✗ | (✗) | |||||
| Adherence review (returned medication) | ✗ | |||||||
| Clinical assessment | ||||||||
| Medical historyb | (✗) | ✗ | ||||||
| Physical examinationb | (✗) | ✗ | ✗ | ✗ | ||||
| Symptom reviewb | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| EQ-5D | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Use of health services | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Laboratory assessment | ||||||||
| Nasopharyngeal swabc | (✗) | ✗ | ✗ | (✗) | ||||
| Haematology | (✗) | (✗) | (✗) | (✗) | ||||
| Biochemistry | (✗) | (✗) | (✗) | (✗) | ||||
| Virology | (✗) | (✗) | (✗) | (✗) | ||||
| Radiological assessment | ||||||||
| Chest radiography | (✗) | (✗) | (✗) | |||||
| Parent-completed diary | ||||||||
| Symptom diary | ✗ | ✗ | ✗ | |||||
| Ancillary subgroup studies | ||||||||
| Stool samplec | ✗ | ✗ | ✗ | ✗ | ||||
Enrolment and randomisation
Following identification, screening and informed consent of eligible patients, baseline information was obtained through interview with the parent/guardian. This included demographic information, such as sex and ethnicity, medical history, including review and duration of symptoms (e.g. cough, temperature and respiratory symptoms), underlying diseases and antibiotic exposure in the preceding 3 months. Details of the physical examination, including weight and vital parameters (e.g. temperature, respiratory rate, heart rate and oxygen saturation in room air), were recorded and a baseline nasopharyngeal swab was obtained.
No additional tests were mandated, but results were collected if tests were performed as part of clinical care, including haematology tests (e.g. haemoglobin, platelet count, leucocyte count, neutrophil count and lymphocyte count), biochemistry tests (e.g. C-reactive protein, procalcitonin and electrolytes), virology [rapid testing for respiratory syncytial virus and influenza A/B (any method)] and chest radiography.
Parents/guardians were provided with trial materials, including a symptom diary, participant information sheet, IMP administration instructions and contact details for the trial team. The symptom diary collected data pertinent to the primary and secondary outcomes and was completed by parents for 14 days following randomisation.
Follow-up
Telephone contact was made with participants on days 3, 7–9, 14–16 and 21–23, with a face-to face visit within 2 days of day 28. At these contacts, primary and secondary end points were reviewed, including additional antibiotic treatment, clinical signs and symptoms, adverse treatment effects and IMP adherence. During face-to-face visits (final or unscheduled) a nasopharyngeal swab was collected, and, if CAP symptoms were ongoing, physical examination findings and physiological parameters were collected. If a hospitable face-to-face visit was not possible for final follow-up, it was attempted by telephone or as a home visit. If this failed, despite reasonable efforts, primary end-point data were sought through contact with the general practitioner (GP) where consent had been given to do so.
If participants required acute clinical assessment for ongoing/re-emerging symptoms during the follow-up period, the treating clinician’s judgement determined if investigations, treatment or hospitalisation was required. On premature discontinuation of IMP, irrespective of reason, parents/guardians were encouraged to remain in follow-up. However, parent/guardian decisions were respected, and if follow-up was stopped prematurely, then data and samples already collected were included in the analysis unless parents/guardians requested otherwise.
Data collection and handling
Data were recorded on paper case report forms and entered onto the CAP-IT database by clinical or research staff at each site. Staff with data entry responsibilities completed standardised database training before being granted access to the database. Data were exported into Stata® (v15.1) (StataCorp LP, College Station, TX, USA) for analysis.
Randomisation
Eligibility was confirmed by CAP-IT site investigators through completion of an eligibility checklist. Patients were randomised simultaneously to each of the two factorial randomisations in a 1 : 1 ratio. Randomisation was stratified by group (PED and ward) according to whether or not they had received any non-trial antibiotics in hospital before being enrolled.
A computer-generated randomisation list was produced by the trial statistician based on random permuted blocks of eight. Each block contained an equal number of the four possible combinations of dose and duration in random order. The IMP supplier packaged the trial medication into kits that were grouped into blocks of eight, in accordance with to the randomisation list specification. Blinded IMP labels were applied to each kit, which contained the kit identifications (IDs). Kit IDs were made up of four numerical digits, the first three of which represented the block ID and fourth specified the kit ID within the block. Blinded randomised blocks of IMP were delivered to trial sites and participants were randomised by dispensing the next sequentially numbered kit within the active block.
Blinding
All treating clinicians, parents/guardians and outcome assessors [including End-Point Review Committee (ERC) members] were blinded to the allocated treatment. The use of placebo, as well as the permuted block randomisation strategy and blinded drug kits, ensured that parents and clinic staff remained blinded to amoxicillin duration and dose.
Access to the randomisation list was restricted to trial statisticians and IMP repackagers, and unblinded data were reviewed confidentially only by the Independent Data Monitoring Committee (IDMC) (annually) and trial statisticians. The Trial Management Team remained blinded until after the trial end and completion of the statistical analysis in accordance with the prespecified statistical analysis plan (SAP).
Unblinding was possible in situations where a treating clinician deemed it necessary, for example in the case of a significant overdose. This could be performed using an emergency unblinding system accessible through the CAP-IT website. Only the treating clinician would then be informed of the child’s allocation, maintaining the blinding of the trial team.
Outcomes
Primary outcome
The primary outcome for CAP-IT was defined as any clinically indicated systemic antibacterial treatment prescribed for respiratory tract infection (including CAP) other than trial medication up to and at week 4 final follow-up (i.e. day 28). Prescription of non-trial medication when the primary reason was (1) illness other than respiratory tract infection, (2) intolerance of or adverse reaction to IMP, (3) parental preference or (4) administrative error did not constitute a primary end point.
An ERC, comprising doctors independent of the Trial Management Group and blinded to randomised allocations, reviewed all cases of a participant being prescribed non-trial systemic antibacterial treatment. The main role of the ERC was to adjudicate, based on all available data, whether or not the primary outcome was met. The ERC classified non-trial systemic antibacterial treatment as being for respiratory tract infection with likelihoods of ‘definitely/probably’, ‘possibly’, ‘unlikely’ or ‘too little information’. Those infections categorised as ‘CAP’, ‘chest infection’ or ‘other respiratory tract infection’ with a treatment likelihood assessment of ‘definitely/probably’ or ‘possibly’ were regarded as fulfilling the primary end point.
Information on additional antibacterial treatments was collected from parents through follow-up telephone contact with parents on days 3, 7, 14 and 21, at the final visit contact and finally through a daily diary completed by parents on days 1–14.
During enrolment, parents were asked to provide consent for the research teams to contact their child’s general practice to collect information regarding antibacterial treatment given during the follow-up period. This additional information supported the ERC in accurately adjudicating events. In addition, this allowed the collection of primary outcome data where contact with participants had been lost prior to completion of the follow-up period.
Changes to primary end point
The primary end-point definition was clarified in protocol v3.0 to specify that ‘systemic antibacterial’ treatments should avoid inclusion of topical antibiotics, which were not of interest. In protocol v4.0, the primary end point was refined further, resulting in the definition in Primary outcome. This definition specified that the systemic antibacterial must be clinically indicated and prescribed for a respiratory tract infection (including CAP), as adjudicated by the ERC.
Secondary outcomes
Secondary outcomes included measures of morbidity, antimicrobial resistance and trial medication adherence.
Morbidity
Morbidity secondary outcomes included severity and duration of parent/guardian-reported CAP symptoms and specified clinical AEs.
The following CAP symptoms were elicited at baseline, in follow-up telephone calls at days 4, 8, 15 and 22 and at the final visit, as well as at unscheduled visits: cough, wet cough (i.e. phlegm), breathing faster (i.e. shortness of breath), wheeze, sleep disturbed by cough, vomiting (including after cough), eating/drinking less and interference with normal activity. Parents/guardians were asked to grade each symptom using the following five categories: (1) not present, (2) slight/little, (3) moderate, (4) bad and (5) severe/very bad. Date of start and resolution were also elicited. Symptoms and their severity (using the same categories) were obtained daily on the symptom diary for 14 days from randomisation.
Information about diarrhoea, skin rash and thrush was collected and graded in the same way as CAP symptoms. In addition, AEs related to the stopping of trial medication or the start of non-trial antibiotics were recorded.
Other AEs meeting the criteria for seriousness [i.e. serious adverse events (SAEs)] were reported within 24 hours of research sites becoming aware of the event. SAEs were classified by system organ class and lower-level term in accordance with the Medical Dictionary for Regulatory Activities (MedDRA®; version 21.1) and were graded using the Division of Aids (DAIDS) Table for Grading the Severity of Adult and Paediatric Adverse Events. 81
Antimicrobial resistance
The antimicrobial resistance secondary end point was defined as phenotypic resistance to penicillin at week 4 measured in S. pneumoniae isolates colonising the nasopharynx. Carriage and resistance of S. pneumoniae isolates were assessed by analysis of nasopharyngeal samples, collected from participants at baseline, at the final visit (i.e. day 29) and at any unscheduled visits during the follow-up period.
Phenotypic penicillin susceptibility was determined for S. pneumoniae isolates by microbroth dilution across a dilution range for penicillin of 0.016–16 mg/l and interpreted in accordance with EUCAST (European Committee on Antimicrobial Susceptibility Testing) clinical break-point tables v10.0 for benzylpenicillin and S. pneumoniae (infections other than meningitis) [i.e. sensitive (MIC ≤ 0.064 mg/l), non-susceptible (MIC 0.125–2 mg/l) or resistant (MIC > 2 mg/l)]. 82 The same approach was taken for amoxicillin susceptibility testing [isolates with MIC ≤ 0.5 mg/l were sensitive and isolates with MIC > 1 mg/l were resistant). S. pneumoniae ATCC® 49619™ (ATCC, Manassas, VA, USA) was used for quality control. 82
Adherence
Data on IMP adherence were elicited during follow-up telephone calls, at the final visit (where follow-up telephone calls were not performed) and at unscheduled visits. At each time point, parents/guardians were asked if IMP had been stopped early, and, if so, the date of the last dose taken and for which of the following reasons: CAP improved/cured, CAP worsened/not improving or gagging/spitting out/refusing. In addition, parents/guardians were asked how many doses of each bottle were either missed or were less than the full prescribed volume.
Sample size
The sample size was based on demonstrating non-inferiority for the primary efficacy end point for each of the duration and dose randomisations. Although inflation factors have been advocated for factorial trials to account for interaction between the interventions, or a reduction in the number of events, this is not necessary if either randomised intervention (dose or duration) has a null effect (i.e. the underlying hypothesis with a non-inferiority design), as marginal analyses can then be conducted.
The expected antibiotic retreatment rate was originally assumed to be 5%. However, data emerging during the enrolment phase suggested that the primary outcome event rate was considerably higher, at approximately 15%. This necessitated a change in the non-inferiority margin, which was increased from 4% to 8%. This is still lower than the European Medicines Agency’s recommendation of a 10% non-inferiority margin for adult CAP trials. 83 Assuming a 15% event rate, 8% non-inferiority margin (on a risk difference scale) assessed against a two-sided 90% confidence interval (CI) and 15% loss to follow-up, the sample size was calculated as 800 children to achieve 90% power.
Statistical methods
Analysis principles
The primary analysis adopted a modified intention-to-treat (ITT) principle, that is it included all patients enrolled and analysed in accordance with the group to which they were randomised, regardless of treatment actually received. One modification to the strict ITT principle prespecified in the trial SAP was the exclusion of randomised patients who did not take any IMP. Owing to the blinded nature of the trial, the risk of introducing bias by exclusion of these patients was considered minimal. A secondary on-treatment analysis was performed that excluded ‘non-adherent’ participants, defined as having taken < 80% of scheduled trial medication, based on (1) all trial medication including placebo and (2) active drug only.
In the primary and secondary analyses, the main effect for each randomisation was estimated by collapsing across levels of the other randomisation factor, supplemented by tests for interaction between the two randomisations and with previous systemic antibacterial exposure. Interaction was assessed on an additive scale.
For continuous variables, the mean (with standard deviation) or median [with interquartile range (IQR)] of absolute values and of changes in absolute values from baseline were reported by scheduled telephone calls/visits and by randomised group.
For binary and categorical variables, differences between groups at particular time points were tested using chi-squared tests (or exact tests, if appropriate). For ordered variables, differences between groups at particular time points were tested using rank tests.
For time-to-event outcomes, the time from baseline to the event date was used, applying Kaplan–Meier estimation. Where participants did not experience an event, data were censored at the date of last review of that event. Differences between groups were tested using a log-rank test.
Formal statistical adjustment for multiple comparisons (particularly pertinent for some of the secondary end points) were not applied, and significance tests should be interpreted in the context of the total number of related comparisons performed.
The primary end point was analysed within a non-inferiority framework, where significance testing has no clear role (with emphasis instead on CIs). Secondary outcomes were analysed within a superiority framework (i.e. assessing the null hypothesis of no difference). All estimates, including differences between randomised groups, are presented with two-sided 90% CIs (rather than the more conventional 95%) to achieve consistency with the reporting of the primary end point.
Primary outcome
The proportion of children meeting the primary end point was obtained from the cumulative incidence at day 28, as estimated by Kaplan–Meier methods (i.e. accounting for the differential follow-up times). Participants with incomplete primary outcome data (e.g. as a result of a missed final visit) were censored at the time of their last contact. In the case of participants who missed the final visit but whose GP confirmed that no additional antibacterials were prescribed during the follow-up period, day 28 was used as the censoring date.
Kaplan–Meier estimates were used to derive the risk difference between the randomised groups for the primary end point, and standard errors and CIs for the risk difference were derived from the estimated standard errors of the individual survival functions.
Lower-dose treatment and shorter-duration treatment were considered ‘non-inferior’ to higher-dose treatment and longer-duration treatment, respectively, if the upper limit of the two-sided 90% CI for the difference in the proportion of children with the primary end point at day 28 was less than the non-inferiority margin of 8%. Although the non-inferiority margin was important to the design of the trial, it is less relevant to its interpretation, which should be based on observed estimates and CIs.
Sensitivity analyses
As described in Primary outcome, the primary analysis included only end points confirmed by the ERC as clinically indicated antibacterial treatment for respiratory tract infection (including CAP). To improve confidence in the primary analysis, the following sensitivity analyses were performed for the primary end point:
-
including all systemic antibacterial treatments other than trial medication regardless of reason and indication
-
including only ERC-adjudicated clinically indicated systemic antibacterial treatment where either CAP or ‘chest infection’ was specified as the reason for this treatment (rather than any respiratory tract infection)
-
as above, but also including, as an end point, all systemic antibacterial treatments for CAP or ‘chest infection’ where the clinical indication was ‘unlikely’, as adjudicated by the ERC
-
disregarding systemic antibacterial prescriptions occurring within the first 3 days from randomisation, as these events cannot be related to the treatment duration randomisation, to allow comparison of shorter and longer treatment.
Subgroup analyses
Two subgroup analyses were performed. The first considered severity of CAP at enrolment to provide reassurance that a potential null effect was not due to dilution arising from inclusion of children with mild disease. The main efficacy analysis was repeated, but included only participants with severe CAP, defined as two or more of the following abnormal signs/symptoms at enrolment: raised respiratory rate (> 37 breaths/minute for children aged 1–2 years; > 28 breaths/minute for children aged 3–5 years), oxygen saturation < 92% in room air and presence of chest retractions.
The second subgroup analysis considered the potential for seasonal changes in infections, by including only primary end points occurring in the two winter seasons spanned by CAP-IT. This was based on Public Health England reports of circulating viruses/bacteria in the winter seasons spanned by CAP-IT.
Community-acquired pneumonia symptoms
The severity of the symptoms (detailed in Morbidity) were reviewed by the number (%) of symptoms in each severity category at each scheduled contact visit and analysed as described for ordered outcomes in Analysis principles.
Duration of a symptom was measured as time from baseline to resolution, defined as the first day the symptom was reported as not present. This was analysed as a time-to-event outcome, as specified in Analysis principles. Where a symptom was not present at enrolment, participants were excluded from the analysis of that symptom.
Clinical adverse events
Solicited clinical AEs, specified in Morbidity, were analysed overall and by randomised arm. Analysis considered total number of events, number of participants with at least one event, the number of participants with at least one new event and event severity. These variables were analysed as described for binary outcomes in Analysis principles.
In addition, the number of participants experiencing at least one SAE were compared as a binary outcome (see Analysis principles).
Antimicrobial resistance
Descriptive analyses of baseline samples were analysed as follows: proportion of samples with positive S. pneumoniae culture, frequency distribution of broth microdilution MIC values and proportion of samples classified as S – susceptible, standard dosing regimen; I – intermediate, increased exposure; and R – resistant (see Secondary outcomes).
S. pneumoniae carriage, determined by tabulation of the proportion of samples with positive S. pneumoniae culture at the final visit by randomisation group, was compared using tests for binary variables, as described in Secondary outcomes. S. pneumoniae culture results at the final visit were cross-tabulated with baseline culture results (including missing values).
For the antimicrobial resistance analysis, a descriptive analysis of the proportion of samples with resistance to penicillin (S – susceptible/I – intermediate/R – resistant categorisation) at the final visit was performed using both cut-off points (penicillin and amoxicillin) described in Secondary outcomes. This analysis was repeated, first, including only samples with a positive S. pneumoniae culture result and, second, including all samples. Randomised groups were compared by tests for binary variables, and cross-tabulation of penicillin resistance at the final visit compared with penicillin resistance at baseline was performed as a descriptive analysis.
Finally, the change in broth microdilution MIC (in patients for whom this was measured at both the baseline and the final visit) was analysed with randomisation group as factors and after adjusting for baseline MIC.
Interim analyses
The trial was reviewed by the CAP-IT IDMC. They met three times over the course of the trial: once at a joint meeting with the Trial Steering Committee (TSC) in June 2017 and twice in strict confidence in January 2018 and January 2019. The IDMC reviewed unblinded safety and efficacy data and made recommendations through correspondence to the TSC following each meeting.
Patient and public involvement
Parents of young children were involved during the development and delivery of CAP-IT. A PPI representative was a member of the TSC, contributing at meetings and in an ad hoc fashion when required. When considering the research question, the trial team were advised by parents that shorter antibiotic courses would be welcomed if equally effective, because of difficulties in giving medicine (due to palatability or challenges with day care and daytime doses). For the same reasons, parents supported the twice-daily dosing of the CAP-IT. Multiple PPI representatives reviewed and provided input on the patient information materials, including the CAP-IT information film, to ensure that they were clear, easy to understand and not off-putting to parents, while still providing sufficient detail to allow informed consent. Valuable input was provided from the PPI representative on the CAP-IT TSC on the plan for dissemination of the CAP-IT results.
Protocol amendments
The CAP-IT protocol v.2.0 was active when recruitment to CAP-IT commenced in January 2017. Two protocol amendments were completed subsequently, with version 3 implemented in September 2017 and version 4 in December 2018. Amendments were largely in relation to selection criteria (see Exclusion criteria) and the SAP (to which three significant updates were made on the basis of accumulating trial data). First, a stratified analysis was originally planned based on the PED and ward groups. This was changed to a joint analysis in protocol version 3 because of significant clinical overlap (see Appendix 1 for more details) Second, the primary end-point definition was made more specific in protocol version 3 and further refined in version 4 (see Changes to primary end point). Finally, the non-inferiority margin was adjusted, as the primary end-point event rate had been substantially underestimated. The trial and all substantial amendments were approved by the London – West London & GTAC Research Ethics Committee (reference 16/LO/0831).
Chapter 3 Results
Participant flow
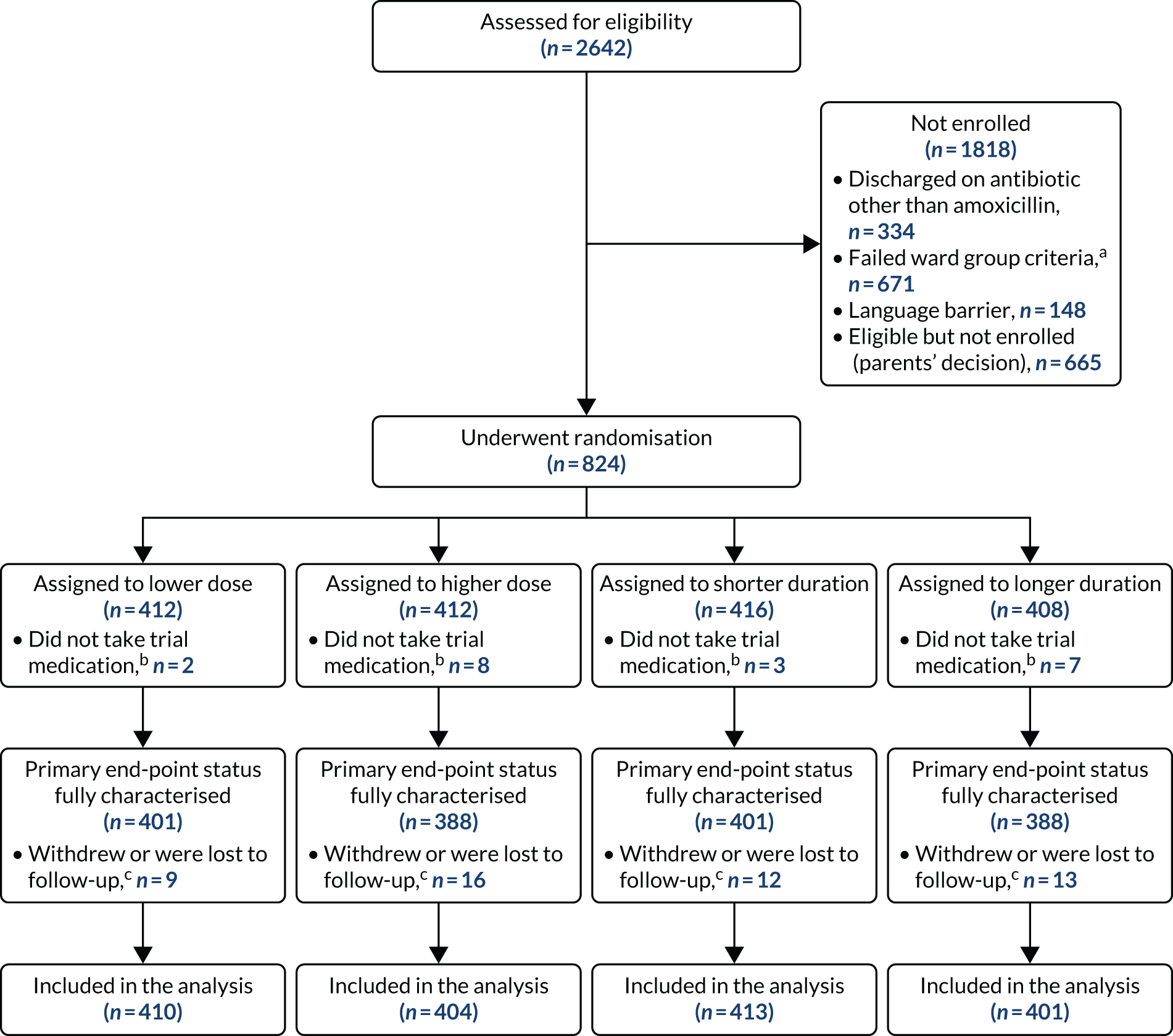
Between 1 February 2017 and 23 April 2019, a total of 2642 children were assessed for eligibility and 824 were randomised. Ten patients were randomised but received no trial medication (owing to, for example, a change of mind by parent/guardian or administrative error) and were, therefore, excluded from the analysis, resulting in an analysis population of 814 patients.
A total of 591 participants had no pre-treatment antibiotic at trial entry. A total of 223 (mainly following admission to assessment units of wards) had received beta-lactam antibiotic pre-treatment for no more than 48 hours. The final follow-up visit occurred on 21 May 2019, which was considered the trial end date.
Six participants were randomised in error but were included in the analysis in accordance with the ITT principle. Of these participants, five did not have all the required symptoms to fulfil the criteria for CAP diagnosis (see Box 1). One patient did not have a cough reported in the previous 96 hours at presentation, two patients did not have a reported fever in the previous 48 hours at presentation and two patients lacked documentation of signs of laboured/difficult breathing and/or focal chest signs at presentation. In one of the final two patients, chest radiography was suggestive of lobar pneumonia, prior to this being added to the inclusion criteria as part of protocol version 4.0, and in the other participant pneumonia was diagnosed on chest radiography, but was documented as patchy infiltrate, which did not fulfil the inclusion criteria. The final patient randomised in error received an antibiotic other than a beta-lactam (clarithromycin) before discharge (Table 3).
| Reason for ineligibility | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Known violation of any inclusion/exclusion criterion | 1 (0.2) | 5 (1.2) | 4 (1.0) | 2 (0.5) | 6 (0.7) |
| No presence of cough | 0 | 1 | 0 | 1 | 1 |
| No presence of fever | 0 | 2 | 2 | 0 | 2 |
| No presence of CAP signs | 0 | 2 | 2 | 0 | 2 |
| Pre-treatment with non-beta-lactams | 1 | 0 | 0 | 1 | 1 |
| Excluded from analysis | 0 | 0 | 0 | 0 | 0 |
Participants were well distributed between arms, with 208 (25.6%) participants receiving 3 days of lower-dose treatment, 202 (24.8%) participants receiving 7 days of lower-dose treatment, 205 (25.2%) participants receiving 3 days of higher-dose treatment and 199 (24.4%) participants receiving 7 days of higher-dose treatment (Figure 3 and Table 4).
FIGURE 3.
A CONSORT (Consolidated Standards of Reporting Trials) flow diagram. a, Inpatient stay > 48 hours and treated with non-beta-lactam antibiotics as inpatients; b, these children have been excluded from all analyses; and c, follow-up included up to time of withdrawal or no further contact.
| Outcome | Treatment arm, n (%) | Total (N = 814), n (%) | |
|---|---|---|---|
| PED (N = 591) | Ward (N = 223) | ||
| Randomisation arm | |||
| Lower dose plus shorter duration | 153 (25.9) | 55 (24.7) | 208 (25.6) |
| Lower dose plus longer duration | 150 (25.4) | 52 (23.3) | 202 (24.8) |
| Higher dose plus shorter duration | 146 (24.7) | 59 (26.5) | 205 (25.2) |
| Higher dose plus longer duration | 142 (24.0) | 57 (25.6) | 199 (24.4) |
| Dose randomisation | |||
| Lower | 303 (51.3) | 107 (48.0) | 410 (50.4) |
| Higher | 288 (48.7) | 116 (52.0) | 404 (49.6) |
| Duration randomisation | |||
| Shorter | 299 (50.6) | 114 (51.1) | 413 (50.7) |
| Longer | 292 (49.4) | 109 (48.9) | 401 (49.3) |
Baseline
Patient characteristics
Baseline patient characteristics were well balanced between the randomisation groups (see Table 4). The median age of participants was 2.5 (IQR 1.6–3.7) years, with a minimum and maximum age of 0.5 and 8.8 years, respectively, and 52% were male (Table 5).
| Characteristic | Treatment arm | Total (N = 814) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Age (years) | |||||
| Median (IQR) | 2.5 (1.6–3.7) | 2.4 (1.6–3.7) | 2.5 (1.7–3.7) | 2.5 (1.5–3.7) | 2.5 (1.6–3.7) |
| Minimum, maximum | 0.5, 8.8 | 0.5, 8.5 | 0.5, 8.5 | 0.5, 8.8 | 0.5, 8.8 |
| Sex, n (%) | |||||
| Male | 210 (51) | 211 (52) | 217 (53) | 204 (51) | 421 (52) |
| Female | 200 (49) | 193 (48) | 196 (47) | 197 (49) | 393 (48) |
| Ethnicity, n (%) | |||||
| White | 275 (67) | 279 (69) | 283 (69) | 271 (68) | 554 (68) |
| Asian or British Asian | 55 (13) | 51 (13) | 53 (13) | 53 (13) | 106 (13) |
| Black or black British | 40 (10) | 36 (9) | 40 (10) | 36 (9) | 76 (9) |
| Other | 40 (10) | 38 (9) | 37 (9) | 41 (10) | 78 (10) |
| Number (%) of households with smokers | 69 (17) | 62 (16) | 61 (15) | 70 (18) | 131 (16) |
Medical history
One-third of participants (30.7%) reported an underlying diagnosis of asthma or use of an asthma inhaler within the past month. The second most common comorbidity (affecting 20% of participants) was eczema, followed by food or drug allergies (9.6%) and hay fever (9.1%). Routine vaccinations had been received by 95% of participants; the remaining 5% either had not had routine vaccinations (3.2%), or were of unknown vaccination status or had been vaccinated outside the UK (1.8%).
Vital parameters and clinical signs
Participant vital parameters were measured at presentation and were similar between randomisation groups (Table 6). The median temperature was 38.1 °C (IQR 37.2–38.8 °C) and median oxygen saturation was 96% (IQR 95–98%). The median number of days for which a child had a cough at presentation was 4 (IQR 2–7) days, and the median number of days for which a child had a temperature was 3 (IQR 1–4) days. The median weight was 13.5 (IQR 11.2–16.4) kg.
| Medical history | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Asthma or inhaler use within past month | 119 (29) | 136 (34) | 125 (30) | 130 (32) | 255 (31) |
| Hay fever | 34 (8) | 40 (10) | 37 (9) | 37 (9) | 74 (9) |
| Food or drug allergy | 38 (9) | 40 (10) | 37 (9) | 41 (10) | 78 (10) |
| Eczema | 84 (20) | 79 (20) | 78 (19) | 85 (21) | 163 (20) |
| Prematurity | 43 (10) | 43 (11) | 51 (12) | 35 (9) | 86 (11) |
| Routine vaccinations? | |||||
| Yes | 388 (95) | 385 (95) | 394 (95) | 379 (95) | 773 (95) |
| No | 14 (3) | 12 (3) | 15 (4) | 11 (3) | 26 (3) |
| Not sure (or vaccinated outside UK) | 8 (2) | 7 (2) | 4 (1) | 11 (3) | 15 (2) |
| Other underlying disease | 37 (9) | 19 (5) | 21 (5) | 35 (9) | 56 (7) |
The most common baseline clinical signs were coryza [affecting 599/814 (73.6%) participants] and chest retractions [affecting 483/814 (59.3%) participants] (Table 7). Other baseline clinical signs were less common (enlarged tonsils or pharyngitis, 22.5%; pallor, 20.9%; nasal flaring, 9.3%, inflamed/bulging tympanic membrane or middle ear effusion, 9%; and stridor, 1.2%).
| Parameter/clinical sign | Treatment arm | Total (N = 814) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Weight (kg), median (IQR) | 13.6 (11.2–16.8) | 13.3 (11.1–16.2) | 13.8 (11.5–16.4) | 13.2 (10.9–16.4) | 13.5 (11.2–16.4) |
| Temperature (°C), median (IQR) | 38.1 (37.3–38.9) | 38.0 (37.2–38.6) | 38.0 (37.1–38.7) | 38.1 (37.3–38.8) | 38.1 (37.2–38.8) |
| Temperature ≥ 38 °C, n (%) | 227 (55) | 214 (53) | 221 (54) | 220 (55) | 441 (54) |
| Heart rate (b.p.m.), median (IQR) | 146 (131–160) | 143 (130–158) | 144 (131–158) | 146 (130–162) | 145 (130–160) |
| Abnormal heart rate,a n (%) | 307 (75) | 271 (67) | 282 (68) | 296 (74) | 578 (71) |
| Respiratory rate (breaths/minute), median (IQR) | 37 (30–44) | 38 (32–44) | 36 (30–43) | 38 (32–45) | 37 (30–44) |
| Abnormal respiratory rate,b n (%) | 270 (66) | 258 (64) | 262 (64) | 266 (67) | 528 (65) |
| Oxygen saturation (%), median (IQR) | 96 (95–98) | 96 (95–98) | 96 (95–98) | 96 (95–98) | 96 (95–98) |
| Abnormal oxygen saturation,c n (%) | 18 (4) | 25 (6) | 18 (4) | 25 (6) | 43 (5) |
| Nasal flaring, n (%) | 33 (8) | 42 (10) | 35 (9) | 40 (10) | 75 (9) |
| Chest retractions, n (%) | 239 (58) | 244 (60) | 239 (58) | 244 (61) | 483 (59) |
| Pallor, n (%) | 82 (20) | 87 (22) | 93 (23) | 76 (19) | 169 (21) |
| Stridor, n (%) | 4 (1) | 6 (1) | 5 (1) | 5 (1) | 10 (1) |
| Inflamed/bulging tympanic membrane or middle ear effusion, n (%) | 37 (9) | 35 (9) | 39 (10) | 33 (8) | 72 (9) |
| Coryza, n (%) | 291 (71) | 308 (76) | 304 (74) | 295 (74) | 599 (74) |
| Enlarged tonsils or pharyngitis, n (%) | 95 (24) | 86 (22) | 92 (22) | 89 (23) | 181 (23) |
Multiple vital parameters and clinical signs differed at presentation between the children previously exposed and unexposed to antibiotics (see Table 7).
Chest examination
Chest examination findings at presentation were reported as absent, bilateral or unilateral. Unilateral findings were present in 691 (85%) participants overall, featuring as crackles/crepitations in 562 (71%) participants, reduced breath sounds in 336 (44%) participants, bronchial breathing in 103 (15%) participants and dullness to percussion in 59 (13%) participants. The proportions of the four chest examination variables were very similar among the randomisation arms (Table 8).
| Chest examination finding | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Dullness to percussion | |||||
| Absent | 194 (86) | 186 (86) | 198 (86) | 182 (86) | 380 (86) |
| Unilateral | 32 (14) | 27 (13) | 31 (13) | 28 (13) | 59 (13) |
| Bilateral | 0 (0) | 3 (1) | 1 (< 1) | 2 (1) | 3 (1) |
| Bronchial breathing | |||||
| Absent | 283 (82) | 263 (82) | 276 (83) | 270 (81) | 546 (82) |
| Unilateral | 53 (15) | 50 (16) | 49 (15) | 54 (16) | 103 (15) |
| Bilateral | 10 (3) | 7 (2) | 8 (2) | 9 (3) | 17 (3) |
| Reduced breath sounds | |||||
| Absent | 202 (52) | 187 (49) | 202 (51) | 187 (50) | 389 (50) |
| Unilateral | 168 (43) | 168 (44) | 174 (44) | 162 (43) | 336 (44) |
| Bilateral | 20 (5) | 26 (7) | 20 (5) | 26 (7) | 46 (6) |
| Crackles/crepitations | |||||
| Absent | 69 (17) | 65 (17) | 71 (18) | 63 (16) | 134 (17) |
| Unilateral | 287 (71) | 275 (70) | 290 (72) | 272 (69) | 562 (71) |
| Bilateral | 48 (12) | 52 (13) | 42 (10) | 58 (15) | 100 (13) |
Parent/guardian-reported community-acquired pneumonia symptoms
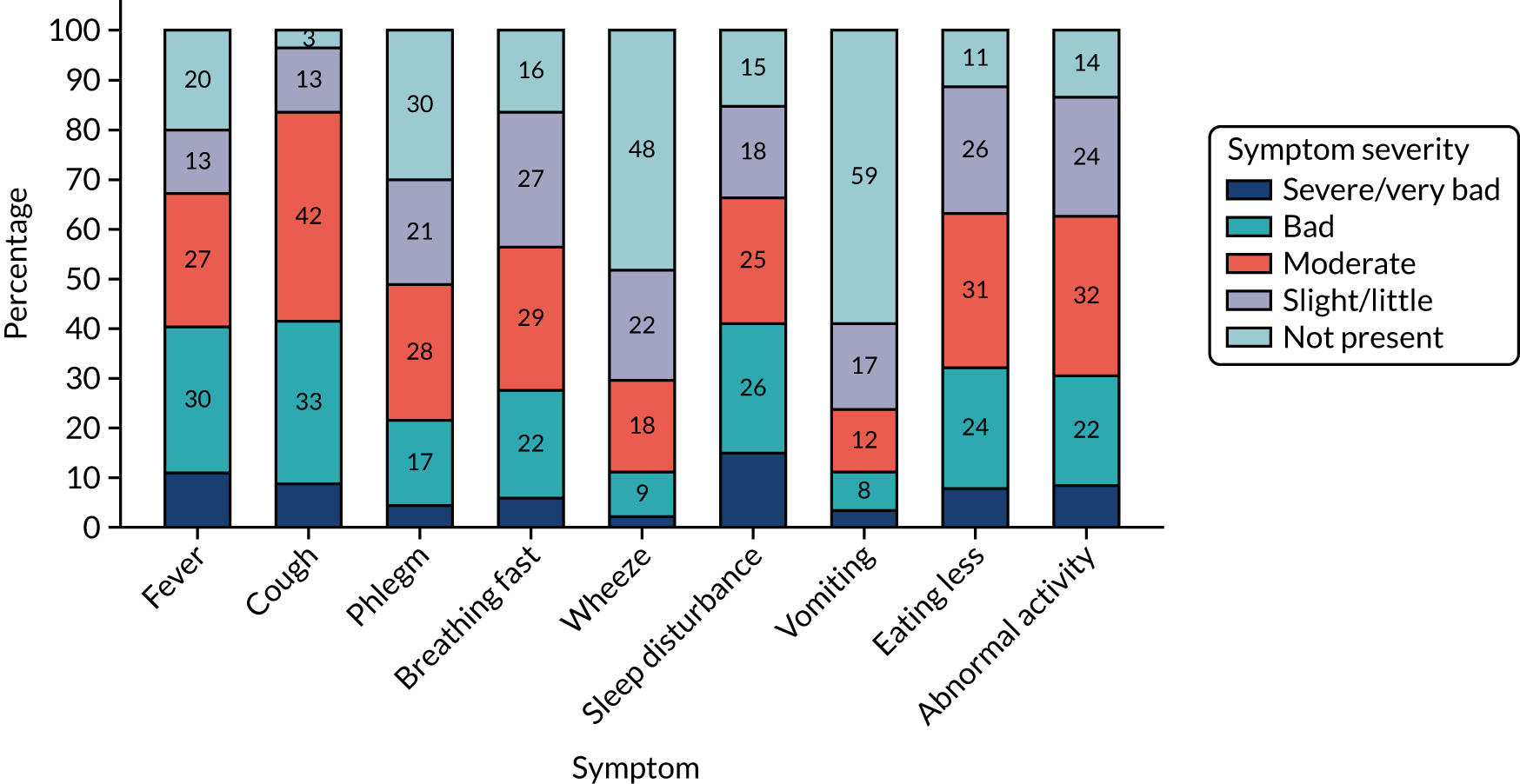
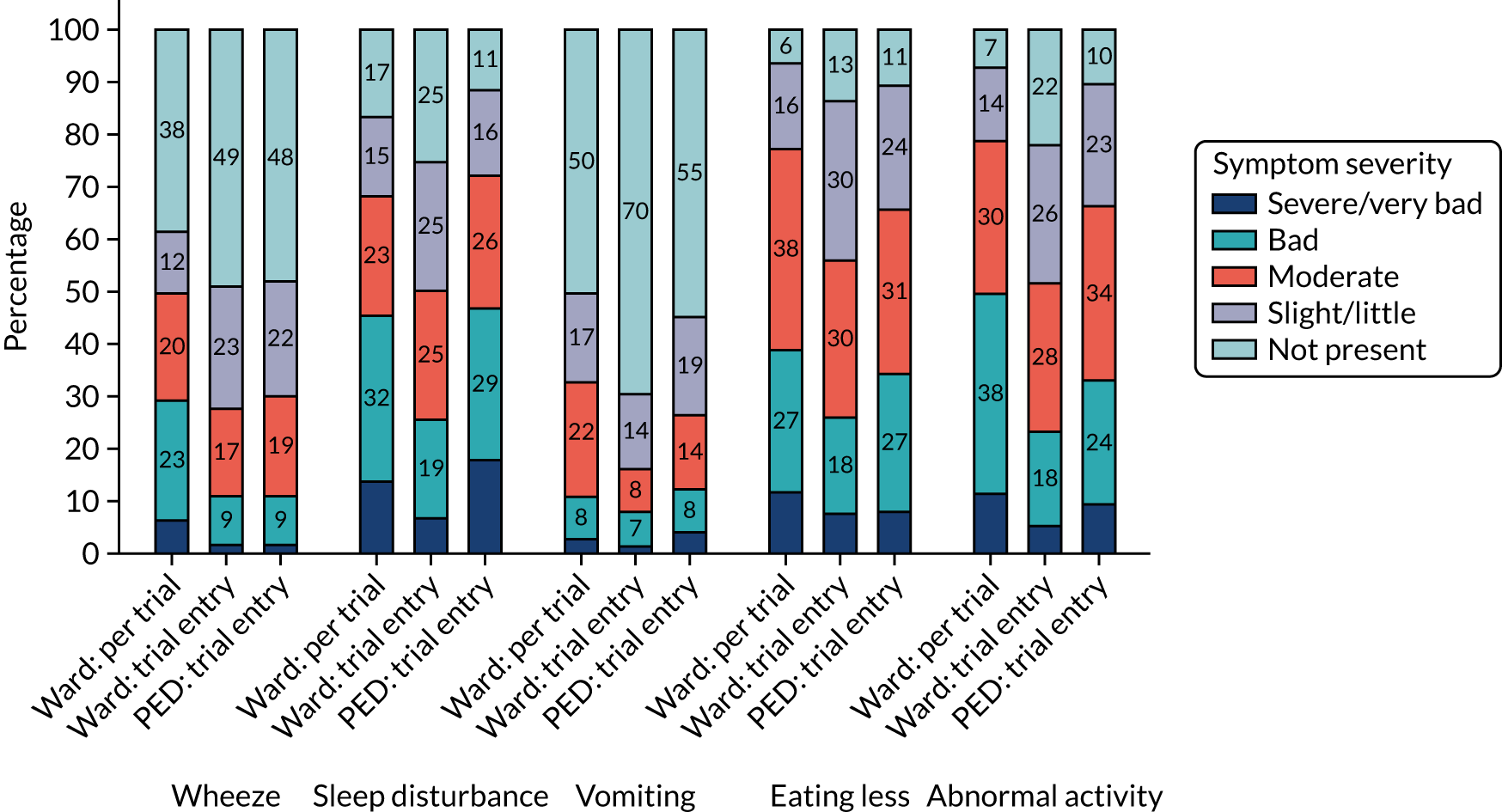
Parent/guardian-reported symptom severity at trial entry is shown in Figure 4. The most common clinical symptom was cough, reported by 96.5% of participants. Fever and fast breathing were reported for 79.6% and 83.5% of participants, respectively, and the least common symptoms at baseline were vomiting and wheeze, reported in 41.1% and 51.8% of participants, respectively. Sleep disturbance, eating less and interference with normal activity were reported in between 80% and 90% of participants.
FIGURE 4.
Symptoms at trial entry.
Clinical symptoms in patients who received in-hospital antibiotics prior to trial entry (i.e. the ward group) were reported by parents/guardians at presentation (pre trial) and at baseline (trial entry). Figures 5 and 6 show parent/guardian-reported clinical symptom severity both pre trial and at trial entry for the ward group and at trial entry only for the PED group. For the ward group, the proportion of participants with presence of symptoms at any level of severity decreased between pre trial and trial entry for all symptoms except wet cough (phlegm). The greatest proportional decrease was for fever, for which the proportion of participants with a severity of slight/little or greater decreased from 87.9% to 50.2%.
FIGURE 5.
Clinical symptoms (i.e. fever, cough, phlegm and breathing fast) at trial entry, by group.
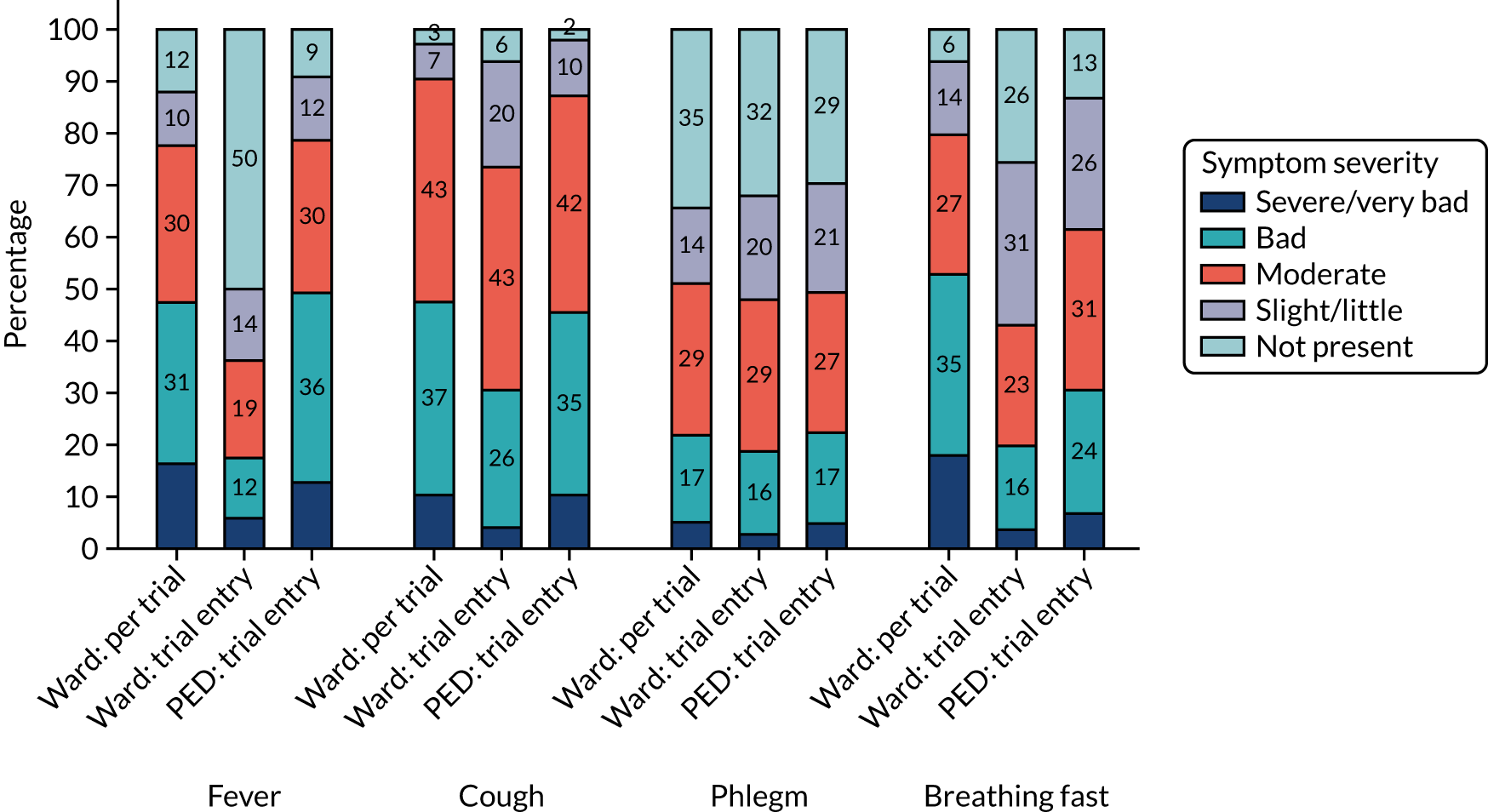
FIGURE 6.
Clinical symptoms (i.e. wheeze, sleep disturbance, vomiting, eating less and abnormal activity) at trial entry, by group.
Community-acquired pneumonia symptoms at trial entry, by stratum, are shown in Appendix 2, Table 27.
Clinical investigations
Clinical investigations, including chest radiography, haematology assessment, biochemistry assessment, blood culture and respiratory samples, were not mandatory in CAP-IT. However, if any of these investigations were undertaken, results were reported.
Chest radiography was the most common investigation and was undertaken in 391 (48%) participants (Table 9). Haematological and biochemical assessments were undertaken in 81 (10%) and 82 (10.1%) participants, respectively, while blood cultures and respiratory specimens were obtained in 41 (5%) and 46 (5.7%) participants, respectively.
| Result of chest radiography | Treatment arm, n (%) | Total (N = 391), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 192) | Higher dose (N = 199) | Shorter duration (N = 196) | Longer duration (N = 195) | ||
| Suggestive of pneumonia: lobar infiltrate | 65 (33.9) | 69 (34.7) | 64 (32.7) | 70 (35.9) | 134 (34.3) |
| Suggestive of pneumonia: patchy infiltrate | 72 (37.5) | 82 (41.2) | 84 (42.9) | 70 (35.9) | 154 (39.4) |
| Unsure if suggestive of pneumonia | 21 (10.9) | 16 (8.0) | 15 (7.7) | 22 (11.3) | 37 (9.5) |
| Other diagnosis | 7 (3.6) | 5 (2.5) | 6 (3.1) | 6 (3.1) | 12 (3.1) |
| No finding/not suggestive of pneumonia | 27 (14.1) | 27 (13.6) | 27 (13.8) | 27 (13.8) | 54 (13.8) |
Of the 46 respiratory samples taken, 44 samples underwent virology assessment and 11 samples underwent bacteriology assessment (Table 10). All 11 of the respiratory samples subjected to bacteriological assessment showed no significant growth.
| Assessment result | Treatment arm, n (%) | Total (N = 44), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 19) | Higher dose (N = 25) | Shorter duration (N = 24) | Longer duration (N = 20) | ||
| Type of respiratory sample for virology | |||||
| Nasopharyngeal | 13 (68) | 21 (84) | 20 (83) | 14 (70) | 34 (77) |
| Oropharyngeal | 6 (32) | 4 (16) | 4 (17) | 6 (30) | 10 (23) |
| Respiratory sample for virology: result | |||||
| Rhinovirus | 5 (26) | 7 (28) | 6 (25) | 6 (30) | 12 (27) |
| Influenza A/B | 1 (5) | 1 (4) | 0 (0) | 2 (10) | 2 (5) |
| Adenovirus | 0 (0) | 1 (4) | 1 (4) | 0 (0) | 1 (2) |
| Rhinovirus plus adenovirus | 2 (11) | 1 (4) | 2 (8) | 1 (5) | 3 (7) |
| Rhinovirus plus enterovirus | 4 (21) | 5 (20) | 5 (21) | 4 (20) | 9 (20) |
| Rhinovirus plus enterovirus plus adenovirus | 0 (0) | 1 (4) | 0 (0) | 1 (5) | 1 (2) |
| Rhinovirus plus enterovirus plus coronavirus | 1 (5) | 0 (0) | 1 (4) | 0 (0) | 1 (2) |
| Human metapneumovirus | 1 (5) | 2 (8) | 2 (8) | 1 (5) | 3 (7) |
| No viral isolate present | 5 (26) | 7 (28) | 7 (29) | 5 (25) | 12 (27) |
Finally, of the 40 blood samples taken for culture, 37 (93%) returned a negative result. The three positive results were considered probably due to contamination, with two identifying as coagulase-negative staphylococci and one identifying as Gram-positive cocci (not further differentiated).
Prior antibiotic exposure
A total of 242 (29.7%) children received antibiotics for up to 48 hours prior to enrolment, of whom 241 received beta-lactam antibiotics and one received a macrolide. Amoxicillin was the most common antibiotic taken prior to trial entry (209/242, 86.4%), followed by co-amoxiclav (20/242, 8.3%). In children receiving antibiotics prior to enrolment, the median number of doses was 2 (IQR 1–3). More than half of children (55%) were enrolled within 12 hours of commencing antibiotic treatment, with 24.8% enrolled within 12–24 hours, 12.4% within 24–36 hours and 7.9% within 36–48 hours (Table 11).
| Prior exposure | Treatment arm | Total (N = 814) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Any systemic antibiotic in last 3 months, n (%) | |||||
| Yes | 64 (16) | 65 (16) | 66 (16) | 63 (16) | 129 (16) |
| No | 346 (84) | 339 (84) | 347 (84) | 338 (84) | 685 (84) |
| Antibiotics received in last 48 hours?, n (%) | |||||
| Yes | 119 (29) | 123 (30) | 123 (30) | 119 (30) | 242 (30) |
| No | 291 (71) | 281 (70) | 290 (70) | 282 (70) | 572 (70) |
| Class of prior antibiotic, n (%) | |||||
| Beta-lactam | 118 (99) | 123 (100) | 123 (100) | 118 (99) | 241 (100) |
| Macrolide | 1 (1) | 0 (0) | 0 (0) | 1 (1) | 1 (< 1) |
| Prior antibiotic, n (%) | |||||
| Amoxicillin | 103 (87) | 106 (86) | 104 (85) | 105 (88) | 209 (86) |
| Benzylpenicillin | 1 (1) | 2 (2) | 1 (1) | 2 (2) | 3 (1) |
| Ceftriaxone | 2 (2) | 4 (3) | 3 (2) | 3 (3) | 6 (2) |
| Cefuroxime | 2 (2) | 0 (0) | 2 (2) | 0 (0) | 2 (1) |
| Clarithromycin | 1 (1) | 0 (0) | 0 (0) | 1 (1) | 1 (< 1) |
| Co-amoxiclav | 9 (8) | 11 (9) | 13 (11) | 7 (6) | 20 (8) |
| Phenoxymethylpenicillin | 1 (1) | 0 (0) | 0 (0) | 1 (1) | 1 (< 1) |
| Number of prior antibiotic doses, median (IQR) | 2 (1–3) | 2 (1–3) | 2 (1–3) | 2 (1–3) | 2 (1–3) |
| Prior antibiotic: route, n (%) | |||||
| Intravenous | 15 (13) | 10 (8) | 17 (14) | 8 (7) | 100 (41) |
| Oral | 103 (87) | 110 (89) | 106 (86) | 107 (90) | 85 (35) |
| Intravenous plus oral | 1 (1) | 3 (2) | 0 (0) | 4 (3) | 28 (12) |
| Duration (hours) of prior antibiotic treatment, n (%) | |||||
| < 12 | 67 (56) | 66 (54) | 68 (55) | 65 (55) | 133 (55) |
| 12–24 | 27 (23) | 33 (27) | 33 (27) | 27 (23) | 60 (25) |
| 24–36 | 13 (11) | 17 (14) | 13 (11) | 17 (14) | 30 (12) |
| 36–48 | 12 (10) | 7 (6) | 9 (7) | 10 (8) | 19 (8) |
Other medical interventions in exposed group
In addition, 54.3% of children in the ward group received supportive measures, including oxygen (49.3%), nasogastric feeds or fluids (2.7%), parenteral fluids (8.5%) and chest physiotherapy (2.7%). Finally, 82.1% of children in the ward group received pharmacological treatment other than antibiotics in hospital, including salbutamol inhalers (58.3%), paracetamol (52.1%), steroids (22.9%), ibuprofen (15.7%) and ipratropium bromide (8.3%).
Follow-up
Of the 814 patients included in the analysis, 642 (79%) completed the final assessment. Where possible, this final assessment was carried out face to face at hospital or at home, but if this proved impossible (e.g. if parents/guardians were unable to attend an appointment), then the assessment was completed by telephone. Overall, 25% of final assessments were performed by telephone, 74% were performed in hospital and 1% were performed at home. In 172 (21%) participants, the final assessment was not conducted with the family. Of these 172 participants, 11 had withdrawn consent and a further 161 could not be contacted. However, 150 of these participants (87%) had provided consent for collection of the primary outcome via hospital and GP records, and primary outcome data were successfully collected in 144 of these participants. This ensured that primary outcome data were available for 786 (97%) participants, and only 28 participants (3%) were considered withdrawn or lost to follow-up (Table 12).
| Final visit and follow-up data | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Attendance | |||||
| Final visit completed | 329 (80) | 313 (77) | 315 (76) | 327 (82) | 642 (79) |
| Previously withdrawn | 8 (2) | 3 (1) | 6 (1) | 5 (1) | 11 (1) |
| Not withdrawn but not completed | 73 (18) | 88 (22) | 92 (22) | 69 (17) | 161 (20) |
| Where/how did final visit take place? | |||||
| Hospital | 242 (74) | 236 (75) | 231 (73) | 247 (76) | 478 (74) |
| Home | 3 (1) | 3 (1) | 3 (1) | 3 (1) | 6 (1) |
| Telephone call | 84 (26) | 74 (24) | 81 (26) | 77 (24) | 158 (25) |
| Consent for further data collection? | |||||
| Yes | 71 (88) | 79 (87) | 87 (89) | 63 (85) | 150 (87) |
| No | 10 (12) | 12 (13) | 11 (11) | 11 (15) | 22 (13) |
| Day 28 data received from GP? | |||||
| Yes | 70 (99) | 74 (94) | 84 (97) | 60 (95) | 144 (96) |
| No | 1 (1) | 5 (6) | 3 (3) | 3 (5) | 6 (4) |
| Final visit status | |||||
| Completed | 329 (80) | 313 (77) | 315 (76) | 327 (82) | 642 (79) |
| Not completed, but GP data received | 70 (17) | 74 (18) | 84 (20) | 60 (15) | 144 (18) |
| Withdrawn/lost | 11 (3) | 17 (4) | 14 (3) | 14 (3) | 28 (3) |
Follow-up data were also collected by telephone at days 3, 7, 14 and 21 (Table 13). Follow-up rates were 88% at day 3, 75% at day 14 and 76% at day 21. A total of 443 (54%) parents/guardians of participants completed all telephone calls and the final visit, with 153 (19%) parents/guardians of participants missing one follow-up visit, 95 (12%) parents/guardians of participants missing two follow-up visits, 51 (6%) parents/guardians of participants missing three follow-up visits and 48 (6%) parents/guardians of participants missing four follow-up visits. Twenty-four (3%) parents/guardians of participants missed all telephone calls and visits.
| Follow-up | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Trial entry | 410 (100) | 404 (100) | 413 (100) | 401 (100) | 814 (100) |
| Day 3 | 355 (87) | 360 (89) | 365 (88) | 350 (87) | 715 (88) |
| Day 7 | 332 (81) | 343 (85) | 342 (83) | 333 (83) | 675 (83) |
| Day 14 | 314 (77) | 299 (74) | 307 (74) | 306 (76) | 613 (75) |
| Day 21 | 315 (77) | 302 (75) | 303 (73) | 314 (78) | 617 (76) |
| Final visit (day 28) | 329 (80) | 313 (77) | 315 (76) | 327 (82) | 642 (79) |
A symptom diary was to be completed daily by parents/guardians for the first 14 days after trial entry. Completed diary data were available for 406 (49.9%) participants and no diary data were available for 227 (27.9%) participants. Parents/guardians were assigned to complete symptom diaries either electronically (42.5%) or on paper (57.5%) using pseudorandomisation. Summary data on diary completion are presented in Table 14.
| Diary completion | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| Diary status | |||||
| Completed: all days | 201 (49.0) | 205 (50.7) | 212 (51.3) | 194 (48.4) | 406 (49.9) |
| Completed: partly | 97 (23.7) | 84 (20.8) | 79 (19.1) | 102 (25.4) | 181 (22.2) |
| No diary data available | 112 (27.3) | 115 (28.5) | 122 (29.5) | 105 (26.2) | 227 (27.9) |
| Number of days completed | |||||
| None | 112 (27.3) | 115 (28.5) | 122 (29.5) | 105 (26.2) | 227 (27.9) |
| 1–4 | 26 (6.3) | 11 (2.7) | 14 (3.4) | 23 (5.7) | 37 (4.5) |
| 5–8 | 27 (6.6) | 32 (7.9) | 33 (8.0) | 26 (6.5) | 59 (7.2) |
| 9–12 | 44 (10.7) | 41 (10.1) | 32 (7.7) | 53 (13.2) | 85 (10.4) |
| 13 | 201 (49.0) | 205 (50.7) | 212 (51.3) | 194 (48.4) | 406 (49.9) |
| No diary data: reason | |||||
| Withdrawal | 7 (6.3) | 2 (1.7) | 5 (4.1) | 4 (3.8) | 9 (4.0) |
| Paper: no final visit | 40 (35.7) | 48 (41.7) | 49 (40.2) | 39 (37.1) | 88 (38.8) |
| Paper: final visit as telephone call | 23 (20.5) | 18 (15.7) | 17 (13.9) | 24 (22.9) | 41 (18.1) |
| Lost/forgot | 21 (18.8) | 19 (16.5) | 24 (19.7) | 16 (15.2) | 40 (17.6) |
| Technical/password issue | 8 (7.1) | 13 (11.3) | 11 (9.0) | 10 (9.5) | 21 (9.3) |
| No time | 4 (3.6) | 6 (5.2) | 6 (4.9) | 4 (3.8) | 10 (4.4) |
| Site error | 0 (0.0) | 1 (0.9) | 1 (0.8) | 0 (0.0) | 1 (0.4) |
| Unknown | 9 (8.0) | 8 (7.0) | 9 (7.4) | 8 (7.6) | 17 (7.5) |
Adherence
A total of 240 (29.5%) participants deviated from the prescribed IMP regimen for reasons including taking fewer doses or a lower volume, taking too many doses or a greater volume, or deviation in timing (Table 15).
| Adherence to trial medication | Treatment arm, n (%) | p-value | Treatment arm, n (%) | p-value | Total (N = 814), n (%) | ||
|---|---|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||||
| Early cessation of trial treatment | |||||||
| Trial treatment completed | 355 (86.6) | 366 (90.6) | 0.10 | 358 (86.7) | 363 (90.5) | 0.015 | 721 (88.6) |
| Early cessation for clinical improvement | 7 (1.7) | 1 (0.2) | 5 (1.2) | 3 (0.7) | 8 (1.0) | ||
| Early cessation for clinical deterioration | 16 (3.9) | 11 (2.7) | 10 (2.4) | 17 (4.2) | 27 (3.3) | ||
| Early cessation for other reason | 32 (7.8) | 26 (6.4) | 40 (9.7) | 18 (4.5) | 58 (7.1) | ||
| Day of last dose of trial medication | |||||||
| Day 0 or 1 | 11 (20) | 4 (11) | 0.62 | 9 (16) | 6 (16) | 0.61 | 15 (16) |
| Day 2 or 3 | 17 (31) | 15 (39) | 16 (29) | 16 (42) | 32 (34) | ||
| Day 4 or 5 | 22 (40) | 15 (39) | 24 (44) | 13 (34) | 37 (40) | ||
| Day 6 or after | 5 (9) | 4 (11) | 6 (11) | 3 (8) | 9 (10) | ||
| Bottles received | |||||||
| Taken bottle A but not bottles B/C | 30 (7.3) | 16 (4.0) | 0.038 | 21 (5.1) | 25 (6.2) | 0.48 | 46 (5.7) |
| Taken bottle A and bottles B/C | 380 (92.7) | 388 (96.0) | 392 (94.9) | 376 (93.8) | 768 (94.3) | ||
| Overall: fewer doses taken than scheduled | |||||||
| Yes | 86 (21.0) | 77 (19.1) | 0.49 | 85 (20.6) | 78 (19.5) | 0.69 | 163 (20.0) |
| No | 324 (79.0) | 327 (80.9) | 328 (79.4) | 323 (80.5) | 651 (80.0) | ||
| Overall: fewer doses or less volume taken than scheduled | |||||||
| Yes | 104 (25.4) | 95 (23.5) | 0.54 | 113 (27.4) | 86 (21.4) | 0.050 | 199 (24.4) |
| No | 306 (74.6) | 309 (76.5) | 300 (72.6) | 315 (78.6) | 615 (75.6) | ||
| Overall: any deviation (including too many doses/volume or timing deviations) | |||||||
| Yes | 128 (31.2) | 107 (26.5) | 0.14 | 133 (32.2) | 102 (25.4) | 0.033 | 235 (28.9) |
| No | 282 (68.8) | 297 (73.5) | 280 (67.8) | 299 (74.6) | 579 (71.1) | ||
For dose randomisation, there was no evidence of an overall difference in adherence deviation between the two arms (p = 0.21). However, a greater proportion of participants in the lower-dose arm (7.3%) than in the higher-dose arm (4%) did not take bottle B/C as prescribed (p = 0.038).
For duration randomisation, 134 (32.4%) participants in the shorter-duration arm deviated, compared with 106 (26.4%) participants in the longer-duration arm (p = 0.06). A greater proportion of participants in the shorter-duration arm (13.3%) than in the longer-duration arm (9.4%) did not complete trial treatment (p = 0.015) (see Table 15).
Primary outcome
End-Point Review Committee results
There were 143 events of non-trial systemic antibacterial treatment in 139 participants (four participants had two events). All events were adjudicated by the ERC (see Chapter 2, Primary outcome) and reasons for starting new non-trial antibacterials are given in Table 16. Of 139 participants, 100 (71.9%) met the criteria for a primary end point (see Table 16). Among the 100 participants who had an event that met the criteria for a primary end point, ‘CAP/chest infection’ was the most common reason for treatment, accounting for 76 (76%) events (see Table 16). The ERC adjudicated 38% of the events as definitely/probably clinically indicated and 62% of the events as possibly indicated (Table 17).
| Reason | Treatment arm (n) | Total (N) (N = 139) | |||
|---|---|---|---|---|---|
| Lower dose (N = 74) | Higher dose (N = 65) | Shorter duration (N = 73) | Longer duration (N = 66) | ||
| CAP/chest infection | 38 | 40 | 40 | 38 | 78 |
| Other respiratory tract infection | 19 | 12 | 18 | 13 | 31 |
| Otitis media | 7 | 3 | 6 | 4 | 10 |
| URTI | 7 | 2 | 4 | 5 | 9 |
| Tonsillitis | 3 | 5 | 5 | 3 | 8 |
| Othera | 2 | 2 | 3 | 1 | 4 |
| Other bacterial infection | 8 | 7 | 9 | 6 | 15 |
| Skin infection | 2 | 2 | 3 | 1 | 4 |
| Urinary tract infection | 2 | 2 | 3 | 1 | 4 |
| Cellulitis | 1 | 2 | 2 | 1 | 3 |
| Scarlet fever | 1 | 1 | 0 | 2 | 2 |
| Nail infection | 1 | 0 | 0 | 1 | 1 |
| Salmonella gastroenteritis | 1 | 0 | 1 | 0 | 1 |
| Other illness/injury | 4 | 2 | 3 | 3 | 6 |
| Appendicitis | 1 | 0 | 1 | 0 | 1 |
| Asthma | 0 | 1 | 0 | 1 | 1 |
| Bronchospasm/asthma | 1 | 0 | 1 | 0 | 1 |
| Dental abscess | 0 | 1 | 1 | 0 | 1 |
| Lymphadenitis | 1 | 0 | 0 | 1 | 1 |
| Prophylaxis | 1 | 0 | 0 | 1 | 1 |
| Intolerance to IMP/AE | 3 | 5 | 5 | 3 | 8 |
| Vomiting | 1 | 4 | 4 | 1 | 5 |
| Diarrhoea | 1 | 0 | 0 | 1 | 1 |
| Rash | 0 | 1 | 0 | 1 | 1 |
| Refusing IMP | 1 | 0 | 1 | 0 | 1 |
| Parental preference | 3 | 0 | 0 | 3 | 3 |
| Pharmacy/administration error | 1 | 1 | 2 | 0 | 2 |
| Primary end-point adjudication result | Treatment arm | Total | |||
|---|---|---|---|---|---|
| Lower dose | Higher dose | Shorter duration | Longer duration | ||
| Patients who started systemic non-trial antibacterials | |||||
| N | 74 | 65 | 73 | 66 | 139 |
| Patients who had a primary end point, n (%) | |||||
| Yes | 51 (69) | 49 (75) | 51 (70) | 49 (74) | 100 (712) |
| No | 23 (31) | 16 (25) | 22 (30) | 17 (26) | 39 (28) |
| Events that met the criteria for primary end point | |||||
| N | 51 | 49 | 51 | 49 | 100 |
| Primary reason for starting new antibacterials, n (%) | |||||
| CAP/chest infection | 37 (73) | 39 (80) | 39 (76) | 37 (76) | 76 (76) |
| Otitis media | 5 (10) | 3 (6) | 4 (8) | 4 (8) | 8 (8) |
| Tonsillitis | 3 (6) | 5 (10) | 5 (10) | 3 (6) | 8 (8) |
| URTI | 5 (10) | 2 (4) | 3 (6) | 4 (8) | 7 (7) |
| Other respiratory tract infection | 1 (2) | 0 (0) | 0 (0) | 1 (2) | 1 (1) |
| Clinical indication, n (%) | |||||
| Definitely/probably | 19 (37) | 19 (39) | 19 (37) | 19 (39) | 38 (38) |
| Possibly | 32 (63) | 30 (61) | 32 (63) | 30 (61) | 62 (62) |
| First new antibiotic, n (%) | |||||
| Amoxicillin | 25 (49) | 24 (49) | 23 (45) | 26 (53) | 49 (49) |
| Amoxicillin (i.v.) | 0 (0) | 1 (2) | 1 (2) | 0 (0) | 1 (1) |
| Azithromycin | 3 (6) | 1 (2) | 2 (4) | 2 (4) | 4 (4) |
| Azithromycin plus amoxicillin (i.v.) | 1 (2) | 0 (0) | 1 (2) | 0 (0) | 1 (1) |
| Cefuroxime | 0 (0) | 1 (2) | 0 (0) | 1 (2) | 1 (1) |
| Cefuroxime plus clarithromycin | 1 (2) | 0 (0) | 1 (2) | 0 (0) | 1 (1) |
| Clarithromycin | 8 (16) | 9 (18) | 13 (25) | 4 (8) | 17 (17) |
| Co-amoxiclav | 5 (10) | 5 (10) | 2 (4) | 8 (16) | 10 (10) |
| Co-amoxiclav plus azithromycin | 2 (4) | 0 (0) | 0 (0) | 2 (4) | 2 (2) |
| Co-amoxiclav (i.v.) | 1 (2) | 0 (0) | 1 (2) | 0 (0) | 1 (1) |
| Erythromycin | 3 (6) | 4 (8) | 3 (6) | 4 (8) | 7 (7) |
| Phenoxymethylpenicillin | 2 (4) | 4 (8) | 4 (8) | 2 (4) | 6 (6) |
| Who prescribed the antibiotic, n (%)a | |||||
| CAP-IT investigator | 3 (6) | 3 (7) | 3 (6) | 3 (7) | 6 (6) |
| Other hospital doctor | 18 (38) | 16 (36) | 17 (36) | 17 (37) | 34 (37) |
| GP | 24 (50) | 25 (56) | 27 (57) | 22 (48) | 49 (53) |
| Other | 3 (6) | 1 (2) | 0 (0) | 4 (9) | 4 (4) |
| Time new antibiotic started, n (%) | |||||
| Days 0–14 | 29 (57) | 25 (51) | 28 (55) | 26 (53) | 54 (54) |
| Days 15–28 | 22 (43) | 24 (49) | 23 (45) | 23 (47) | 46 (46) |
The most commonly prescribed antibacterial was oral amoxicillin, which was prescribed in 49 (49%) participants who met the criteria for a primary end point. Oral clarithromycin and co-amoxiclav accounted for 17% and 10% of prescriptions for participants who met the criteria for a primary end point, respectively, and erythromycin, phenoxymethylpenicillin and azithromycin accounted for 7%, 6% and 4%, respectively.
Analysis of primary end point
Overall
Overall, 100 participants in the analysis population (n = 814) met the criteria for a primary end point during the follow-up period (i.e. a cumulative proportion of 12.5%, 90% CI 10.7% to 14.6%, as estimated with Kaplan–Meier methods).
Dose randomisation
The observed number of primary end points was similar in the lower-dose arm (n = 51, 12.6%) and in the higher-dose arm (n = 49, 12.4%). The estimated risk difference at day 28 was 0.2% (90% CI –3.7% to 4.0%), meeting the criterion for non-inferiority (Figure 7).
FIGURE 7.
Kaplan–Meier curve for primary end point: dose randomisation.
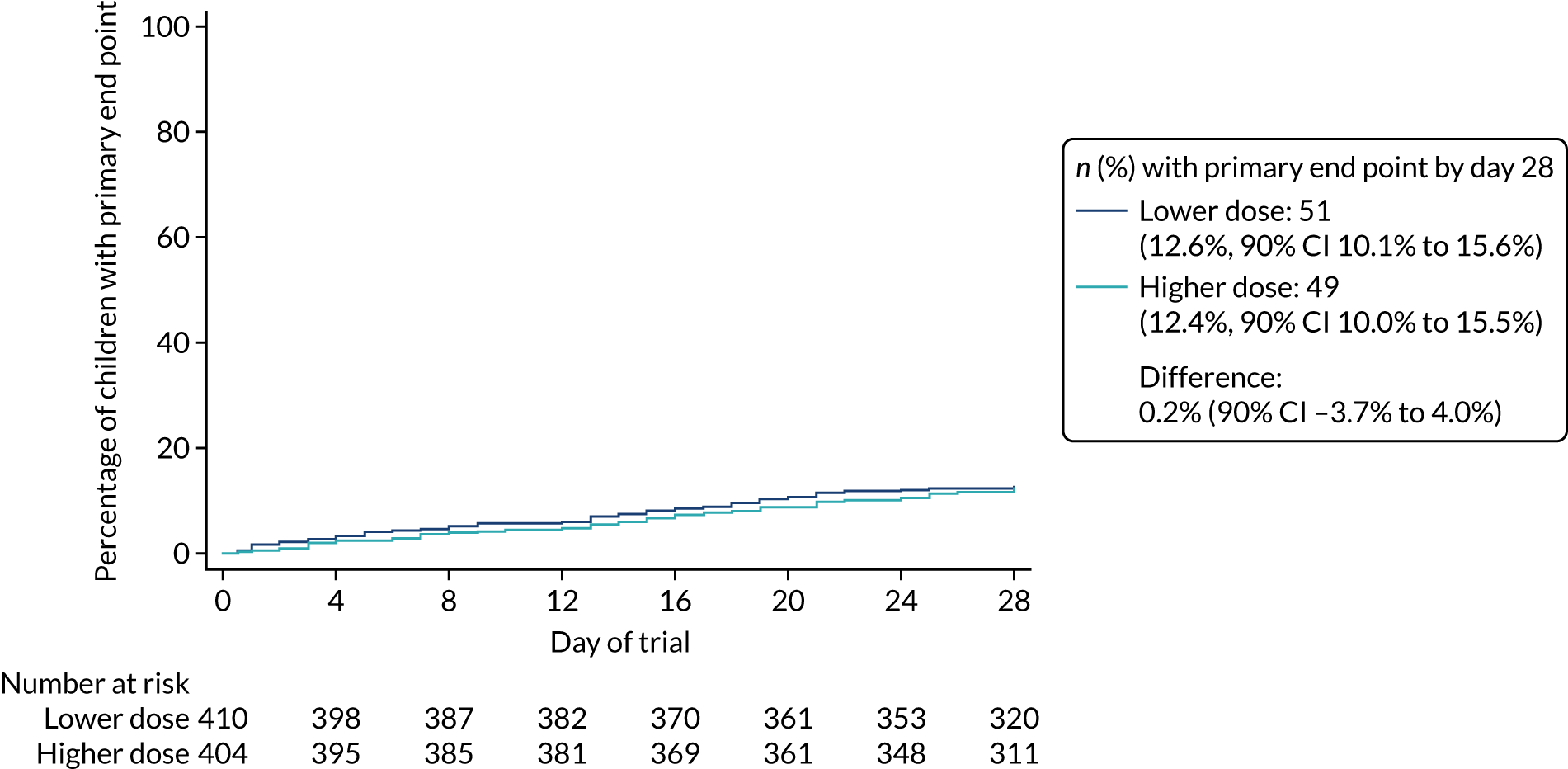
Duration randomisation
A total of 51 (12.5%) participants experienced a primary end point in the shorter-duration arm and 49 (12.5%) participants experienced a primary end point in the longer-duration arm. The estimated risk difference at day 28 was 0.1% (90% CI –3.8% to 3.9%), again satisfying the non-inferiority criterion (Figure 8).
FIGURE 8.
Kaplan–Meier curve for primary end point: duration randomisation.
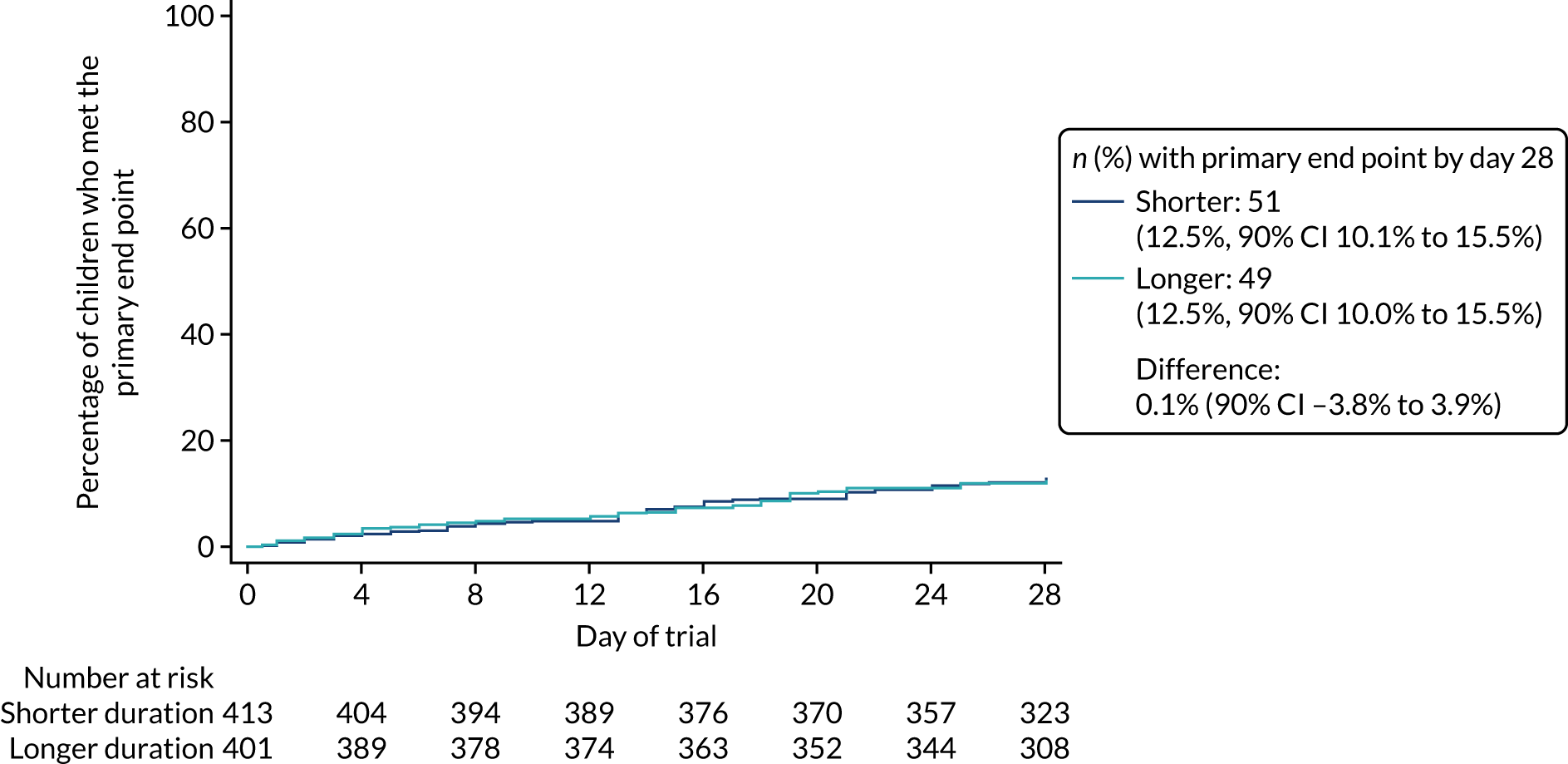
Interaction effects
The outcomes for the analyses of interaction effects between the two randomisations (i.e. dose and duration), between pre-exposure to antibiotics and dose randomisation and between pre-exposure to antibiotics and duration randomisation are shown in Figures 9–11, respectively.
FIGURE 9.
Kaplan–Meier curve for analysis of interaction between the two randomisations.
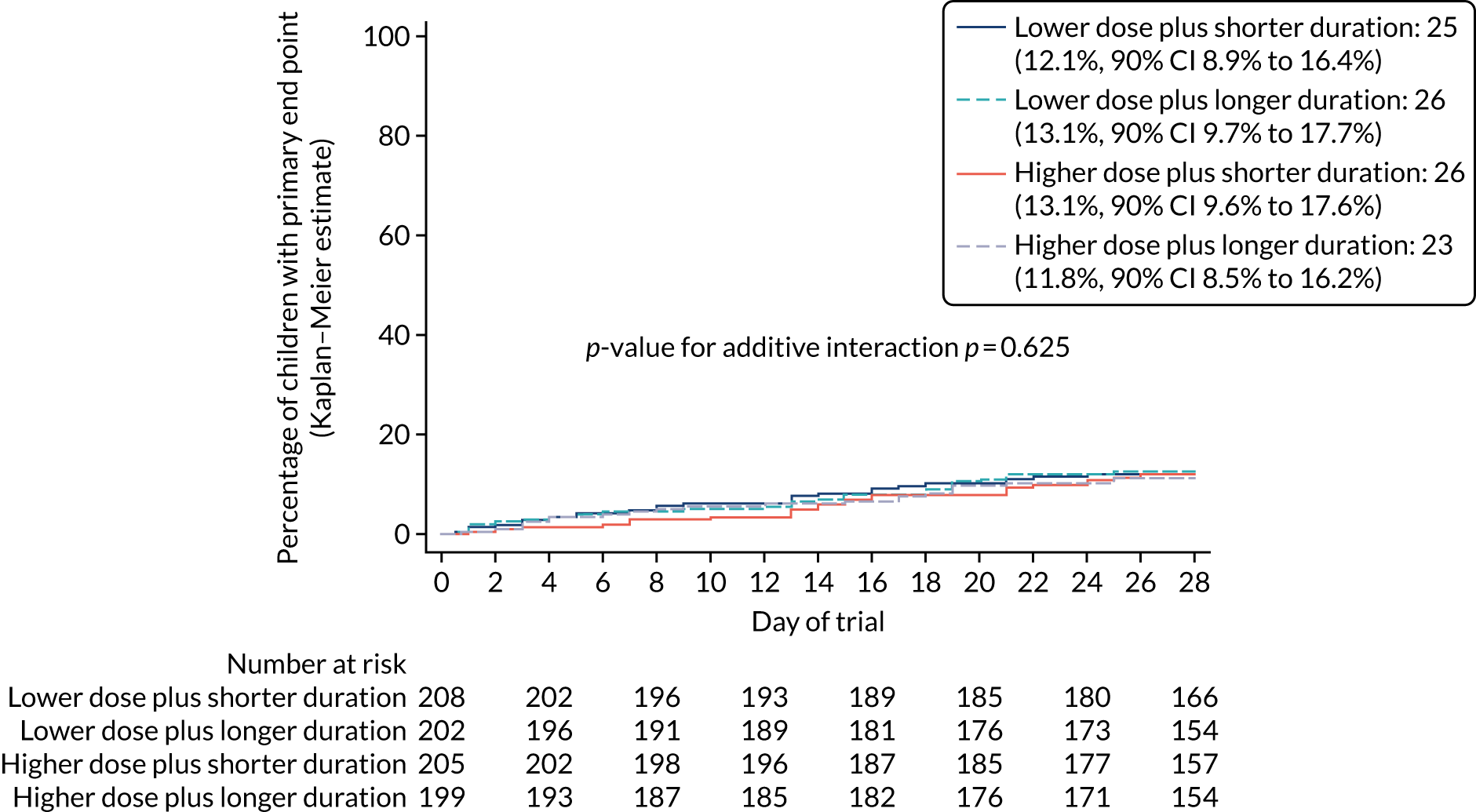
FIGURE 10.
Kaplan–Meier curve for analysis of interaction between pre-exposure with antibiotics and dose randomisation.
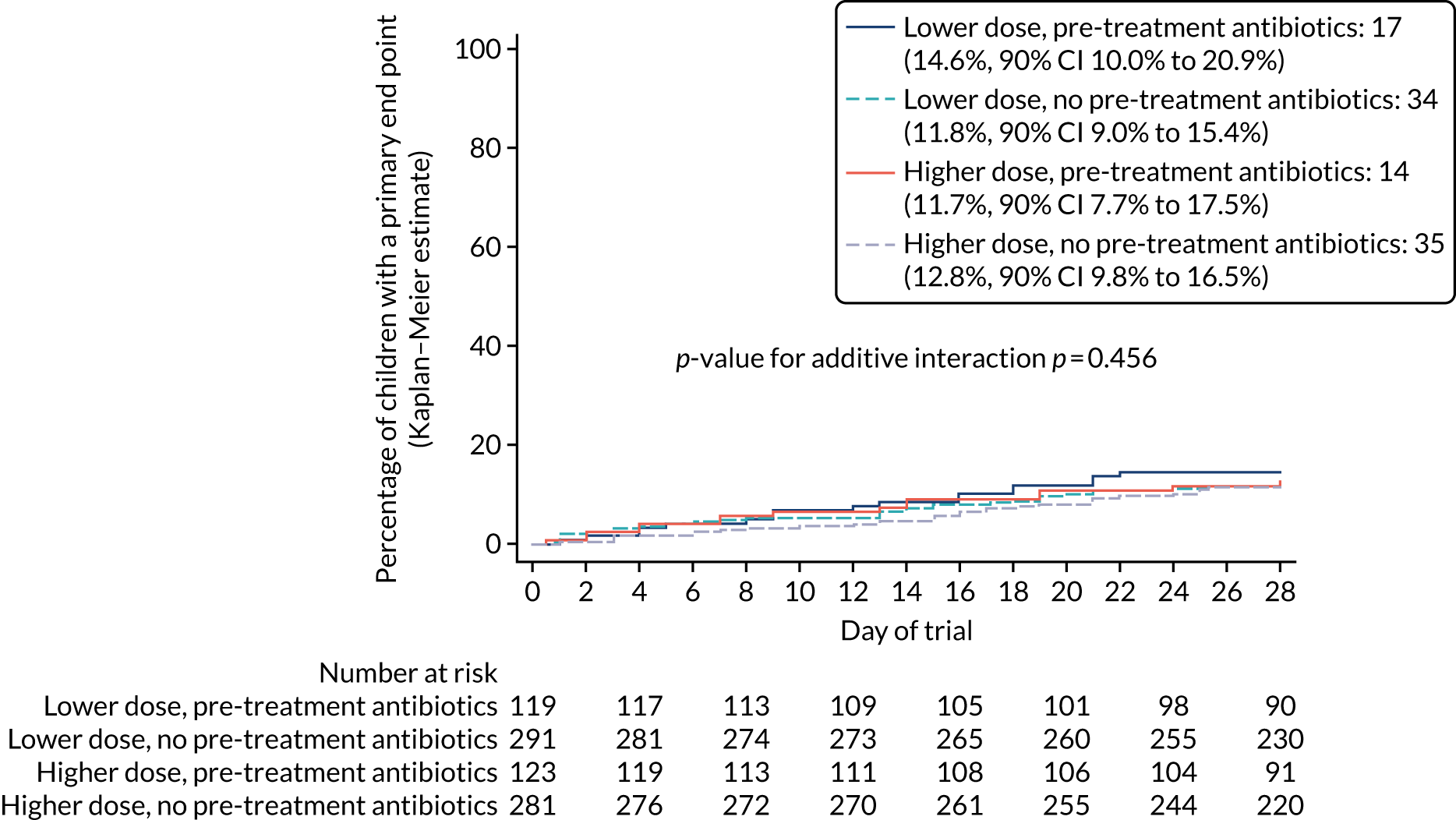
FIGURE 11.
Kaplan–Meier curve for analysis of interaction between pre-exposure with antibiotics and duration randomisation.
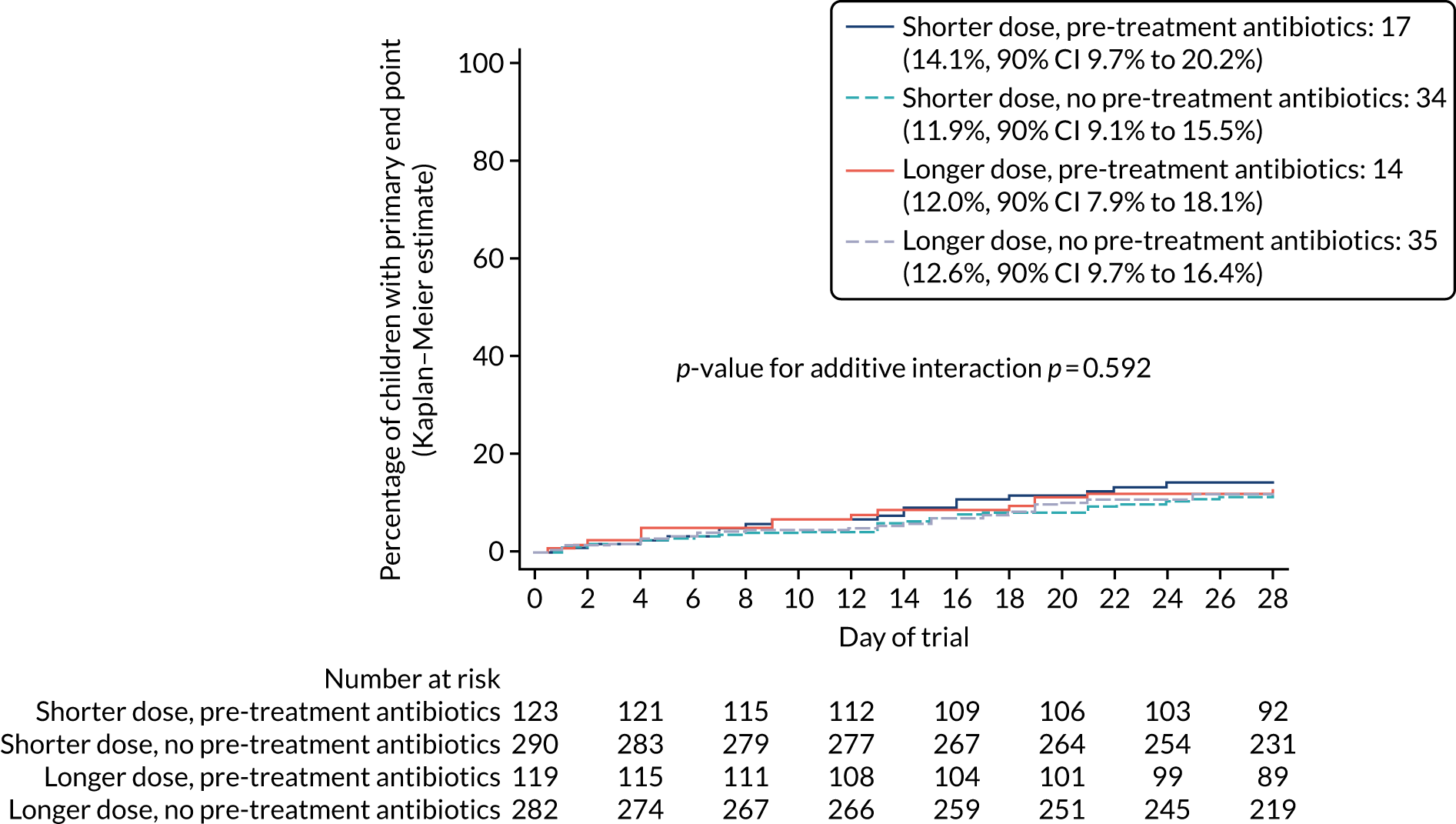
There was no evidence of an interaction between either of the two randomisation arms (p = 0.625), between the dose randomisation arm and pre-exposure to antibiotics (p = 0.456) or between the duration randomisation arm and pre-exposure to antibiotics (p = 0.592). This justifies analysis of the ‘main effects’ for the two randomisations (see Figures 7 and 8).
Primary end-point sensitivity analyses
The results of the sensitivity and subgroup analyses are summarised in Figures 12 and 13. Non-inferiority was demonstrated for all sensitivity analyses for both dose and duration comparisons.
FIGURE 12.
Forest plot summarising sensitivity and subgroup analyses outcomes in terms of difference in proportions of retreatment by day 28 for the dose randomisation.
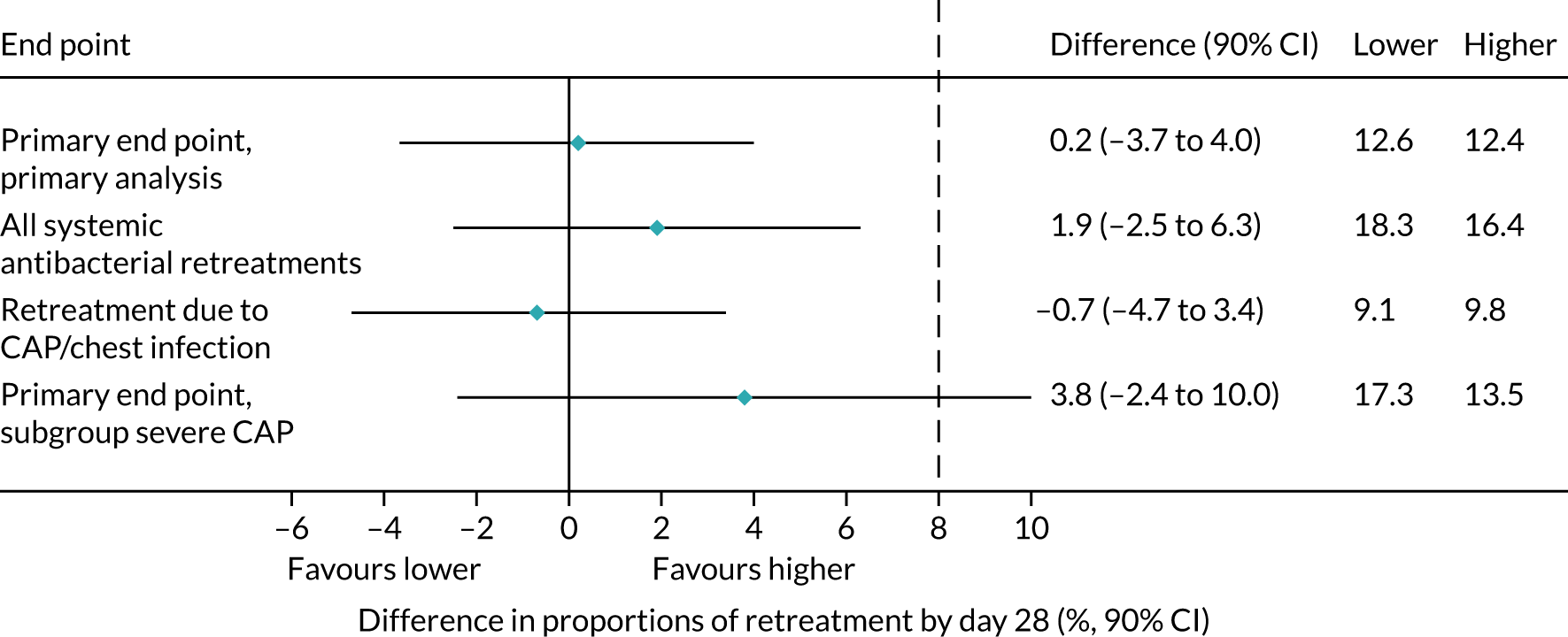
FIGURE 13.
Forest plot summarising sensitivity and subgroup analyses outcomes in terms of difference in proportions of retreatment by day 28 for the duration randomisation.
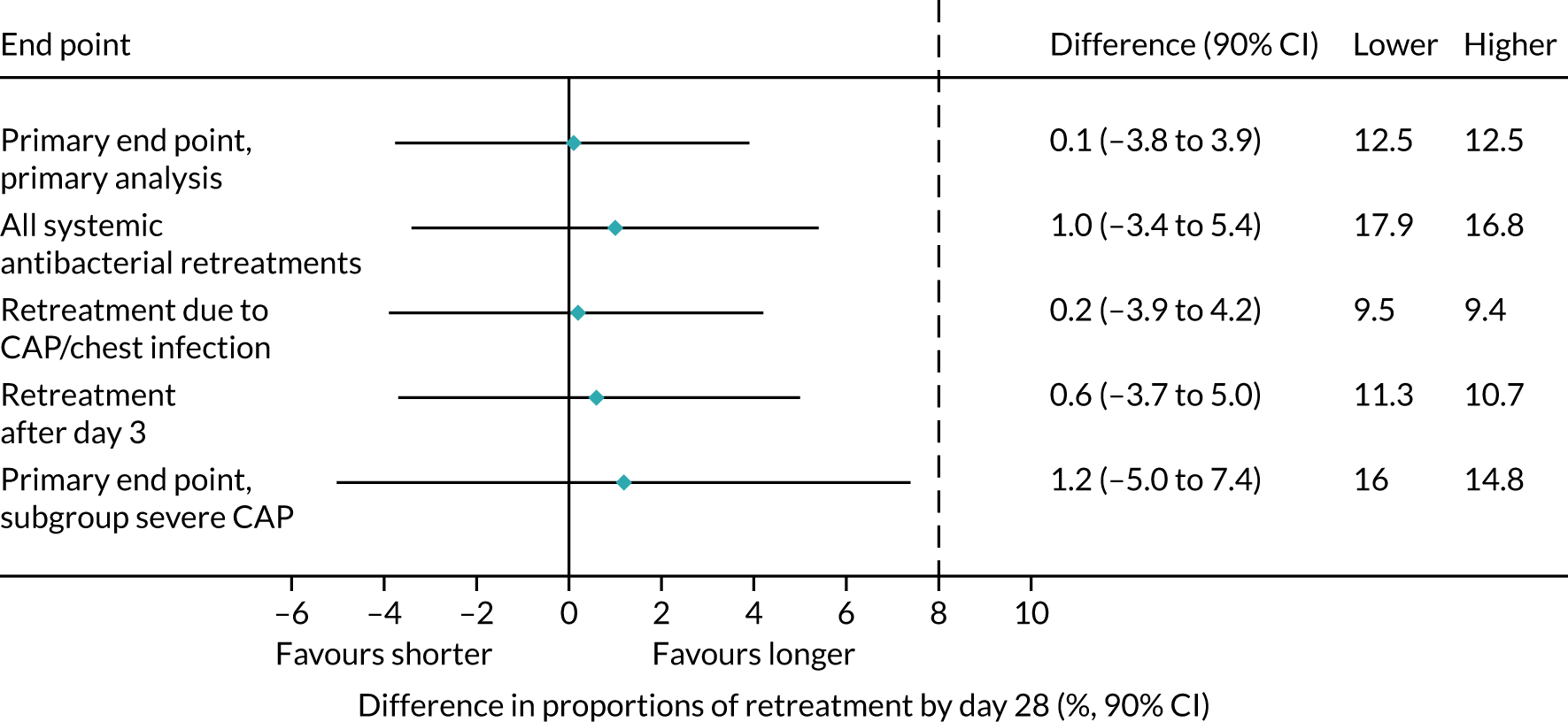
All systemic antibacterial treatments
The first sensitivity analysis repeated the primary analysis and considered all systemic antibacterial treatments other than trial medication, regardless of reason and indication. The total number of participants experiencing an end point in this analysis was 139 of 814 participants (17.4%, 90% CI 15.3% to 19.7%).
For the dose comparison, the estimated risk difference at day 28 was 1.9% (90% CI –2.5% to 6.3%). For the duration comparison, the estimated risk difference at day 28 was 1.0% (90% CI –3.4% to 5.4%). For both comparisons, the upper limit of the 90% CI was less than the non-inferiority margin of 8%, supporting the observations of the primary end-point analysis.
Treatment events for community-acquired pneumonia/chest infection
In a second sensitivity analysis, only those treatment events for which the clinical indication was adjudicated by the ERC to be CAP/chest infection were included. The total number of participants experiencing an end point in this analysis was 76 out of 814 (9.4%, 90% CI 7.9% to 11.3%).
For the dose comparison, the estimated risk difference at day 28 was –0.7% (90% CI –4.7% to 3.4%). For the duration comparison, the estimated risk difference at day 28 was 0.2% (90% CI –3.9% to 4.2%). As for the first sensitivity analysis, for both comparisons the upper limit of the 90% CI was less than the non-inferiority margin, supporting the observations of the primary end-point analysis.
All treatment events for community-acquired pneumonia/chest infection
A third sensitivity analysis considered treatment events for which the clinical indication was adjudicated by the ERC to be CAP/chest infection, including those adjudicated ‘unlikely’ to be clinically indicated. The number of participants experiencing an end point in this analysis was 78 of 814 participants (9.7%, 90% CI 8.1% to 11.6%).
For the dose comparison, the estimated risk difference at day 28 was –0.7% (90% CI –4.8% to 3.4%). For the duration comparison, the estimated risk difference at day 28 was 0.2% (90% CI –3.9% to 4.3%). For both comparisons, the upper limit of the 90% CI was less than the non-inferiority margin, supporting the observations of the primary end-point analysis.
Only treatment events started after the first 3 days (duration randomisation)
A final sensitivity analysis considered only ERC-adjudicated primary end points when non-trial antibacterial treatment was started after the first 3 days. This assessment was relevant for the duration randomisation only, and the estimated risk difference at day 28 was 0.6% (90% CI –3.7% to 5.0%). Non-inferiority was demonstrated, with the upper CI (5.0%) less than the non-inferiority margin of 8%, supporting the observations of the primary end-point analysis.
On-treatment analyses
The on-treatment analyses gave very similar results to the primary analysis. For both the dose and the duration comparison, the upper 90% CI limit of the estimated difference at day 28 was lower than the non-inferiority margin of 8% for both definitions of non-adherence (see Appendix 3, Figures 21–24).
Subgroup analyses
Participants with severe community-acquired pneumonia
This a priori subgroup analysis repeated the primary analysis, limited to participants defined as having severe CAP. Table 18 shows the total number (%) of participants with each abnormality by randomisation group. Only 155 (19%) participants had none of these abnormalities at presentation; 291 (35.7%) participants had one, 341 (41.9%) had two and 27 (3.3%) had three. A total of 368 (45.2%) participants were included in the subgroup analysis.
| Abnormality | Treatment arm, n (%) | p-value | Treatment arm, n (%) | p-value | Total (N = 814), n (%) | ||
|---|---|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||||
| Chest retractions | 239 (58.4) | 244 (60.4) | 0.57 | 239 (58.0) | 244 (60.8) | 0.41 | 483 (59.4) |
| Oxygen saturation < 92% | 18 (4.4) | 25 (6.2) | 0.25 | 18 (4.4) | 25 (6.3) | 0.23 | 43 (5.3) |
| High respiratory rate | 270 (65.9) | 258 (64.3) | 0.65 | 262 (63.7) | 266 (66.5) | 0.41 | 528 (65.1) |
| Number of abnormalities | 0.62 | 0.47 | |||||
| 0 | 75 (18.3) | 80 (19.8) | 82 (19.9) | 73 (18.2) | 155 (19.0) | ||
| 1 | 155 (37.8) | 136 (33.7) | 154 (37.3) | 137 (34.2) | 291 (35.7) | ||
| 2 | 168 (41.0) | 173 (42.8) | 166 (40.2) | 175 (43.6) | 341 (41.9) | ||
| 3 | 12 (2.9) | 15 (3.7) | 11 (2.7) | 16 (4.0) | 27 (3.3) | ||
| > 1 | 180 (43.9) | 188 (46.5) | 0.45 | 177 (42.9) | 191 (47.6) | 0.17 | 368 (45.2) |
In total, 56 (15.4%) participants experienced a primary end point. There was no significant difference between the arms for either the dose comparison (p = 0.283) or the duration comparison (p = 0.821). For duration randomisation, the estimated risk difference at day 28 was 1.2% (90% CI –5.0% to 7.4%) (Figure 14). For dose randomisation, the estimated difference at day 28 was 3.8% (90% CI –2.4% to 10.0%). This is consistent with no effect, although the 90% CI crossed the non-inferiority margin (Figure 15).
FIGURE 14.
Kaplan–Meier curve for severe CAP subgroup primary analysis for duration randomisation.
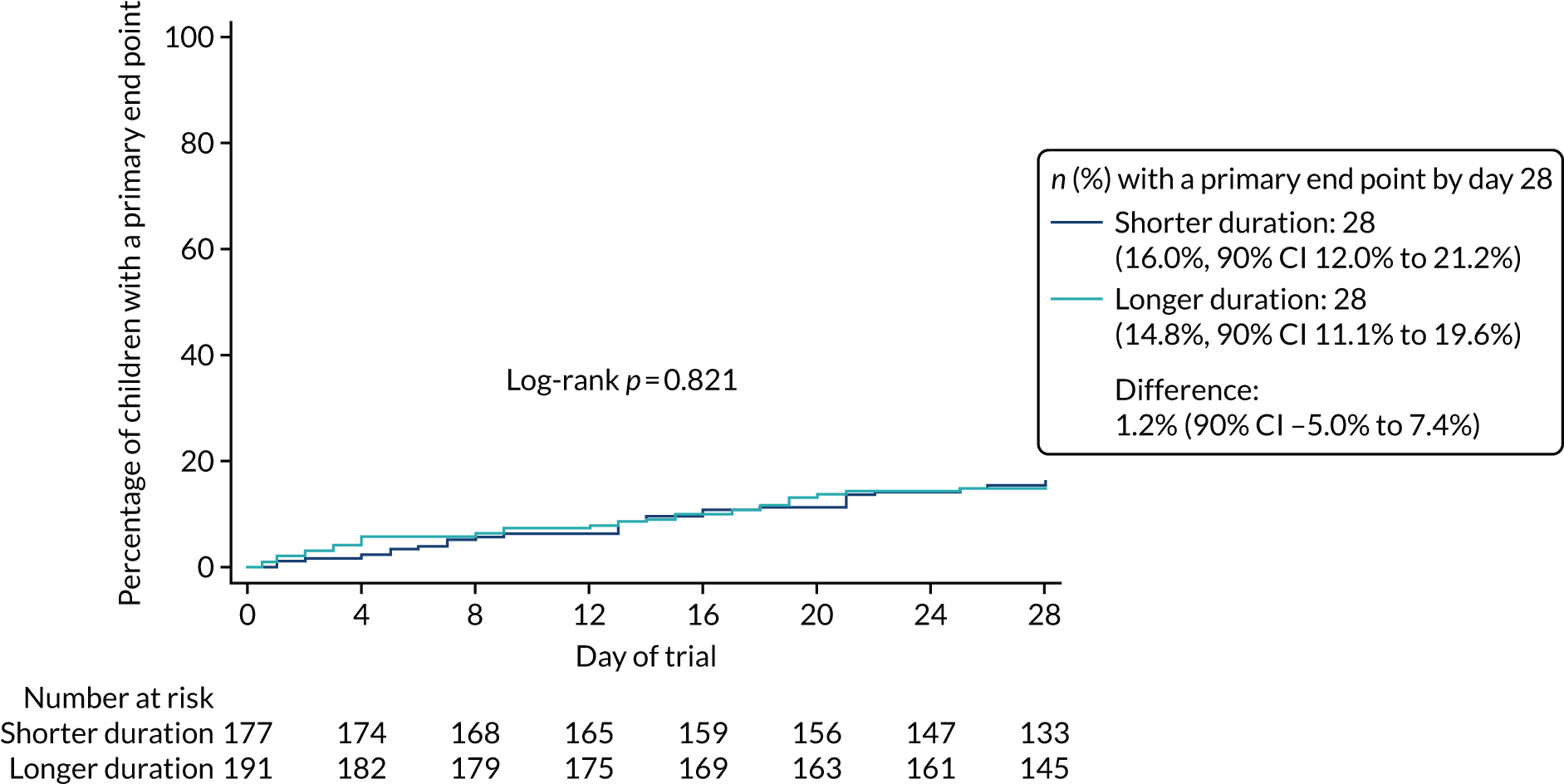
FIGURE 15.
Kaplan–Meier curve for severe CAP subgroup primary analysis for dose randomisation.
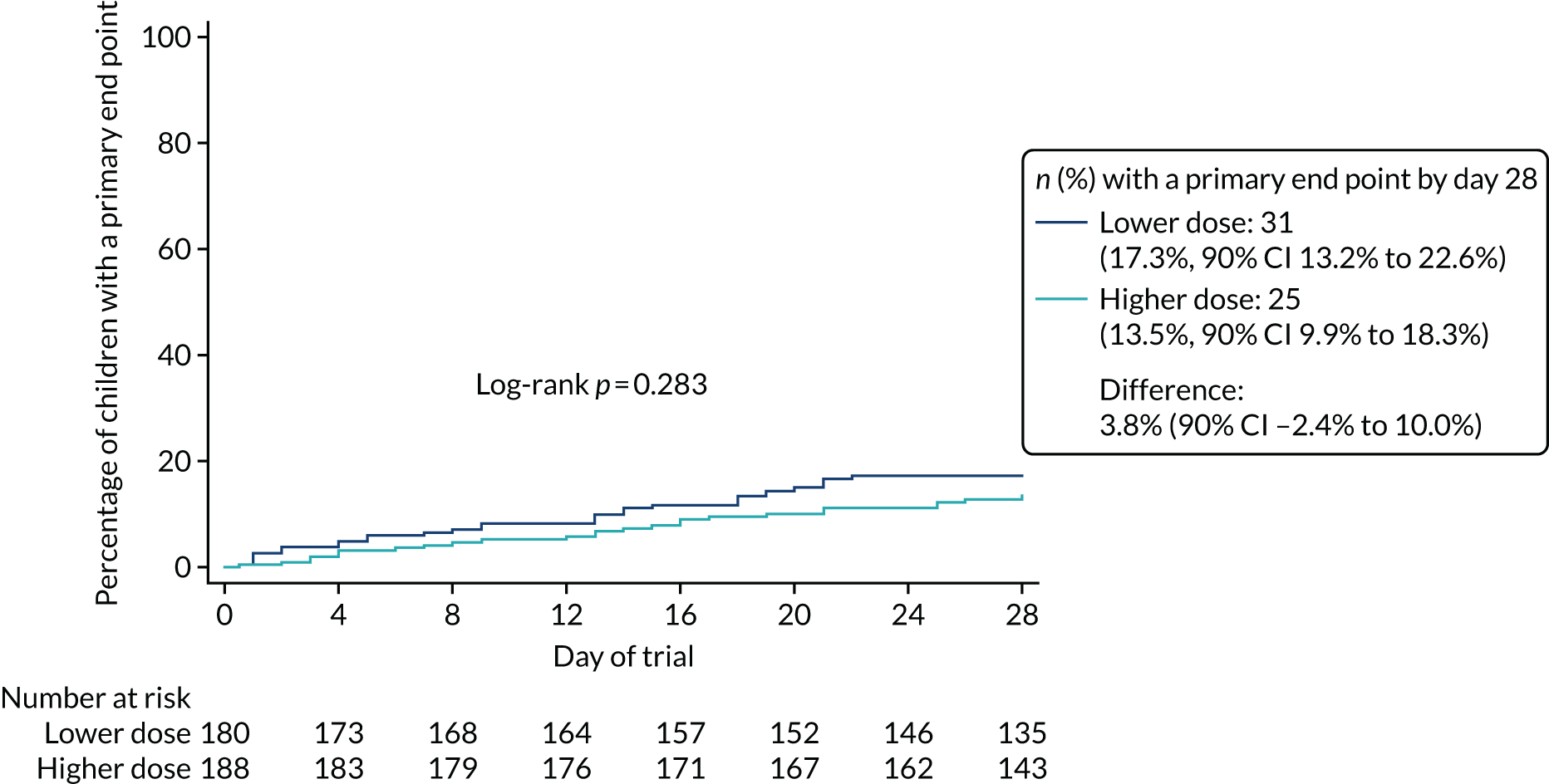
Seasonal effect
A further a priori planned subgroup analysis repeated the primary analysis, but including only events occurring during the two winter periods spanned by CAP-IT (i.e. 2017/18 and 2018/19), based on Public Health England reports of circulating viruses/bacteria.
The overall event rate in 2017/18 was 14.1% and 12.2% in 2018/19 (p = 0.515). There was no evidence of an interaction with either the duration or dose randomisations (p = 0.848 and p = 0.677, respectively).
Streptococcus pneumoniae carriage and resistance
Carriage and resistance to penicillin of S. pneumoniae isolates were assessed by analysis of nasopharyngeal samples collected from participants at baseline, at the final visit and at any unscheduled visits during follow-up.
Availability of nasopharyngeal culture results
Of the 814 participants in the analysis population, 647 (79%) had a nasopharyngeal sample taken at baseline and 437 (54%) had a sample taken at the final visit. There were 376 (46%) participants who had both a baseline and final visit sample taken, 271 (33%) who had just a baseline sample taken and 61 (7%) who had just a final visit sample taken. The remaining 106 (13%) participants did not have a sample taken. In addition, 28 (4%) participants had a sample taken at an unscheduled visit and four participants had samples taken at two unscheduled visits (1%) (Table 19).
| Culture result | Group, n (%) | p-value | Total (N = 814), n (%) | |
|---|---|---|---|---|
| PED (N = 591) | Ward (N = 223) | |||
| Baseline culture available | 474 (80) | 173 (78) | 0.41 | 647 (79) |
| Final visit culture available | 316 (53) | 121 (54) | 0.84 | 437 (54) |
| If final visit happened hospital, at home | 316 (89) | 121 (92) | 0.25 | 437 (90) |
| Summary availability | ||||
| None | 75 (13) | 31 (14) | 0.84 | 106 (13) |
| Both baseline and final visit | 274 (46) | 102 (46) | 376 (46) | |
| Baseline only | 200 (34) | 71 (32) | 271 (33) | |
| Final visit only | 42 (7) | 19 (9) | 61 (7) | |
| Unscheduled visit: number of culture samples available | ||||
| 0 | 490 (95) | 186 (97) | 0.37 | 676 (95) |
| 1 | 22 (4) | 6 (3) | 28 (4) | |
| 2 | 4 (1) | 0 (1) | 4 (1) | |
Streptococcus pneumoniae carriage
Overall, 272 of 647 (42%) baseline samples and 129 of 437 (30%) final visit samples were culture positive for S. pneumoniae. Of the participants with a culture result at both baseline and final visit, 70 of 376 (19%) were positive for S. pneumoniae at both visits, 100 of 376 (27%) were positive at baseline only and 41 of 376 (11%) were positive at the final visit only. The remaining 165 (44%) sample cultures were negative at both visits (Table 20).
| S. pneumoniae carriage | Treatment arm, n (%) | p-value | Treatment arm, n (%) | p-value | Total, n (%) | ||
|---|---|---|---|---|---|---|---|
| Lower dose | Higher dose | Shorter duration | Longer duration | ||||
| Baseline: positive | 133 (41) | 139 (43) | 132 (42) | 140 (42) | 272 (42) | ||
| Final visit: positive | 66 (29) | 63 (30) | 0.98 | 65 (32) | 64 (28) | 0.35 | 129 (30) |
| Summary: pneumococcal carriagea | |||||||
| Never | 93 (48) | 72 (40) | 76 (44) | 89 (43) | 165 (44) | ||
| Baseline only | 46 (24) | 54 (30) | 39 (23) | 61 (30) | 100 (27) | ||
| Final visit only | 21 (11) | 20 (11) | 20 (12) | 21 (10) | 41 (11) | ||
| Both | 34 (18) | 36 (20) | 36 (21) | 34 (17) | 70 (19) | ||
Streptococcus pneumoniae penicillin non-susceptibility
No penicillin-resistant pneumococcal isolates were identified in CAP-IT. Penicillin non-susceptibility was detected in 45 of 647 (7%) baseline samples providing a culture result (either positive or negative) (17% of S. pneumoniae-positive samples) and in 21 (5%) samples taken at the final visit and providing a culture result (either positive or negative) (16% of S. pneumoniae-positive samples). Of participants with positive or negative culture results at both baseline and final visit, 23 (6%) had pneumococcal penicillin non-susceptibility identified in the baseline sample only, 11 (3%) participants had pneumococcal penicillin non-susceptibility identified in in the final visit sample only and seven (2%) participants had pneumococcal penicillin non-susceptibility identified in both baseline and final visit samples. In the remaining 335 (89%) participants, penicillin resistance was not identified in either sample culture.
There was no evidence of a difference between the lower- and higher-dose randomisation groups in the penicillin non-susceptibility of isolates cultured from either baseline or final visit samples, or between the shorter- and longer-duration randomisation groups in the penicillin non-susceptibility of isolates cultured from baseline samples (see Table 21). The proportion of pneumococcal isolates cultured from final visit samples that were penicillin non-susceptible was slightly higher in the shorter-duration group (n = 14, 7% of all samples, 22% of S. pneumoniae-positive samples) than in the longer-duration group [n = 7, 3% of all samples (p = 0.063), 11% of S. pneumoniae-positive samples (p = 0.10)]. This pattern was also found when the analysis was limited to participants with a positive culture result for S. pneumoniae (excluding all samples with a negative culture result), with penicillin non-susceptibility detected in 22% (n = 14) of samples taken from participants in the shorter-duration arm and in 11% (n = 7) of samples from participants in the longer-duration arm (p = 0.10).
Streptococcus pneumoniae amoxicillin resistance/non-susceptibility
Amoxicillin non-susceptibility or resistance was detected in S. pneumoniae isolates cultured from seven (2%) baseline samples with a culture result (either positive or negative) and in four final visit (1%) samples with a culture result (either positive or negative). Among participants for whom culture results (positive or negative) were available for both baseline and final visit samples, amoxicillin resistance was detected in isolates cultured from the baseline sample only in the case of one (< 1%) participant, in isolates cultured from the final visit sample only in the case of two (1%) participants and in isolates from both samples in the case of one (< 1%) participant. In the remaining 361 (99%) participants, neither amoxicillin non-susceptibility nor resistance was identified in any samples.
There was no evidence of a difference between the lower- and higher-dose groups in the amoxicillin non-susceptibility of isolates cultured from either baseline or final visit sample cultures, or between the shorter- and longer-duration groups groups in the amoxicillin non-susceptibility of isolates cultured from either baseline or final visit samples (Table 21). This was also found when the analysis of amoxicillin non-susceptibility was limited to participants whose samples provided a positive culture result for S. pneumoniae (excluding all samples with a negative culture result).
| Penicillin and amoxicillin resistance/non-susceptibility | Treatment arm, n (%) | p-value | Treatment arm, n (%) | p-value | ||
|---|---|---|---|---|---|---|
| Lower dose | Higher dose | Shorter duration | Longer duration | |||
| Penicillin non-susceptibility at baseline | ||||||
| No | 302 (92) | 299 (93) | 0.59 | 293 (92) | 308 (93) | 0.65 |
| Yes | 25 (8) | 21 (7) | 24 (8) | 22 (7) | ||
| Penicillin non-susceptibility at the final visit | ||||||
| No | 212 (95) | 204 (96) | 0.58 | 191 (93) | 225 (97) | 0.063 |
| Yes | 12 (5) | 9 (4) | 14 (7) | 7 (3) | ||
| Penicillin non-susceptibility: summarya | ||||||
| Never | 175 (90) | 166 (91) | 0.79 | 151 (88) | 190 (93) | 0.29 |
| Baseline only | 10 (5) | 9 (5) | 9 (5) | 10 (5) | ||
| Final visit only | 6 (3) | 3 (2) | 6 (4) | 3 (1) | ||
| Both baseline and final visit | 3 (2) | 4 (2) | 5 (3) | 2 (1) | ||
| Amoxicillin resistance/non-susceptibility at baseline | ||||||
| No | 318 (98) | 311 (99) | 0.27 | 309 (99) | 320 (98) | 0.28 |
| Yes | 5 (2) | 2 (1) | 2 (1) | 5 (2) | ||
| Amoxicillin resistance/non-susceptibility at the final visit | ||||||
| No | 218 (99) | 210 (99) | 0.97 | 199 (99) | 229 (99) | 0.89 |
| Yes | 2 (1) | 2 (1) | 2 (1) | 2 (1) | ||
| Amoxicillin resistance/non-susceptibility: summarya | ||||||
| Never | 185 (99) | 176 (99) | 0.26 | 162 (99) | 199 (99) | 0.56 |
| Baseline only | 1 (1) | 0 (0) | 0 (0) | 1 (< 1) | ||
| Final visit only | 0 (0) | 2 (1) | 1 (1) | 1 (< 1) | ||
| Both baseline and final visit | 1 (1) | 0 (0) | 1 (1) | 0 (0) | ||
Community-acquired pneumonia symptoms
Parent/guardian-reported symptom data were elicited at follow-up telephone calls and through parental/guardian completion of a daily diary up to day 14. The proportion of participants for whom parent/guardian-reported symptom data from any source were available reduced from days 3 (93%), 7 (88%), 14 (83%) and 21 (76%) to day 28 (75%) (Figure 16).
FIGURE 16.
Availability of symptom data over time, by data source. a, No data available because telephone call/visit did not happen or no data were reported.
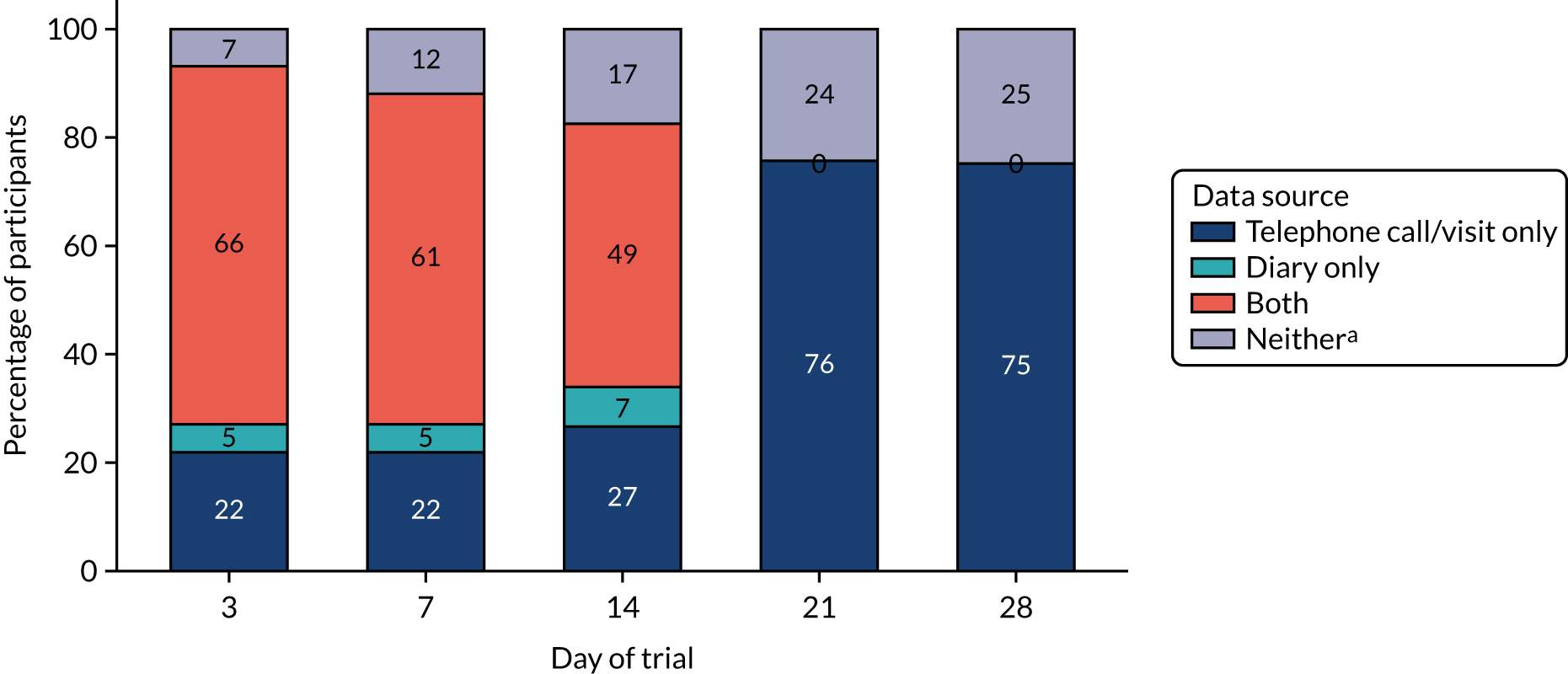
Time to resolution of community-acquired pneumonia symptoms: overall
Severity was elicited for nine CAP symptoms, each of which was analysed separately in terms of time to resolution. As multiple comparisons were performed, the p-value from each individual analysis needs to be interpreted cautiously. Participants were included in the analysis only if a symptom was present at trial entry. Cough had the longest median time to resolution (11 days), followed by the related symptom wet cough (phlegm) (6 days). An estimated 20% of participants still had cough symptoms at day 28 (Figure 17). Vomiting and fever both resolved rapidly, in a median of 1 day and 2 days, respectively. The remaining symptoms had a median time to resolution of between 3 and 5 days.
FIGURE 17.
Kaplan–Meier curves for time to symptom resolution across all randomisation arms. Participants excluded if symptom not present at enrolment.
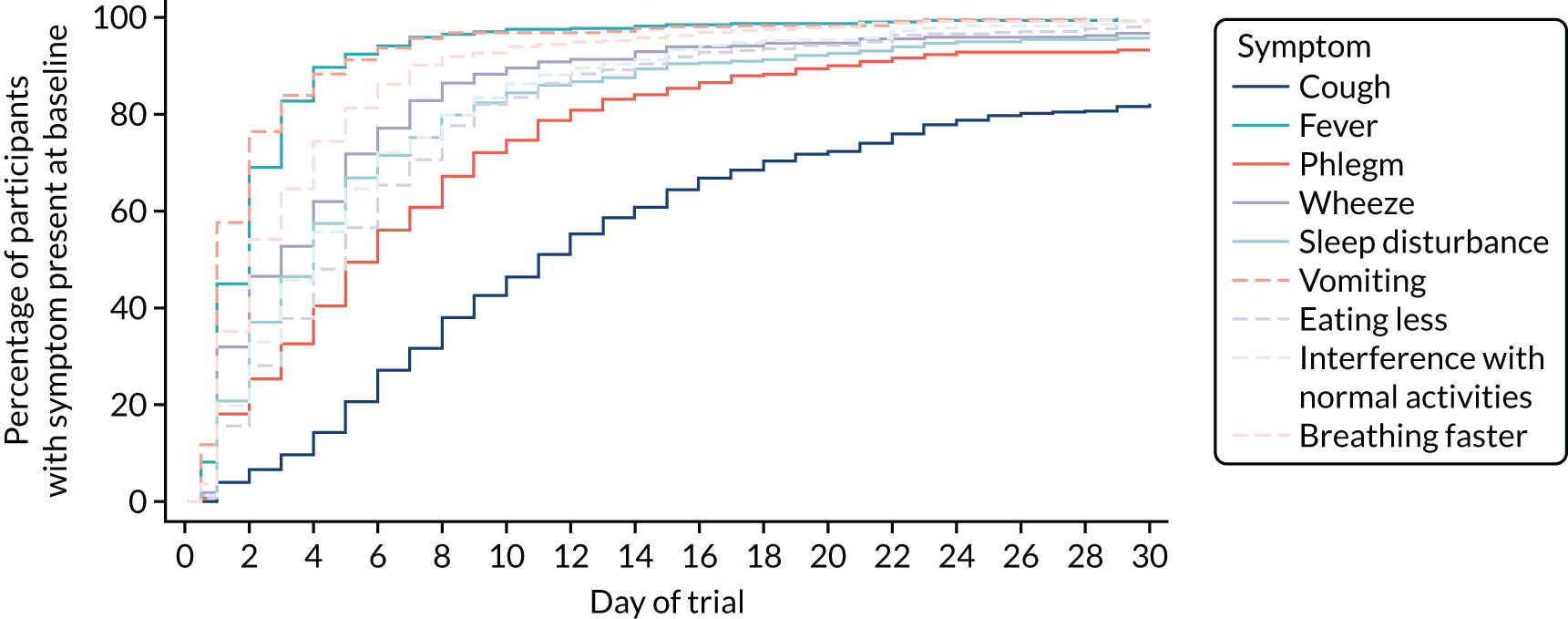
Time to resolution of community-acquired pneumonia symptoms: dose randomisation
There was no significant difference between participants receiving lower and higher doses in time to resolution of any of the nine symptoms (log-rank p > 0.05).
Time to resolution of community-acquired pneumonia symptoms: duration randomisation
For duration randomisation, there was no significant difference between groups for seven symptoms (log-rank p > 0.05). However, there was a difference in time to resolution of cough (p = 0.040) and sleep disturbed by cough (p = 0.026), with a significantly faster time to resolution in the longer-duration arm in both cases (Figures 18 and 19).
FIGURE 18.
Kaplan–Meier curve for time to resolution of cough in the duration treatment arms. Participants excluded if symptom not present at enrolment.
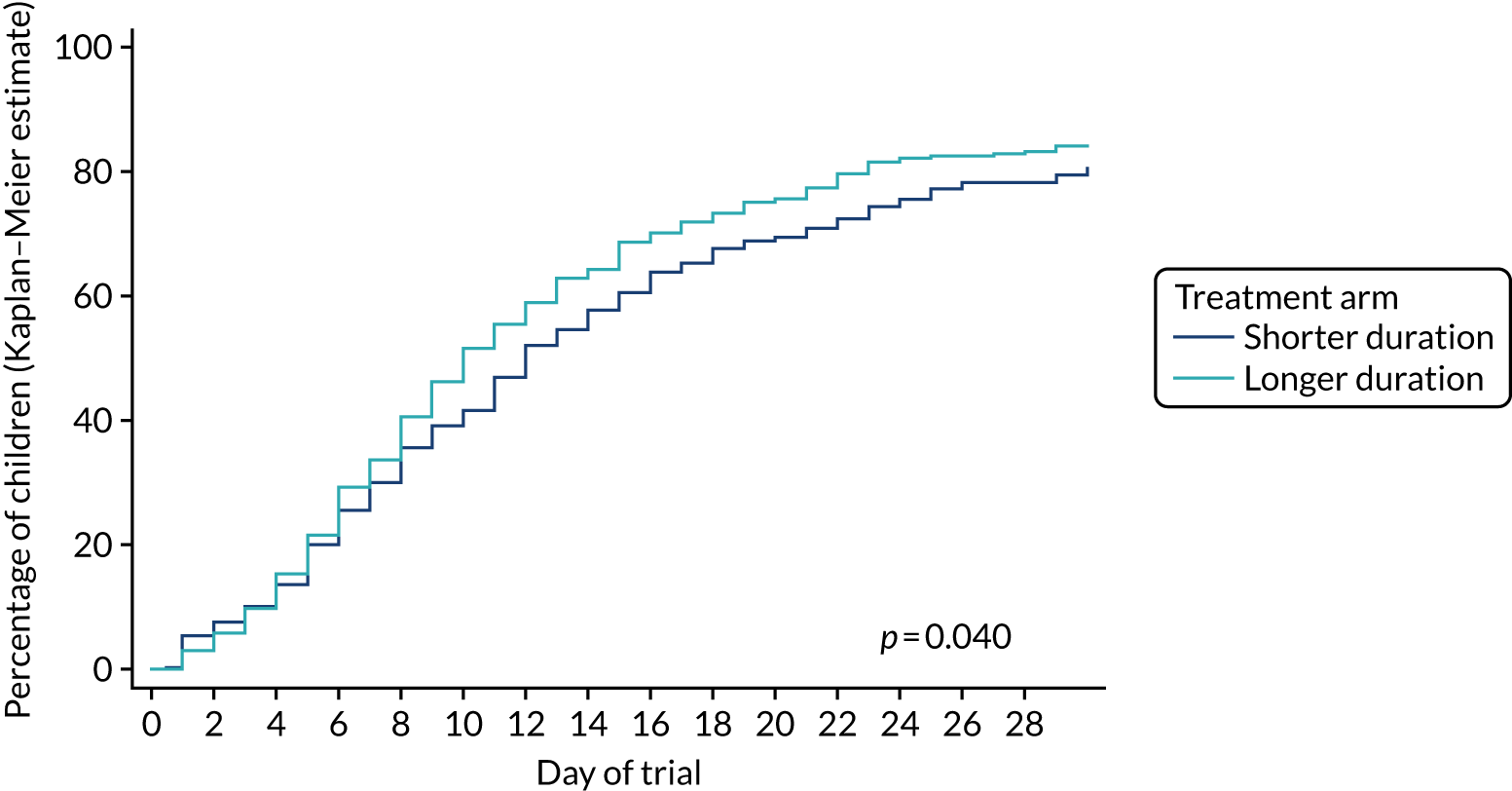
FIGURE 19.
Kaplan–Meier curve for time to resolution of sleep disturbed by cough in the duration treatment arms. Participants excluded if symptom not present at enrolment.
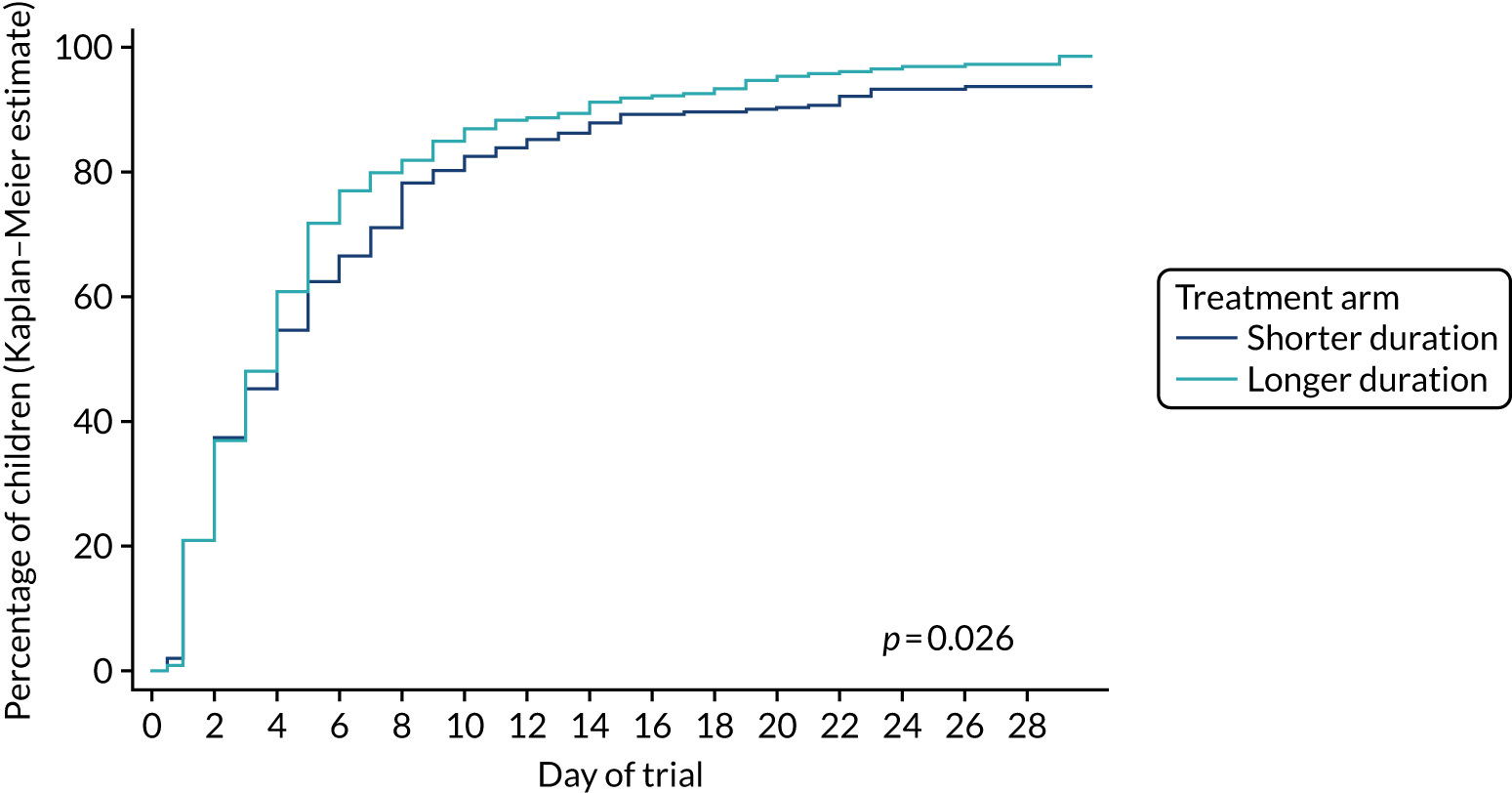
Sensitivity analysis for duration randomisation
As symptom resolution within the first 3 days from randomisation cannot, by definition, be related to the treatment duration randomisation, a prespecified sensitivity analysis was performed, changing the time origin to day 4 for the comparison of shorter and longer treatment.
Log-rank tests were repeated and the same pattern was observed as in the main analyses. Participants in the shorter-duration arm had a significantly longer time to resolution of cough (p = 0.039) and sleep disturbed by cough (p = 0.031) than participants in the longer-duration arm. There was no evidence of a significant difference between the two duration arms in time to resolution of the remaining seven symptoms.
Adverse events
Serious adverse events
In total, 43 (5.3%) participants experienced a SAE, one participant (0.1%) experienced a serious adverse reaction (SAR) and no participants experienced a suspected unexpected SAR. There was no evidence of differences between proportions of participants experiencing a SAE in any of the dose or duration treatment arms (Table 22).
| SAE summary | Treatment arm, n (%) | p-value | Treatment arm, n (%) | p-value | Total (N = 814), n (%) | ||
|---|---|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||||
| Number of SAEs per participant | |||||||
| 0 | 387 (94.4) | 384 (95.0) | 0.67 | 388 (93.9) | 383 (95.5) | 0.32 | 771 (94.7) |
| 1 | 23 (5.6) | 20 (5.0) | 25 (6.1) | 18 (4.5) | 43 (5.3) | ||
| SAR confirmed | 0 (0.0) | 1 (0.2) | 0.50 | 1 (0.2) | 0 (0.0) | 1.00 | 1 (0.1) |
| SUSAR confirmed | 0 | 0 | 0 | 0 | 0 | ||
Of the 43 SAEs, 42 (98%) were hospitalisations and one (2%) (exacerbation of asthma, unrelated to the trial medication) was classified as life-threatening (Table 23), necessitating intubation and transfer of the patient to a paediatric intensive care unit. Respiratory events were the most common diagnoses, accounting for 35 of 43 SAEs (81%), of which 16 (37%) were classified as respiratory distress, eight (19%) were lower respiratory tract infection and five (12%) were pneumonia; the remaining six were asthma (n = 3, 7%), bronchiolitis (n = 2, 5%) and influenza (n = 1, 2%). Most SAEs occurred between days 1 and 4 (n = 29, 67%).
| SAE details | Treatment arm, n (%) | Total (N = 43), n (%) | |||
|---|---|---|---|---|---|
| Shorter duration (N = 25) | Longer duration (N = 18) | Lower dose (N = 23) | Higher dose (N = 20) | ||
| Type of SAE | |||||
| Life-threatening | 0 | 1 (6) | 0 | 1 (5) | 1 (2) |
| Hospitalisation | 25 (100) | 17 (94) | 23 (100) | 19 (95) | 42 (98) |
| Body system | |||||
| Dermatological | 1 (4) | 1 (6) | 1 (4) | 1 (5) | 2 (5) |
| Cyanosis peripheral | 0 | 1 (6) | 1 (4) | 0 | 1 (2) |
| Herpes simplex oral | 1 (4) | 0 | 0 | 1 (5) | 1 (2) |
| Gastrointestinal | 4 (16) | 0 | 2 (9) | 2 (10) | 4 (9) |
| Coffee ground vomiting | 1 (4) | 0 | 0 | 1 (5) | 1 (2) |
| Epiploic appendagitis | 1 (4) | 0 | 1 (4) | 0 | 1 (2) |
| Salmonella gastroenteritis | 1 (4) | 0 | 1 (4) | 0 | 1 (2) |
| Vomiting | 1 (4) | 0 | 0 | 1 (5) | 1 (2) |
| Neurological | 1 (4) | 1 (6) | 2 (9) | 0 | 2 (5) |
| Cerebellar tumour | 0 | 1 (6) | 1 (4) | 0 | 1 (2) |
| Febrile seizure | 1 (4) | 0 | 1 (4) | 0 | 1 (2) |
| Respiratory | 19 (76) | 16 (89) | 18 (78) | 17 (85) | 35 (81) |
| Asthma | 1 (4) | 2 (11) | 0 | 3 (15) | 3 (7) |
| Bronchiolitis | 2 (8) | 0 | 2 (9) | 0 | 2 (5) |
| Influenza | 1 (4) | 0 | 1 (4) | 0 | 1 (2) |
| Lower respiratory tract infection viral | 1 (4) | 7 (39) | 3 (13) | 5 (25) | 8 (19) |
| Pneumonia | 2 (8) | 3 (17) | 5 (22) | 0 | 5 (12) |
| Respiratory distress | 12 (48) | 4 (22) | 7 (30) | 9 (45) | 16 (37) |
| Trial study day of hospitalisationa | |||||
| Days 0–3 | 20 (80) | 9 (50) | 16 (70) | 13 (65) | 29 (67) |
| Days 4–7 | 0 | 2 (11) | 1 (4) | 1 (5) | 2 (5) |
| Days 8–14 | 2 (8) | 1 (6) | 1 (4) | 2 (10) | 3 (7) |
| Days 15–21 | 0 | 2 (11) | 0 | 2 (10) | 2 (5) |
| Days 22–28 | 3 (12) | 4 (22) | 5 (22) | 2 (10) | 7 (16) |
| Event grade | |||||
| 1 | 11 (44) | 4 (22) | 9 (39) | 6 (30) | 15 (35) |
| 2 | 6 (24) | 9 (50) | 7 (30) | 8 (40) | 15 (35) |
| 3 | 8 (32) | 3 (17) | 6 (26) | 5 (25) | 11 (26) |
| 4 | 0 | 2 (11) | 1 (4) | 1 (5) | 2 (5) |
| Relationship to trial medication | |||||
| Not related | 20 (80) | 16 (89) | 19 (83) | 17 (85) | 36 (84) |
| Unlikely | 5 (20) | 2 (11) | 4 (17) | 3 (15) | 7 (16) |
| Possibly | 0 | 0 | 0 | 0 | 0 |
| Probably | 0 | 0 | 0 | 0 | 0 |
| Definitely | 0 | 0 | 0 | 0 | 0 |
| Expected of trial medication | |||||
| Expected | 7 (29) | 0 | 5 (22) | 2 (11) | 7 (17) |
| Unexpected | 17 (71) | 18 (100) | 18 (78) | 17 (89) | 35 (83) |
| Action on trial medication | |||||
| None | 16 (64) | 8 (44) | 10 (43) | 14 (70) | 24 (56) |
| Treatment delayed | 1 (4) | 0 | 1 (4) | 0 | 1 (2) |
| Treatment stopped | 8 (32) | 10 (56) | 12 (52) | 6 (30) | 18 (42) |
| Started new antibiotic during SAE? | 12 (48) | 15 (83) | 16 (70) | 11 (55) | 27 (63) |
| Event status at the end of follow-up | |||||
| Resolved | 24 (96) | 17 (94) | 21 (91) | 20 (100) | 41 (95) |
| Ongoing at study exit | 1 (4) | 1 (6) | 2 (9) | 0 | 2 (5) |
The SAR was a diagnosis of vomiting, originally classified by the site investigator as unlikely to be related to the IMP. However, on clinical review by the Trial Management Team, it was felt that the SAE could be related to the IMP and the event was, therefore, reclassified as a SAR.
Specified clinical adverse events (diarrhoea, thrush and skin rash)
Presence and severity of diarrhoea, thrush and skin rash were elicited from parents at trial entry and throughout follow-up. Diarrhoea was the most common clinical AE and was present in 345 (43.6%) participants after baseline. Skin rash was present in 193 (24.4%) participants and oral thrush in 57 (7.2%) participants after baseline. For both dose and duration randomisations, there was no evidence of a difference between the randomised arms in terms of overall prevalence of diarrhoea and oral thrush after baseline (Table 24). For skin rash, there was some evidence of a difference between shorter- and longer-duration arms in terms of prevalence after baseline, with the number of participants ever having skin rash after baseline being 106 (27.4%) in the longer-duration arm and 87 (21.5%) in the shorter-duration arm (p = 0.055).
| Prevalence of diarrhoea, oral thrush and skin rash | Treatment arm, n (%) | Total (N = 814), n (%) | |||
|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||
| First diarrhoea after baselinea | |||||
| No | 276 (78.0) | 252 (76.4) | 259 (75.1) | 269 (79.4) | 528 (77.2) |
| Yes | 78 (22.0) | 78 (23.6) | 86 (24.9) | 70 (20.6) | 156 (22.8) |
| p-value | p = 0.62 | p = 0.18 | |||
| New diarrhoea after baseline or worse than at baseline | |||||
| No | 303 (75.6) | 288 (73.8) | 296 (73.3) | 295 (76.2) | 591 (74.7) |
| Yes | 98 (24.4) | 102 (26.2) | 108 (26.7) | 92 (23.8) | 200 (25.3) |
| p-value | p = 0.58 | p = 0.34 | |||
| Ever diarrhoea after baseline | |||||
| No | 234 (58.2) | 213 (54.6) | 217 (53.7) | 230 (59.3) | 447 (56.4) |
| Yes | 168 (41.8) | 177 (45.4) | 187 (46.3) | 158 (40.7) | 345 (43.6) |
| p-value | p = 0.31 | p = 0.11 | |||
| First thrush after baselineb | |||||
| No | 386 (96.3) | 381 (96.0) | 390 (96.8) | 377 (95.4) | 767 (96.1) |
| Yes | 15 (3.7) | 16 (4.0) | 13 (3.2) | 18 (4.6) | 31 (3.9) |
| p-value | p = 0.83 | p = 0.33 | |||
| New thrush after baseline or worse than at baseline | |||||
| No | 385 (96.0) | 374 (95.9) | 390 (96.5) | 369 (95.3) | 759 (96.0) |
| Yes | 16 (4.0) | 16 (4.1) | 14 (3.5) | 18 (4.7) | 32 (4.0) |
| p-value | p = 0.94 | p = 0.40 | |||
| Ever thrush after baseline | |||||
| No | 374 (93.3) | 360 (92.3) | 379 (93.8) | 355 (91.7) | 734 (92.8) |
| Yes | 27 (6.7) | 30 (7.7) | 25 (6.2) | 32 (8.3) | 57 (7.2) |
| p-value | p = 0.60 | p = 0.26 | |||
| First rash after baselinec | |||||
| No | 310 (86.6) | 317 (86.8) | 329 (88.4) | 298 (84.9) | 627 (86.7) |
| Yes | 48 (13.4) | 48 (13.2) | 43 (11.6) | 53 (15.1) | 96 (13.3) |
| p-value | p = 0.92 | p = 0.16 | |||
| New rash after baseline or worse than at baseline | |||||
| No | 348 (86.8) | 331 (84.9) | 354 (87.6) | 325 (84.0) | 679 (85.8) |
| Yes | 53 (13.2) | 59 (15.1) | 50 (12.4) | 62 (16.0) | 112 (14.2) |
| p-value | p = 0.44 | p = 0.14 | |||
| Ever rash after baseline | |||||
| No | 307 (76.6) | 291 (74.6) | 317 (78.5) | 281 (72.6) | 598 (75.6) |
| Yes | 94 (23.4) | 99 (25.4) | 87 (21.5) | 106 (27.4) | 193 (24.4) |
| p-value | p = 0.52 | p = 0.055 | |||
In addition, when considering skin rash severity during the treatment period only, there was evidence of a difference between the shorter- and longer-duration arms. Participants in the longer-duration arm experienced greater skin rash severity than participants in the shorter-duration arm at days 3 (p = 0.019) and 7 (p = 0.005) (Figure 20). There was no evidence of a difference between dose randomisation arms in terms of skin rash severity during the treatment period, and there was no evidence of a difference between the dose and duration randomisation arms in severity of diarrhoea or oral thrush during the treatment period (see Table 24).
FIGURE 20.
Skin rash severity during treatment period: duration randomisation.
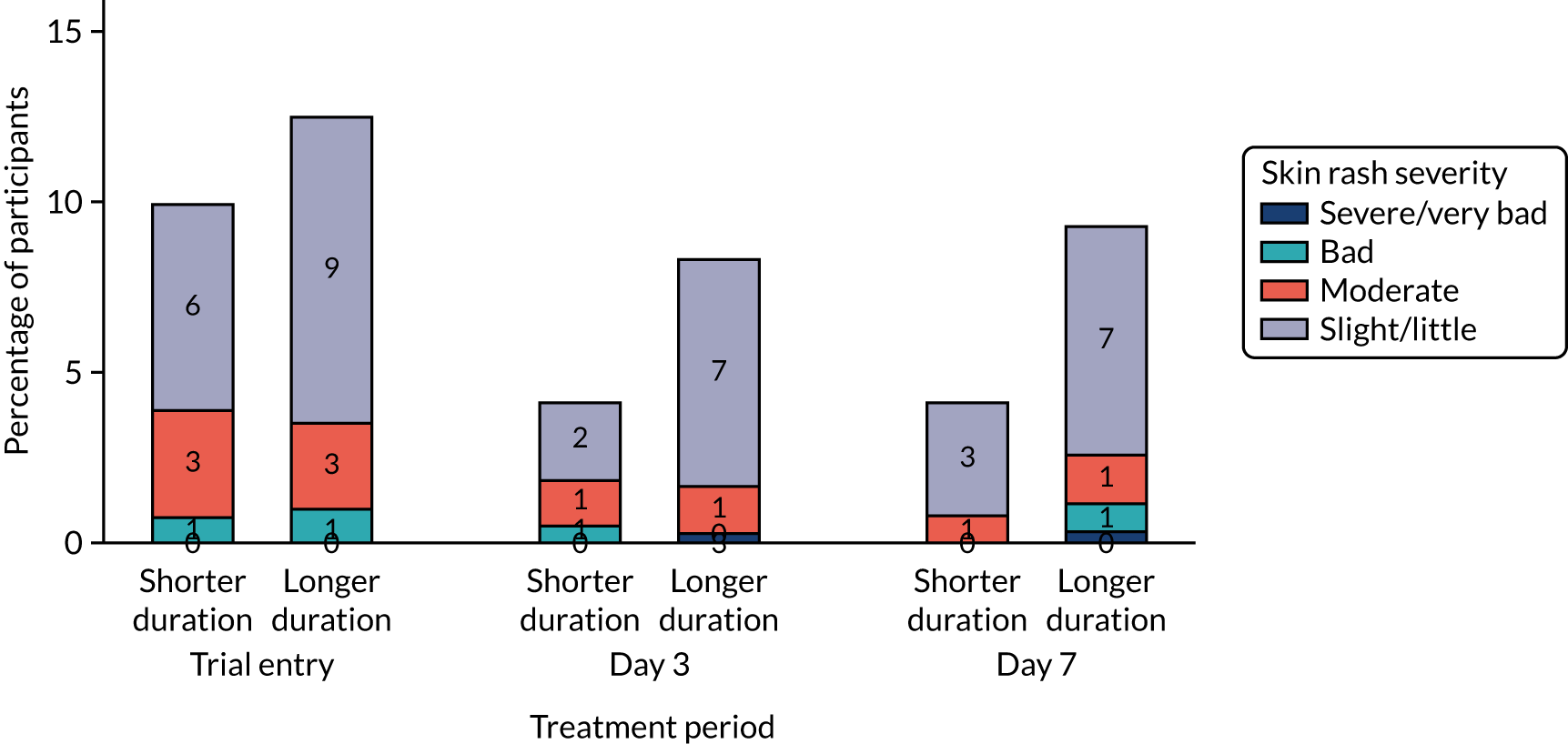
Health-care services
Utilisation of health-care services was unrelated to randomisation arm. Hospital admissions and visits to the ED without admission were reported in 46 (5.7%) and 43 (5.3%) participants, respectively, whereas a larger proportion of participants reported using any health-care service (n = 304, 37.3%) (Table 25).
| Health-care service utilisation | Treatment arm, n (%) | p-value | Treatment arm, n (%) | p-value | Total (N = 814), n (%) | ||
|---|---|---|---|---|---|---|---|
| Lower dose (N = 410) | Higher dose (N = 404) | Shorter duration (N = 413) | Longer duration (N = 401) | ||||
| Ever admitted to hospital? | |||||||
| Yes | 24 (5.9) | 22 (5.4) | 0.80 | 27 (6.5) | 19 (4.7) | 0.27 | 46 (5.7) |
| No | 386 (94.1) | 382 (94.6) | 386 (93.5) | 382 (95.3) | 768 (94.3) | ||
| Visited ED (without admission)? | |||||||
| Yes | 21 (5.1) | 22 (5.4) | 0.84 | 18 (4.4) | 25 (6.2) | 0.23 | 43 (5.3) |
| No | 389 (94.9) | 382 (94.6) | 395 (95.6) | 376 (93.8) | 771 (94.7) | ||
| Ever used any other health-care service? | |||||||
| Yes | 149 (36.3) | 155 (38.4) | 0.55 | 152 (36.8) | 152 (37.9) | 0.75 | 304 (37.3) |
| No | 261 (63.7) | 249 (61.6) | 261 (63.2) | 249 (62.1) | 510 (62.7) | ||
Daily activities and child care
Data on daily activities and child care were available from parent/guardian-completed diaries for 441 participants (Table 26). No differences in reported disruption to daily activities and child care were found between randomisation arms. In total, 73.9% of participants reported that the child had missed school, day care or nursery, and the median number of days missed was 4 (IQR 0–6) days. In addition, 63.8% of parents reported missing work, with a median of 3 (IQR 0–5) days missed, and 34.9% of parents reported requiring additional care for the child.
| Daily activity/child care | Treatment arm | p-value | Treatment arm | p-value | Total (N = 441) | ||
|---|---|---|---|---|---|---|---|
| Lower dose (N = 298) | Higher dose (N = 289) | Shorter duration (N = 291) | Longer duration (N = 296) | ||||
| Child missed school, day care or nursery: ever, n (%) | |||||||
| Yes | 152 (71.0) | 174 (76.7) | 0.18 | 159 (72.3) | 167 (75.6) | 0.43 | 326 (73.9) |
| No | 62 (29.0) | 53 (23.3) | 61 (27.7) | 54 (24.4) | 115 (26.1) | ||
| Days child missed school, day care or nursery, median (IQR) | 4 (0–5) | 4 (2–6) | 0.14 | 4 (0–6) | 4 (2–6) | 0.62 | 4 (0–6) |
| Parent missed work: ever, n (%) | |||||||
| Yes | 128 (64.0) | 136 (63.6) | 0.92 | 127 (62.9) | 137 (64.6) | 0.71 | 264 (63.8) |
| No | 72 (36.0) | 78 (36.4) | 75 (37.1) | 75 (35.4) | 150 (36.2) | ||
| Days parent missed work, median (IQR) | 3 (0–4) | 3 (0–5) | 0.43 | 3 (0–4) | 3 (0–5) | 0.20 | 3 (0–5) |
| Parent missed other activities: ever, n (%) | |||||||
| Yes | 50 (33.6) | 56 (33.7) | 0.97 | 53 (34.2) | 53 (33.1) | 0.84 | 106 (33.7) |
| No | 99 (66.4) | 110 (66.3) | 102 (65.8) | 107 (66.9) | 209 (66.3) | ||
| Days parent missed other activities: cumulative, median (IQR) | 0 (0–4) | 0 (0–4) | 0.88 | 0 (0–5) | 0 (0–4) | 0.50 | 0 (0–4) |
| Additional care needed for child: ever, n (%) | |||||||
| Yes | 73 (36.3) | 72 (33.5) | 0.54 | 73 (34.9) | 72 (34.8) | 0.98 | 145 (34.9) |
| No | 128 (63.7) | 143 (66.5) | 136 (65.1) | 135 (65.2) | 271 (65.1) | ||
| Days additional care needed for child: cumulative, median (IQR) | 0 (0–3) | 0 (0–3) | 0.54 | 0 (0–3) | 0 (0–3) | 0.83 | 0 (0–3) |
Chapter 4 Discussion
We investigated the impact of dose and duration of amoxicillin treatment for uncomplicated CAP in children discharged from hospital after assessment in a PED, or after a short stay on an assessment unit or inpatient ward. Regarding duration, we focused on oral amoxicillin treatment after discharge rather than total treatment duration, given that discharge home is a key time point for clinical decision-making, as close monitoring of the child will no longer be possible. In this population, we found a 3-day treatment course of amoxicillin to be non-inferior to a 7-day course, and a lower total daily dose to be non-inferior to a higher dose, in terms of antibiotic retreatment for respiratory tract infection within 28 days.
Limitations
In contrast to the majority of trials addressing optimal antibiotic treatment duration and dose of a single drug for childhood pneumonia, CAP-IT was conducted in a high-income setting where the expected mortality, even from moderate to severe CAP, is low. We selected antibiotic retreatment for respiratory tract infection during a follow-up period of 28 days as a clinically relevant and ascertainable event with limited risk of bias in a placebo-controlled trial. 84
To further guard against bias, an independent ERC, comprising experienced clinicians, adjudicated all antibiotic retreatments during the trial period, regarding the reason (i.e. respiratory tract infection or other) and clinical indication. Of note, the primary end point could be ascertained in 97% of CAP-IT participants either at final follow-up or through contact with the GP. Therefore, we consider the impact of loss to follow-up negligible.
We aimed to exclude children in whom antibiotics would not be expected to have any beneficial effect, primarily those likely to have obstructive airway disease only. However, a mixed picture was common for hospitalised children, with 16% of children receiving either salbutamol or steroids during their hospital stay. Mostly, this affected children with pre-existing hyper-reactive airway disease, and treatment was discontinued in a majority of cases by the time children were discharged home. Compared with the 48% bronchodilator use observed in the most recent UK paediatric pneumonia audit85 the use of salbutamol or steroids was low in CAP-IT, indicating that there was a strong clinical suspicion of CAP likely to benefit from antibiotics in enrolled children.
We observed no relevant impact of either amoxicillin duration or dose on pneumococcal penicillin non-susceptibility at 28 days, but did not assess pneumococcal resistance at other time points. We did not obtain end-of-treatment samples on all children for resistance analysis for several reasons. First, an additional face-to-face visit would have been a major barrier to participation for many families. Second, penicillin colonisation rates at, or shortly after, the end of antibiotic treatment are expected to be very low, whereas significant recolonisation or regrowth was expected (and observed) by 28 days. Finally, we considered penicillin-resistant pneumococcal colonisation at final follow-up to be the most relevant population- and individual-level resistance marker, as children colonised at this time point could transmit resistant pneumococci to others and would be at higher risk of potentially more difficult to treat respiratory tract infections in the future. 86
Generalisability
Children were enrolled in the trial based on clinically diagnosed pneumonia requiring antibiotic treatment with amoxicillin, and are typical of children treated for pneumonia with amoxicillin in PEDs. We included children discharged from hospital within 48 hours of admission for observation or initial clinical management, as hospital stays for acute respiratory tract infections, including pneumonia, are mostly of very short duration. 87,88 Data from the pilot phase confirmed that these children could be considered part of the same spectrum of disease as those discharged directly home from the ED. Only 13% of screened children were not approached because of physician preference for an antibiotic other than amoxicillin at discharge. This is in keeping with guidance suggesting that amoxicillin is used as the first-line antibiotic for oral treatment of uncomplicated childhood pneumonia in the community.
We excluded children with complicated pneumonia requiring prolonged hospitalisation, and those receiving non-beta-lactam treatment. Our findings, therefore, cannot be directly generalised to more severely ill children or those treated for atypical pneumonia. However, it is highly likely that our observations are relevant to children with mild to moderate pneumonia seen in primary care, who would be treated with oral amoxicillin at home. In primary care, the acuity of disease is generally lower and a lower rate of pneumonia likely to benefit from antibiotic treatment is expected.
No nasopharyngeal penicillin-resistant pneumococcal isolates were observed in the trial, either at baseline or at final follow-up, which is consistent with reported low penicillin resistance levels in northern Europe. 89 Therefore, our findings for the effectiveness of lower compared with higher amoxicillin dose, and impact on resistance, may be of limited generalisability to children with pneumonia in other high-income settings with higher pneumococcal penicillin resistance prevalence.
Twice-daily dosing of amoxicillin in line with WHO and other international recommendations was used in CAP-IT, rather than administration in three daily doses, as recommended by the BNFc. This was selected as it is known to maximise adherence, which would be particularly important in children allocated to the lower-dose and shorter-duration arms. In addition, patient representatives involved in the design phase indicated this approach to be particularly family friendly, as an additional mid-day dose is difficult to give to children who attend day care. Consequently, our findings, especially for antimicrobial resistance outcomes, may not be generalisable to children being treated with a thrice-daily amoxicillin regimen. However, participants in CAP-IT had rates of antibiotic retreatment and secondary or rehospitalisation similar to those described in observational studies conducted in settings with standard administration of amoxicillin in three doses. 41,87,90,91
Interpretation
To the best of our knowledge, few head-to-head comparisons of the same antibiotic in different dosing or duration regimens have been conducted in children being treated for pneumonia. Most of the existing literature reports on trials conducted in low- and middle-income settings prior to the widespread availability of PCVs and in an era with lower pneumococcal penicillin resistance. 92,93 Two recent relevant trials94,95 conducted in Malawi investigated 3-day compared with 5-day amoxicillin treatment and 3-day amoxicillin treatment compared with placebo in young children with non-severe pneumonia and not infected with human immunodeficiency virus. In summary, 3-day treatment at a dose corresponding to the higher total daily dose in CAP-IT was found to be non-inferior to 5-day treatment for early treatment failure, but this was not the case for placebo compared with 3-day treatment. The same trial identified the number needed to treat for children with non-severe fast-breathing pneumonia to be 33. These trials used high-sensitivity, but low-specificity eligibility criteria appropriate for a high-mortality setting. Evidence specific to high-income settings is lacking and has led some guideline-setting bodies to question the generalisability of findings from large trials in low- or middle-income countries to high-income settings. The persisting evidence gap for children identified as having pneumonia applying higher specificity clinical criteria in high-income settings has now been addressed by CAP-IT.
A relatively high retreatment rate of 12.5% was observed in the CAP-IT cohort. This is consistent with similarly high retreatment rates in primary care reported in large observational studies, but has not previously been described for children with CAP seen in EDs or discharged from hospital after a short stay. Similarly, the secondary or rehospitalisation rate of around 5% was similar to that described for children with pneumonia in observational studies.
We observed remarkably similar retreatment rates for respiratory tract infections between the 3- and 7-day treatment durations, despite 2-day slower resolution of mild cough, on average, in the shorter-duration arm. We did not identify any differences between the lower- and higher-dose treatment arms. Antibiotic retreatment for respiratory tract infection during the follow-up period could be related to true failure of the initial treatment or could be linked to persistent symptoms unlikely to be responsive to amoxicillin because they were mainly triggered by a viral (co-)infection or new respiratory tract infection episodes.
Children and parents in the 3-day randomisation arm were not reported to have spent a longer time away from day care or school and work, making it unlikely that cough had a major impact on children’s usual routines. Slightly longer time to symptom resolution in placebo arms or placebo-controlled shorter-duration arms has been reported for acute otitis media. 96 However, it is unclear how children being mildly symptomatic for longer is weighed against the benefits of shorter treatment by children and their families. When symptoms are minor, shorter treatment is likely to be a key factor in allowing children to return to usual activities and will maximise adherence. 97,98
Antimicrobial resistance was a key secondary outcome in CAP-IT. Colonisation by penicillin-non-susceptible pneumococci at 28 days was similar for both randomisation arms. In general, the observed prevalence of pneumococcal penicillin non-susceptibility and the complete absence of penicillin-resistant pneumococci was in line with the UK being a low-resistance setting. Pneumococcal penicillin resistance alone is unlikely to reflect the full impact of amoxicillin dose and duration on the child nasopharyngeal microflora, including the presence of resistance genotypes. Next-generation sequencing approaches could provide in-depth information about differential changes in the microbiome and resistome with higher or lower amoxicillin dose and shorter or longer treatment duration. However, the interpretation of such analyses is likely to be complex, and will need to take account of the interactions between different pneumococcal subpopulations, as well as between pneumococci and other bacteria in a densely populated niche. An analysis of nasopharyngeal samples obtained in CAP-IT using next-generation sequencing approaches is ongoing.
Several other trials have generated results that complement CAP-IT findings, or are expected to in the near future. In the UK, this includes the primary care-based ARTIC PC (Antibiotics for lower Respiratory Tract Infection in Children presenting in Primary Care) study,99 a randomised placebo-controlled trial investigating the benefit of a 7-day course of oral amoxicillin in children with possible lower respiratory tract infection (but not considered to have pneumonia clinically). The SAFER (Short-Course Antimicrobial Therapy for Pediatric Community-Acquired Pneumonia) trial100 in Canada and SCOUT-CAP (Short Course Outpatient Therapy of Community Acquired Pneumonia) study in the USA both target children presenting to EDs but not admitted to hospital, the former comparing 5- and 10-day treatment courses with amoxicillin and the latter a selection of beta-lactams. The SCOUT-CAP study is expected to report on results at the end of 2021. Finally, a Canadian open-label RCT (NCT03031210) is investigating twice-compared with thrice-daily amoxicillin dosing in children treated for pneumonia. The total daily dose in this trial corresponds to the higher total daily dose investigated in CAP-IT.
Implications
For clinical practice, CAP-IT supports routine use of shorter, 3-day, oral amoxicillin courses at current doses for children presenting to hospital with uncomplicated clinically diagnosed CAP for community-based treatment after discharge from acute care. A slightly longer time to resolution of mild cough can be expected in children treated for 3 days, compared with children treated for 7 days.
For research, existing systematic reviews and meta-analyses should be updated to include CAP-IT and other high-income setting trials. A series of relevant trials includes studies already completed or about to complete. Their inclusion, for example in existing Cochrane reviews, would ensure that key reference systematic reviews are relevant globally.
The question of the comparison between two and three times daily dosing of amoxicillin needs to be addressed. However, this may best be tackled by modelling and simulation based on high-quality pharmacokinetic data analysed using modern pharmacometric approaches. Such data are needed from a variety of settings, including low/high prevalence of pneumococcal penicillin resistance, varying pneumococcal vaccine coverage and low-, middle- and high-income settings characterised by varying prevalence of important covariates, such as malnutrition and obesity. Data from adults suggest that gut amoxicillin absorption may be saturable, limiting the expected utility of high-dose regimens. 101
A proportion of children screened for CAP-IT were identified to be ineligible because the managing clinician was planning treatment with an antibiotic other than amoxicillin. Trial data supporting the use of macrolides (targeting atypical pathogens) or alternative beta-lactams, such as amoxicillin/clavulanate (co-amoxiclav, targeting Gram-negative respiratory pathogens producing beta-lactamases), are lacking.
Chapter 5 Conclusions
-
For children presenting to acute care settings with uncomplicated, clinically diagnosed, moderate or moderate to severe CAP who can be managed at home, there is no evidence to suggest that a longer 7-day treatment course of oral amoxicillin offers any advantage over a shorter 3-day course, in terms of antibiotic retreatment for respiratory tract infection within 4 weeks. Therefore, the trial supports routine use of 3-day oral amoxicillin courses after discharge from hospital in this population.
-
Slightly longer time to resolution of mild cough was observed in children treated for 3 days than in those treated for 7 days. Given the advantages of a shorter duration of treatment for adherence and the observed declining adherence during treatment days 4–7 in the trial, a 3-day course of oral amoxicillin nonetheless appears preferable. This would have the added benefit of greater harmonisation of antibiotic treatment duration guidance between low-/middle-income and high-income settings.
-
Similarly, we found that lower total daily doses of oral amoxicillin were non-inferior to higher daily doses, in terms of antibiotic retreatment for respiratory tract infection within 4 weeks. Dosing regimens were also similar in terms of impact on pneumococcal antimicrobial resistance and safety.
-
Of note, a weight-banded approach was used for dose selection, resulting in less variability in total daily dose compared with an age-banded approach (as is used in the UK in clinical practice). Based on the age-banded approach, both doses studied in CAP-IT are expected to be prescribed in the UK because of variations in weight within broad age bands.
-
Either (lower and higher) total daily dose is feasible to deliver in high-income settings where amoxicillin suspensions of different concentrations are available and are prescribed in preference to solid child-appropriate formulations (i.e. solid forms that are liquid on ingestion or become liquid on administration). As a result, moving between lower and higher total daily doses does not result in greater volumes per dose for treated children.
-
However, the situation is different in low- and middle-income settings, where the preferred formulation is dispersible tablets. The lowest-concentration child-appropriate solid formulation supported by the United Nations International Children's Emergency Fund (UNICEF) and WHO contains 250 mg of amoxicillin in a non-divisible dispersible tablet. Administration of this tablet twice a day to young infants (weighing 4–10 kg) gives a wide dose range of 50 (10 kg) to 125 (4 kg) mg/kg per day, with many children expected to receive doses in the higher dose range of CAP-IT. CAP-IT results did not identify any clinically relevant disadvantages to using higher doses; therefore, supporting the continued use of existing dispersible tablets.
-
We did not formally compare twice- with thrice-daily dosing. However, we note that children in CAP-IT had good clinical outcomes, with antibiotic retreatment rates and secondary or re-admission rates similar to those described for children with acute lower respiratory tract infections in observational studies in the UK where amoxicillin treatment would generally be given three times daily.
Acknowledgements
We would like to thank all of the patients, their families and the staff from the participating centres for their vital contributions to CAP-IT.
Trial Steering Committee
Elizabeth Molyneux (chairperson), Chris Butler, Alan Smyth and Catherine Pritchard (PPI).
Independent Data Monitoring Committee
Tim Peto (chairperson), Simon Cousens and Stuart Logan.
Independent End-Point Review Committee
Alasdair Bamford (chairperson), Anna Turkova, Anna Goodman and Felicity Fitzgerald.
Trial Management Group
Mike Sharland (chairperson), Julia Bielicki, Mark Lyttle, Colin Powell, Paul Little, Saul Faust, Adam Finn, Julie Robotham, Mandy Wan, David Dunn, Wolfgang Stöhr, Lynda Harper, Sam Barratt, Nigel Klein, Louise Rogers, Elia Vitale and Diana Gibb.
Investigator sites
Alder Hey Children’s Hospital NHS Foundation Trust, Liverpool: Dan Hawcutt (principal investigator), Matt Rotheram, Liz D Lee and Rachel Greenwood-Bibby.
Birmingham Children’s Hospital: Stuart Hartshorn (principal investigator), Deepthi Jyothish, Louise Rogers and Juliet Hopkins.
Bristol Royal Hospital for Children: Mark Lyttle (principal investigator), Stefania Vergnano, Lucie Aplin, Pauline Jackson, Gurnie Kaur, Beth Pittaway, Michelle Ross, Sarah Sheedy, Alice Smith and Yesenia Tanner.
Chelsea and Westminster Hospital NHS Foundation Trust, London: James Ross (principal investigator), Poonam Patel, Nabila Burney, Hannah Fletcher, Jaime Carungcong and Kribashnie Nundlall.
City Hospitals Sunderland NHS Foundation Trust: Niall Mullen (principal investigator), Rhona McCrone and Paul P Corrigan.
Countess of Chester Hospital: Susie Holt (principal investigator), John Gibbs, Caroline Burchett, Caroline Lonsdale and Sarah De-Beger.
Darlington Memorial Hospital: Godfrey Nyamugunuduru (principal investigator), John Furness (former principal investigator) and Dawn Eggington.
Derbyshire Children’s Hospital, Derby: Gisela Robinson (principal investigator), Lizzie Starkey and Mel Hayman.
Evelina London Children’s Hospital: Anastasia Alcock (principal investigator), Dani Hall (former principal investigator), Ronny Cheung, Alyce B Sheedy and Mohammad Ahmad.
James Cook University Hospital, Middlesbrough: Arshid Murad (principal investigator), Katherine Jarman and Joanna Green.
John Radcliffe Hospital, Oxford: Tanya Baron (principal investigator), Chris Bird (former principal investigator I), Shelley Segal and Sally Beer.
King’s College Hospital, London: Fleur Cantle (principal investigator), Hannah Eastman, Raine Astin-Chamberlain, Paul Riozzi and Hannah Cotton.
Leeds Children’s Hospital: Sean O’Riordan (principal investigator), Alice Downes, Marjorie Allen and Louise Conner.
Leicester Royal Infirmary: Damian Roland (principal investigator), Srini Bandi and Rekha Patel.
Nottingham University Hospitals NHS Trust: Chris Gough (principal investigator), Megan McAulay and Louise Conner.
Southport and Ormskirk Hospital NHS Trust: Sharryn Gardner (principal investigator), Zena Haslam and Moira Morrison.
Our Lady’s Children’s Hospital, Crumlin: Michael Barrett (principal investigator) and Madeleine Niermeyer.
Royal Alexandra Children’s Hospital, Brighton: Emily Walton (principal investigator), Akshat Kapur and Vivien Richmond.
Royal Hospital for Children, Glasgow: Steven Foster (principal investigator), Ruth Bland, Ashleigh Neil, Barry Milligan and Helen Bannister.
Royal London Hospital: Ben Bloom (principal investigator), Ami Parikh, Olivia Boulton and Imogen Skene.
Royal Manchester Children’s Hospital: Katherine Potier (principal investigator), Fiona Poole and Jill L Wilson.
Sheffield Children’s NHS Foundation Trust: Judith Gilchrist (principal investigator), Noreen West, Jayne Evans and Julie Morecombe.
St George’s Hospital, London: Paul Heath (principal investigator), Yasser Iqbal, Malte Kohns, Elena Stefanova and Elia Vitale.
St Mary’s Hospital, London: Ian Maconochie (principal investigator), Suzanne Laing and Rikke Jorgensen.
University Hospital Lewisham: Maggie Nyirenda (principal investigator), Anastasia Alcock, Samia Pilgrim and Emma Gardiner.
University Hospital of Wales, Cardiff: Jeff Morgan (principal investigator), Colin VE Powell (former principal investigator), Jennifer Muller and Gail Marshall.
Southampton Children’s Hospital: Katrina Cathie (principal investigator), Jane Bayreuther, Ruth Ensom and Emily Cornish.
Contributions of authors
Sam Barratt (https://orcid.org/0000-0003-3265-9486) (Clinical Trials Manager) contributed to compiling data and drafting the report.
Julia A Bielicki (https://orcid.org/0000-0002-3902-5489) (Consultant in Paediatric Infectious Diseases) drafted the report.
David Dunn (https://orcid.org/0000-0003-1836-4446) (Professor of Medical Statistics) performed statistical analyses.
Saul N Faust (https://orcid.org/0000-0003-3410-7642) (Professor of Paediatric Immunology and Infectious Diseases) contributed to drafting the report.
Adam Finn (https://orcid.org/0000-0003-1756-5668) (Professor of Paediatrics) performed resistance analyses.
Lynda Harper (https://orcid.org/0000-0003-3430-0127) (Clinical Project Manager) contributed to drafting the report.
Pauline Jackson (https://orcid.org/0000-0001-6461-953X) (Paediatric Nurse) contributed to drafting the report.
Mark D Lyttle (https://orcid.org/0000-0002-8634-7210) (Consultant in Paediatric Emergency Medicine) contributed to drafting the report.
Colin VE Powell (https://orcid.org/0000-0001-8181-875X) (Consultant in Paediatric Emergency Medicine) contributed to drafting the report.
Louise Rogers (https://orcid.org/0000-0002-5037-847X) (Clinical Research Sister) contributed to drafting the report.
Damian Roland (https://orcid.org/0000-0001-9334-5144) (Consultant in Paediatric Emergency Medicine) contributed to drafting the report.
Wolfgang Stöhr (https://orcid.org/0000-0002-6533-2888) (Senior Medical Statistician) performed statistical analyses.
Kate Sturgeon (https://orcid.org/0000-0003-2209-141X) (Senior Research Nurse) contributed to drafting the report.
Elia Vitale (https://orcid.org/0000-0001-5750-8436) (Senior Paediatric Clinical Research Nurse) contributed to drafting the report.
Mandy Wan (https://orcid.org/0000-0001-8802-0425) (Paediatric Clinical Trials Pharmacist) contributed to drafting the report.
Diana M Gibb (https://orcid.org/0000-0002-9738-5490) (Professor, Epidemiology) drafted the report.
Mike Sharland (https://orcid.org/0000-0001-8626-8291) (Professor of Paediatric Infectious Diseases) drafted the report.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Lyttle MD, Bielicki JA, Barratt S, Dunn D, Finn A, Harper L, et al. Efficacy, safety and impact on antimicrobial resistance of duration and dose of amoxicillin treatment for young children with Community-Acquired Pneumonia: a protocol for a randomIsed controlled Trial (CAP-IT). BMJ Open 2019;9. https://doi.org/10.1136/bmjopen-2019-029875.
- Holstiege J, Schink T, Molokhia M, Mazzaglia G, Innocenti F, Oteri A, et al. Systemic antibiotic prescribing to paediatric outpatients in 5 European countries: a population-based cohort study. BMC Pediatr 2014;14. https://doi.org/10.1186/1471-2431-14-174.
- Sturkenboom MC, Verhamme KM, Nicolosi A, Murray ML, Neubert A, Caudri D, et al. Drug use in children: cohort study in three European countries. BMJ 2008;337. https://doi.org/10.1136/bmj.a2245.
- Vaz LE, Kleinman KP, Raebel MA, Nordin JD, Lakoma MD, Dutta-Linn MM, et al. Recent trends in outpatient antibiotic use in children. Pediatrics 2014;133:375-85. https://doi.org/10.1542/peds.2013-2903.
- Grijalva CG, Nuorti JP, Griffin MR. Antibiotic prescription rates for acute respiratory tract infections in US ambulatory settings. JAMA 2009;302:758-66. https://doi.org/10.1001/jama.2009.1163.
- Hersh AL, Shapiro DJ, Pavia AT, Shah SS. Antibiotic prescribing in ambulatory pediatrics in the United States. Pediatrics 2011;128:1053-61. https://doi.org/10.1542/peds.2011-1337.
- Cevey-Macherel M, Galetto-Lacour A, Gervaix A, Siegrist CA, Bille J, Bescher-Ninet B, et al. Etiology of community-acquired pneumonia in hospitalized children based on WHO clinical guidelines. Eur J Pediatr 2009;168:1429-36. https://doi.org/10.1007/s00431-009-0943-y.
- Bielicki J, Doerholt K, Sharland M. The Effect of Age Banding on Daily Dose of Amoxicillin-Based Treatments of Community Acquired Pneumonia n.d.
- Sands R, Shanmugavadivel D, Stephenson T, Wood D. Medical problems presenting to paediatric emergency departments: 10 years on. Emerg Med J 2012;29:379-82. https://doi.org/10.1136/emj.2010.106229.
- Oostenbrink R, Thompson M, Lakhanpaul M, Steyerberg EW, Coad N, Moll HA. Children with fever and cough at emergency care: diagnostic accuracy of a clinical model to identify children at low risk of pneumonia. Eur J Emerg Med 2013;20:273-80. https://doi.org/10.1097/MEJ.0b013e32835771fd.
- Gill PJ, Goldacre MJ, Mant D, Heneghan C, Thomson A, Seagroatt V, et al. Increase in emergency admissions to hospital for children aged under 15 in England, 1999–2010: national database analysis. Arch Dis Child 2013;98:328-34. https://doi.org/10.1136/archdischild-2012-302383.
- Brousseau DC, Nimmer MR, Yunk NL, Nattinger AB, Greer A. Nonurgent emergency-department care: analysis of parent and primary physician perspectives. Pediatrics 2011;127:e375-81. https://doi.org/10.1542/peds.2010-1723.
- NHS Digital . Hospital Episode Statistics n.d. www.hscic.gov.uk/hes (accessed 26 September 2020).
- Koshy E, Murray J, Bottle A, Sharland M, Saxena S. Impact of the seven-valent pneumococcal conjugate vaccination (PCV7) programme on childhood hospital admissions for bacterial pneumonia and empyema in England: national time-trends study, 1997–2008. Thorax 2010;65:770-4. https://doi.org/10.1136/thx.2010.137802.
- Honkinen M, Lahti E, Osterback R, Ruuskanen O, Waris M. Viruses and bacteria in sputum samples of children with community-acquired pneumonia. Clin Microbiol Infect 2012;18:300-7. https://doi.org/10.1111/j.1469-0691.2011.03603.x.
- McEllistrem MC, Adams JM, Patel K, Mendelsohn AB, Kaplan SL, Bradley JS, et al. Acute otitis media due to penicillin-nonsusceptible Streptococcus pneumoniae before and after the introduction of the pneumococcal conjugate vaccine. Clin Infect Dis 2005;40:1738-44. https://doi.org/10.1086/429908.
- Casey JR, Pichichero ME. Changes in frequency and pathogens causing acute otitis media in 1995-2003. Pediatr Infect Dis J 2004;23:824-8. https://doi.org/10.1097/01.inf.0000136871.51792.19.
- Public Health England, Department of Health and Social Care . Immunisation Against Infectious Disease n.d. www.gov.uk/government/collections/immunisation-against-infectious-disease-the-green-book (accessed 25 September 2020).
- Screening and Immunisations Team, Health and Social Care Information Centre . NHS Immunisation Statistics, England – 2012–13 n.d. https://digital.nhs.uk/data-and-information/publications/statistical/nhs-immunisation-statistics/nhs-immunisation-statistics-england-2012-13 (accessed 3 August 2021).
- Foster D, Walker AS, Paul J, Griffiths D, Knox K, Peto TE, et al. Reduction in invasive pneumococcal disease following implementation of the conjugate vaccine in the Oxfordshire region, England. J Med Microbiol 2011;60:91-7. https://doi.org/10.1099/jmm.0.023135-0.
- van Gageldonk-Lafeber AB, Bogaerts MA, Verheij RA, van der Sande MA. Time trends in primary-care morbidity, hospitalization and mortality due to pneumonia. Epidemiol Infect 2009;137:1472-8. https://doi.org/10.1017/S0950268809002258.
- Harris M, Clark J, Coote N, Fletcher P, Harnden A, McKean M, et al. British Thoracic Society guidelines for the management of community acquired pneumonia in children: update 2011. Thorax 2011;66:ii1-23. https://doi.org/10.1136/thoraxjnl-2011-200598.
- Courtoy I, Lande AE, Turner RB. Accuracy of radiographic differentiation of bacterial from nonbacterial pneumonia. Clin Pediatr 1989;28:261-4. https://doi.org/10.1177/000992288902800604.
- Swingler GH. Radiologic differentiation between bacterial and viral lower respiratory infection in children: a systematic literature review. Clin Pediatr 2000;39:627-33. https://doi.org/10.1177/000992280003901101.
- McCarthy PL, Spiesel SZ, Stashwick CA, Ablow RC, Masters SJ, Dolan TF. Radiographic findings and etiologic diagnosis in ambulatory childhood pneumonias. Clin Pediatr 1981;20:686-91. https://doi.org/10.1177/000992288102001101.
- Neuman MI, Hall M, Hersh AL, Brogan TV, Parikh K, Newland JG, et al. Influence of hospital guidelines on management of children hospitalized with pneumonia. Pediatrics 2012;130:e823-30. https://doi.org/10.1542/peds.2012-1285.
- Hayward G, Thompson M, Hay AD. What factors influence prognosis in children with acute cough and respiratory tract infection in primary care?. BMJ 2012;345. https://doi.org/10.1136/bmj.e6212.
- Hay AD, Fahey T, Peters TJ, Wilson A. Predicting complications from acute cough in pre-school children in primary care: a prospective cohort study. Br J Gen Pract 2004;54:9-14.
- Chorazy ML, Lebeck MG, McCarthy TA, Richter SS, Torner JC, Gray GC. Polymicrobial acute respiratory infections in a hospital-based pediatric population. Pediatr Infect Dis J 2013;32:460-6. https://doi.org/10.1097/INF.0b013e31828683ce.
- Van den Bruel A, Haj-Hassan T, Thompson M, Buntinx F, Mant D. European Research Network on Recognising Serious Infection investigators . Diagnostic value of clinical features at presentation to identify serious infection in children in developed countries: a systematic review. Lancet 2010;375:834-45. https://doi.org/10.1016/S0140-6736(09)62000-6.
- Craig JC, Williams GJ, Jones M, Codarini M, Macaskill P, Hayen A, et al. The accuracy of clinical symptoms and signs for the diagnosis of serious bacterial infection in young febrile children: prospective cohort study of 15 781 febrile illnesses. BMJ 2010;340. https://doi.org/10.1136/bmj.c1594.
- Van den Bruel A, Bartholomeeusen S, Aertgeerts B, Truyers C, Buntinx F. Serious infections in children: an incidence study in family practice. BMC Fam Pract 2006;7. https://doi.org/10.1186/1471-2296-7-23.
- Aujesky D, Fine MJ. The pneumonia severity index: a decade after the initial derivation and validation. Clin Infect Dis 2008;47:133-9. https://doi.org/10.1086/591394.
- Welte T. Risk factors and severity scores in hospitalized patients with community-acquired pneumonia: prediction of severity and mortality. Eur J Clin Microbiol Infect Dis 2012;31:33-47. https://doi.org/10.1007/s10096-011-1272-4.
- Reed C, Madhi SA, Klugman KP, Kuwanda L, Ortiz JR, Finelli L, et al. Development of the Respiratory Index of Severity in Children (RISC) score among young children with respiratory infections in South Africa. PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0027793.
- Scott JA, Wonodi C, Moïsi JC, Deloria-Knoll M, DeLuca AN, Karron RA, et al. The definition of pneumonia, the assessment of severity, and clinical standardization in the Pneumonia Etiology Research for Child Health study. Clin Infect Dis 2012;54:109-16. https://doi.org/10.1093/cid/cir1065.
- Tiewsoh K, Lodha R, Pandey RM, Broor S, Kalaivani M, Kabra SK. Factors determining the outcome of children hospitalized with severe pneumonia. BMC Pediatr 2009;9. https://doi.org/10.1186/1471-2431-9-15.
- Lozano JM. Epidemiology of hypoxaemia in children with acute lower respiratory infection. Int J Tuberc Lung Dis 2001;5:496-504.
- Fu LY, Ruthazer R, Wilson I, Patel A, Fox LM, Tuan TA, et al. Brief hospitalization and pulse oximetry for predicting amoxicillin treatment failure in children with severe pneumonia. Pediatrics 2006;118:e1822-30. https://doi.org/10.1542/peds.2005-2673.
- Webb C, Ngama M, Ngatia A, Shebbe M, Morpeth S, Mwarumba S, et al. Treatment failure among Kenyan children with severe pneumonia – a cohort study. Pediatr Infect Dis J 2012;31:e152-7. https://doi.org/10.1097/INF.0b013e3182638012.
- Currie CJ, Berni E, Jenkins-Jones S, Poole CD, Ouwens M, Driessen S, et al. Antibiotic treatment failure in four common infections in UK primary care 1991–2012: longitudinal analysis. BMJ 2014;349. https://doi.org/10.1136/bmj.g5493.
- Cornford C, Morgan M, Ridsdale L. Why do mothers consult when their children cough?. Fam Pract 1993;10:193-6. https://doi.org/10.1093/fampra/10.2.193.
- Kai J. What worries parents when their preschool children are acutely ill, and why: a qualitative study. BMJ 1996;313:983-6. https://doi.org/10.1136/bmj.313.7063.983.
- Kuzujanakis M, Kleinman K, Rifas-Shiman S, Finkelstein JA. Correlates of parental antibiotic knowledge, demand, and reported use. Ambul Pediatr 2003;3:203-10. https://doi.org/10.1367/1539-4409(2003)003<0203:COPAKD>2.0.CO;2.
- Hay AD, Wilson A, Fahey T, Peters TJ. The duration of acute cough in pre-school children presenting to primary care: a prospective cohort study. Fam Pract 2003;20:696-705. https://doi.org/10.1093/fampra/cmg613.
- Atkinson M, Lakhanpaul M, Smyth A, Vyas H, Weston V, Sithole J, et al. Comparison of oral amoxicillin and intravenous benzyl penicillin for community acquired pneumonia in children (PIVOT trial): a multicentre pragmatic randomised controlled equivalence trial. Thorax 2007;62:1102-6. https://doi.org/10.1136/thx.2006.074906.
- Thompson M, Vodicka TA, Blair PS, Buckley DI, Heneghan C, Hay AD. TARGET Programme Team . Duration of symptoms of respiratory tract infections in children: systematic review. BMJ 2013;347. https://doi.org/10.1136/bmj.f7027.
- Esposito S, Cohen R, Domingo JD, Pecurariu OF, Greenberg D, Heininger U, et al. Antibiotic therapy for pediatric community-acquired pneumonia: do we know when, what and for how long to treat?. Pediatr Infect Dis J 2012;31:e78-85. https://doi.org/10.1097/INF.0b013e318255dc5b.
- Bradley JS, Byington CL, Shah SS, Alverson B, Carter ER, Harrison C, et al. The management of community-acquired pneumonia in infants and children older than 3 months of age: clinical practice guidelines by the Pediatric Infectious Diseases Society and the Infectious Diseases Society of America. Clin Infect Dis 2011;53:e25-76. https://doi.org/10.1093/cid/cir531.
- World Health Organization (WHO) . Pocket Book of Hospital Care for Children 2013.
- National Institute for Health and Care Excellence . Pneumonia (Community-Acquired): Antimicrobial Prescribing 2019. www.nice.org.uk/guidance/ng138/chapter/Recommendations#choice-of-antibiotic (accessed 25 September 2020).
- Calbo E, Garau J. Application of pharmacokinetics and pharmacodynamics to antimicrobial therapy of community-acquired respiratory tract infections. Respiration 2005;72:561-71. https://doi.org/10.1159/000089567.
- Appelbaum PC. Microbiological and pharmacodynamic considerations in the treatment of infection due to antimicrobial-resistant Streptococcus pneumoniae. Clin Infect Dis 2000;31:29-34. https://doi.org/10.1086/314057.
- Vilas-Boas AL, Fontoura MS, Xavier-Souza G, Araújo-Neto CA, Andrade SC, Brim RV, et al. Comparison of oral amoxicillin given thrice or twice daily to children between 2 and 59 months old with non-severe pneumonia: a randomized controlled trial. J Antimicrob Chemother 2014;69:1954-9. https://doi.org/10.1093/jac/dku070.
- BNF for Children 2012–2013. London: BMJ Group, Pharmaceutical Press and RCPCH Publications; 2012.
- Bielicki JA, Barker CIS, Saxena S, Wong ICK, Long PF, Sharland M. Not too little, not too much: problems of selecting oral antibiotic dose for children. BMJ 2015;351. https://doi.org/10.1136/bmj.h5447.
- Lassi ZS, Das JK, Haider SW, Salam RA, Qazi SA, Bhutta ZA. Systematic review on antibiotic therapy for pneumonia in children between 2 and 59 months of age. Arch Dis Child 2014;99:687-93. https://doi.org/10.1136/archdischild-2013-304023.
- Lassi ZS, Imdad A, Bhutta ZA. Short course versus long course intravenous therapy of the same antibiotic for severe community-acquired pneumonia in children aged two months to 59 months. Cochrane Database Syst Rev 2017;10. https://doi.org/10.1002/14651858.CD008032.pub3.
- The British Society for Antimicrobial Chemotherapy . Resistance Surveillance Project – Respiratory Data n.d. www.bsacsurv.org/reports/respiratory/ (accessed 28 August 2020).
- Kessel AS, Sharland M. The new UK antimicrobial resistance strategy and action plan. BMJ 2013;346. https://doi.org/10.1136/bmj.f1601.
- European Committee on Antimicrobial Susceptibility Testing . Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 5.0, 2015 2015.
- Samore MH, Magill MK, Alder SC, Severina E, Morrison-De Boer L, Lyon JL, et al. High rates of multiple antibiotic resistance in Streptococcus pneumoniae from healthy children living in isolated rural communities: association with cephalosporin use and intrafamilial transmission. Pediatrics 2001;108:856-65. https://doi.org/10.1542/peds.108.4.856.
- Regev-Yochay G, Raz M, Dagan R, Porat N, Shainberg B, Pinco E, et al. Nasopharyngeal carriage of Streptococcus pneumoniae by adults and children in community and family settings. Clin Infect Dis 2004;38:632-9. https://doi.org/10.1086/381547.
- Dunais B, Pradier C, Carsenti H, Sabah M, Mancini G, Fontas E, et al. Influence of child care on nasopharyngeal carriage of Streptococcus pneumoniae and Haemophilus influenzae. Pediatr Infect Dis J 2003;22:589-92. https://doi.org/10.1097/01.inf.0000073203.88387.eb.
- Lietzau S, Raum E, von Baum H, Marre R, Brenner H. Household contacts were key factor for children’s colonization with resistant Escherichia coli in community setting. J Clin Epidemiol 2007;60:1149-55. https://doi.org/10.1016/j.jclinepi.2007.01.016.
- Costelloe C, Metcalfe C, Lovering A, Mant D, Hay AD. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ 2010;340. https://doi.org/10.1136/bmj.c2096.
- Raum E, Lietzau S, von Baum H, Marre R, Brenner H. Changes in Escherichia coli resistance patterns during and after antibiotic therapy: a longitudinal study among outpatients in Germany. Clin Microbiol Infect 2008;14:41-8. https://doi.org/10.1111/j.1469-0691.2007.01841.x.
- Chung A, Perera R, Brueggemann AB, Elamin AE, Harnden A, Mayon-White R, et al. Effect of antibiotic prescribing on antibiotic resistance in individual children in primary care: prospective cohort study. BMJ 2007;335. https://doi.org/10.1136/bmj.39274.647465.BE.
- Guillemot D, Carbon C, Balkau B, Geslin P, Lecoeur H, Vauzelle-Kervroëdan F, et al. Low dosage and long treatment duration of beta-lactam: risk factors for carriage of penicillin-resistant Streptococcus pneumoniae. JAMA 1998;279:365-70. https://doi.org/10.1001/jama.279.5.365.
- Duffy MA, Hernandez-Santiago V, Orange G, Davey PG, Guthrie B. Trimethoprim prescription and subsequent resistance in childhood urinary infection: multilevel modelling analysis. Br J Gen Pract 2013;63:e238-43. https://doi.org/10.3399/bjgp13X665198.
- Hillier S, Roberts Z, Dunstan F, Butler C, Howard A, Palmer S. Prior antibiotics and risk of antibiotic-resistant community-acquired urinary tract infection: a case-control study. J Antimicrob Chemother 2007;60:92-9. https://doi.org/10.1093/jac/dkm141.
- Schrag SJ, Peña C, Fernández J, Sánchez J, Gómez V, Pérez E, et al. Effect of short-course, high-dose amoxicillin therapy on resistant pneumococcal carriage: a randomized trial. JAMA 2001;286:49-56. https://doi.org/10.1001/jama.286.1.49.
- Samore MH, Tonnerre C, Hannah EL, Stoddard GJ, Borotkanics RJ, Haddadin B, et al. Impact of outpatient antibiotic use on carriage of ampicillin-resistant Escherichia coli. Antimicrob Agents Chemother 2011;55:1135-41. https://doi.org/10.1128/AAC.01708-09.
- Tam VH, Louie A, Fritsche TR, Deziel M, Liu W, Brown DL, et al. Impact of drug-exposure intensity and duration of therapy on the emergence of Staphylococcus aureus resistance to a quinolone antimicrobial. J Infect Dis 2007;195:1818-27. https://doi.org/10.1086/518003.
- Drusano GL, Liu W, Brown DL, Rice LB, Louie A. Impact of short-course quinolone therapy on susceptible and resistant populations of Staphylococcus aureus. J Infect Dis 2009;199:219-26. https://doi.org/10.1086/595739.
- Martinez MN, Papich MG, Drusano GL. Dosing regimen matters: the importance of early intervention and rapid attainment of the pharmacokinetic/pharmacodynamic target. Antimicrob Agents Chemother 2012;56:2795-805. https://doi.org/10.1128/AAC.05360-11.
- Jumbe N, Louie A, Leary R, Liu W, Deziel MR, Tam VH, et al. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J Clin Invest 2003;112:275-85. https://doi.org/10.1172/JCI16814.
- Opatowski L, Mandel J, Varon E, Boelle P-Y, Temime L, Guillemot D. Antibiotic dose impact on resistance selection in the community: a mathematical model of beta-lactams and Streptococcus pneumoniae dynamics. Antimicrob Agents Chemother 2010;54:2330-7. https://doi.org/10.1128/AAC.00331-09.
- Schechner V, Temkin E, Harbarth S, Carmeli Y, Schwaber MJ. Epidemiological interpretation of studies examining the effect of antibiotic usage on resistance. Clin Microbiol Rev 2013;26:289-307. https://doi.org/10.1128/CMR.00001-13.
- Lyttle MD, O’Sullivan R, Hartshorn S, Bevan C, Cleugh F, Maconochie I. PERUKI . Pediatric Emergency Research in the UK and Ireland (PERUKI): developing a collaborative for multicentre research. Arch Dis Child 2014;99:602-3. https://doi.org/10.1136/archdischild-2013-304998.
- Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, U.S. Department of Health and Human Services . Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events n.d. https://rsc.niaid.nih.gov/sites/default/files/daids-ae-grading-table-v2-nov2014.pdf (accessed 28 September 2020).
- European Committee on Antimicrobial Susceptibility Testing . Växjö: European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters Version 10.0, 2020 2020.
- Committee for Medicinal Products for Human Use . Guideline on the Evaluation of Medicinal Products Indicated for Treatment of Bacterial Infections, Rev. 3 n.d. www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-evaluation-medicinal-products-indicated-treatment-bacterial-infections-revision-3_en.pdf (accessed 3 August 2021).
- Bradley JS, McCracken GH. Unique considerations in the evaluation of antibacterials in clinical trials for pediatric community-acquired pneumonia. Clin Infect Dis 2008;47:241-8. https://doi.org/10.1086/591410.
- Legg J, Rampton C. BTS 2016 17 Paediatric Pneumonia Audit Report – 15 January 2018 2018.
- Tyrstrup M, Melander E, Hedin K, Beckman A, Mölstad S. Children with respiratory tract infections in Swedish primary care; prevalence of antibiotic resistance in common respiratory tract pathogens and relation to antibiotic consumption. BMC Infect Dis 2017;17. https://doi.org/10.1186/s12879-017-2703-3.
- Brogan TV, Hall M, Williams DJ, Neuman MI, Grijalva CG, Farris RW, et al. Variability in processes of care and outcomes among children hospitalized with community-acquired pneumonia. Pediatr Infect Dis J 2012;31:1036-41. https://doi.org/10.1097/INF.0b013e31825f2b10.
- Al-Mahtot M, Barwise-Munro R, Wilson P, Turner S. Changing characteristics of hospital admissions but not the children admitted – a whole population study between 2000 and 2013. Eur J Pediatr 2018;177:381-8. https://doi.org/10.1007/s00431-017-3064-z.
- European Centre for Disease Prevention and Control . Surveillance of Antimicrobial Resistance in Europe 2018 n.d. www.ecdc.europa.eu/sites/default/files/documents/surveillance-antimicrobial-resistance-Europe-2018.pdf (accessed 28 September 2020).
- Florin TA, French B, Zorc JJ, Alpern ER, Shah SS. Variation in emergency department diagnostic testing and disposition outcomes in pneumonia. Pediatrics 2013;132:237-44. https://doi.org/10.1542/peds.2013-0179.
- Neuman MI, Hall M, Gay JC, Blaschke AJ, Williams DJ, Parikh K, et al. Readmissions among children previously hospitalized with pneumonia. Pediatrics 2014;134:100-9. https://doi.org/10.1542/peds.2014-0331.
- Pakistan Multicentre Amoxycillin Short Course Therapy (MASCOT) Pneumonia Study Group . Clinical efficacy of 3 days versus 5 days of oral amoxicillin for treatment of childhood pneumonia: a multicentre double-blind trial. Lancet 2002;360:835-41. https://doi.org/10.1016/S0140-6736(02)09994-4.
- Agarwal G, Awasthi S, Kabra SK, Kaul A, Singhi S, Walter SD. ISCAP Study Group . Three day versus five day treatment with amoxicillin for non-severe pneumonia in young children: a multicentre randomised controlled trial. BMJ 2004;328. https://doi.org/10.1136/bmj.38049.490255.DE.
- Ginsburg AS, Mvalo T, Nkwopara E, McCollum ED, Phiri M, Schmicker R, et al. Amoxicillin for 3 or 5 days for chest-indrawing pneumonia in Malawian children. N Engl J Med 2020;383:13-2. https://doi.org/10.1056/NEJMoa1912400.
- Ginsburg AS, Mvalo T, Nkwopara E, McCollum ED, Ndamala CB, Schmicker R, et al. Placebo vs. amoxicillin for nonsevere fast-breathing pneumonia in Malawian children aged 2 to 59 months: a double-blind, randomized clinical noninferiority trial. JAMA Pediatr 2019;173:21-8. https://doi.org/10.1001/jamapediatrics.2018.3407.
- Hoberman A, Paradise J, Rockette HE, Kearney DH, Bhatnagar S, Shope TR, et al. Shortened antimicrobial treatment for acute otitis media in young children. N Engl J Med 2016;375:2446-56. https://doi.org/10.1056/NEJMoa1606043.
- Kardas P. Patient compliance with antibiotic treatment for respiratory tract infections. J Antimicrob Chemother 2002;49:897-903. https://doi.org/10.1093/jac/dkf046.
- Llor C, Hernández S, Bayona C, Moragas A, Sierra N, Hernández M, et al. A study of adherence to antibiotic treatment in ambulatory respiratory infections. Int J Infect Dis 2013;17:e168-72. https://doi.org/10.1016/j.ijid.2012.09.012.
- Little P, Francis NA, Stuart B, O’Reilly G, Thompson N, Becque T, et al. Antibiotics for lower respiratory tract infection in children presenting in primary care in England (ARTIC PC): a double-blind, randomised, placebo-controlled trial. Lancet 2021. https://doi.org/10.1016/S0140-6736(21)01431-8.
- Pernica JM, Harman S, Kam AJ, Carciumaru R, Vanniyasingam T, Crawford T, et al. Short-course antimicrobial therapy for pediatric community-acquired pneumonia: the SAFER randomized clinical trial. JAMA Pediatr 2021;175:475-82. https://doi.org/10.1001/jamapediatrics.2020.6735.
- de Velde F, de Winter BC, Koch BC, van Gelder T, Mouton JW. COMBACTE-NET consortium . Non-linear absorption pharmacokinetics of amoxicillin: consequences for dosing regimens and clinical breakpoints. J Antimicrob Chemother 2016;71:2909-17. https://doi.org/10.1093/jac/dkw226.
Appendix 1 Details of main protocol amendment: joint analysis of paediatric emergency department and ward groups
Initially, PED and ward groups were treated as separate strata because of (1) an expected higher severity of CAP in the ward group, (2) the expected differences in prior receipt of antibiotic for current episode having an impact on the duration of treatment analysis and (3) the need for different trial procedures (i.e. consent process, enrolment and additional data capture during the inpatient period for the ward group). However, based on the pilot phase, the following key aspects emerged and formed the basis for the joint analysis of PED and ward groups. First, in a substantial proportion of participating hospitals, children were first seen in a paediatric assessment unit before either being formally admitted or discharged. This made the distinction between PED and ward less relevant, especially as many paediatric assessment units admitted children for up to 48 hours. Second, although clinical signs and symptoms at presentation to ED were (as expected) worse, on average, in ward children than in PED children, considerable overlap in the two distributions was observed. Third, the duration of prior antibiotic exposure in the ward group was much shorter than anticipated (< 12 hours, 54%; < 24 hours, 75%). Finally, there was no evidence of a difference between the primary end-point rate between PED and ward groups.
Appendix 2 Community-acquired pneumonia symptoms at trial entry by strata
| Stratum | Strata, n (%) | p-value | Total (N = 814), n (%) | |
|---|---|---|---|---|
| PED (N = 591) | Ward (N = 223) | |||
| Fever | ||||
| Not present | 54 (9.2) | 111 (49.8) | < 0.001 | 165 (20.3) |
| Slight/little | 71 (12.0) | 31 (13.9) | 102 (12.5) | |
| Moderate | 175 (29.7) | 42 (18.8) | 217 (26.7) | |
| Bad | 215 (36.4) | 26 (11.7) | 241 (29.6) | |
| Severe/very bad | 75 (12.7) | 13 (5.8) | 88 (10.8) | |
| Cough | ||||
| Not present | 14 (2.4) | 14 (6.3) | < 0.001 | 28 (3.4) |
| Slight/little | 61 (10.3) | 45 (20.2) | 106 (13.0) | |
| Moderate | 246 (41.7) | 96 (43.0) | 342 (42.1) | |
| Bad | 208 (35.3) | 59 (26.5) | 267 (32.8) | |
| Severe/very bad | 61 (10.3) | 9 (4.0) | 70 (8.6) | |
| Wet cough (phlegm) | ||||
| Not present | 174 (29.5) | 72 (32.3) | 0.58 | 246 (30.3) |
| Slight/little | 125 (21.2) | 44 (19.7) | 169 (20.8) | |
| Moderate | 159 (26.9) | 65 (29.1) | 224 (27.6) | |
| Bad | 103 (17.5) | 36 (16.1) | 139 (17.1) | |
| Severe/very bad | 29 (4.9) | 6 (2.7) | 35 (4.3) | |
| Breathing faster (shortness of breath) | ||||
| Not present | 77 (13.1) | 57 (25.6) | < 0.001 | 134 (16.5) |
| Slight/little | 151 (25.6) | 70 (31.4) | 221 (27.2) | |
| Moderate | 182 (30.8) | 52 (23.3) | 234 (28.8) | |
| Bad | 140 (23.7) | 36 (16.1) | 176 (21.6) | |
| Severe/very bad | 40 (6.8) | 8 (3.6) | 48 (5.9) | |
| Wheeze | ||||
| Not present | 283 (48.0) | 109 (48.9) | 0.95 | 392 (48.2) |
| Slight/little | 129 (21.9) | 52 (23.3) | 181 (22.3) | |
| Moderate | 112 (19.0) | 37 (16.6) | 149 (18.3) | |
| Bad | 56 (9.5) | 21 (9.4) | 77 (9.5) | |
| Severe/very bad | 10 (1.7) | 4 (1.8) | 14 (1.7) | |
| Sleep disturbed by cough | ||||
| Not present | 67 (11.4) | 56 (25.1) | < 0.001 | 123 (15.2) |
| Slight/little | 95 (16.2) | 55 (24.7) | 150 (18.5) | |
| Moderate | 151 (25.7) | 55 (24.7) | 206 (25.4) | |
| Bad | 170 (28.9) | 42 (18.8) | 212 (26.1) | |
| Severe/very bad | 105 (17.9) | 15 (6.7) | 120 (14.8) | |
| Vomiting (including after cough) | ||||
| Not present | 324 (54.9) | 155 (69.5) | 0.003 | 479 (58.9) |
| Slight/little | 110 (18.6) | 32 (14.3) | 142 (17.5) | |
| Moderate | 83 (14.1) | 18 (8.1) | 101 (12.4) | |
| Bad | 49 (8.3) | 15 (6.7) | 64 (7.9) | |
| Severe/very bad | 24 (4.1) | 3 (1.3) | 27 (3.3) | |
| Eating/drinking less | ||||
| Not present | 63 (10.7) | 30 (13.5) | 0.073 | 93 (11.4) |
| Slight/little | 140 (23.7) | 68 (30.5) | 208 (25.6) | |
| Moderate | 184 (31.2) | 67 (30.0) | 251 (30.9) | |
| Bad | 157 (26.6) | 41 (18.4) | 198 (24.4) | |
| Severe/very bad | 46 (7.8) | 17 (7.6) | 63 (7.7) | |
| Interference with normal activity | ||||
| Not present | 61 (10.3) | 49 (22.0) | < 0.001 | 110 (13.5) |
| Slight/little | 136 (23.1) | 59 (26.5) | 195 (24.0) | |
| Moderate | 198 (33.6) | 63 (28.3) | 261 (32.1) | |
| Bad | 140 (23.7) | 40 (17.9) | 180 (22.1) | |
| Severe/very bad | 55 (9.3) | 12 (5.4) | 67 (8.2) | |
Appendix 3 On-treatment analysis of the primary end point
The on-treatment analyses of the primary end point excluded participants who took < 80% of trial medication as scheduled (e.g. when patients missed two doses of medication, when a smaller volume of medication was taken). When patients switched from medication to non-trial antibiotics because of deterioration this was not regarded as non-adherence. For each randomised comparison, non-adherence was analysed in two ways: (1) based on all trial medication including placebo and (2) based on active drug only (Figures 21–24).
FIGURE 21.
Dose randomisation: participants who took at least 80% of all trial medication, including placebo.
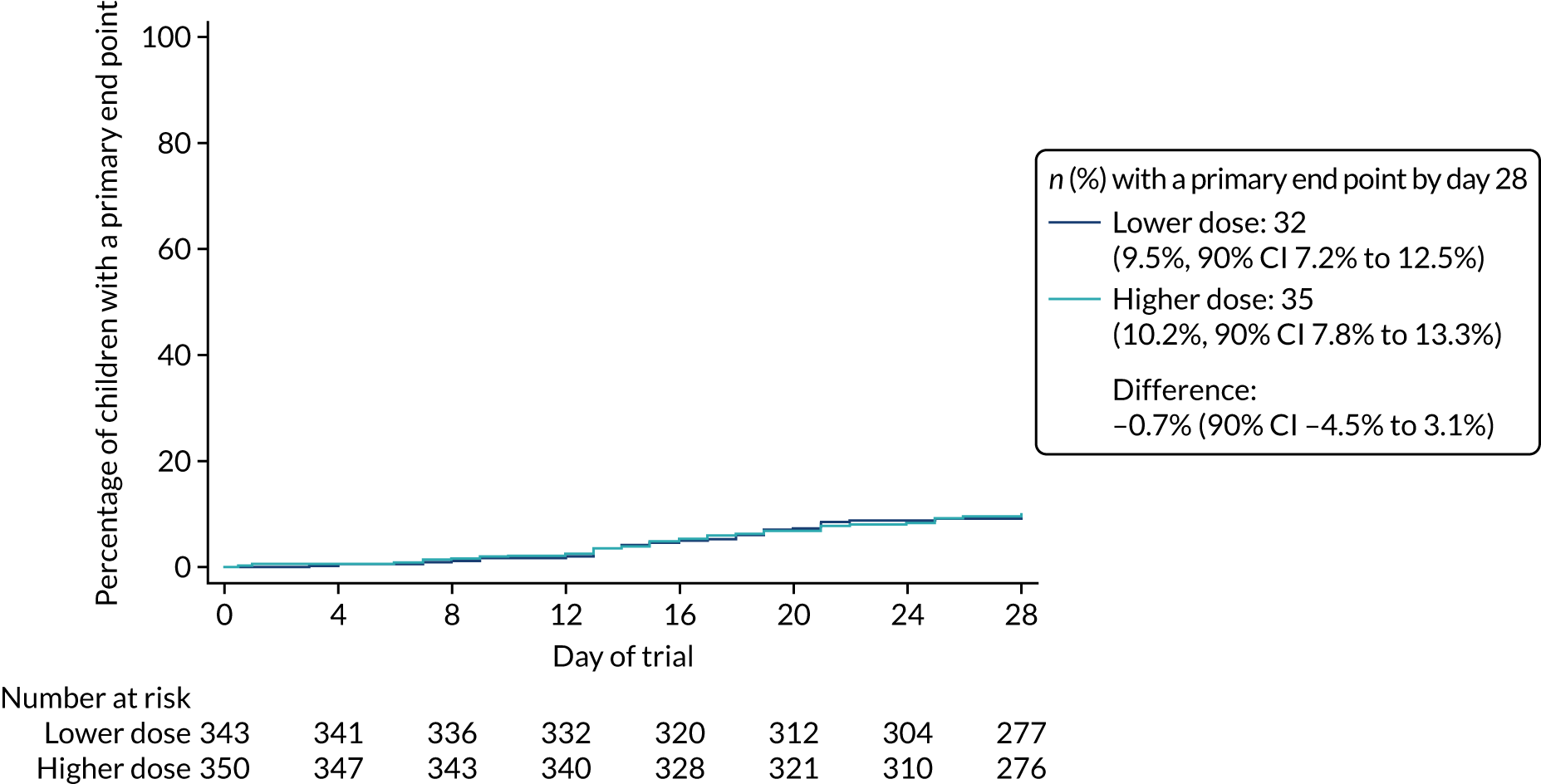
FIGURE 22.
Dose randomisation: participants who took at least 80% of active trial drug.
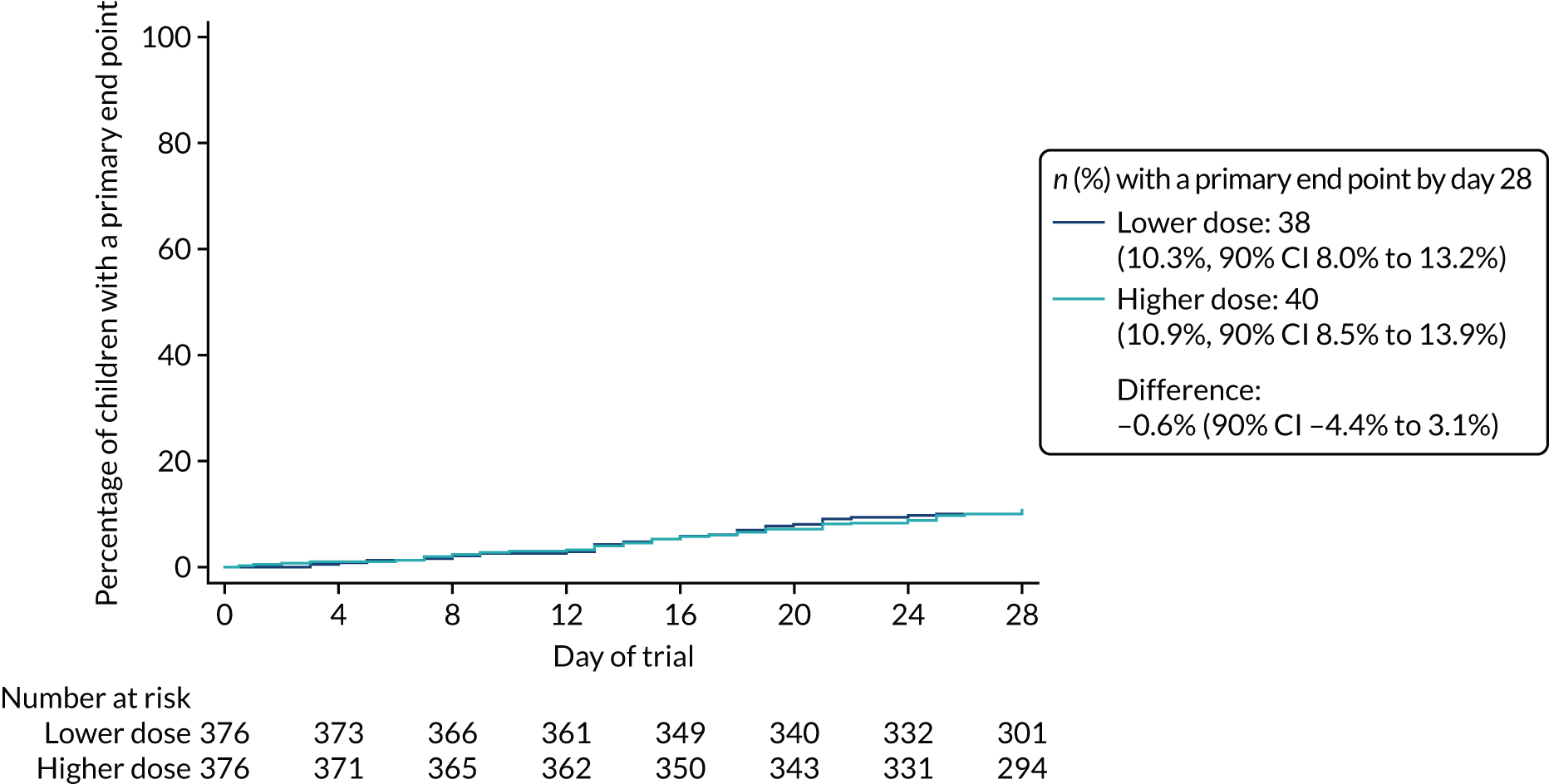
FIGURE 23.
Duration randomisation: participants who took at least 80% of all trial medication, including placebo.
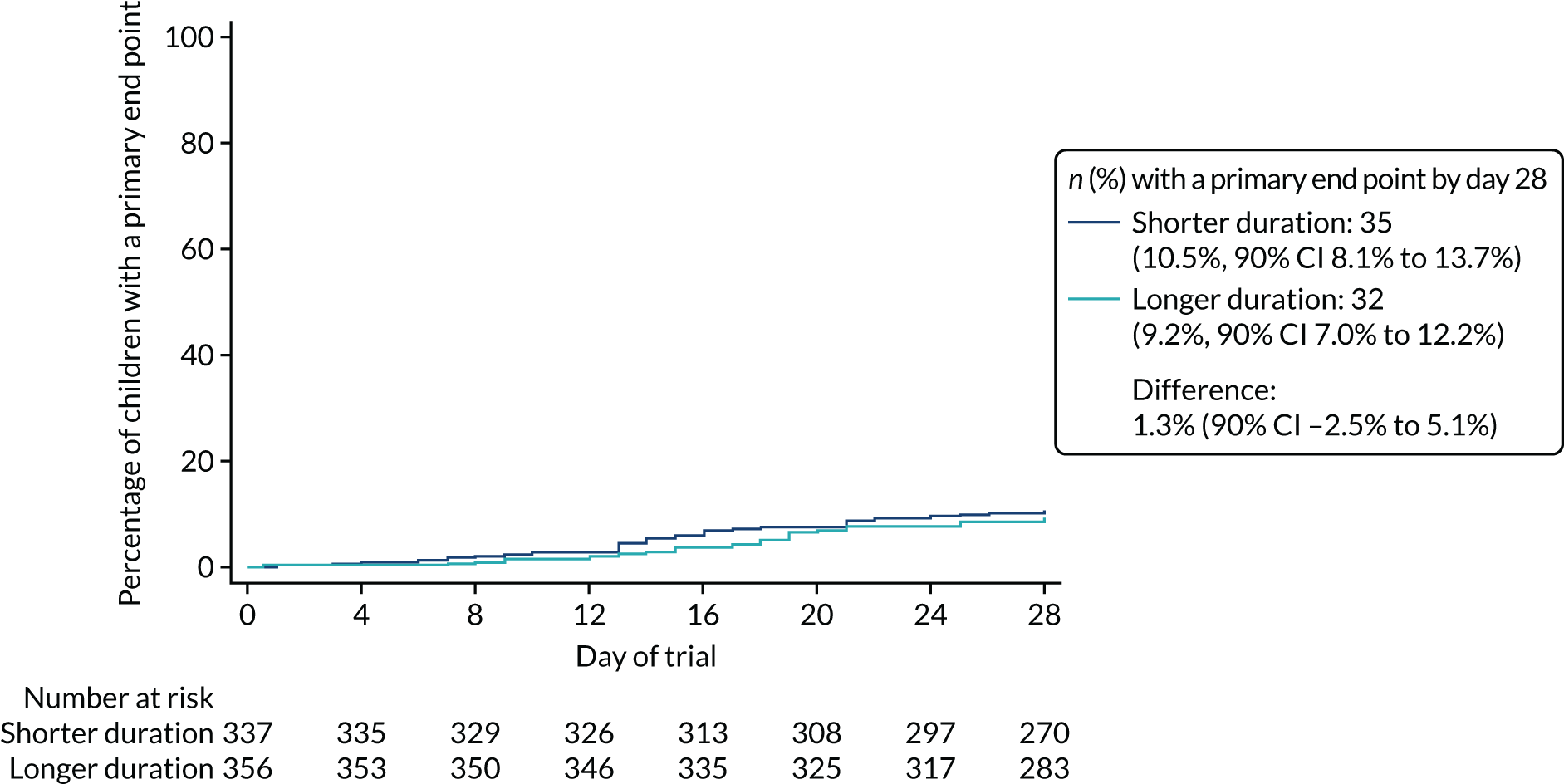
FIGURE 24.
Duration randomisation: participants who took at least 80% of active trial drug.
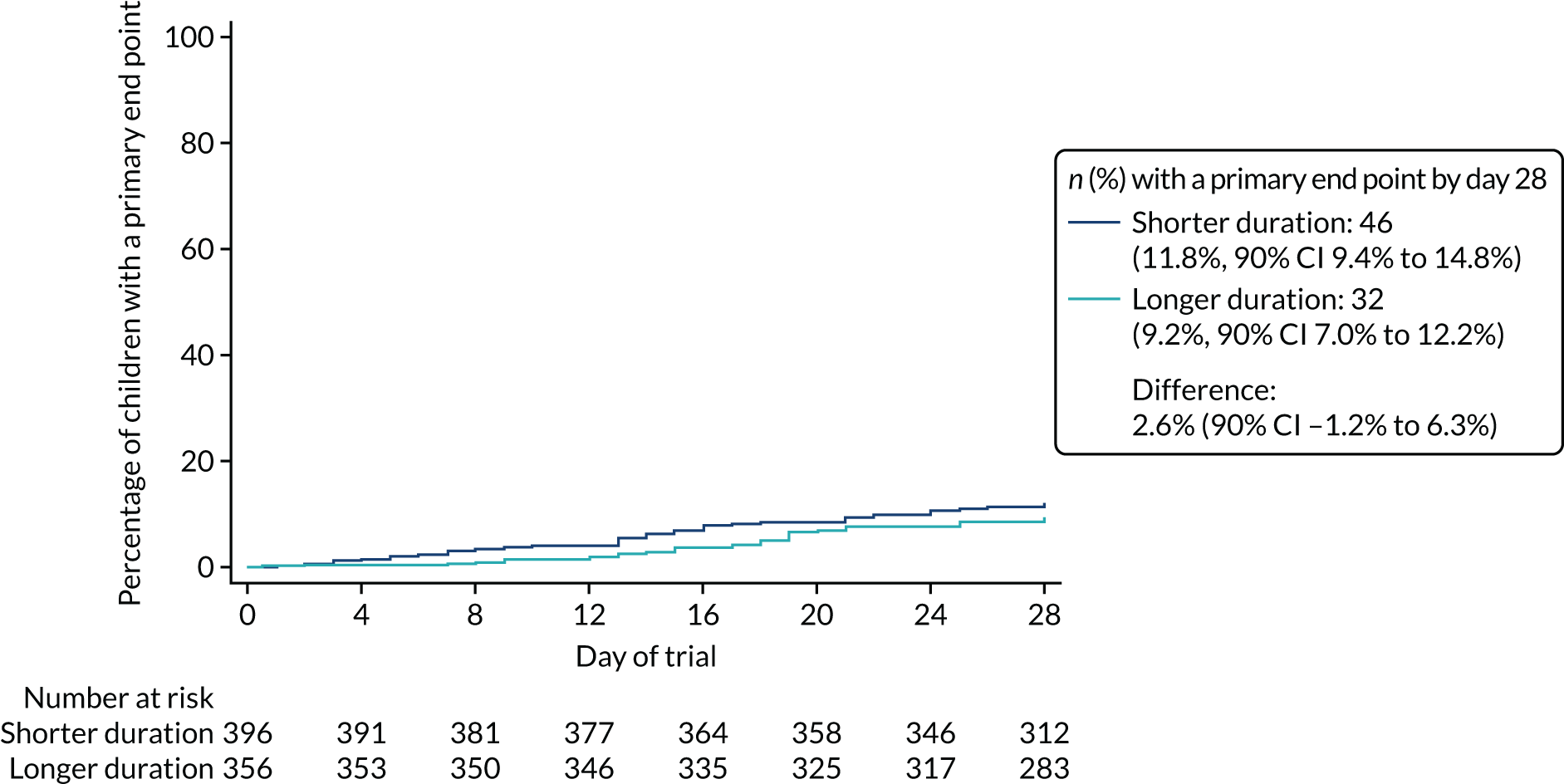
List of abbreviations
- AE
- adverse event
- BNFc
- British National Formulary for Children
- BTS
- British Thoracic Society
- CAP
- community-acquired pneumonia
- CAP-IT
- Community-Acquired Pneumonia: a protocol for a randomIsed controlled Trial
- CI
- confidence interval
- ED
- emergency department
- ERC
- End-Point Review Committee
- GP
- general practitioner
- ID
- identification
- IDMC
- Independent Data Monitoring Committee
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ITT
- intention to treat
- MIC
- minimal inhibitory concentration
- PCV
- pneumococcal conjugate vaccine
- PED
- paediatric emergency department
- PPI
- patient and public involvement
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SAR
- serious adverse reaction
- T > MIC
- time spent above the minimum inhibitory concentration
- TSC
- Trial Steering Committee
- WHO
- World Health Organization