

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 08/56/04. The contractual start date was in June 2009. The draft report began editorial review in December 2018 and was accepted for publication in August 2021. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2022 Jayne et al. This work was produced by Jayne et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2022 Jayne et al.
Chapter 1 Introduction
Parts of this chapter have been reproduced with permission from the PEXIVAS protocol version 1.0. 1
Scientific background
Granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) are syndromes of primary systemic vasculitis that are associated with anti-neutrophil cytoplasm antibodies (ANCA). Together, these syndromes are grouped as ANCA-associated vasculitis (AAV). The prevalence of AAV is estimated at 14–30 per 100,000 in England. 2 If left untreated, AAV has a universally poor prognosis, with mortality rates approaching 100% within 5 years. 3 The introduction of treatment regimens based on cyclophosphamide (CYC) and glucocorticoids (GCs) has transformed AAV from a rapidly fatal disease to one of chronic morbidity and reduced survival, often preceded by end-stage renal disease (ESRD).
Plasma exchange (PLEX), a method of rapidly removing potentially pathogenic ANCA and other mediators of inflammation and coagulation, has shown promise as an adjunctive therapy in AAV to improve early disease control and improve rates of renal recovery in severe disease. GCs are the standard of care for the treatment of AAV. High doses of GCs early in disease undeniably reduce disease activity because of their anti-inflammatory and immunosuppressive properties, but they also increase the risk of infection, particularly in the elderly and in the presence of uraemia. 4–7 To our knowledge, there are no randomised trial data to guide GC dosing.
There is a need for therapies with reduced toxicity that improve disease control. Defining the role of therapies that are already in use but are invasive, expensive and unproven is a priority in AAV research. PEXIVAS (Plasma Exchange In VASculitis) was a randomised controlled trial that tested two interventions in a 2 × 2 factorial design (standard care and PLEX compared with standard care alone, and a standard-dose GC regimen compared with a reduced-dose GC regimen) to address these issues.
Current treatment options and target population
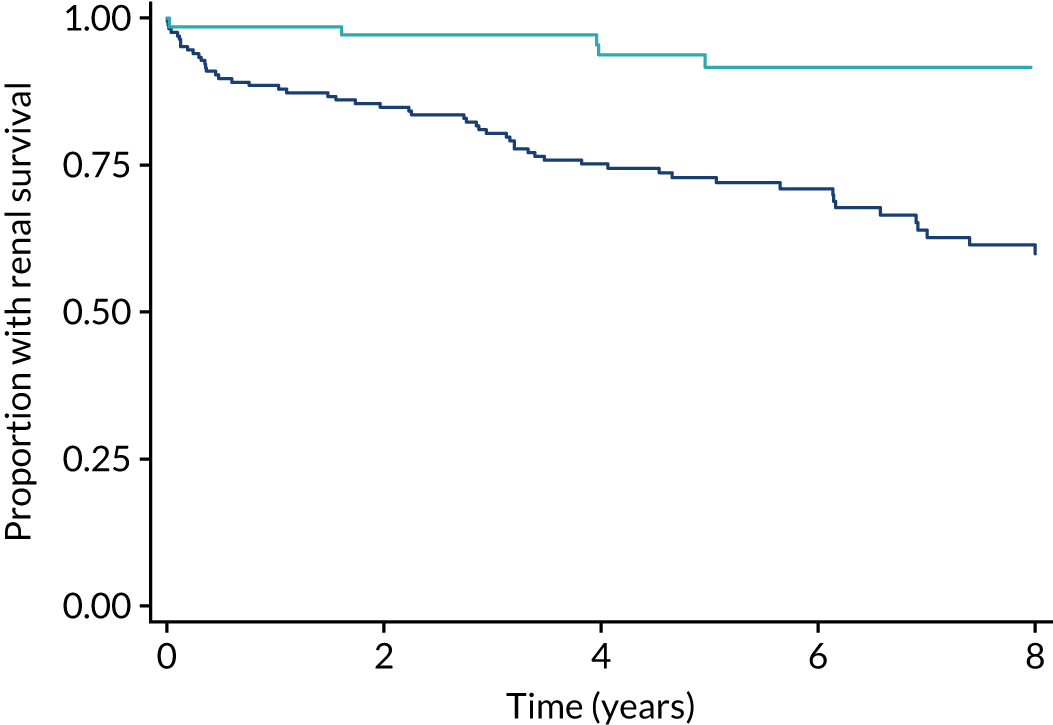
Current standard treatment regimens still have poor outcomes. Unselected cohorts with AAV now demonstrate 5-year renal survival (defined as the composite of ESRD or death) of 60–70%. 8,9 In patients with vital-organ-threatening disease (e.g. kidneys or lungs), renal survival is worse than in patients with other types of disease. Long-term follow-up data from three completed randomised controlled trials by the European Vasculitis Study Group (EUVAS) involving 285 patients with an estimated glomerular filtration rate (eGFR) of > 50 ml/minute/1.73 m2 of body weight demonstrated that 5-year ESRD-free survival was only 54%, and the median time to renal failure or death was 6 years, despite the exclusion of patients with lung haemorrhage (Figure 1). In the subgroup of patients with a creatinine level of > 500, and a glomerular filtration rate (GFR) of < 50 ml/minute/1.73 m2, a group traditionally thought to have a favourable prognosis, 33% died or developed ESRD by 5 years (Figure 2). Additionally, patients with lung haemorrhage, who were excluded from these studies, have a mortality rate of up to 50% in the first year.
FIGURE 1.
Differences in long-term renal survival (ESRD or death) on the basis of eGFR at baseline for patients with AAV enrolled in three randomised trials. The dark blue line represents patients with an eGFR of ≤ 50 ml/minute/1.73 m2 body weight; the light blue line represents patients with an eGFR of > 50 ml/minute/1.73 m2 body weight. Reproduced with permission from the PEXIVAS protocol, version 1.0. 1
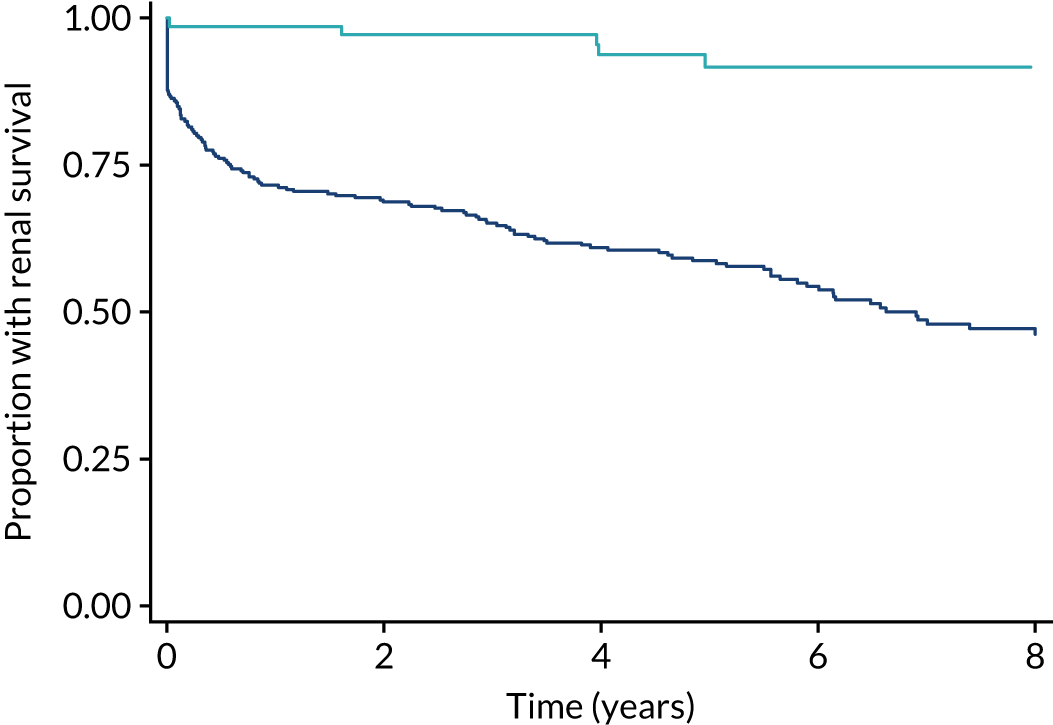
FIGURE 2.
Differences in long-term renal survival (ESRD or death) on the basis of eGFR at baseline for patients with AAV enrolled in two randomised trials with a baseline creatinine level of < 500 µmol/l. The dark blue line represents patients with an eGFR of ≤ 50 ml/minute/1.73 m2 body weight; the light blue line represents patients with an eGFR of > 50 ml/minute/1.73 m2 body weight. Reproduced with permission from the PEXIVAS protocol, version 1.0. 1
Poor outcomes in AAV are attributed to both ineffective therapies and complications of the standard treatments (CYC and GC). Approximately 20% of patients either do not have adequate disease control or are intolerant of their induction or remission treatment. 10–12 An additional 50% of patients will have relapsing AAV over the subsequent 5 years. Inadequate disease control is associated with increased immunosuppressive medication and, thus, increased risk of treatment-related toxicity, progressive organ scarring and death. Additionally, between 25% and 50% of patients with severe AAV experience a severe infection within the first 12 months of treatment, and the most frequently cited causes of death are infection or uncontrolled vasculitis. 13,14 Treatment regimens that minimise toxicity and infections and also provide adequate disease control are, therefore, needed.
Evidence for plasma exchange in anti-neutrophil cytoplasm antibody-associated vasculitis
Plasma exchange involves the extracorporeal separation of plasma from blood cells, and then the return of the cells to the body with a plasma substitute. In this way, harmful plasma constituents are removed. Early studies of PLEX in rapidly progressive glomerulonephritis predominantly due to AAV have had mixed results. 15–18 These studies had heterogeneous treatment regimens, small sample sizes and short follow-up periods.
The Methylprednisolone versus Plasma Exchange (MEPEX) trial examined the effect of PLEX on renal recovery for patients with renal failure due to AAV. 14 This trial compared PLEX with intravenous (i.v. ) methylprednisolone as an addition to standard therapy for 137 incident patients with severe AAV, which manifested as a creatinine level of > 500 µmol/l or dialysis dependency at presentation, and demonstrated an absolute reduction in the development of ESRD of 24% [95% confidence interval (CI) 6.5% to 41%] after 12 months for patients treated with PLEX. There was no demonstrable difference in the mortality rate at 12 months between those treated with PLEX and those treated with i.v. methylprednisolone (there was a mortality rate of 25% in both groups). Long-term results from MEPEX did not demonstrate a statistically significant difference between the treatment groups in terms of the composite of ESRD or death (p = 0.57) (Figure 3).
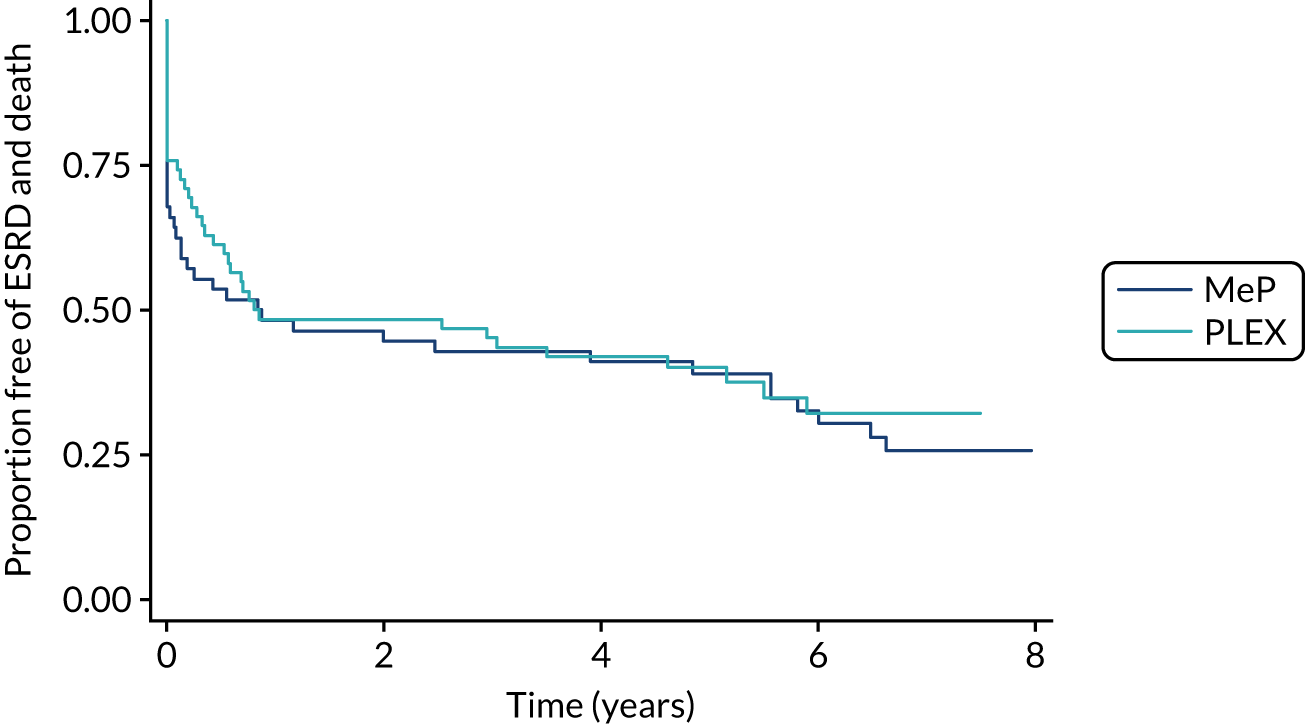
The role of PLEX in patients with less severe renal dysfunction at the time of presentation is even more unclear. Exploratory work found that patients with a renal biopsy demonstrating active lesions were the most likely to benefit from PLEX. 19 In patients who do not have advanced, chronic renal injuries, the rapid disease control afforded by PLEX may prevent renal (or other vital organ) scarring and, thus, the cascade of glomerulosclerosis and hyperfiltration that perpetuates renal injury.
Plasma exchange is also widely used for patients with lung haemorrhage due to AAV. This practice comes from cohort data in AAV and experience with anti-glomerular basement membrane disease, but has never been rigorously tested and, in contemporary cohort data, appears effective in selected subgroups of patients with lung haemorrhage by improving survival. 20 However, PLEX has the potential to exacerbate haemorrhage through the removal of clotting factors and increase the risk of infection through antibody removal. Therefore, its use in this indication demands critical appraisal.
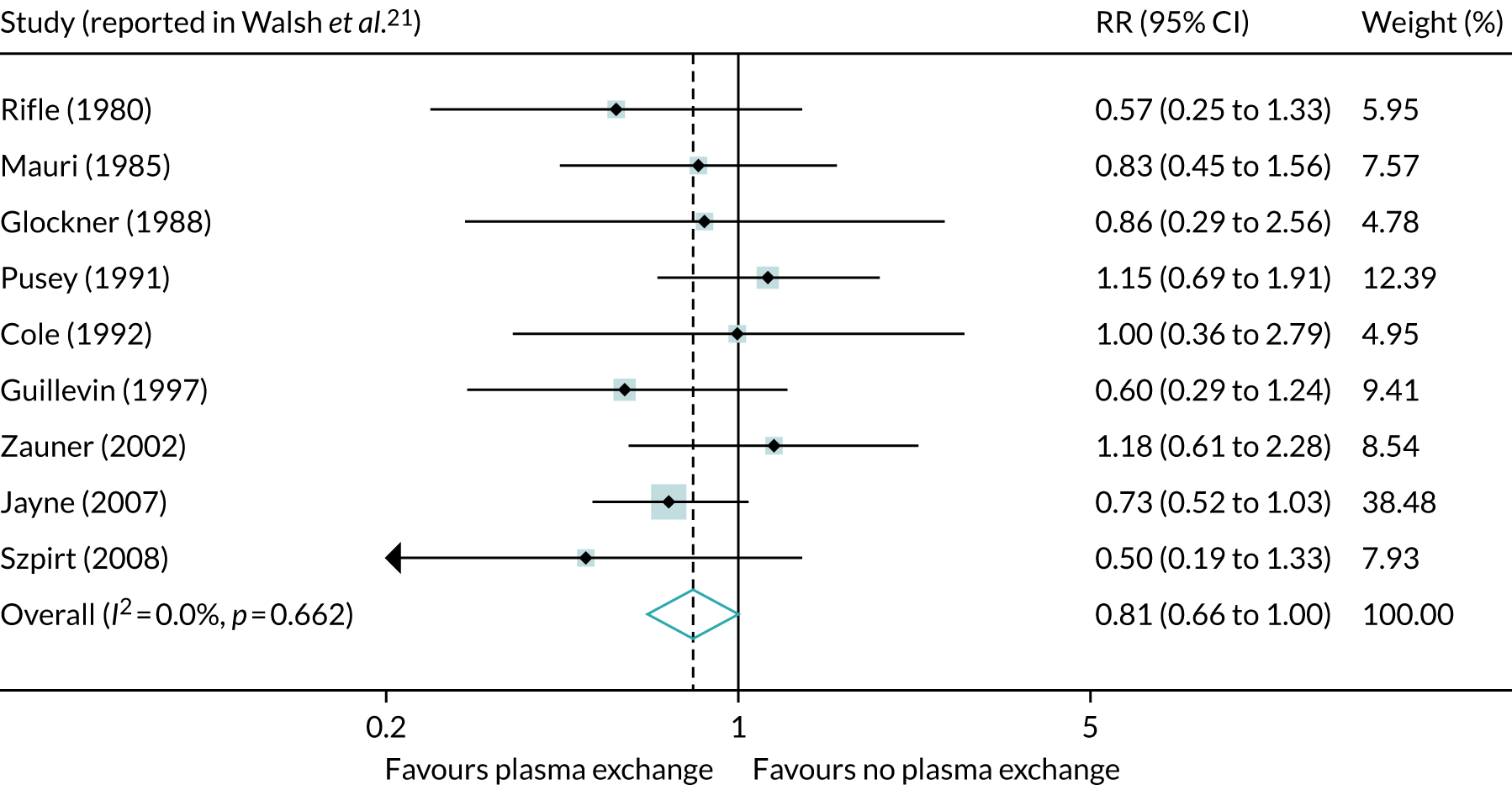
A systematic review21 of PLEX in AAV identified nine randomised studies. The study populations were skewed towards severe renal dysfunction and often included diseases other than AAV. A meta-analysis21 of the nine trials that reported death and ESRD outcomes showed a beneficial reduction of dialysis dependency [relative risk (RR) 0.64, 95% CI 0.47 to 0.88], but no beneficial reduction of mortality (RR 1.01, 95% CI 0.71 to 1.43). When considering the composite end point of death or dialysis, the RR was 0.81 (95% CI 0.66 to 1.00) (Figure 4).
FIGURE 4.
Meta-analysis of nine studies, including 387 patients, examining the effect of adjunctive PLEX on the end point of ESRD or death in patients with rapidly progressive glomerulonephritis. Reproduced with permission from the PEXIVAS protocol, version 1.0. 1
The treatment of anti-neutrophil cytoplasm antibody-associated vasculitis with glucocorticoids
High-dose oral GCs were the standard of care for the treatment of AAV on the basis of cohort data3 prior to the widespread use of cytotoxic medications and strategies for earlier diagnosis with ANCA testing. There is a complex relationship between GC dose and its effects on the immune system when it is used as an immunosuppressive rather than as an anti-inflammatory agent. 4 There is also an increasing trend to reduce GC doses to mitigate their toxicity but maintain efficacy, a trend supported by laboratory evidence of a ceiling effect of GC dosing with respect to anti-inflammatory properties. 5 When combined with cytotoxic medications, high-dose GCs may increase treatment-related toxicity, but add little to therapeutic efficacy.
Infections in AAV are most common in the first 2 months of treatment, which is when GC doses are highest. Although this relationship is confounded by disease activity and co-treatment with CYC, it is important to note that infection rates fall in parallel with the decreasing GC dose despite the maintenance of constant immunosuppression. Dose-dependent increases in infections are also observed in rheumatoid arthritis and lupus nephritis. 6,7 Furthermore, high cumulative doses of GCs are associated with osteoporosis, infections, cardiovascular disease and gastrointestinal bleeding. 6,7 Despite the association of higher GC doses and adverse events, and despite the widespread use of GCs, there is a paucity of literature to guide the optimal dosing of GCs in AAV.
Clinical trials in anti-neutrophil cytoplasm antibody-associated vasculitis
A clinical trial conducted by the Medical Research Council from the late 1950s22 of cortisone in polyarteritis nodosa, a form of systemic vasculitis, and funded by the Medical Research Council, found an increase in rate of survival at 1 year, with the benefit lost by the second year because of GC-related complications. Eligibility for subsequent trials was influenced by prevalent classification systems, and the first trials of AAV, including GPA and MPA, occurred in the 1990s. These trials were either single centre, with around 50 subjects, or international, with EUVAS launching a sequence of randomised controlled trials that aimed to harmonise therapies across patient subgroups defined by disease extent and severity. 23 The trials were open label, but used central randomisation and stratification, and developed an approach to disease assessment based on scoring tools for activity [Birmingham Vasculitis Activity Score/Wegener’s Granulomatosis version (BVAS/WG)], all-cause damage [Vasculitis Damage Index (VDI)] and quality of life. Sample sizes were 100–160 patients, with end points based around disease remission or relapse. In the 2000s, North American investigators conducted two academic trials, with industry support, of newer biologic agents: etanercept (WGET24) and rituximab (RAVE25). These were multicentre and double blind and included 150–200 patients, with RAVE leading to the licensing of rituximab for AAV, which remains the only licensed drug for this indication. Pharmaceutical company trials began in the late 1990s with international, multicentre, Phase II studies of anisperimus, deoxyspergualin, belimumab (Benlysta; GlaxoSmithKline, Dublin, Ireland) and avacopan (Thaneos; ChemoCentryx Inc., San Carlos, CA, USA). 26 These adopted the disease assessment tools developed by the EUVAS group. 27,28 Only one study,29 with avacopan, has proceeded to Phase III. A North American Consortium [Vasculitis Clinical Research Consortium (VCRC)] now co-ordinates a portfolio of clinical studies. The vasculitis research experience, which covers over 20 years of multiple international centres and close working between EUVAS and VCRC, provided the background for the PEXIVAS study.
Nature and delivery of the intervention
Plasma exchange was developed for the removal of pathogenic plasma constituents, such as antibodies, in the 1970s. It requires the extracorporeal separation of plasma from blood, discarding of plasma and replacement with a compatible fluid, such as an albumin solution. The modalities include plasma filtration, in which plasma proteins are filtered but larger blood components are retained, and centrifugation, in which blood components are separated by density. Centrifugation is also used to extract platelet and white cell fractions, and leads to a more complete removal of macromolecules and cell microparticles than filtration. Both modalities remove coagulation factors and immunoglobulin, and some form of coagulation monitoring, with factor replacement for high-risk individuals, is employed. Because there is a distribution of most soluble plasma constituents throughout the extracellular volume, PLEX is administered in daily or alternate-day procedures, allowing the re-equilibration of components between procedures. In this way, five to seven procedures of one to two plasma volumes will remove a plasma constituent, such as immunoglobulin G (IgG), by 80–90%. The extracorporeal circuit requires i.v. access, and usually involves the central cannulation of the internal jugular, subclavian or femoral veins, with attendant risks of haemorrhage, thrombosis, infection and pneumothorax. Volume replacement with albumin or plasma is expensive; carries risks associated with blood products, including supply shortages and availability; and can cause allergy, anaphylaxis, hypocalcaemia and transfusion-associated acute lung injury (TRALI).
Rationale for research
Circumstantial clinical data and robust experimental evidence support a pathogenic role for ANCA, but other plasma constituents, including platelet and endothelial microparticles, complement and coagulation factors, and cytokines, are also involved in the pathology of vasculitis. 30 PLEX has been used as a treatment for vasculitis since before the discovery of ANCA, and available evidence indicates a potential role in improving the rate of renal recovery in severe renal vasculitis and reducing the mortality rate of lung haemorrhage due to vasculitis. 15 In view of the expense and risks associated with PLEX, there is a need for higher-quality evidence to guide treatment decisions. Although GCs are a central component of vasculitis treatment protocols, their associated toxicity contributes to the morbidity, incapacity and mortality rate of vasculitis. Better evidence is required to optimise the balance between efficacy and safety in GC dosing.
Aims and objectives
Primary objectives
The PEXIVAS trial aimed to determine the clinical efficacy of PLEX in addition to immunosuppressive therapy and GCs with respect to reducing the risk of death and ESRD. It also aimed to determine whether or not a reduced-dose GC regimen was non-inferior to a standard-dose regimen with respect to death and ESRD.
Secondary objectives
Secondary objectives of the trial were to determine the clinical efficacy of PLEX and reduced-dose GCs for sustained remission of vasculitis, death and ESRD as separate outcomes; and to determine the safety of PLEX and reduced-dose GCs, assessed using serious adverse event (SAE) and serious infection rates. PEXIVAS also investigated the impact of PLEX and reduced-dose GCs on health-related quality of life.
Chapter 2 Trial design and methods
Parts of this chapter have been reproduced with permission from the PEXIVAS protocol version 1.0. 1
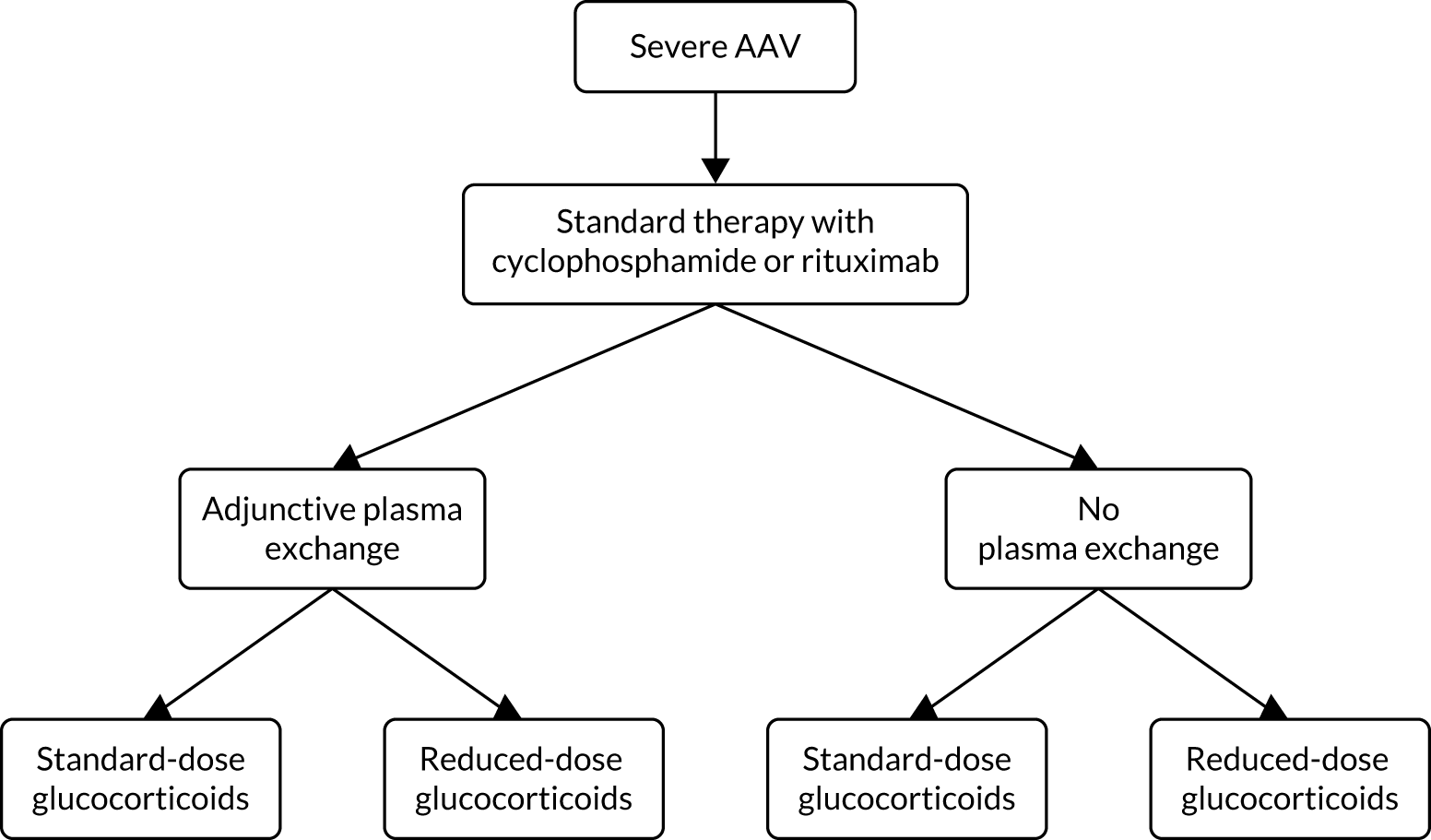
PEXIVAS was an international, multicentre, open-label factorial design randomised control trial in GPA and MPA examining the effect of adjunctive PLEX on the composite end point of death and ESRD, and the effect of a reduced-dose GC regimen compared with a standard-dose GC regimen on the composite end point.
Eligible patients were randomised to one of four groups:
-
PLEX and standard-dose GC
-
PLEX and reduced-dose GC
-
no PLEX and standard-dose GC
-
no PLEX and reduced-dose GC.
Randomisation to each intervention was in a 1 : 1 ratio, stratified by the other intervention. Patients were randomised to receive adjunctive PLEX or no PLEX, and randomised to receive either a standard or reduced GC dose (Figure 5). All patients received standard immunosuppressive induction therapy with either CYC or rituximab. The primary outcome of the trial was a composite of all-cause mortality or ESRD.
FIGURE 5.
General schema of randomisation. Reproduced with permission from the PEXIVAS protocol, version 1.0. 1
Number of centres and participants
PEXIVAS recruited from 95 centres in Europe, North America, Japan and Australia/New Zealand and aimed to recruit 500 patients with AAV over 5 years, which was extended to 700 patients over 7 years because of a lower than anticipated event rate.
Methods to protect against bias
Participants were allocated to the interventions in a 1 : 1 ratio by a central randomisation facility, utilising a computerised minimisation algorithm. The algorithm was not made available to investigators. Equal numbers of patients were to receive PLEX or no PLEX, and standard-dose GCs or reduced-dose GCs. Allocation followed a minimisation scheme with the following strata:
-
severity of renal disease at presentation [requiring dialysis or a creatinine level of ≥ 500 µmol/l (5.6 mg/dl) vs. < 500 µmol/l]
-
age (< 60 vs. ≥ 60 years)
-
ANCA-binding specificity [proteinase 3 (PR3) vs. myeloperoxidase (MPO)]
-
severity of lung haemorrhage (no haemorrhage, haemorrhage with blood oxygen saturation > 85% on room air, or haemorrhage with blood oxygen saturation ≤ 85% on room air or ventilated)
-
planned induction immunosuppression therapy to be used [i.v. CYC vs. oral CYC vs. rituximab].
Plasma exchange is an invasive procedure, generally requiring the placement of a large central venous catheter and the use of a large, complex device, with additional monitoring and nursing care. Therefore, it is difficult to blind patients or physicians to this treatment allocation. There was the potential for treating physicians to alter their treatment on the basis of knowing whether or not the patient would receive PLEX. This was partially controlled by randomly determining the GC regimen and predetermining the immunosuppressive regimen. GC dosing was also not blinded and patients and physicians had knowledge of the allocation group. Provision of the 1-year GC regimen in advance and regular adherence checks were used to minimise risk of deviation from the regimen.
Study duration and evaluations
Each patient was followed to a common study close-out, with a planned minimum follow-up of 12 months. With a predicted recruitment period of 7 years, the maximum duration of follow-up was almost 8 years. Evaluation time points were at baseline, at 2, 4, 8, 12 and 26 weeks (induction of remission period), and then at 9 and 12 months and every 6 months thereafter (maintenance of remission period). Disease activity was assessed at each evaluation time point using the BVAS/WG. 27 The BVAS/WG is a list of 35 items, grouped into 10 organ systems and scored if an item in the list currently reflects active vasculitis. Items are categorised as major or minor and are scored 3 if major and 1 if minor. The total score is the number of weighted items at each evaluation. The Short Form questionnaire-36 items (SF-36) and EuroQoL-5 Dimensions (EQ-5D) were also assessed at baseline, at 12, 26 and 52 weeks and then every 6 months. ‘All-cause’ damage was assessed using the Combined Damage Assessment Index (CDA) at baseline, at 12 and 26 weeks and then every 6 months. 28 The CDA records 114 items of irreversible damage in 18 categories. An item has to be present for at least 3 months to be scored on the CDA. Once scored, an item remains scored for the trial duration. The score is the non-weighted total of the items checked at each evaluation.
Trial end points
The primary end point was the composite of all-cause mortality or ESRD. ESRD was defined as the requirement for renal replacement therapy (haemodialysis or peritoneal dialysis) for at least 12 consecutive weeks or the receipt of a renal transplant.
The secondary end points were:
-
sustained remission (i.e. remission defined as a BVAS/WG of 0, that occurred within the 6 months after randomisation and lasted without relapse until 12 months after randomisation)
-
all-cause mortality
-
ESRD
-
SAEs
-
serious infections, defined as an infectious syndrome that required i.v. antibiotics or hospitalisation for treatment
-
SF-36 physical composite score and mental composite score
-
EQ-5D index score.
End points were ascertained at study assessments.
Treatments
Plasma exchange
Plasma exchange consisted of seven exchanges within 14 days of randomisation of at least 60 ml/kg (based on body weight) per session using albumin (3–5% depending on local availability, with or without crystalloid) as a replacement solution. The minimum replacement solution volume was 3000 ml. The following parameters were determined in accordance with local practice:
-
modality, centrifugation or filter separation technique (double filtration apheresis was not permitted)
-
anticoagulation by citrate or heparin
-
vascular access by central venous catheter, or peripheral access
-
monitoring of coagulation parameters and immunoglobulin levels
-
reduction of PLEX dose for PLEX-related complications.
Renal biopsy was avoided on the day of PLEX to minimise bleeding risk. Local practice was followed for patients with active or recent bleeding, including lung haemorrhage. This could include fresh-frozen plasma at the end of the exchange. Additional PLEX outside the protocol could be considered for refractory disease after discussion with the trial medical monitor.
Glucocorticoids
All patients received between 1000 and 3000 mg (total dose) of i.v. methylprednisolone as one to three daily pulses in the 14 days prior to or after entry into the trial; the dose was determined by the local physician. Oral GC therapy consisted of non-enteric-coated prednisone or prednisolone at equivalent doses, depending on patient weight categories (Table 1).
| Week | Standard | Reduced | ||||
|---|---|---|---|---|---|---|
| < 50 kg | 50–75 kg | > 75 kg | < 50 kg | 50–75 kg | > 75 kg | |
| Pulse | Pulse | Pulse | Pulse | Pulse | Pulse | |
| 1 | 50 | 60 | 75 | 50 | 60 | 75 |
| 2 | 50 | 60 | 75 | 25 | 30 | 40 |
| 3–4 | 40 | 50 | 60 | 20 | 25 | 30 |
| 5–6 | 30 | 40 | 50 | 15 | 20 | 25 |
| 7–8 | 25 | 30 | 40 | 12.5 | 15 | 20 |
| 9–10 | 20 | 25 | 30 | 10 | 12.5 | 15 |
| 11–12 | 15 | 20 | 25 | 7.5 | 10 | 12.5 |
| 13–14 | 12.5 | 15 | 20 | 6 | 7.5 | 10 |
| 15–16 | 10 | 10 | 15 | 5 | 5 | 7.5 |
| 17–18 | 10 | 10 | 15 | 5 | 5 | 7.5 |
| 19–20 | 7.5 | 7.5 | 10 | 5 | 5 | 5 |
| 21–22 | 7.5 | 7.5 | 7.5 | 5 | 5 | 5 |
| 23–52 | 5 | 5 | 5 | 5 | 5 | 5 |
| > 52 | Investigators’ local practice | Investigators’ local practice | ||||
Immunosuppressive therapy
All patients received induction therapy with either CYC or rituximab. CYC was administered for 13–26 weeks, using either daily oral or pulsed i.v. routes, with dose reduction for age and renal impairment (Tables 2 and 3). Modifications to dose and frequency were made for intolerance or leucopenia. For patients in the PLEX group receiving i.v. CYC, the next PLEX was at least 24 hours after the i.v. dose. For patients receiving PLEX and daily oral CYC, CYC was given following PLEX. Concomitant use of mesna was optional.
| Time (weeks) | Pulse number | Dose (mg/kg) |
|---|---|---|
| 0 | 1 | 15 |
| 2 | 2 | 15 |
| 4 | 3 | 15 |
| 7 | 4 | 15 |
| 10 | 5 | 15 |
| 13 | 6 | 15 |
| 16 | 7 | 15 |
| 19 | 8 | 15 |
| 22 | 9 | 15 |
| 25 | 10 | 15 |
| Age | Oral CYC | i.v. CYC | ||
|---|---|---|---|---|
| eGFR (ml/minute/1.73 m2) | eGFR (ml/minute/1.73 m2) | |||
| > 30 | ≤ 30 | > 30 | ≤ 30 | |
| < 60 | 2 | 1.5 | 15 | 12.5 |
| 60–70 | 1.5 | 1.25 | 12.5 | 10 |
| > 70 | 1.25 | 1 | 10 | 7.5 |
Rituximab was administered at a dose of 375 mg/m2/week ×4, with the first dose within 14 days of trial entry. Because rituximab as an antibody is removed by PLEX, at least two of the four doses were given after the end of the PLEX course. Prophylaxis against infusion reactions employed 100 mg of i.v. hydrocortisone and an antihistamine agent before the first rituximab infusion, and local guidelines were followed for subsequent infusions.
Remission-maintenance immunosuppressive therapy
Patients who had completed at least 13 weeks of CYC treatment and achieved a clinical remission (i.e. a BVAS/WG of 0) were converted to maintenance therapy with azathioprine at a target dose of 2 mg/kg/day (rounded to the nearest 25 mg/day). Those who had an intolerance to azathioprine received methotrexate or mycophenolate mofetil at their physician’s discretion.
Treatment of resistant disease
Resistant disease was defined as active AAV that did not improve or worsened despite administration of the allocated induction of remission therapy. A repeated course of i.v. methylprednisolone (1000–3000 mg total dose) or high-dose oral prednisone/prednisolone (1 mg/kg/day for 1 week) was administered in accordance with local practice.
Treatment of relapse
A major relapse was new or worsened disease activity that occurred after remission that involved a major BVAS/WG item. Treatment with CYC or rituximab, additional doses of i.v. methylprednisolone (up to 3000 mg) or an increase in oral prednisolone was recommended for a major relapse. A minor relapse was new or worsening disease activity that did not involve a major BVAS/WG item and was treated with up to 20 mg/day of prednisone or prednisolone for a maximum of 14 days.
Prophylactic therapies
Prophylaxis against GC-induced osteoporosis, in accordance with local guidelines, and Pneumocystis jirovecii pneumonia infection with sulfamethoxazole-trimethoprim was recommended for at least 6 months.
Eligibility
Inclusion criteria
Inclusion required:
-
a diagnosis of new or relapsing GPA or MPA consistent with the Chapel-Hill consensus definitions
-
PR3-ANCA or MPO-ANCA positivity
-
severe vasculitis defined as either –
-
renal involvement characterised by renal biopsy demonstrating focal necrotising glomerulonephritis or urine haematuria/cellular casts and proteinuria, and an eGFR of < 50 ml/minute/1.73 m2
-
pulmonary haemorrhage due to active vasculitis, defined using a compatible chest X-ray or computerised tomography (CT) scan (diffuse pulmonary infiltrates), with no alternative explanation, and at least one of:
-
alveolar haemorrhage on bronchoscopic examination
-
increasingly bloody returns with bronchoalveolar lavage
-
observed haemoptysis
-
unexplained anaemia (< 10 g/dl)
-
documented drop in haemoglobin (> 1 g/dl) from < 10 g/dl
-
an increased diffusing capacity of carbon monoxide.
-
-
-
informed consent from patient or a surrogate decision-maker.
In some participating countries, permission from ethics committees was granted to use deferred consent for enrolling a patient until a legal representative became available to consent on their behalf or the patient was capable of consenting.
Exclusion criteria
-
A diagnosis of vasculitis other than GPA or MPA.
-
A positive serum result for anti-glomerular basement membrane antibody test or renal biopsy demonstrating linear glomerular immunoglobulin deposition.
-
Receipt of dialysis for > 21 days immediately prior to randomisation, or prior renal transplant.
-
Age ≤ 15 years (or age < 18 years at centres that do not treat paediatric patients).
-
Pregnant at time of study entry.
-
Treatment with more than one i.v. dose of CYC and/or > 14 days of oral CYC and/or > 14 days of prednisone/prednisolone (> 30 mg/day) and/or more than one dose of rituximab within the 28 days immediately prior to randomisation.
-
A comorbidity or condition that, in the opinion of the investigator, precluded the use of CYC/rituximab, GCs or PLEX, or that absolutely mandated the use of PLEX.
-
PLEX in the 3-month period prior to randomisation.
Sample size
Sample size estimation
The sample size was event driven to detect a hazard ratio (HR) of 0.64 (PLEX vs. no PLEX), with 80% power and a two-sided alpha of 0.05. Protocol versions 1.05 and 2.0 estimated a required sample size of 500, predicting 164 events over the study period, which was equivalent to an absolute difference in the risk of the primary end point at 5 years of 12%, i.e. 44% in the no-PLEX group, compared with 38% in the PLEX group (overall risk reduction of 38%). This sample size estimate assumed a median time to ESRD or death of 5 years on the basis of previous extended follow-up studies in randomised trials of AAV of a similar severity to those targeted in this study (see Figure 1). A review of the PEXIVAS event rates in 2014 indicated a 2-year event rate of 24% and predicted an overall 5-year event rate of 30–35%. Improvements in death and ESRD have been recently reported in registry studies. To obtain the required number of events, the sample size was increased to 700 patients, allowing a 10% loss to follow-up or crossover between treatment groups. It was planned for the trial to enrol 700 patients over a 7-year period to observe at least 160 events. These calculations assumed no significant interaction between the two treatment factors. Although this absolute risk appears larger than is often clinically significant, the expensive and invasive nature of the primary intervention, PLEX, warranted a relatively large effect size. Additionally, this effect size is close to the estimated effect of PLEX in the meta-analysis of prior studies (i.e. 80% power to detect a RR reduction of 27% with our sample size compared with a RR reduction of 20% in the meta-analysis).
Although this effect size appeared reasonable to detect for PLEX, it was unlikely that a reduction in GCs would result in a 12% absolute risk reduction of death or dialysis. It was expected that 25% of patients would experience a severe infection based on prior studies. A sample size of > 700 patients allowed 80% power to detect at least a 10% absolute risk reduction in severe infections (RR reduction of severe infection by 40%), which would be a finding of clinical significance. In terms of the non-inferiority hypothesis, a sample size of 700 patients would allow > 80% power to ensure that the reduced-dose GC regimen resulted in an increase in ESRD or death by no more than 11% (one-sided alpha of 0.05).
Statistical methods
Interim analyses
Annual interim analyses of efficacy and safety data were undertaken and reported to the independent Data Monitoring Committee (DMC)/Data Safety Monitoring Board (DSMB). The Haybittle–Peto approach was used, whereby all interim analyses used a difference of 3 standard errors (approximately p = 0.002) as a stopping guideline.
Primary end-point analyses
The primary outcome was a composite of all-cause mortality or ESRD. The primary comparison groups were composed of those randomised to PLEX and those randomised to no PLEX (comparison 1) and those randomised to a reduced-dose GC regimen and those randomised to a standard-dose GC regimen (comparison 2).
The comparison of PLEX and no PLEX was based on the intention-to-treat (ITT) analysis population using a time-to-event analysis (time from randomisation to death and/or ESRD). The primary outcome was compared between treatment groups using survival analysis methods. Kaplan–Meier survival curves were constructed for visual presentation of time-to-event comparisons. A Cox proportional hazards model was fitted to obtain an adjusted HR and 95% CI. The analyses were adjusted for both treatment group parameters and all of the minimisation variables. Any patient randomised to the PLEX group who had received at least one PLEX within 14 days of randomisation was considered compliant. In addition, if any patient randomised to the PLEX group died within 14 days of randomisation and did not receive any PLEX, they were classed as compliant with PLEX based on the intention to undergo PLEX. Any patient randomised to the no-PLEX group who received any PLEX was considered non-compliant.
The comparison of reduced-dose and standard-dose GC was based on a non-inferiority hypothesis, with a non-inferiority margin of an 11% absolute risk increase. An ITT analysis can increase the risk of falsely claiming non-inferiority. 31 Therefore, for the assessment of whether or not a reduced-dose GC regimen was non-inferior to a standard-dose GC regimen with respect to the primary outcome (only), a per-protocol analysis was undertaken for the primary analysis. The absolute risks in each group reaching the primary outcome using the binary outcome of death and/or ESRD were calculated using the complete follow-up data. A binomial model was fitted to obtain the adjusted risk difference and 90% CI, adjusting for both treatment group parameters and all of the minimisation variables. If the 90% CI excluded an 11% increase, then non-inferiority was claimed. In addition to the absolute risk difference, the adjusted HR and 95% CI from the Cox proportional hazards model using the ITT analysis population was also calculated. Compliance with the GC regimen was defined for patients randomised to the reduced-dose GC group as receiving ≤ 130% of the cumulative oral dose of the reduced-dose GC regimen in the first 6 months of therapy. Compliance for patients randomised to the standard-dose GC group was defined as receiving ≥ 70% of the cumulative oral dose of the standard-dose GC regimen in the first 6 months of therapy.
It was expected a priori that no interaction between treatments would be identified, but if one was found, this was considered a chance finding. Therefore, although a test for interaction was undertaken, it was interpreted with caution, and a test for interaction was carried out for the primary outcome only.
There was a risk of patients randomised to the no-PLEX group crossing over to the PLEX group, and vice versa, although this practice was discouraged. Similarly, there was a risk that patients may receive a dose of GC appreciably different from the dose to which they were allocated. The primary ITT and per-protocol analyses dealt with these crossovers in a conservative manner (bias to the null). Sensitivity analyses were performed to explore the potential of these crossovers to reduce the true magnitude of treatment effects. These analyses included per-protocol and ITT analyses (for the PLEX and GC comparisons, respectively).
Secondary end-point analyses
All analyses of secondary end points for both the PLEX and no-PLEX groups and the reduced-dose versus standard-dose GC comparisons used the ITT analysis population.
Sustained remission
Disease activity was analysed in terms of sustained remission. Participants were considered to have achieved sustained remission if they had a BVAS/WG of zero (complete remission) within 26 weeks of randomisation and maintained a BVAS/WG of zero without evidence of relapse from complete remission until at least 52 weeks after randomisation. The number and percentage of participants who achieved a sustained remission were calculated for each group and an adjusted RR and 95% CI were estimated using a log-binomial regression model.
Death and end-stage renal disease
Death and ESRD were analysed as separate end points using survival analysis methods identical to those described for the primary composite end point for the PLEX versus no-PLEX comparison.
Serious adverse events
The number and percentage of participants experiencing at least one SAE were analysed as categorical binary (yes/no) variables, and adjusted RRs and 95% CIs were estimated using a log-binomial model. A more complex model of SAE occurrences was constructed utilising SAE as a count variable. The number of SAEs that a participant experienced was analysed using a Poisson regression or negative binomial regression model (if there was evidence of overdispersion), with an offset for the length of time that the participant was in the trial included in the model, to obtain an adjusted incidence rate ratio (IRR) and 95% CI.
Serious infections
The rate of serious infections was assessed for the first year of the trial and at trial end. The number of serious infections that a participant experienced was analysed using a Poisson regression model or a negative binomial regression model (if there was evidence of over dispersion), with an offset for the length of time that the participant was in the trial included in the model, to obtain an adjusted IRR and 95% CI.
Quality-of-life measures
Quality-of-life data, obtained using the SF-36 and EQ-5D, were analysed using mixed-effect repeated-measures models with the two treatment group parameters, the minimisation variables and the baseline scores included as covariates in the model. Time was included as a continuous variable in the model. In the initial model, a treatment by time cross-term was included; if this was not statistically significant, it was considered that the treatment effect was constant over time, and models without the treatment by time cross-term were fitted.
Subgroup analyses
Several a priori subgroup analyses were planned with respect to the end point of time to death and/or ESRD using the ITT analysis populations for both PLEX and no PLEX and the reduced-dose versus standard-dose GC comparisons. The subgroups were defined as each of the variables included in the randomisation minimisation variables (see Methods to protect against bias). The effects of these subgroups were examined by including the relevant subgroup-by-treatment interaction term in the Cox proportional hazards model for each subgroup analysis to explore whether or not there was evidence that the treatment effects (PLEX vs. no PLEX, or reduced-dose vs. standard-dose GCs) differed across the subgroup strata.
Exploratory analyses
Owing to the possibility that the investigational treatments may have a considerable effect on early mortality and renal function, analyses of the primary outcome (the composite of death and ESRD) were performed after censoring data at the 12-month follow-up.
To assess the efficacy of PLEX for autoantibody removal, the proportion of patients who were ANCA negative at 2 and 4 weeks after trial entry was assessed for each treatment comparison.
Because lung haemorrhage is the major cause of early death in AAV, we performed an additional post hoc subgroup analysis for severity of lung haemorrhage for the outcome of mortality at any stage during the trial for the two randomisations.
All analyses were adjusted for the two treatment group parameters, the minimisation variables (i.e. age, ANCA subtype, severity of renal failure, presence and severity of lung haemorrhage and planned type of induction immunosuppression therapy) and baseline values where available (e.g. the quality-of-life analyses). The no-PLEX and standard-dose GC groups were used as the reference groups. All estimates of differences between groups are presented with two-sided 95% CIs (unless otherwise specified), with a p-value of < 0.05 considered statistically significant, unless otherwise stated. No corrections for multiple tests were made. The statistical analyses were undertaken in SAS® (SAS Institute Inc., Cary, NC, USA) and Stata® (StataCorp LP, College Station, TX, USA).
Chapter 3 Results
Recruitment information
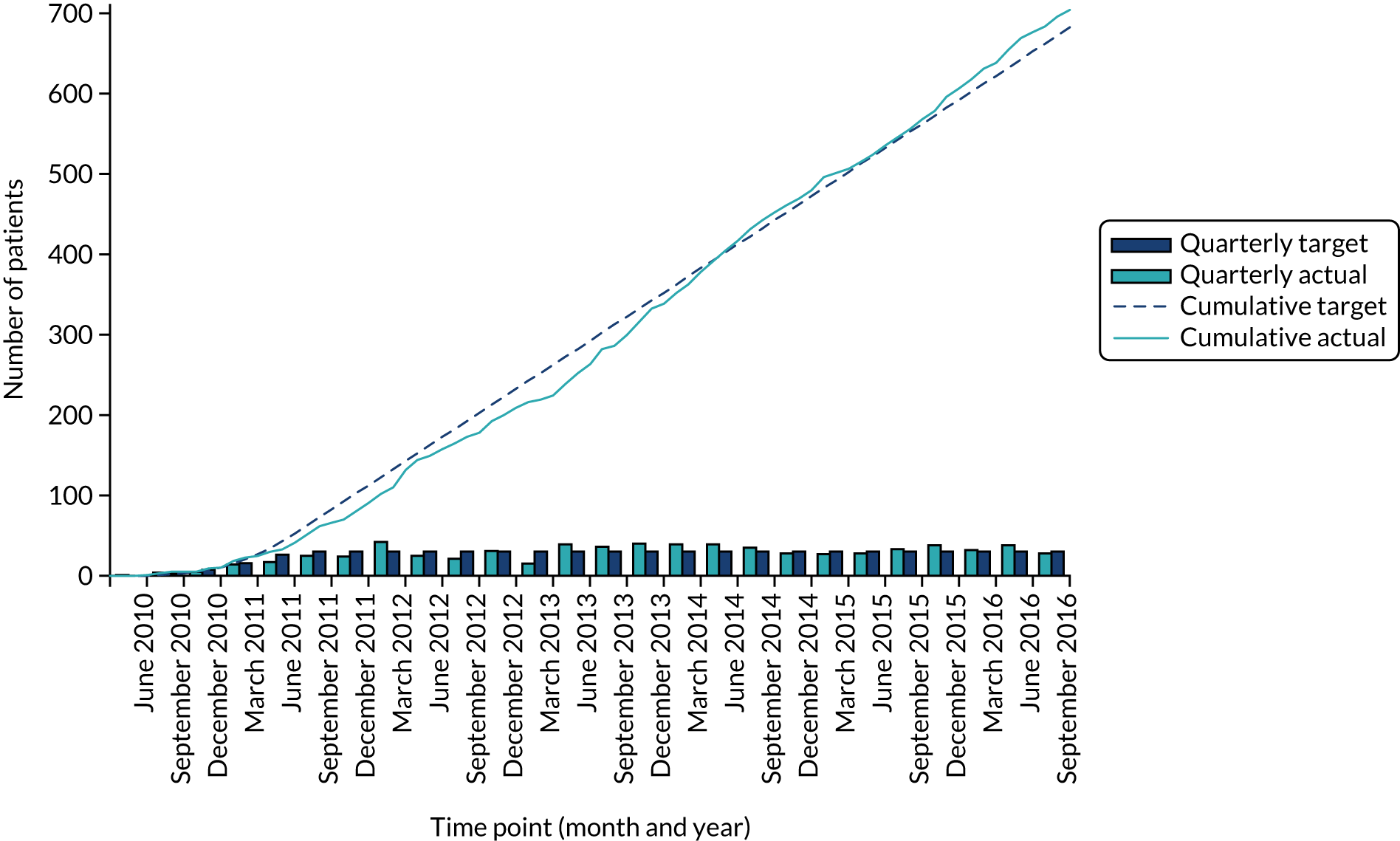
A total of 704 participants were recruited by 95 international sites across 10 European Union member states, Australia, Canada, Japan, Mexico, the USA and New Zealand, from July 2010 to September 2016 (Figures 6 and 7 and Table 4). Patients were followed to a common close-out in July 2017, with a median follow-up of 2.9 years.
FIGURE 6.
PEXIVAS participating sites. Reproduced with permission from Google Maps (Google Inc., Mountain View, CA, USA). © 2018 Google.
FIGURE 7.
Actual vs. expected recruitment rates.
| Country | Centres (n) | Patients recruited (n) |
|---|---|---|
| Canada | 9 | 191 |
| UK | 19 | 179 |
| Australia | 17 | 94 |
| Denmark | 4 | 57 |
| France | 17 | 52 |
| USA | 8 | 39 |
| Italy | 2 | 26 |
| Japan | 5 | 12 |
| Czechia | 1 | 10 |
| New Zealand | 4 | 10 |
| Sweden | 3 | 9 |
| Norway | 2 | 8 |
| Mexico | 1 | 7 |
| Poland | 1 | 7 |
| Spain | 1 | 2 |
| Belgium | 1 | 1 |
| Total | 95 | 704 |
Flow of participants in the trial
Of the 704 patients recruited to the trial, 352 were randomised to both the PLEX group and no-PLEX group, and 353 and 351 were randomised to the reduced-dose GC group and standard-dose GC group, respectively. The protocol compliance was 96% for the PLEX group and 92% for the no-PLEX group. Seventeen (4.8%) patients were withdrawn or lost to follow-up prior to reaching ESRD or death in the PLEX group, compared with 11 (3.1%) in the no-PLEX group. Of those who received PLEX, the modality was centrifugation in 60% and filtration in 40%. The compliance rates with the allocated GC regimen were 94% and 93% for the reduced-dose GC regimen and the standard-dose GC regimen, respectively, with 11 (3.1%) and 17 (4.8%) patients being withdrawn or lost to follow-up prior to reaching ESRD or death in the reduced-dose GC group and the standard-dose GC group, respectively (Figures 8 and 9).
FIGURE 8.
The Consolidated Standards of Reporting Trials (CONSORT) flow diagram for PLEX randomisation.
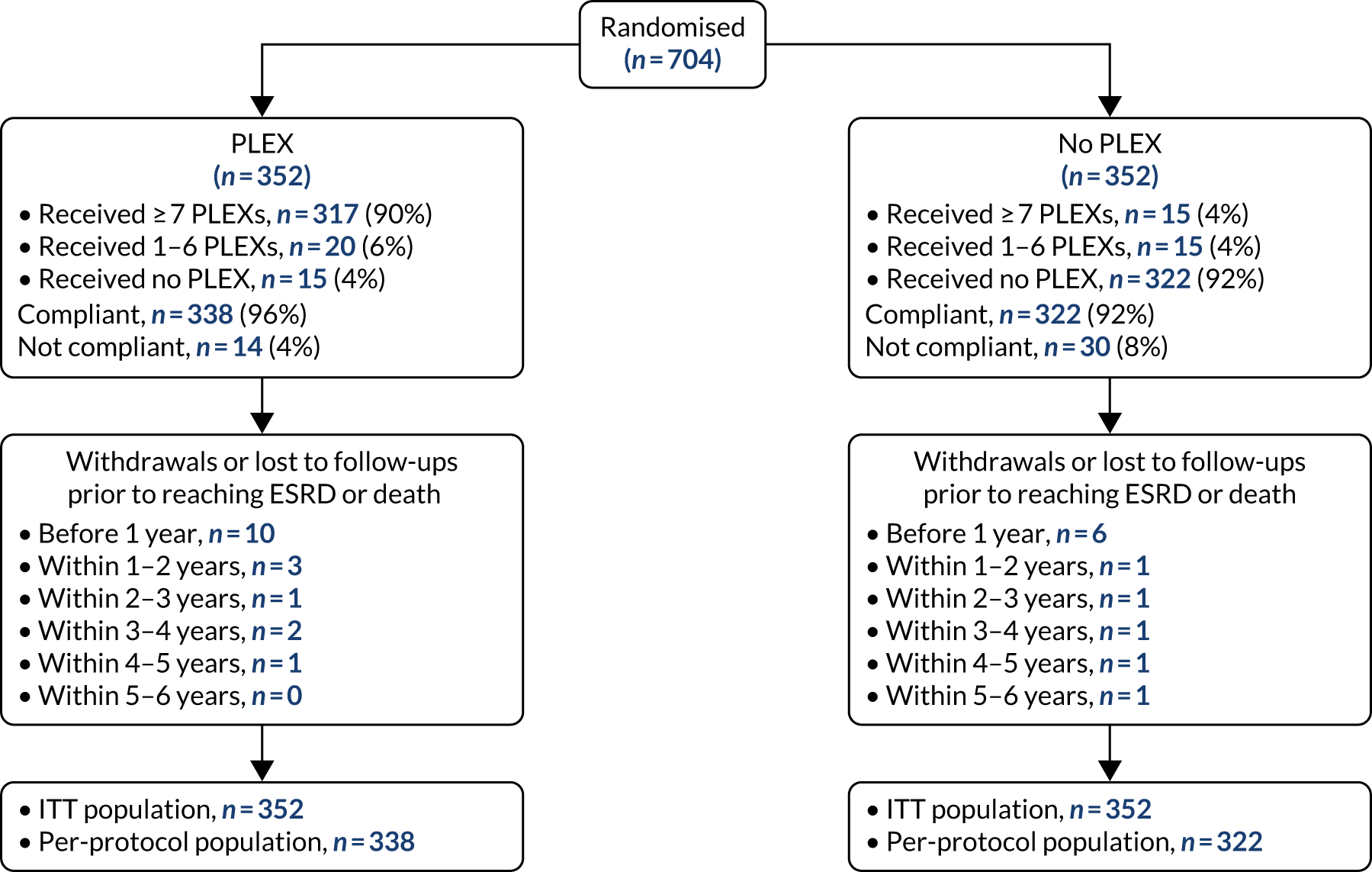
FIGURE 9.
The CONSORT flow diagram for GC randomisation.
Baseline comparability
The randomisation groups were well balanced for baseline variables, in line with the minimisation strategy. The baseline demographic and disease-related data are summarised in Table 5 according to treatment allocation.
| Variable | PLEX (N = 352) | No PLEX (N = 352) | Reduced-dose GCs (N = 353) | Standard-dose GCs (N = 351) | Total (N = 704) |
|---|---|---|---|---|---|
| Age, mean (SD) | 62.8 (14.4) | 63.5 (13.7) | 63.3 (14.2) | 63.1 (13.9) | 63.2 (14) |
| Gender, n (%) | |||||
| Male | 203 (57.7) | 194 (55.1) | 197 (55.8) | 200 (57) | 397 (56.4) |
| Female | 149 (42.3) | 158 (44.9) | 156 (44.2) | 151 (43) | 307 (43.6) |
| Ethnicity, n (%) | |||||
| South Asian | 7 (2) | 15 (4.3) | 7 (2) | 15 (4.3) | 22 (3.1) |
| Chinese | 1 (0.3) | 5 (1.4) | 2 (0.6) | 4 (1.1) | 6 (0.9) |
| Japanese | 6 (1.7) | 6 (1.7) | 8 (2.3) | 4 (1.1) | 12 (1.7) |
| Other Asian | 0 (0) | 2 (0.6) | 0 (0) | 2 (0.6) | 2 (0.3) |
| Arab | 0 (0) | 1 (0.3) | 1 (0.3) | 0 (0) | 1 (0.1) |
| Black African | 4 (1.1) | 2 (0.6) | 2 (0.6) | 4 (1.1) | 6 (0.9) |
| Mixed-race African | 0 (0) | 1 (0.3) | 0 (0) | 1 (0.3) | 1 (0.1) |
| European | 281 (79.8) | 279 (79.3) | 278 (78.8) | 282 (80.3) | 560 (79.5) |
| Native North/South American | 16 (4.5) | 7 (2) | 15 (4.2) | 8 (2.3) | 23 (3.3) |
| Latin American | 5 (1.4) | 5 (1.4) | 6 (1.7) | 4 (1.1) | 10 (1.4) |
| Other black | 5 (1.4) | 2 (0.6) | 1 (0.3) | 6 (1.7) | 7 (1) |
| Other | 24 (6.8) | 26 (7.4) | 30 (8.5) | 20 (5.7) | 50 (7.1) |
| Missing | 3 (0.9) | 1 (0.3) | 3 (0.8) | 1 (0.3) | 4 (0.6) |
| Medical history, n (%) | |||||
| Hypertension (yes) | 171 (48.6) | 187 (53.1) | 179 (50.7) | 179 (51) | 358 (50.9) |
| Ischaemic heart disease (yes) | 34 (9.7) | 34 (9.7) | 34 (9.6) | 34 (9.7) | 68 (9.7) |
| Diabetes (yes) | 52 (14.8) | 57 (16.2) | 43 (12.2) | 66 (18.8) | 109 (15.5) |
| Pulmonary disease (yes) | 65 (18.5) | 60 (17) | 57 (16.1) | 68 (19.4) | 125 (17.8) |
| Cerebrovascular disease (yes) | 16 (4.5) | 18 (5.1) | 15 (4.2) | 19 (5.4) | 34 (4.8) |
| Peripheral vascular disease (yes) | 11 (3.1) | 10 (2.8) | 12 (3.4) | 9 (2.6) | 21 (3) |
| Venous thromboembolic disease (yes) | 14 (4) | 16 (4.5) | 15 (4.2) | 15 (4.3) | 30 (4.3) |
| Malignancy (yes) | 25 (7.1) | 35 (9.9) | 35 (9.9) | 25 (7.1) | 60 (8.5) |
| Smoking (yes) | 64 (18.2) | 70 (19.9) | 60 (17) | 74 (21.1) | 134 (19.0) |
| Prior history of AAV (yes) | 35 (9.9) | 28 (8.0) | 34 (9.6) | 29 (8.3) | 63 (8.9) |
| ANCA, n (%) | |||||
| PR3 positive | 143 (40.6) | 143 (40.6) | 143 (40.5) | 143 (40.7) | 286 (40.6) |
| MPO positive | 209 (59.4) | 209 (59.4) | 210 (59.5) | 208 (59.3) | 418 (59.4) |
| ESR (mm/hour), n (%) | 163 (46.3) | 178 (50.6) | 181 (51.3) | 160 (45.6) | 341 (48.4) |
| Median (IQR) | 74 (48–102) | 72.5 (50–101) | 71 (47–99) | 78 (51–106) | 74 (49–101) |
| CRP (mg/l), n (%) | 280 (79.5) | 296 (84.1) | 286 (81) | 290 (82.6) | 576 (81.8) |
| Median (IQR) | 50.9 (13.8–122.8) | 42.1 (14–97.2) | 44.6 (13–117) | 45.5 (14–98) | 45 (14–110.5) |
| Haemoglobin (g/l), n (%) | 348 (98.9) | 349 (99.1) | 349 (98.9) | 348 (99.1) | 697 (99) |
| Median (IQR) | 94 (83–105) | 95 (85–105) | 95 (84–105) | 95 (84.5–105) | 95 (84–105) |
| Creatinine level at randomisation, n (%) | 349 (99.1) | 351 (99.7) | 350 (99.2) | 350 (99.7) | 700 (99.4) |
| Median (IQR) | 327 (206–491) | 335.9 (209–495) | 320 (190–480) | 335 (219–502) | 327.4 (206–494.5) |
| < 500 µmol/l, n (%) | 251 (71.3) | 248 (70.5) | 251 (71.1) | 248 (70.7) | 499 (70.9) |
| ≥ 500 µmol/l or on dialysis, n (%) | 101 (28.7) | 104 (29.5) | 102 (28.9) | 103 (29.3) | 205 (29.1) |
| On dialysis, n (%) | 66 (18.8) | 74 (21) | 67 (19) | 73 (20.8) | 140 (19.9) |
| Lung haemorrhage, n (%) | |||||
| No haemorrhage | 257 (73) | 256 (72.7) | 257 (72.8) | 256 (72.9) | 513 (72.9) |
| Not severe | 64 (18.2) | 66 (18.8) | 65 (18.4) | 65 (18.5) | 130 (18.5) |
| Severe | 31 (8.8) | 30 (8.5) | 31 (8.8) | 30 (8.5) | 61 (8.7) |
| Treatment prior to randomisation | |||||
| i.v. methylprednisolone (mg), n (%) | 259 (73.6) | 260 (73.9) | 264 (74.8) | 255 (72.6) | 519 (73.7) |
| Median (IQR) | 1500 (1000–3000) | 1500 (1000–3000) | 1500 (1000–3000) | 1500 (1000–3000) | 1500 (1000–3000) |
| i.v. CYC (mg), n (%) | 55 (15.6) | 46 (13.1) | 52 (14.7) | 49 (14) | 101 (14.3) |
| Median (IQR) | 900 (600–1000) | 892.5 (750–1000) | 923.5 (750–1000) | 830 (600–1000) | 900 (700–1000) |
| Oral CYC (mg) , n (%) | 64 (18.2) | 64 (18.2) | 55 (15.6) | 73 (20.8) | 128 (18.2) |
| Median (IQR) | 100 (75–137.5) | 100 (75–125) | 100 (75–125) | 100 (75–150) | 100 (75–125) |
| Rituximab (mg), n (%) | 16 (4.5) | 11 (3.1) | 17 (4.8) | 10 (2.8) | 27 (3.8) |
| Median (IQR) | 740 (675–925) | 700 (600–850) | 750 (700–950) | 675 (600–750) | 730 (650–850) |
| AAV organ involvement,a n (%) | |||||
| Cutaneous | 37 (10.5) | 39 (11.1) | 34 (9.6) | 42 (12) | 76 (10.8) |
| Mucous membranes/eyes | 30 (8.5) | 36 (10.2) | 30 (8.5) | 36 (10.3) | 66 (9.4) |
| Ear, nose and throat | 95 (27) | 103 (29.3) | 98 (27.8) | 100 (28.5) | 198 (28.1) |
| Cardiovascular | 6 (1.7) | 4 (1.1) | 5 (1.4) | 5 (1.4) | 10 (1.4) |
| Gastrointestinal | 2 (0.6) | 2 (0.6) | 1 (0.3) | 3 (0.9) | 4 (0.6) |
| Pulmonary | 145 (41.2) | 149 (42.3) | 147 (41.6) | 147 (41.9) | 294 (41.8) |
| Renal | 342 (97.2) | 349 (99.1) | 346 (98) | 345 (98.3) | 691 (98.2) |
| Nervous system | 37 (10.5) | 25 (7.1) | 33 (9.3) | 29 (8.3) | 62 (8.8) |
| Other | 61 (17.3) | 59 (16.8) | 59 (16.7) | 61 (17.4) | 120 (17) |
| BVAS/WG | |||||
| Mean (SD) | 9.3 (3.5) | 9.3 (3.5) | 9.1 (3.4) | 9.5 (3.6) | 9.3 (3.5) |
| Median (IQR) | 9 (7–11) | 9 (7–11) | 9 (7–11) | 9 (7–11) | 9 (7–11) |
| BVAS/WG major items | |||||
| Mean (SD) | 7.1 (2.7) | 6.9 (2.5) | 6.8 (2.6) | 7.2 (2.6) | 7 (2.6) |
| Median (IQR) | 6 (6–9) | 6 (6–9) | 6 (6–9) | 6 (6–9) | 6 (6–9) |
Missing data
Data return was good for the first year, then progressively reduced over the course of the trial. Illustrative data for follow-up forms confirmed a 99% return in the first year, falling to 93% after 4 years. Where forms were not available, primary end-point data (i.e. data on death or ESRD) were returned for all patients remaining in the trial (Table 6).
| Follow-up assessment point | Forms expected (n) | Forms returned (n) | Form return rate (%) |
|---|---|---|---|
| 2 weeks | 686 | 677 | 99 |
| 4 weeks | 672 | 666 | 99 |
| 8 weeks | 662 | 655 | 99 |
| 12 weeks | 655 | 650 | 99 |
| 26 weeks | 642 | 633 | 99 |
| 39 weeks | 633 | 618 | 98 |
| 52 weeks | 622 | 614 | 99 |
| 18 months | 556 | 544 | 98 |
| 24 months | 477 | 466 | 98 |
| 30 months | 422 | 407 | 96 |
| 36 months | 363 | 344 | 95 |
| 42 months | 292 | 270 | 92 |
| 48 months | 226 | 210 | 93 |
| 54 months | 173 | 159 | 92 |
| 60 months | 137 | 126 | 92 |
| 66 months | 87 | 72 | 83 |
| 72 months | 45 | 35 | 78 |
| 78 months | 18 | 13 | 72 |
| 84 months | 5 | 3 | 60 |
Primary outcome
The primary outcome was a composite of all-cause mortality or ESRD.
Plasma exchange versus no plasma exchange
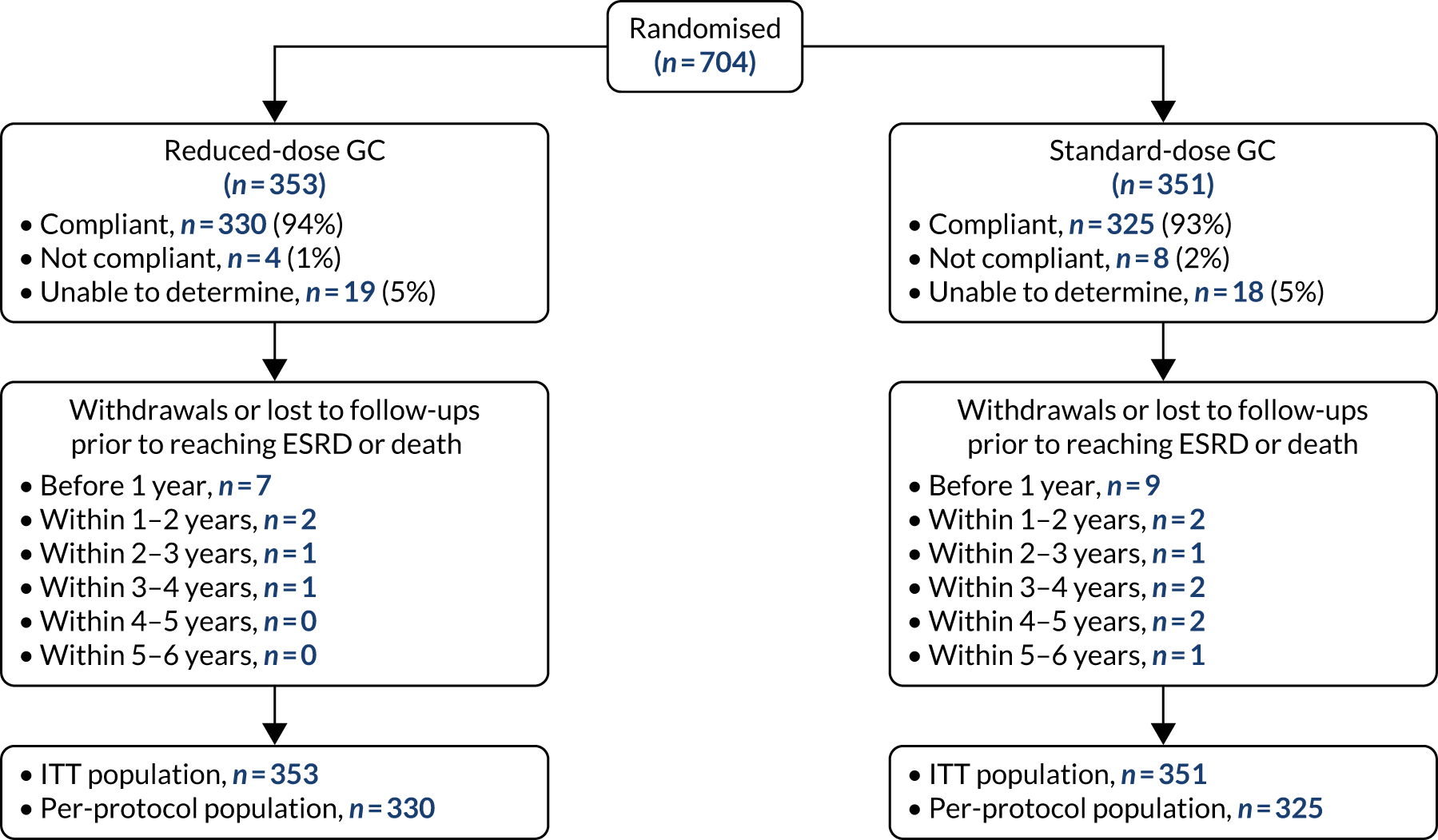
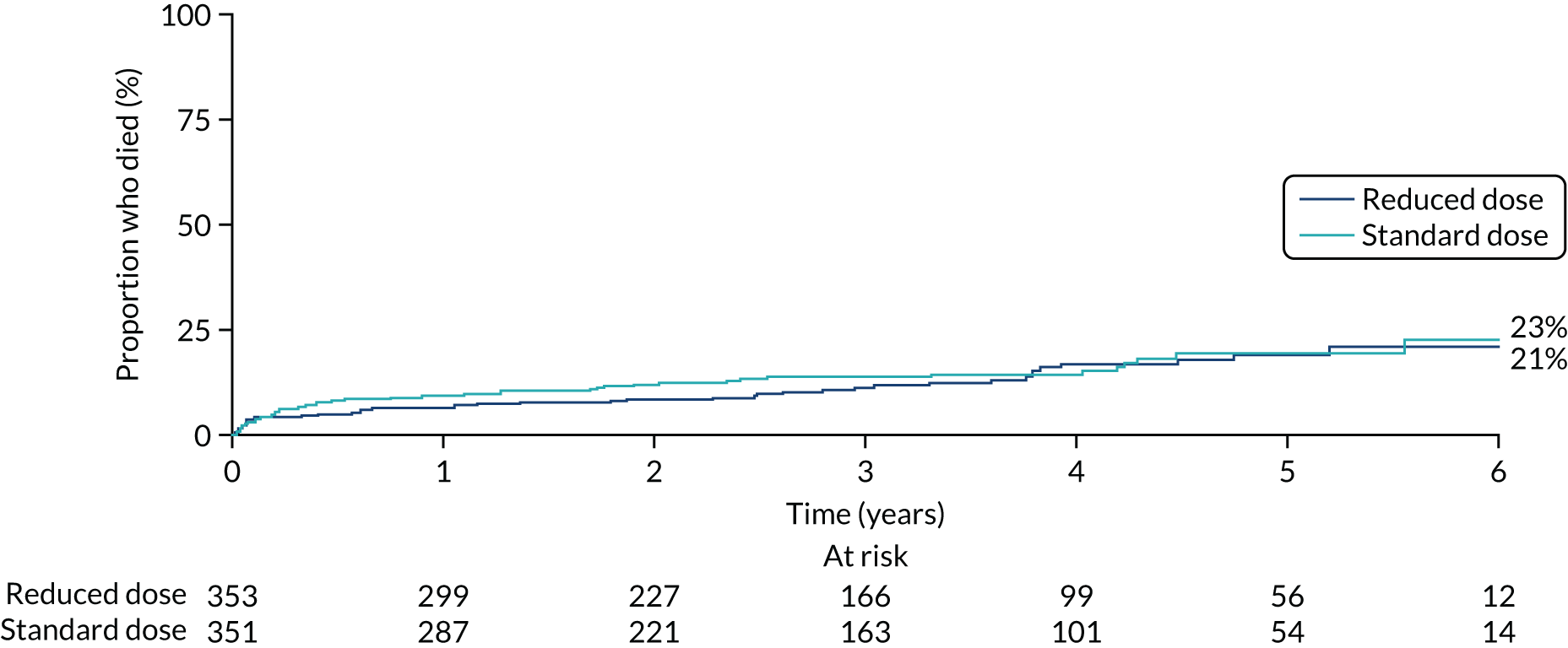
The primary comparison of PLEX versus no PLEX was based on the ITT analysis population, using a time-to-event analysis (i.e. time from randomisation to death or ESRD). A total of 100 patients in the PLEX group and 109 patients in the no-PLEX group reached the primary end point (HR 0.86, 95% CI 0.65 to 1.13; p = 0.3) (Table 7 and Figure 10). There was no evidence of a treatment interaction (p = 0.7). The per-protocol analysis produced similar results (HR 0.85, 95% CI 0.64 to 1.13; p = 0.3) (see Table 7).
| ITT population | PLEX (N = 352) | No PLEX (N = 352) | HRa (95% CI) | p-value | Interaction p-value |
|---|---|---|---|---|---|
| Event, n (%) | |||||
| No | 252 (72) | 243 (69) | 0.86 (0.65 to 1.13) | 0.268 | 0.722 |
| Yes | 100 (28) | 109 (31) | |||
| Per-protocol population | PLEX (N = 338) | No PLEX (N = 322) | HRa (95% CI) | p-value | Interaction p-value |
| Event, n (%) | |||||
| No | 243 (72) | 223 (69) | 0.85 (0.64 to 1.13) | 0.266 | |
| Yes | 95 (28) | 99 (31) | |||
FIGURE 10.
Kaplan–Meier plot for PLEX vs. no PLEX (primary outcome: ITT).
Reduced-dose glucocorticoids versus standard-dose glucocorticoids regimen
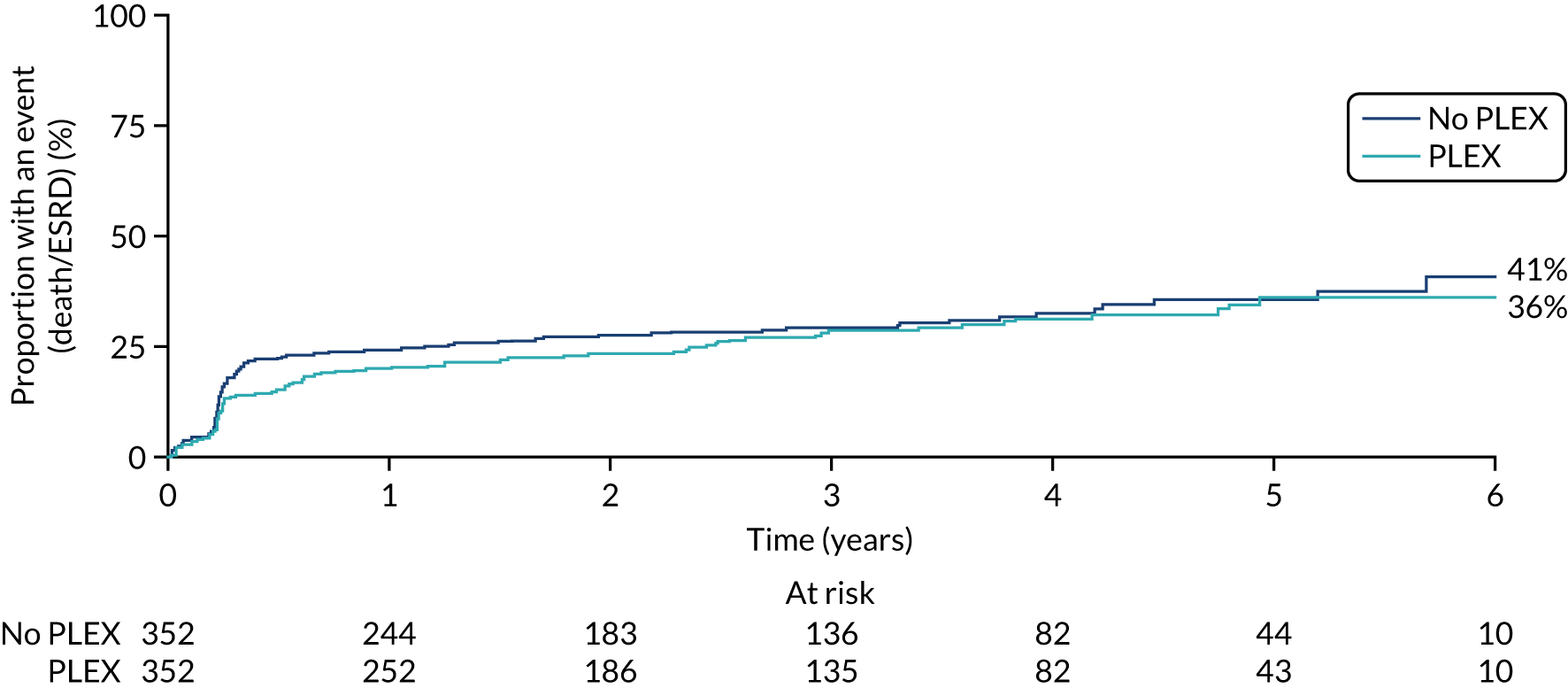
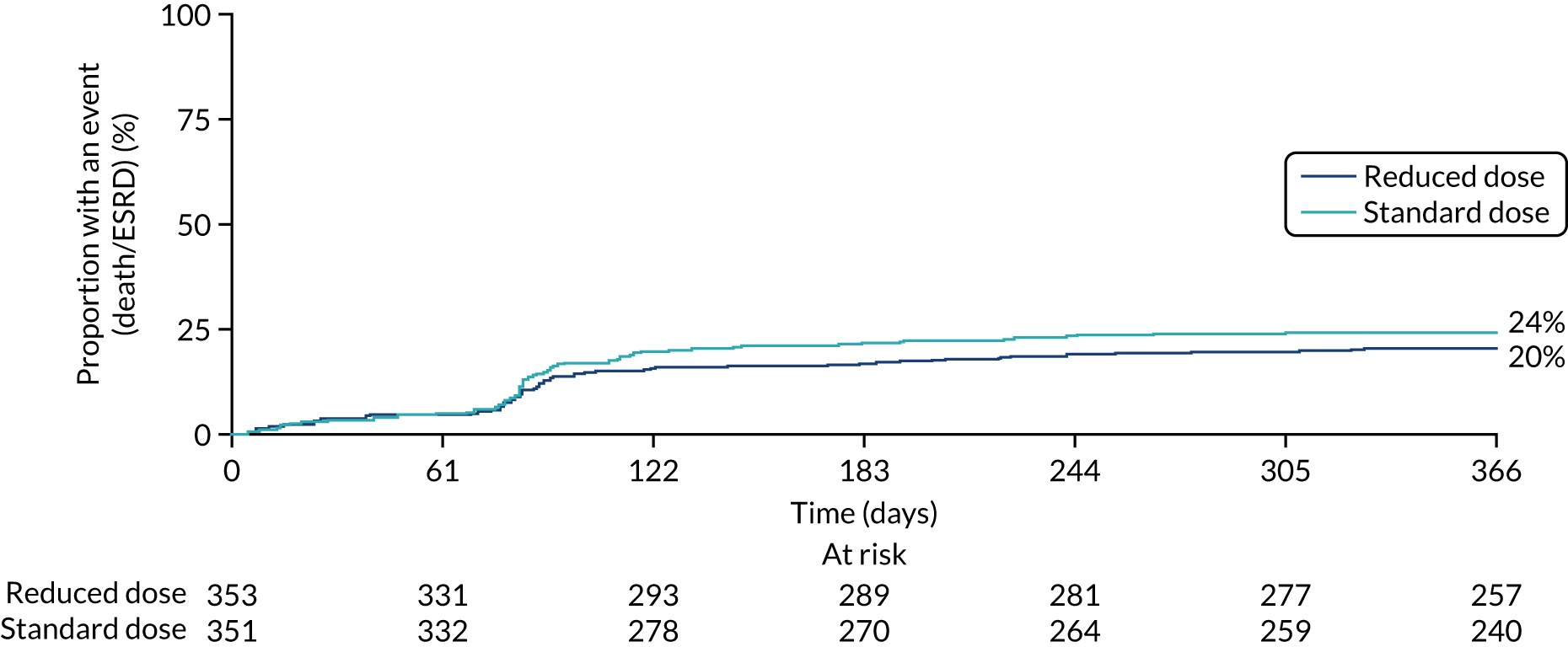
Overall, 107 patients in the reduced-dose GCs group and 102 patients in the standard-dose GCs group reached the primary end point. The comparison of reduced-dose GCs and standard-dose GCs was a non-inferiority hypothesis, with a non-inferiority margin of an 11% absolute risk increase, expressed as the reduced-dose GCs group relative to the standard-dose GCs group. This analysis was based on the GC per-protocol analysis population, with a binary outcome, using a binomial model. In the per-protocol analysis population, 92 of the 330 patients in the reduced-dose GCs group and 83 of 325 patients in the standard-dose GCs group reached the primary end point. The binomial model including both of the treatment group parameters and the minimisation variables did not converge, so a partially adjusted model was fitted that included the two treatment group parameters only. The absolute risk difference between the groups was 0.023 (90% CI –0.034 to –0.08; p = 0.5). This met our predefined non-inferiority criteria for the reduced-dose GC regimen. An analysis of time to death or ESRD using the ITT population was also undertaken (HR 1.00, 95% CI 0.76 to 1.31; p = 0.998) (Table 8 and Figure 11).
| ITT population | Reduced-dose GCs (N = 353) | Standard-dose GCs (N = 351) | HRa (95% CI) | p-value | Interaction p-value |
|---|---|---|---|---|---|
| Event, n (%) | |||||
| No | 246 (70) | 249 (71) | 1.00 (0.76 to 1.31) | 0.998 | 0.722 |
| Yes | 107 (30) | 102 (29) | |||
| Per-protocol population | Reduced-dose GCs (N = 330) | Standard-dose GCs (N = 325) | Risk differenceb (90% CI) | p-value | Interaction p-value |
| Event, n (%) | |||||
| No | 238 (72) | 242 (74) | 0.023 (–0.034 to 0.08) | 0.507 | |
| Yes | 92 (28) | 83 (26) | |||
FIGURE 11.
Kaplan–Meier plot for reduced-dose GC vs. standard-dose GC (primary outcome: ITT).
Secondary outcomes
All secondary outcomes were analysed as per the ITT principle.
Sustained remission
There was no difference in the proportion of patients who achieved sustained remission between the PLEX group and the no-PLEX group, or between the reduced-dose GC group and the standard-dose GC group (Table 9).
| Sustained remission, n (%) | PLEX (N = 352) | No PLEX (N = 352) | RRa (95% CI) | p-value |
|---|---|---|---|---|
| No | 152 (43) | 155 (44) | 1.01 (0.89 to 1.15) | 0.887 |
| Yes | 200 (57) | 197 (56) | ||
| Sustained remission, n (%) | Reduced-dose GCs (N = 353) | Standard-dose GCs (N = 351) | RRa (95% CI) | p-value |
| No | 149 (42) | 158 (45) | 1.04 (0.92 to 1.19) | 0.505 |
| Yes | 204 (58) | 193 (55) |
All-cause mortality
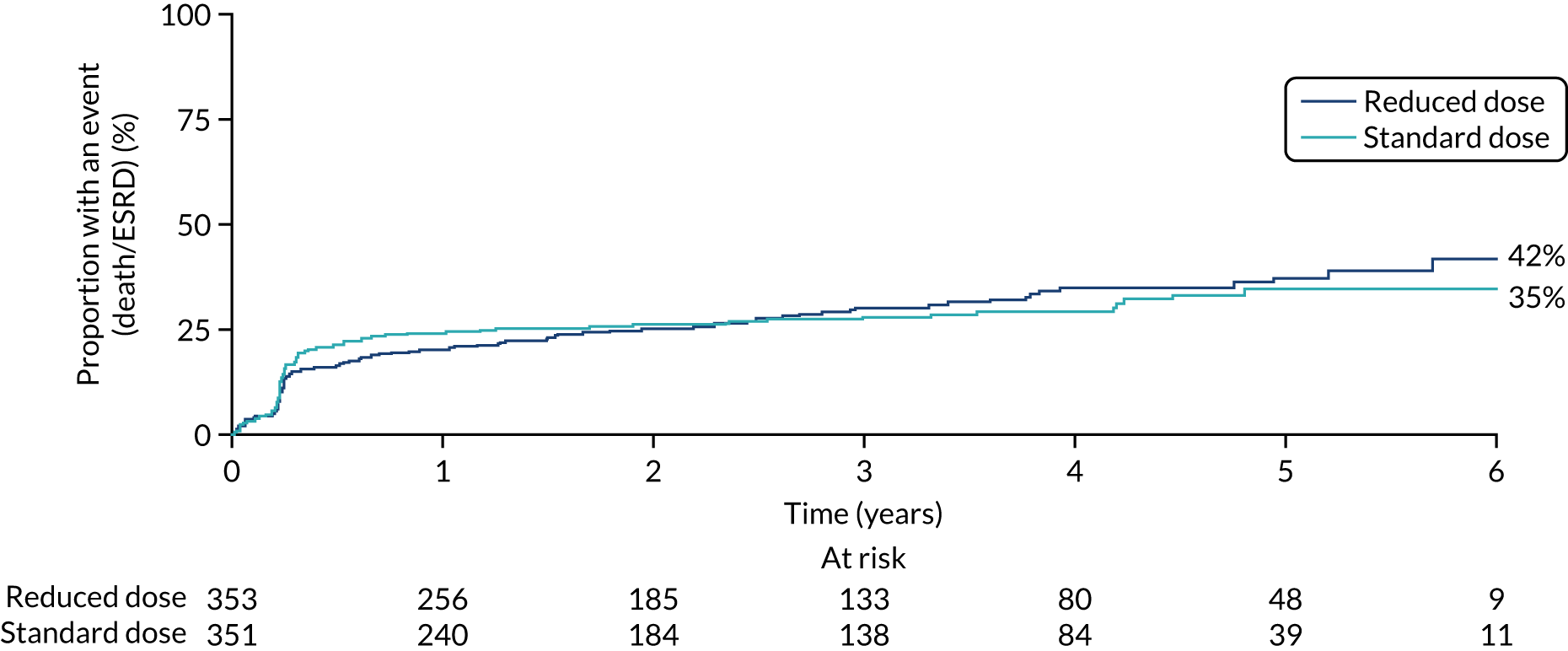
There was no difference in the mortality rate between the PLEX group and the no-PLEX group, or between the reduced-dose GCs group and the standard-dose GCs group (Table 10, and Figures 12 and 13). The reasons for death are shown in Table 11. The ‘other’ category was the most frequently used, representing 46 different causes; infection was the most common single cause, with 20 events. No comparisons of causes of death were made.
| Death | PLEX (N = 352), n (%) | No PLEX (N = 352), n (%) | HRa (95% CI) | p-value |
|---|---|---|---|---|
| No | 306 (87) | 299 (85) | 0.87 (0.58 to 1.29) | 0.482 |
| Yes | 46 (13) | 53 (15) | ||
| Death | Reduced-dose GCs (N = 353), n (%) | Standard-dose GCs (N = 351), n (%) | HRa (95% CI) | p-value |
| No | 307 (87) | 298 (85) | 0.78 (0.53 to 1.17) | 0.231 |
| Yes | 46 (13) | 53 (15) |
FIGURE 12.
Kaplan–Meier plot for PLEX vs. no PLEX (death).
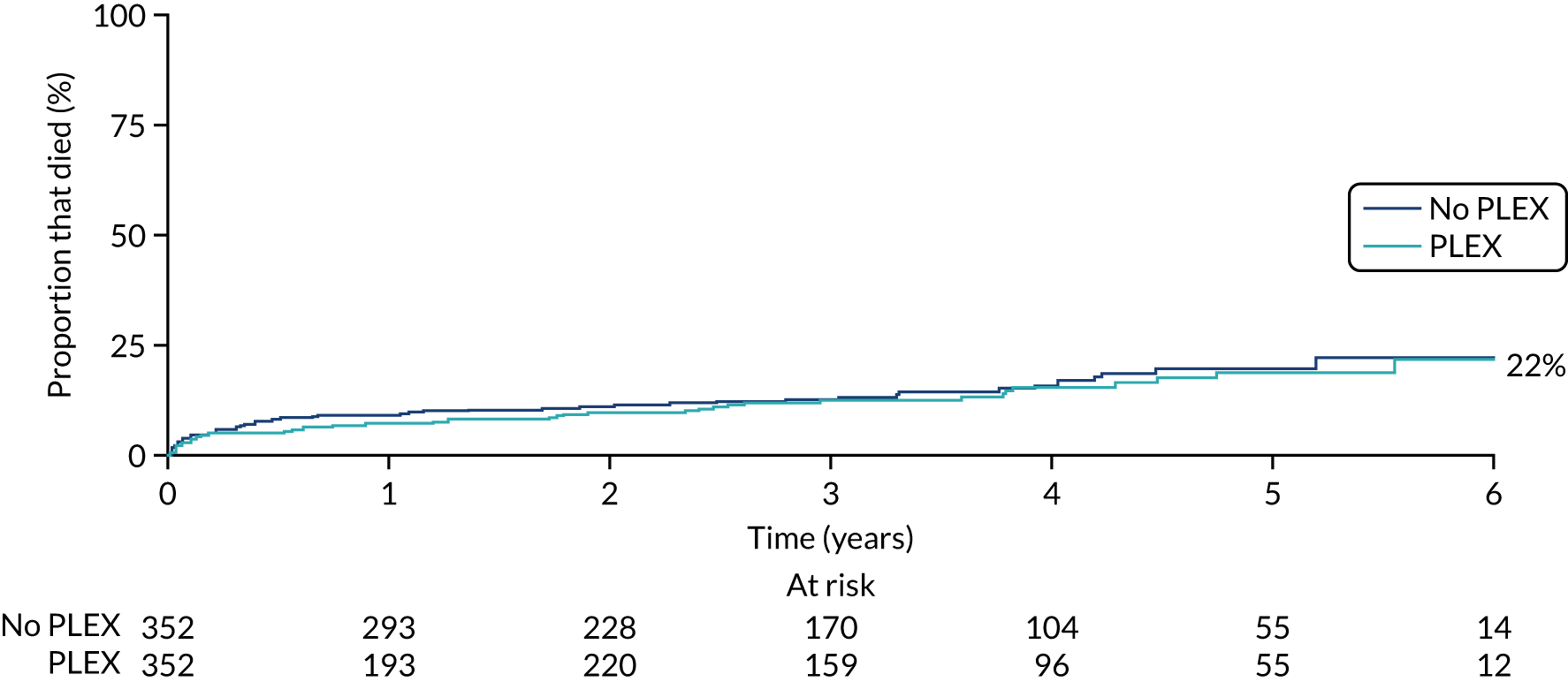
FIGURE 13.
Kaplan–Meier plot for reduced-dose GCs vs. standard-dose GCs (death).
| Reason for death | PLEX | No PLEX | Reduced-dose GCs | Standard-dose GCs | Total |
|---|---|---|---|---|---|
| Infection | 11 | 9 | 7 | 13 | 20 |
| Pulmonary haemorrhage | 0 | 3 | 2 | 1 | 3 |
| Pulmonary embolus | 1 | 2 | 2 | 1 | 3 |
| Withdrawal of renal replacement therapy | 1 | 6 | 4 | 3 | 7 |
| Acute myocardial infarction | 2 | 1 | 0 | 3 | 3 |
| Sudden death (cause unknown) | 3 | 4 | 4 | 3 | 7 |
| Cerebrovascular disease | 2 | 2 | 3 | 1 | 4 |
| Cancer | 3 | 3 | 2 | 4 | 6 |
| Other (46 causes identified) | 23 | 23 | 22 | 24 | 46 |
| Total | 46 | 53 | 46 | 53 | 99 |
End-stage renal disease
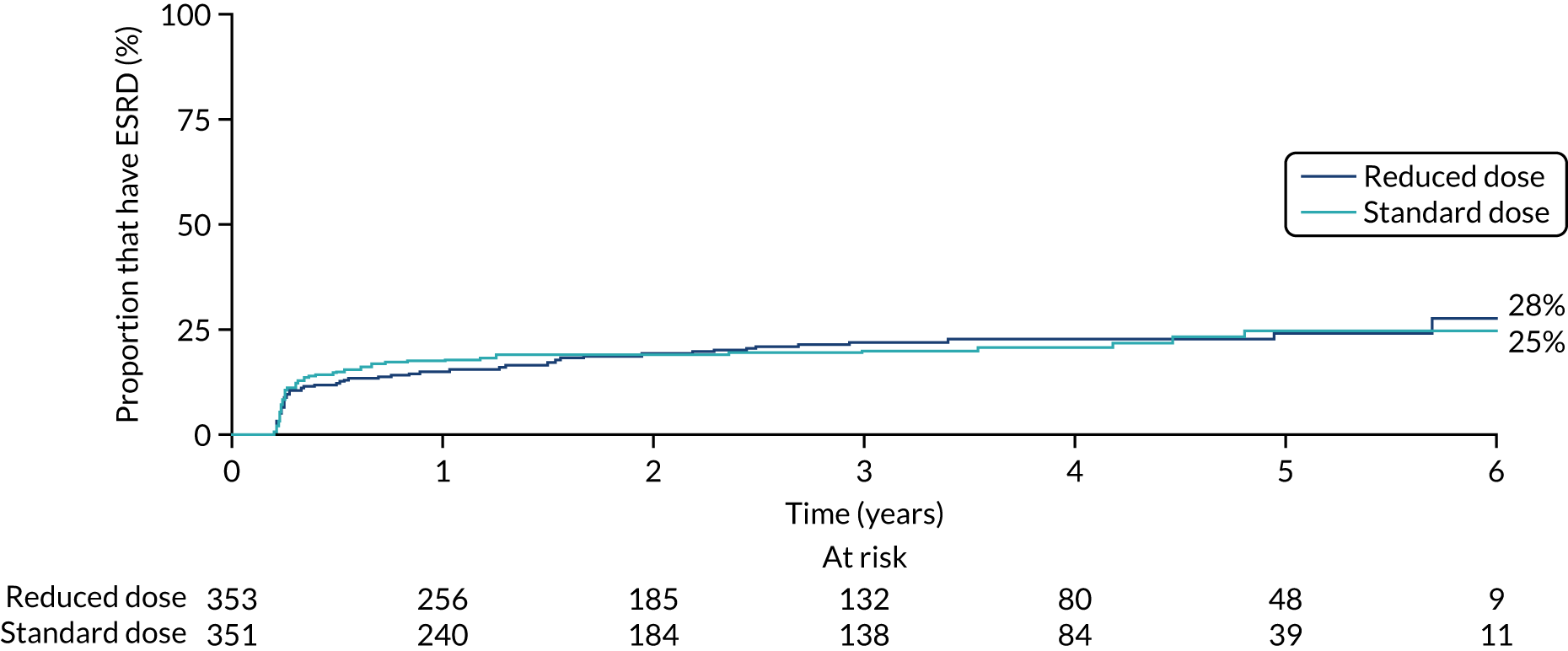
No differences were seen in ESRD between the PLEX group and the no-PLEX group or between the reduced-dose GCs group and the standard-dose GCs group (Table 12, and Figures 14 and 15).
| ESRD | PLEX (N = 352), n (%) | No PLEX (N = 352), n (%) | HRa (95% CI) | p-value |
|---|---|---|---|---|
| No | 285 (81) | 281 (80) | 0.81 (0.57 to 1.13) | 0.208 |
| Yes | 67 (19) | 71 (20) | ||
| ESRD | Reduced-dose GCs (N = 353), n (%) | Standard-dose GCs (N = 351), n (%) | HRa (95% CI) | p-value |
| No | 283 (80) | 283 (81) | 0.96 (0.68 to 1.34) | 0.795 |
| Yes | 70 (20) | 68 (19) |
FIGURE 14.
Kaplan–Meier plot for PLEX vs. no PLEX (ESRD).
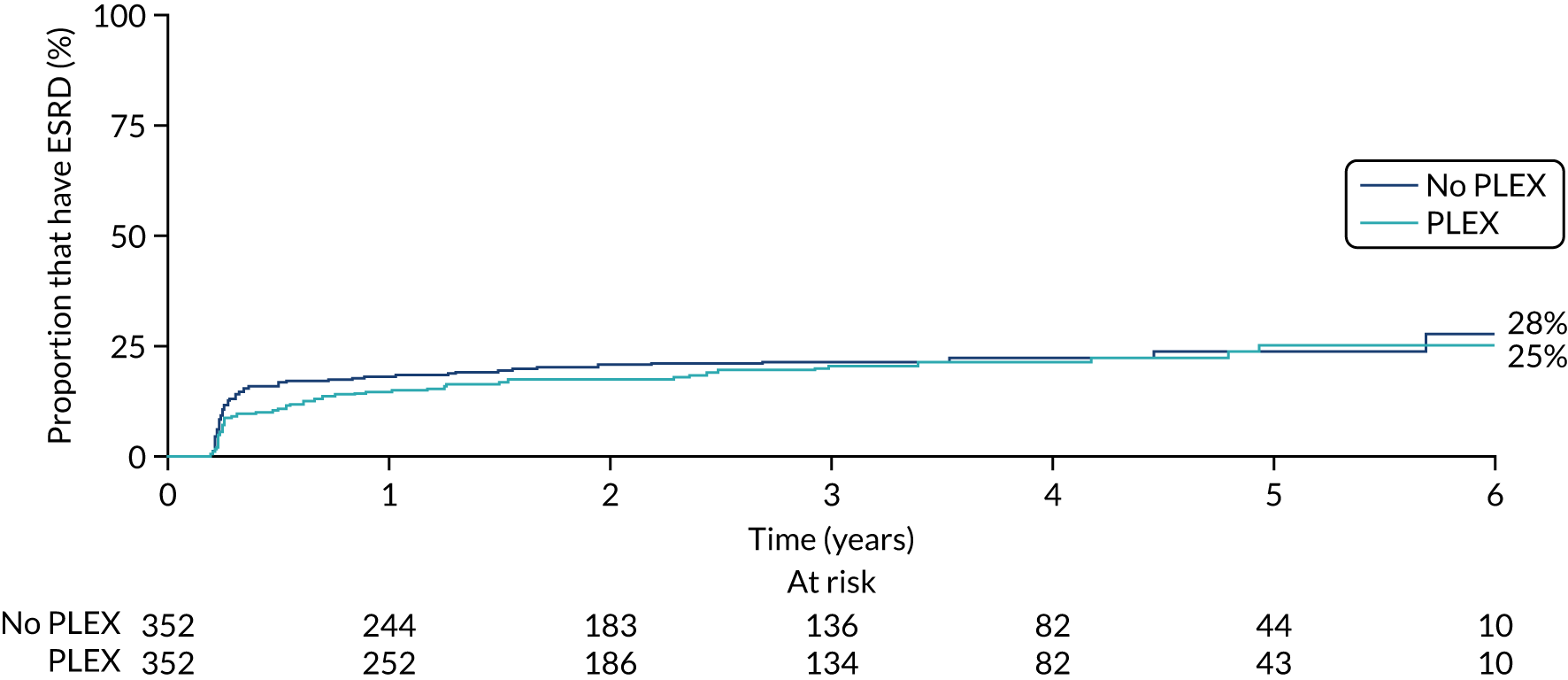
FIGURE 15.
Kaplan–Meier plot for reduced-dose GCs vs. standard-dose GCs (ESRD).
Serious adverse events
There were 1191 SAEs, reported in 449 of the 704 randomised patients, with infection being the most common SAE (423 infections in 250 patients), followed by cardiovascular events (176 events in 124 patients) (Tables 13 and 14). There was no statistically significant difference in the proportion of patients experiencing SAEs between the PLEX group (64%) and the no-PLEX group (64%) (Table 15), or between the reduced-dose GCs group (65%) and the standard-dose GCs dose group (62%) (Table 16).
| SAE category | PLEX (N = 352) | No PLEX (N = 352) | Total (N = 704) | |||
|---|---|---|---|---|---|---|
| Events (n) | Patients with event, n (%) | Events (n) | Patients with event, n (%) | Events (n) | Patients with event, n (%) | |
| Cardiovascular | 104 | 69 (20) | 72 | 55 (16) | 176 | 124 (18) |
| Endocrine | 10 | 9 (3) | 3 | 3 (1) | 13 | 12 (2) |
| Gastrointestinal | 45 | 34 (10) | 55 | 39 (11) | 100 | 73 (10) |
| Haematological | 28 | 25 (7) | 18 | 16 (5) | 46 | 41 (6) |
| Infection | 227 | 136 (39) | 196 | 114 (32) | 423 | 250 (36) |
| Renal | 50 | 41 (12) | 53 | 36 (10) | 103 | 77 (11) |
| Surgery | 19 | 16 (5) | 15 | 13 (4) | 34 | 29 (4) |
| Vasculitis relapse | 28 | 23 (7) | 33 | 32 (9) | 61 | 55 (8) |
| Other | 119 | 89 (25) | 116 | 79 (22) | 235 | 168 (24) |
| Total SAEs | 630 | 224 (64) | 561 | 225 (64) | 1191 | 449 (64) |
| SAE category | Reduced-dose GC (N = 353) | Standard-dose GC (N = 351) | Total (N = 704) | |||
|---|---|---|---|---|---|---|
| Events (n) | Patients with event, n (%) | Events (n) | Patients with event, n (%) | Events (n) | Patients with event, n (%) | |
| Cardiovascular | 104 | 68 (19) | 72 | 56 (16) | 176 | 124 (18) |
| Endocrine | 5 | 4 (1) | 8 | 8 (2) | 13 | 12 (2) |
| Gastrointestinal | 60 | 43 (12) | 40 | 30 (9) | 100 | 73 (10) |
| Haematological | 24 | 22 (6) | 22 | 19 (5) | 46 | 41 (6) |
| Infection | 203 | 119 (34) | 220 | 131 (37) | 423 | 250 (36) |
| Renal | 69 | 50 (14) | 34 | 27 (8) | 103 | 77 (11) |
| Surgery | 14 | 14 (4) | 20 | 15 (4) | 34 | 29 (4) |
| Vasculitis relapse | 36 | 32 (9) | 25 | 23 (7) | 61 | 55 (8) |
| Other | 130 | 91 (26) | 105 | 77 (22) | 235 | 168 (24) |
| Total SAEs | 645 | 231 (65) | 546 | 218 (62) | 1191 | 449 (64) |
| SAEs | PLEX (N = 352) | No PLEX (N = 352) | RRa (95% CI) | p-value |
|---|---|---|---|---|
| Patients with at least one SAE, n (%) | ||||
| No | 128 (36) | 127 (36) | 1.00 (0.90 to 1.12) | 0.991 |
| Yes | 224 (64) | 225 (64) | ||
| IRRb (95% CI) | ||||
| Total SAEs (n) | 630 | 561 | 1.21 (0.96 to 1.52) | 0.104 |
| SAEs | Reduced-dose GCs (N = 353) | Standard-dose GCs (N = 351) | RRa (95% CI) | p-value |
|---|---|---|---|---|
| Patients with at least one SAE, n (%) | ||||
| No | 122 (35) | 133 (38) | 1.05 (0.94 to 1.17) | 0.354 |
| Yes | 231 (65) | 218 (62) | ||
| IRRb (95% CI) | ||||
| Total SAEs (n) | 645 | 546 | 0.95 (0.75 to 1.20) | 0.665 |
Serious infections
The rate of serious infections was assessed both for the first year of the trial and at trial end. Because the difference in treatment in the PLEX intervention was in the first 2 weeks and the difference in regimen in the GC intervention was in the first 6 months, it was expected that any difference in the outcome measure would be larger in the first year than across the duration of the trial. At 1 year, the number of serious infections was larger in the PLEX group (173 serious infections reported in 119 patients) than in the no-PLEX group (149 serious infections reported in 93 patients), but this difference was not statistically significant (IRR 1.16, 95% CI 0.87 to 1.56; p = 0.317). There were fewer serious infections in the reduced-dose GCs group (142 serious infections reported in 96 patients) than in the standard-dose GCs group (180 serious infections reported in 116 patients), and there was a statistically significant difference between the GC groups (IRR 0.69, 95% CI 0.52 to 0.93; p = 0.016).
At trial end, there remained no statistically significant difference in the rate of serious infections between the PLEX group and the no-PLEX group (IRR 1.16, 95% CI 0.87 to 1.55; p = 0.323) (Table 17), and the difference in the rate of serious infections in the reduced-dose group and the standard-dose GCs group at trial end was borderline insignificant (IRR 0.76, 95% CI 0.57 to 1.01; p = 0.058) (Table 18).
| Serious infections | PLEX (N = 352) | No PLEX (N = 352) | IRRa (95% CI) | p-value |
|---|---|---|---|---|
| Patients with at least one serious infection at 1 year, n (%) | ||||
| No | 233 (66) | 259 (74) | – | – |
| Yes | 119 (34) | 93 (26) | – | – |
| Total serious infections at 1 year (n) | 173 | 149 | 1.16 (0.87 to 1.56) | 0.317 |
| Patients with at least one serious infection at trial end, n (%) | ||||
| No | 203 (58) | 221 (63) | – | – |
| Yes | 149 (42) | 131 (37) | – | – |
| Total serious infections at trial end (n) | 268 | 245 | 1.16 (0.87 to 1.55) | 0.323 |
| Serious infections | Reduced-dose GCs (N = 353) | Standard-dose GCs (N = 351) | IRRa (95% CI) | p-value |
|---|---|---|---|---|
| Patients with at least one serious infection at 1 year, n (%) | ||||
| No | 257 (73) | 235 (67) | – | – |
| Yes | 96 (27) | 116 (33) | – | – |
| Total serious infections at 1 year (n) | 142 | 180 | 0.69 (0.52 to 0.93) | 0.016 |
| Patients with at least one serious infection at trial end, n (%) | ||||
| No | 219 (62) | 205 (58) | – | – |
| Yes | 134 (38) | 146 (42) | – | – |
| Total serious infections at trial end (n) | 250 | 263 | 0.76 (0.57 to 1.01) | 0.058 |
Quality of life
SF-36
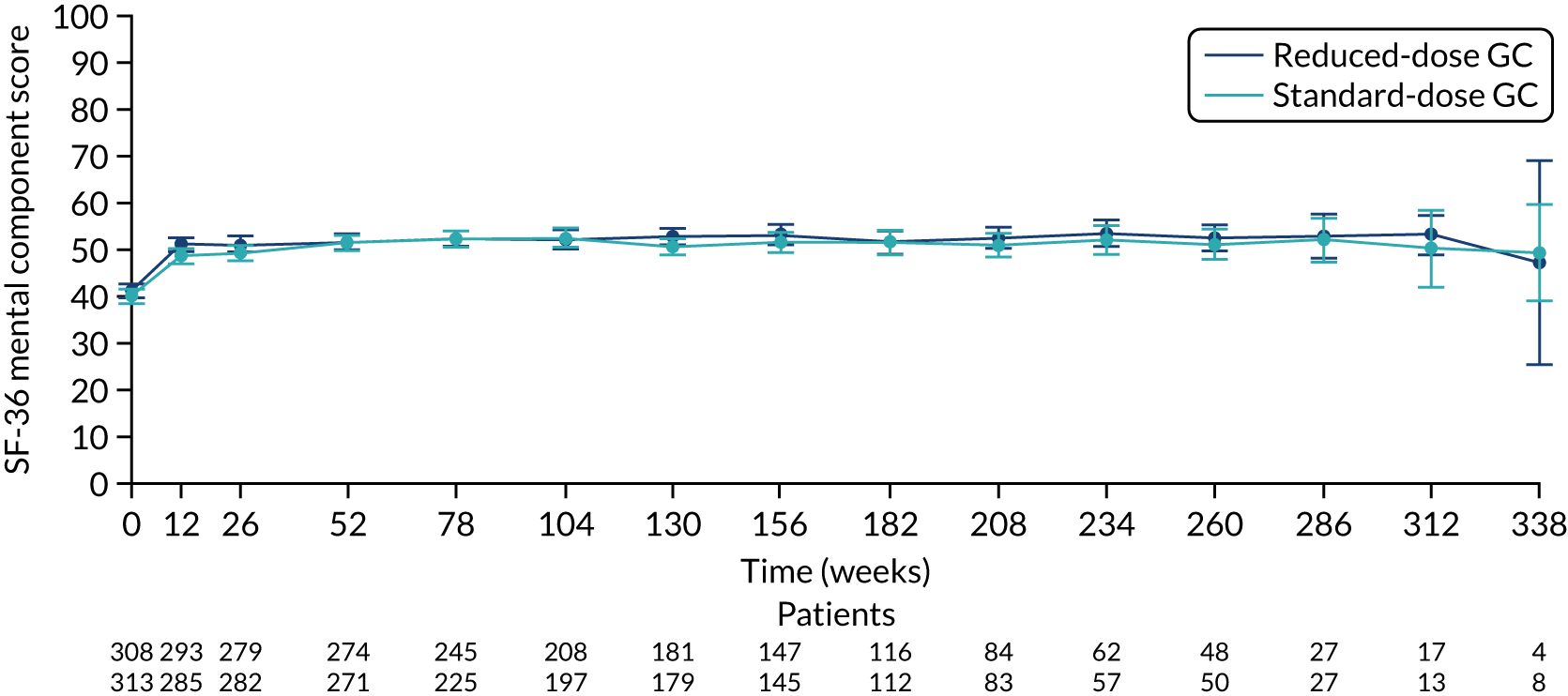
There was no difference between the PLEX group and the no-PLEX group, or between the reduced-dose GC group and the standard-dose GC group for the mental component score, or between the PLEX group and the no-PLEX group for the physical component score. For the reduced-dose GCs vs. standard-dose GCs comparison, there was evidence of a treatment-by-time interaction for the physical component score (p < 0.001). The SF-36 physical component scores increased over time, and the slopes diverged at a rate of –0.015 points per week (95% CI –0.023 to –0.008; p < 0.001) (Table 19 and Figures 16–19).
| Analysis | Mean differencea (95% CI) | p-value |
|---|---|---|
| PLEX vs. no PLEX | ||
| SF-36 physical component scoreb | 1.073 (–0.458 to 2.605) | 0.169 |
| SF-36 mental component scoreb | 0.545 (–0.665 to 1.755) | 0.377 |
| Reduced-dose GCs vs. standard-dose GCs | ||
| SF-36 physical component score | ||
| Reduced-dose GCs vs. standard-dose GCs | 2.087 (0.442 to 3.732) | 0.013 |
| Reduced-dose GCs × time (weeks) | –0.015 (–0.023 to –0.008) | < 0.001 |
| Time (weeks) | 0.007 (0.002 to 0.0126) | 0.004 |
| SF-36 mental component score | ||
| Reduced-dose GCs vs. standard-dose GCsb | 0.971 (–0.240 to 2.182) | 0.116 |
FIGURE 16.
The SF-36 physical component scores for PLEX vs. no PLEX. SF-36 score ranges from 0 to 100, with lower scores indicating greater disability. Values are means and 95% CIs.
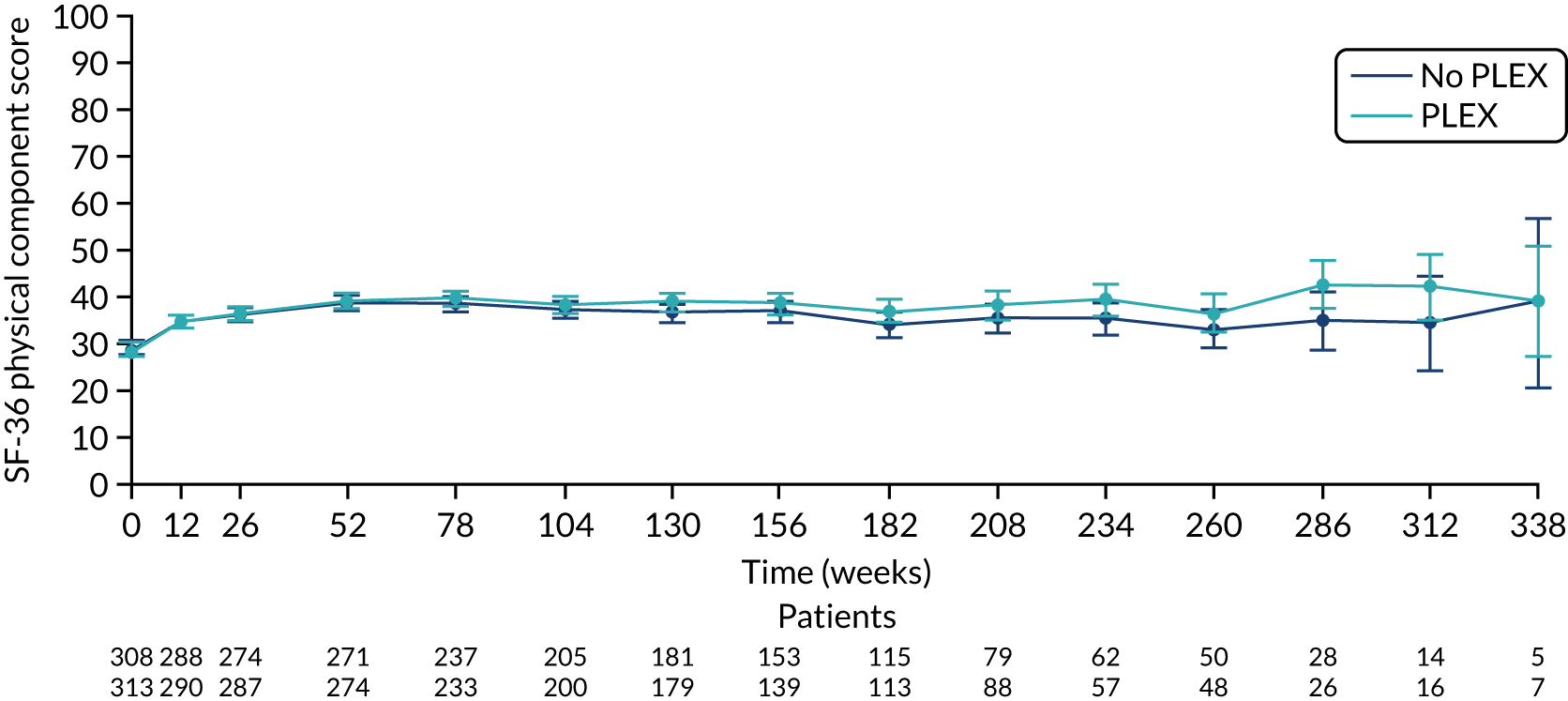
FIGURE 17.
The SF-36 mental component scores for PLEX vs. no PLEX. SF-36 score ranges from 0 to 100, with lower scores indicating greater disability. Values are means and 95% CIs.
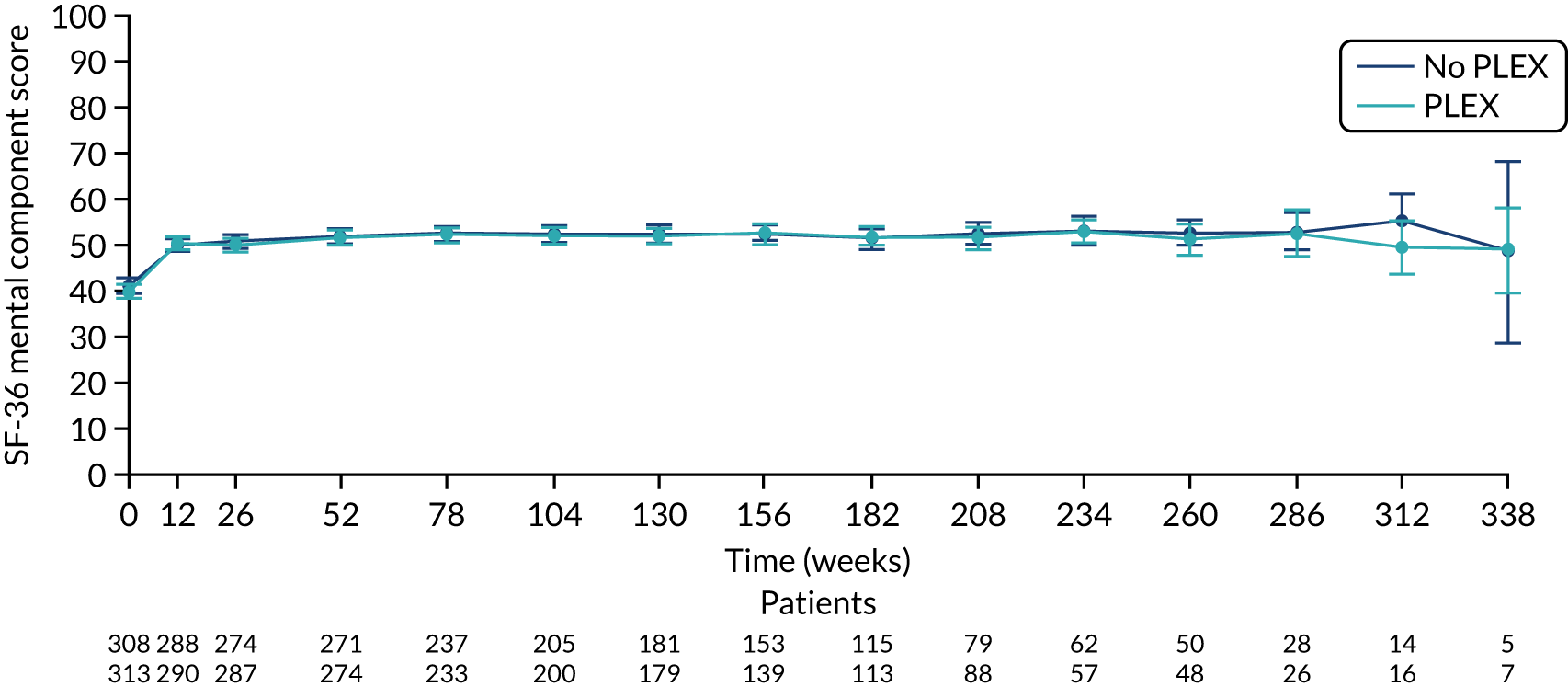
FIGURE 18.
The SF-36 physical component score for reduced-dose GCs vs. standard-dose GCs. SF-36 score ranges from 0 to 100, with lower scores indicating greater disability. Values are means and 95% CIs.
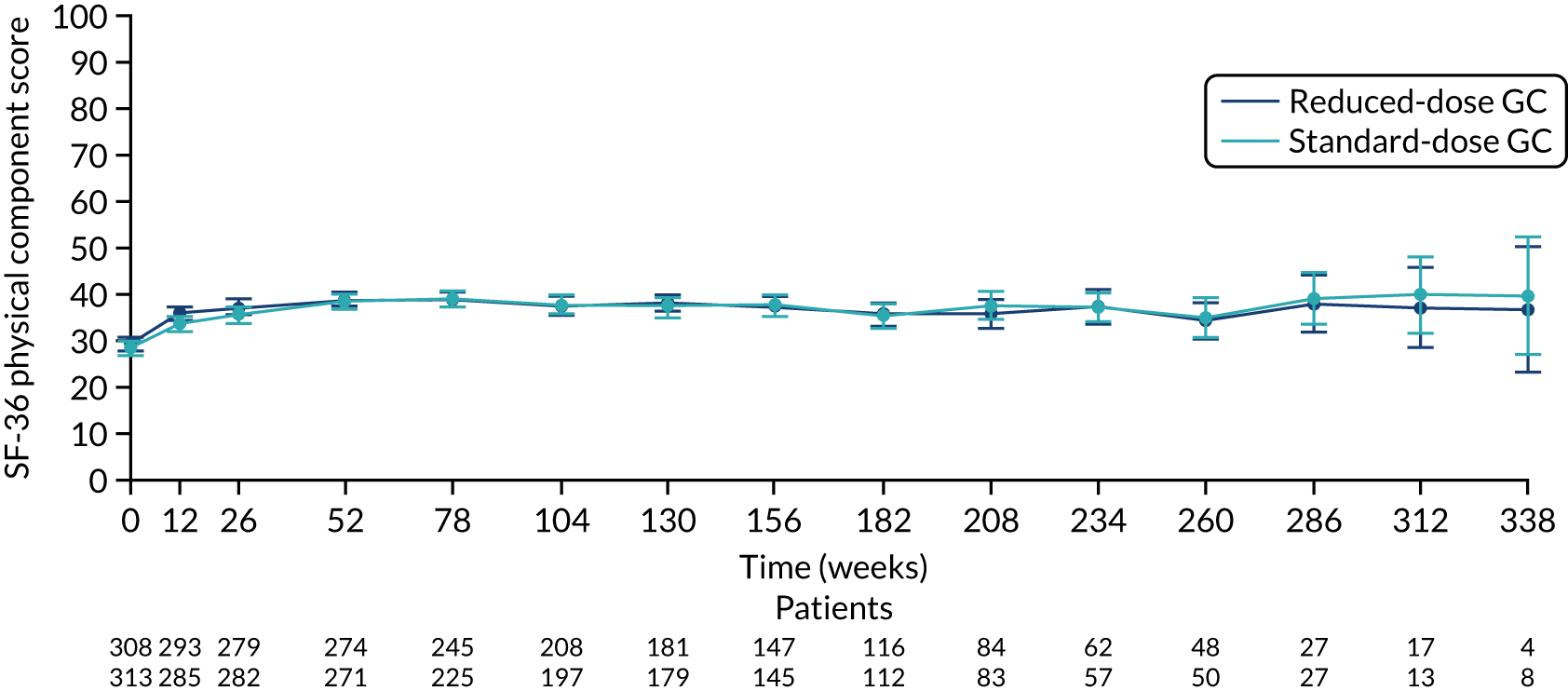
FIGURE 19.
The SF-36 mental component score for reduced-dose GCs vs. standard-dose GCs. SF-36 score ranges from 0 to 100, with lower scores indicating greater disability. Values are means and 95% CIs.
EQ-5D
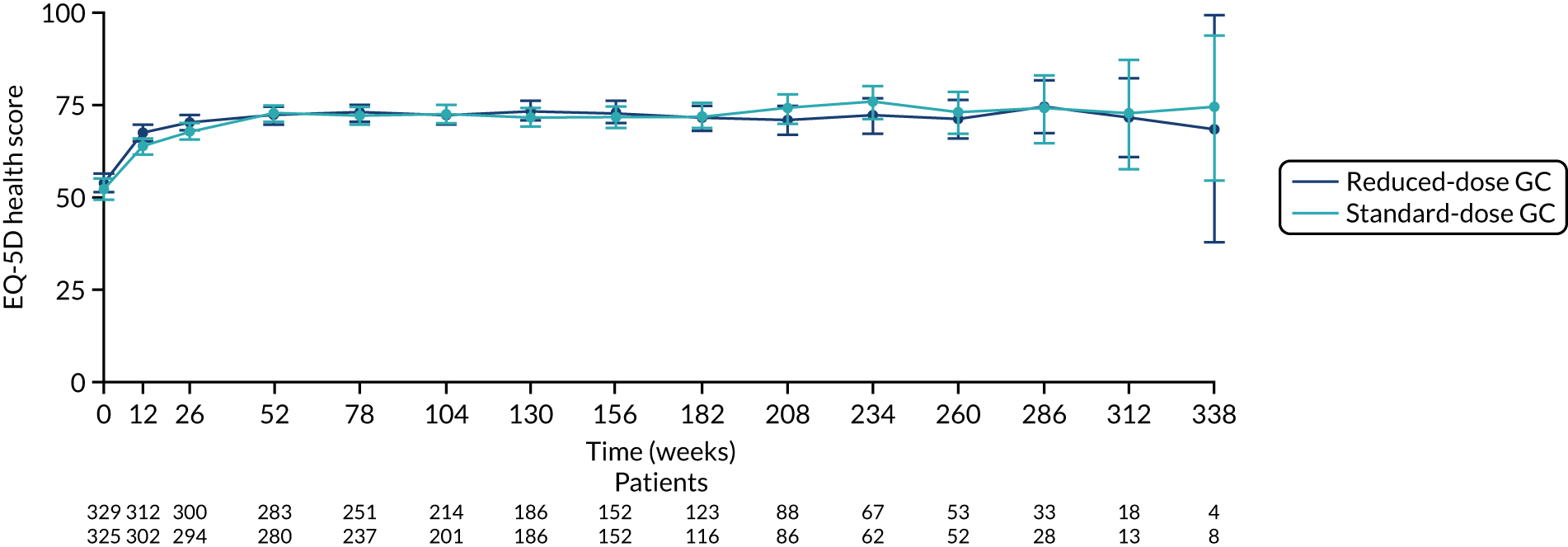
There was no difference between the PLEX group and the no-PLEX group for either the EQ-5D index score or the EQ-5D health score. However, for the reduced-dose GCs versus standard-dose GCs comparison, there was some evidence of a treatment-by-time interaction for both the EQ-5D index score and health score. For the EQ-5D index score, the scores decreased over time and the slopes diverged at a rate of –0.0002 points per week (95% CI –0.0003 to –0.00003; p = 0.021). The EQ-5D health score increased over time and the slopes diverged at a rate of –0.019 points per week (95% CI –0.031 to –0.007; p = 0.002) (Table 20 and Figures 20–23).
| Analysis | Mean differencea (95% CI) | p-value |
|---|---|---|
| PLEX vs. no PLEX | ||
| EQ-5D index scoreb | 0.0219 (–0.007 to 0.051) | 0.14 |
| EQ-5D health scoreb | 1.035 (–1.087 to 3.157) | 0.339 |
| Reduced-dose GCs vs. standard-dose GCs | ||
| EQ-5D index score | ||
| Reduced-dose GCs vs. standard-dose GCs | 0.029 (–0.003 to 0.06) | 0.075 |
| Reduced-dose GCs × time (weeks) | –0.0002 (–0.0003 to –0.00003) | 0.021 |
| Time (weeks) | –000001 (–0.00011 to 0.00011) | 0.988 |
| EQ-5D health score | ||
| Reduced-dose GCs vs. standard-dose GCs | 1.712 (–0.636 to 4.061) | 0.153 |
| Reduced-dose GCs × time (weeks) | –0.019 (–0.031 to –0.007) | 0.002 |
| Time (weeks) | 0.0255 (0.0168 to 0.0341) | < 0.001 |
FIGURE 20.
The EQ-5D index score for PLEX vs. no PLEX. EQ-5D index score ranges from –0.59 to 1, with a score of 1 implying perfect health. Values are means and 95% CIs.
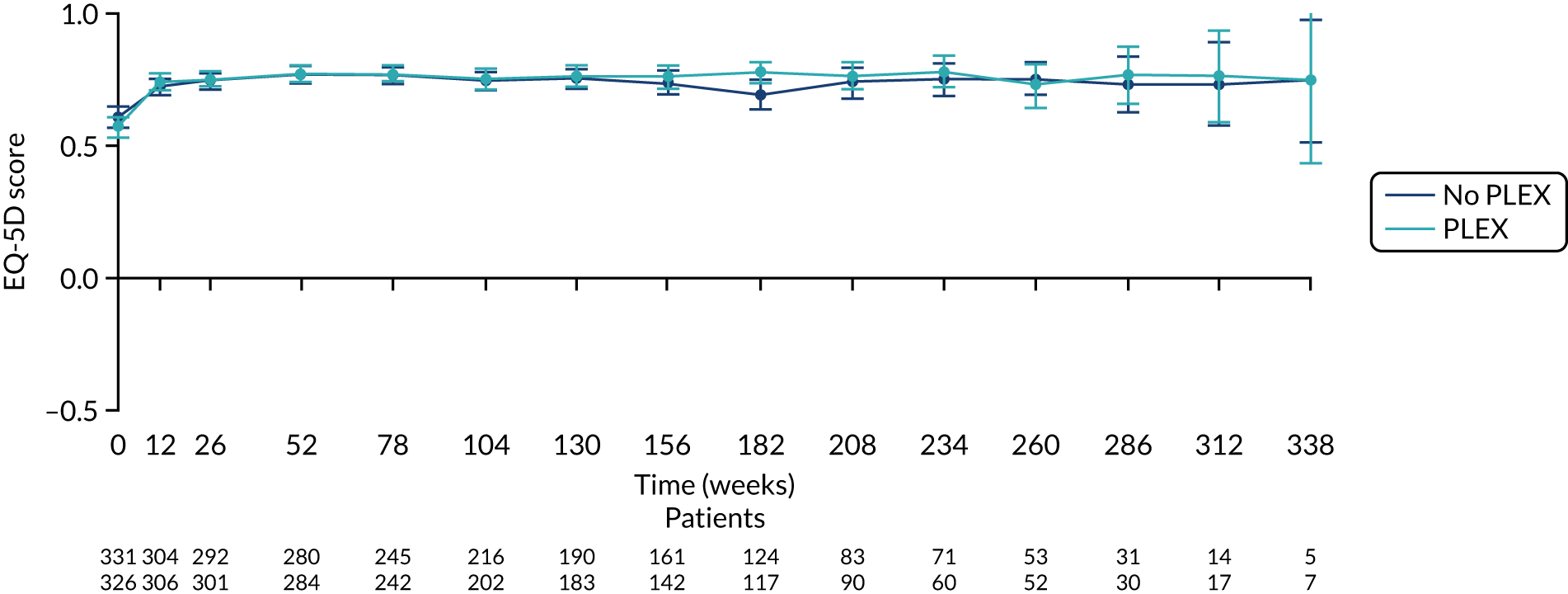
FIGURE 21.
The EQ-5D health score for PLEX vs. no PLEX. EQ-5D health score ranges from 0 to 100, with higher scores indicating better health. Values are means and 95% CIs.
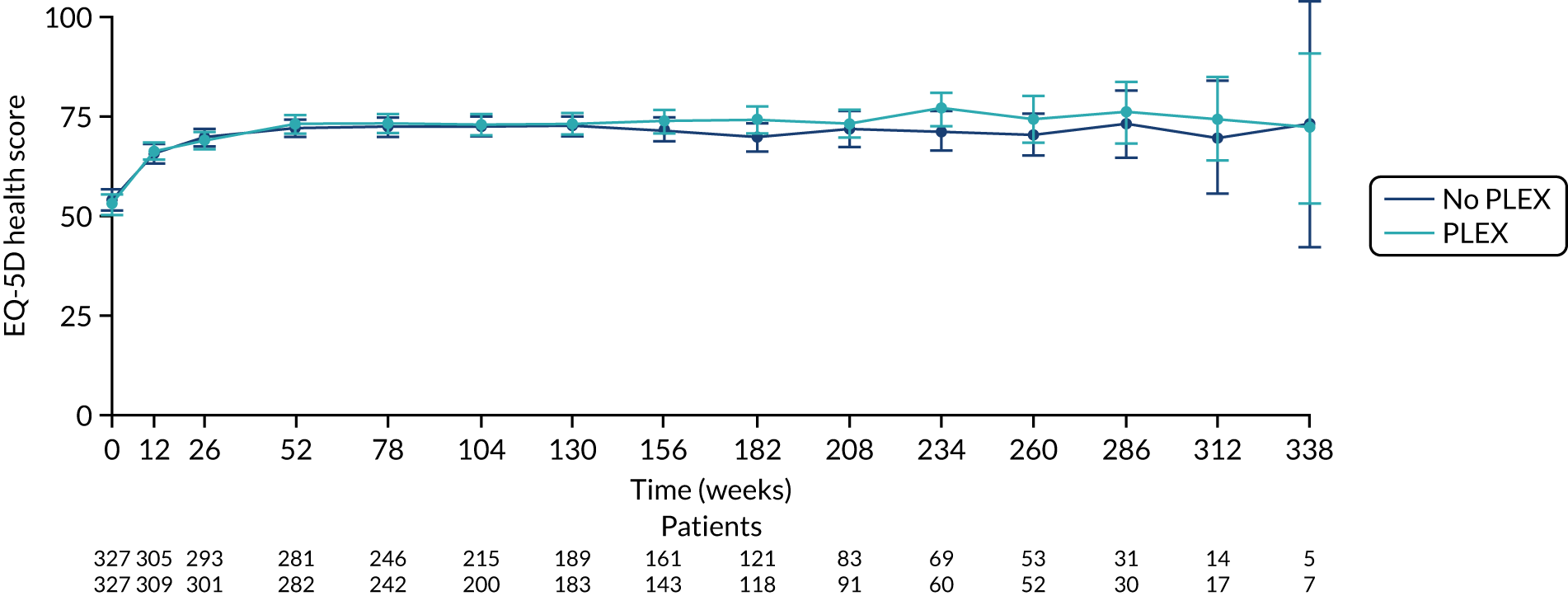
FIGURE 22.
The EQ-5D index score for reduced-dose GCs vs. standard-dose GCs. EQ-5D index score ranges from –0.59 to 1, with a score of 1 implying perfect health. Values are means and 95% CIs.
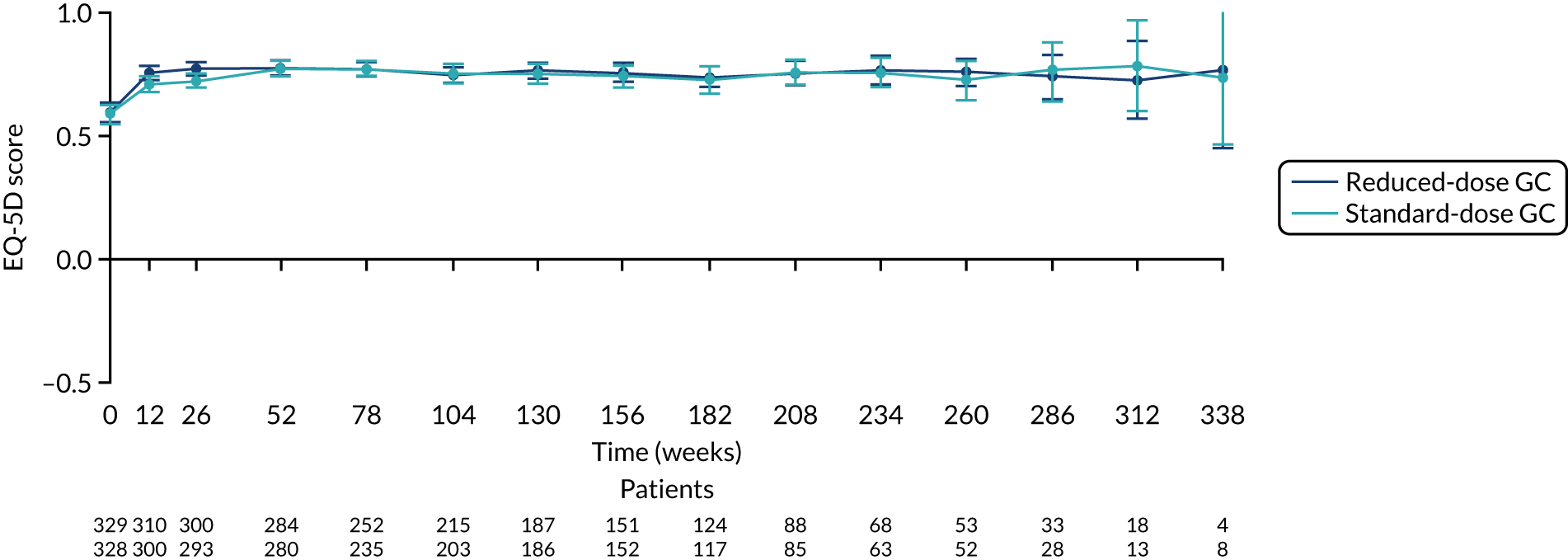
FIGURE 23.
The EQ-5D health score for reduced-dose GCs vs. standard-dose GCs. EQ-5D health score ranges from 0 to 100, with higher scores indicating better health. Values are means and 95% CIs.
Subgroup analysis
Several a priori subgroup analyses were planned with respect to the primary outcome (i.e. time to death or ESRD) using the ITT analysis populations for both the PLEX vs. no-PLEX comparison and the reduced-dose GCs vs. standard-dose GCs comparison. There was no evidence that the treatment effect differed across any of the subgroups (Table 21).
| Subgroup analysis | Total events (n) | PLEX vs. no PLEX | Total events (n) | Reduced-dose GCs vs. standard-dose GCs | ||||
|---|---|---|---|---|---|---|---|---|
| PLEX (N = 100) | No PLEX (N = 109) | HRa (95% CI) | Interaction p-value | Reduced-dose (N = 107) | Standard-dose (N = 102) | HRa (95% CI) | Interaction p-valueb | |
| Age (years), n/N | ||||||||
| < 60 | 35/114 | 28/116 | 1.20 (0.73 to 1.97) | 0.126 | 31/116 | 32/114 | 0.86 (0.52 to 1.41) | 0.488 |
| ≥ 60 | 65/238 | 81/236 | 0.75 (0.54 to 1.04) | 76/237 | 70/237 | 1.06 (0.76 to 1.48) | ||
| Severity of renal disease at presentation, n/N | ||||||||
| Creatinine level of < 500 µmol/l | 45/251 | 46/248 | 0.98 (0.65 to 1.48) | 0.384 | 50/251 | 41/248 | 1.24 (0.82 to 1.88) | 0.171 |
| Requiring dialysis or a creatinine level of ≥ 500 µmol/l | 55/101 | 63/104 | 0.77 (0.53 to 1.11) | 57/102 | 61/103 | 0.85 (0.59 to 1.22) | ||
| ANCA, n/N | ||||||||
| PR3-ANCA | 30/143 | 35/143 | 0.84 (0.51 to 1.36) | 0.907 | 31/143 | 34/143 | 0.82 (0.50 to 1.34) | 0.334 |
| MPO-ANCA | 70/209 | 74/209 | 0.87 (0.62 to 1.21) | 76/210 | 68/208 | 1.10 (0.79 to 1.53) | ||
| Severity of lung haemorrhage, n/N | ||||||||
| No haemorrhage | 77/257 | 75/256 | 0.95 (0.69 to 1.31) | 0.486 | 75/257 | 77/256 | 0.94 (0.68 to 1.29) | 0.736 |
| Haemorrhage, blood O2 saturation > 85% on room air | 15/64 | 21/66 | 0.64 (0.33 to 1.24) | 20/65 | 16/65 | 1.16 (0.60 to 2.26) | ||
| Haemorrhage, blood O2 saturation ≤ 85% on room air or ventilated | 8/31 | 13/30 | 0.67 (0.28 to 1.64) | 12/31 | 9/30 | 1.25 (0.52 to 3.03) | ||
| Induction immunosuppression therapy to be used, n/N | ||||||||
| i.v. CYC | 56/177 | 59/177 | 0.79 (0.55 to 1.14) | 0.792 | 58/179 | 57/175 | 0.84 (0.58 to 1.21) | 0.199 |
| Oral CYC | 34/120 | 35/121 | 0.98 (0.61 to 1.57) | 35/120 | 34/121 | 1.08 (0.67 to 1.73) | ||
| Rituximab | 10/55 | 15/54 | 0.87 (0.38 to 1.96) | 14/54 | 11/55 | 1.86 (0.83 to 4.14) | ||
Exploratory analyses
Primary end point at 1 year
A prespecified exploratory analysis was undertaken for the primary outcome (i.e. death or ESRD) based on the ITT analysis population, with the data censored at 1 year. In this analysis, all patients were censored at 365 days even if they had follow-up beyond 1 year, and any events (i.e. death or ESRD) that occurred after 1 year were not included (Table 22, and Figures 24 and 25). Censoring the data at 1 year reduced the HR for PLEX versus no PLEX from 0.86 to 0.77, and reduced the HR for reduced-dose GCs versus standard-dose GCs from 1.00 to 0.80, with neither result reaching statistical significance.
| Events (ESRD and/or death) | PLEX (N = 352) | No PLEX (N = 352) | HRa (95% CI) |
|---|---|---|---|
| Event, n (%) | |||
| No | 282 (80) | 267 (76) | 0.77 (0.56 to 1.06) |
| Yes | 70 (20) | 85 (24) | |
| Events (ESRD and/or death) | Reduced-dose GCs (N = 353) | Standard-dose GCs (N = 351) | HRa (95% CI) |
| Event, n (%) | |||
| No | 282 (80) | 267 (76) | 0.80 (0.58 to 1.10) |
| Yes | 71 (20) | 84 (24) | |
FIGURE 24.
Kaplan–Meier plot for PLEX vs. no PLEX (primary outcome censoring at 1 year).
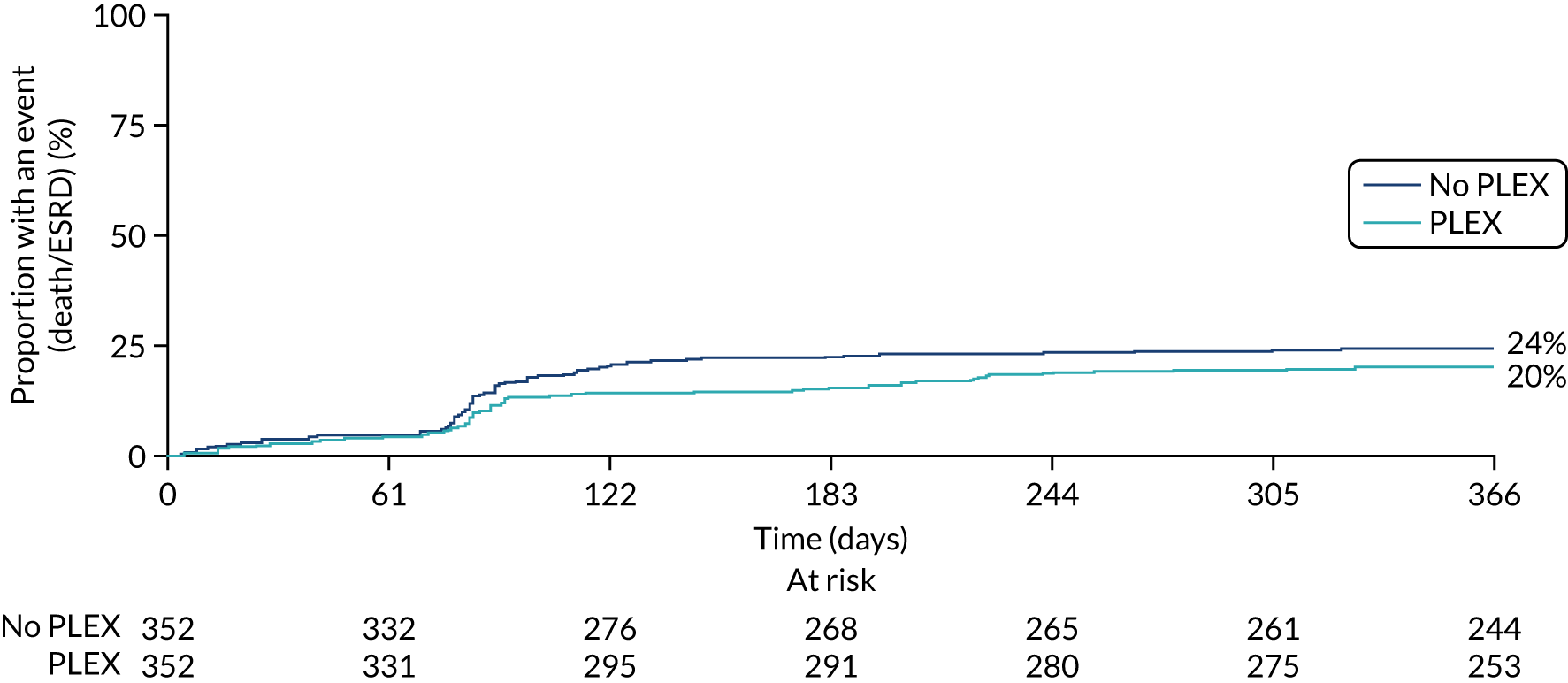
FIGURE 25.
Kaplan–Meier plot for reduced-dose GC vs. standard-dose GC (primary outcome censoring at 1 year: ITT).
Change in anti-neutrophil cytoplasm antibody levels
Another exploratory analysis investigated changes in ANCA levels and the impact of randomisation. Because PLEX is designed to reduce antibody levels, it was expected that more patients in the PLEX group would become ANCA negative than in the no-PLEX group. A maximal difference was expected after the PLEX course at 2 weeks. Numerically, more patients with PR3- or MPO-ANCA became ANCA negative at 2 and 4 weeks in the PLEX group than in the no-PLEX group (at 2 weeks: PR3-ANCA, 40% vs. 32% and MPO-ANCA, 34% vs. 22%; at 4 weeks: PR3-ANCA, 39% vs. 33% and MPO-ANCA 31% vs. 24%, respectively). There was little or no difference between the two GC groups. No comparative statistics were performed (Tables 23–26).
| Anti-PR3 at 2 weeks | PLEX (N = 340), n (%) | No PLEX (N = 341), n (%) | Reduced-dose GCs (N = 341), n (%) | Standard-dose GCs (N = 340), n (%) |
|---|---|---|---|---|
| Abnormally positive | 59 (17) | 80 (24) | 79 (23) | 60 (18) |
| Negative | 135 (40) | 110 (32) | 123 (36) | 122 (36) |
| Not measured | 145 (43) | 149 (44) | 138 (41) | 156 (46) |
| Missing | 1 (< 1) | 2 (< 1) | 1 (< 1) | 2 (< 1) |
| Anti-MPO at 2 weeks | PLEX (N = 340), n (%) | No PLEX (N = 341), n (%) | Reduced-dose GCs (N = 341), n (%) | Standard-dose GCs (N = 340), n (%) |
|---|---|---|---|---|
| Abnormally positive | 81 (24) | 106 (31) | 96 (28) | 91 (27) |
| Negative | 115 (34) | 74 (22) | 103 (30) | 86 (25) |
| Not measured | 142 (42) | 160 (47) | 141 (42) | 161 (48) |
| Missing | 2 (< 1) | 1 (< 1) | 1 (< 1) | 2 (< 1) |
| Anti-PR3 at 4 weeks | PLEX (N = 333), n (%) | No PLEX (N = 334), n (%) | Reduced-dose GCs (N = 334), n (%) | Standard-dose GCs (N = 333), n (%) |
|---|---|---|---|---|
| Abnormally positive | 50 (15) | 71 (21) | 69 (21) | 52 (15) |
| Negative | 129 (39) | 111 (33) | 111 (33) | 129 (39) |
| Not measured | 154 (46) | 152 (46) | 154 (46) | 152 (46) |
| Missing | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Anti-MPO at 4 weeks | PLEX (N = 333), n (%) | No PLEX (N = 334), n (%) | Reduced-dose GCs (N = 334), n (%) | Standard-dose GCs (N = 333), n (%) |
|---|---|---|---|---|
| Abnormally positive | 78 (23) | 94 (28) | 84 (25) | 88 (26) |
| Negative | 102 (31) | 81 (24) | 93 (28) | 90 (27) |
| Not measured | 152 (46) | 159 (48) | 156 (47) | 155 (47) |
| Missing | 1 (< 1) | 0 (0) | 1 (< 1) | 0 (0) |
Lung haemorrhage and mortality
Lung haemorrhage is the major cause of early death in AAV and was an inclusion criterion for PEXIVAS, being present in 191 patients. Because death due to lung haemorrhage occurs early, a post hoc analysis of death for this subgroup was performed for the PLEX and GC randomisations. Twelve patients with lung haemorrhage at randomisation died in the PLEX group, compared with 20 patients in the no-PLEX group (no lung haemorrhage, HR 1.07, 95% CI 0.66 to 1.73; lung haemorrhage without hypoxia, HR 0.65, 95% CI 0.23 to 1.86; and lung haemorrhage with hypoxia, HR 0.48, 95% CI 0.18 to 1.32). There was no evidence that the treatment effect differed according to severity of lung haemorrhage at trial entry (p = 0.322) (Table 27). There were no significant differences between reduced and standard GC dosing for this end point.
| Subgroup analysis for death | Total deaths, n/N | PLEX vs. no PLEX | Total events, n/N | Reduced-dose GCs vs. standard-dose GCs | ||||
|---|---|---|---|---|---|---|---|---|
| PLEX (N = 46) | No PLEX (N = 53) | HRa (95% CI) | Interaction p-valueb | Reduced dose (N = 46) | Standard dose (N = 53) | HRa (95% CI) | Interaction p-valueb | |
| Severity of lung haemorrhage | ||||||||
| No haemorrhage | 34/257 | 33/256 | 1.07 (0.66 to 1.73) | 0.322 | 29/257 | 38/256 | 0.69 (0.42 to 1.12) | 0.453 |
| Haemorrhage: blood O2 saturation of > 85% on room air | 6/64 | 9/66 | 0.65 (0.23 to 1.86) | 7/65 | 8/65 | 0.75 (0.27 to 2.10) | ||
| Haemorrhage: blood O2 saturation of ≤ 85% on room air or ventilated | 6/31 | 11/30 | 0.48 (0.18 to 1.32) | 10/31 | 7/30 | 1.39 (0.52 to 3.73) | ||
Chapter 4 Discussion and conclusions
Summary of findings
This trial had two primary objectives, which were as follows: to determine whether PLEX delayed the time to death or ESRD for patients with severe AAV, and to assess whether or not a reduced-dose oral GC regimen was non-inferior to a standard-dose regimen for the occurrence of death or ESRD and was safer for patients with severe AAV. PEXIVAS did not demonstrate a benefit of PLEX in the treatment of severe AAV when combined with immunosuppressive and GC therapy. PLEX was not shown to delay the time to death or ESRD for the whole patient cohort; the effect was not different for any of the predefined patient subgroups over the whole trial, at 12 months, or for death or ESRD separately. PEXIVAS was powered to detect a relatively large benefit of PLEX; a smaller benefit for death or ESRD cannot be excluded. The reduced-dose oral GC regimen was shown to be non-inferior to the standard-dose GC regimen for the outcome of death or ESRD, and was associated with fewer serious infections.
PEXIVAS and outcomes of severe anti-neutrophil cytoplasm antibody-associated vasculitis
Previous studies have determined that the presence of renal dysfunction or diffuse alveolar haemorrhage are adverse predictors for death and ESRD in AAV. 32 A GFR of 50 ml/minute/1.73 m2 at diagnosis separated patients into those with near-normal risks for these events and those with a 50% risk of an event by 5 years. 33 PLEX has been used to try and improve outcomes for patients with these adverse prognostic features. It has also been shown that GCs contribute to the risk of SAEs and death; however, there is no high quality evidence to determine optimal dosing. PEXIVAS was designed to both test the value of PLEX and to improve the dosing of GCs for patients with this disease. The recruited population had the expected baseline characteristics for patients with severe disease: patients’ mean age was 63 years, and 59% of patients had MPO-ANCA, 29% had a serum creatinine level of > 500 µmol/l or required dialysis, and 27% had lung haemorrhage. The frequency of death and ESRD seen in PEXIVAS was lower than predicted from previous studies, with 138 patients developing ESRD, 99 dying and 209 (30%) having the composite end point after a median of nearly 3 years’ follow-up. 14,33 This led to an increase in the sample size from 500 to 700 patients to obtain the predicted number of events. Previous reports have highlighted improvements in the outcome of severe AAV, but these reports have suggested earlier diagnosis and initiation of definitive therapy as a cause of improvement. 34 It is possible that refinements to CYC dosing, the introduction of rituximab and improved supportive care have had a benefit, in particular by reducing the rate of treatment-associated death. However, outcomes for patients with AAV remain poor and are worse than the outcomes for common malignancies (e.g. breast, prostate, colon) in the same age group; SAE rates are high, with treatment-related events, including infections, being the most common categories.
Effectiveness of plasma exchange
A meta-analysis of previous PLEX studies in AAV reported a reduction in the incidence of ESRD, no consistent mortality benefit and a borderline effect on the composite of death and/or ESRD. 21 One study claimed that the benefit was limited to those with the most severe renal disease at presentation, a serum creatinine level of > 500 µmol/l and who were already on renal supportive therapy. 17 The MEPEX study, to the best of our knowledge, had been the largest previous study of plasma exchange in AAV, with 137 patients, and restricted enrolment to those with a serum creatinine level of > 500 µmol/l. 14 It demonstrated a 20% reduction in the incidence rate of ESRD in survivors at 1 year (from 60% to 40%), but had a high 1-year mortality rate (27%) and no mortality benefit with PLEX. 14 PEXIVAS did not find the predicted size of benefit (0.64) on the composite outcome of ESRD or death by ITT or per-protocol analysis, or by censoring the follow-up at 12 months. Of note is the fact that no differences in response were observed between the PLEX and no-PLEX groups for subgroups defined by a baseline serum creatinine of > or < 500 µmol/l. However, there remains the possibility of a smaller magnitude of benefit that might be clinically meaningful or financially relevant.
Concerning the components of the primary outcome separately, previous studies have observed a high early mortality rate in severe AAV (> 25% at 1 year), with no benefit seen in the PLEX group in the MEPEX trial. 14 Overall, the mortality rate in PEXIVAS was 13% in the PLEX group and 15% in the no-PLEX group, which was considerably lower than the predicted rate, with no benefit for PLEX. Because the causes of death are varied, with infection being the most common, it is, perhaps, unsurprising that PLEX did not have an impact on the mortality rate. PLEX might be expected to reduce the number of deaths attributable to vasculitis, especially lung haemorrhage, but only three deaths with this cause were recorded. In the longer term, if PLEX had a major effect on ESRD rates, the lower survival rate of patients with ESRD might also have been a factor. However, the lower than predicted rates of ESRD in PEXIVAS and the lack of a major reduction in risk with PLEX made this an unlikely contributor to any difference.
Results from previous randomised controlled trials vary regarding the benefit of PLEX on rates of renal recovery for patients with a creatinine level of < 500 µmol/l, but generally found a stronger treatment effect with the subgroup presenting with a creatinine level of > 500 µmol/l or with immediate requirement for dialysis. 35 PEXIVAS found no major effect of PLEX on ESRD and no differences between the subgroups defined by presenting creatinine levels. There appeared to be an early separation in the rate of development of ESRD in favour of PLEX, with a loss of difference over time. Further studies of ESRD rates and changes in GFR at 6- and 12-month end points are needed to explore this separation.
Because the pathogenesis of pulmonary capillaritis is similar to the glomerular pathology of AAV, PLEX has also been considered for diffuse alveolar haemorrhage. This manifestation of vasculitis is associated with early death due to respiratory failure. 36 Observational studies from the 1990s reported a 50% in-hospital mortality rate if assisted ventilation was required, but survival has improved over the past 20 years because of better intensive care techniques and adjustments to immunosuppressive and GC medication. 37,38 No prospective study of PLEX in diffuse alveolar haemorrhage has been performed, and patients requiring ventilator support were excluded from previous trials. The criteria developed for PEXIVAS that defined the severity of haemorrhage were novel and required radiological evidence of bilateral pulmonary infiltrates with either arterial hypoxia or evidence of lung bleeding. The performance of these criteria in PEXIVAS merits further study and validation. A total of 191 patients had diffuse alveolar haemorrhage, and almost all also met the GFR criteria for inclusion. No benefit of PLEX was seen in the lung haemorrhage subgroups for the composite of death and ESRD, and a post hoc analysis of death at any time also showed no significant benefit. However, a previous study found no impact on death due to non-severe lung haemorrhage, and that it is patients who are hypoxic at presentation who might have the most to gain from this intervention. 39 Among the 61 patients in this category there were six (among 31 patients) deaths in those receiving PLEX and 11 (among 30 patients) deaths in those not receiving PLEX. Further analysis of the timing and cause of these deaths is required before a useful effect of PLEX in this subgroup can be ruled out.
Plasma exchange is designed to reduce the components in plasma that contribute to the pathogenesis of vasculitis. It was introduced in autoimmunity to remove pathogenic autoantibodies being applied in antiglomerular basement membrane disease, another cause of severe glomerulonephritis, before ‘pauci-immune’ necrotising glomerulonephritis, subsequently associated with ANCA. 15 The pathogenic role of ANCA has been demonstrated for MPO-ANCA in an animal model, but controversy persists as to how relevant this is for the human version of the disease. 40 Other possible plasma constituents that are known to be harmful and can be removed by PLEX are endothelial, platelet and neutrophil microparticles; complement components; cytokines; and chemokines. PLEX also removes coagulation factors, and perturbed thrombosis contributes to the pathogenesis of vasculitis. No study in AAV has titrated the dose of PLEX against a biomarker, such as ANCA binding level. The selected dose of seven exchanges was the same as that used for MEPEX, and also reflects the median from a survey of PEXIVAS investigators. The dose results in an 80–90% reduction in circulating IgG, and ANCA is largely composed of the IgG isotype. Diffuse alveolar haemorrhage has been associated with ANCA of the immunoglobulin M (IgM) isotype and IgM is more efficiently removed by PLEX, further supporting a mechanistic role for PLEX in this presentation. 41 All patients in PEXIVAS were ANCA positive at entry and approximately 10% more had become ANCA negative in the PLEX group than in the no-PLEX group at 2 weeks. A subsequent analysis will assess the value of PLEX in ANCA reduction and look for any associations with clinical efficacy.
A concern in the design of PEXIVAS was the no-PLEX patients receiving PLEX, in part because they are seriously ill and physicians want to use all available therapies, and in part because some PEXIVAS investigators had greater prior belief in the clinical efficacy of PLEX than others. Compliance was surprisingly good, with 90% of the PLEX group receiving the protocol-defined dose and 96% receiving some PLEX. The reasons for not receiving PLEX in the PLEX treatment group were, typically, concomitant events, such as myocardial infarction or death. Only 8% of the no-PLEX group received PLEX, usually for progressive, refractory disease. As a result, there were no major differences between the ITT and per-protocol analyses. PLEX is administered by either plasma filtration or centrifugation. The latter has potential advantages, as it removes macromolecular structures that may not be filtered and it causes less activation of inflammatory components. Around 60% of patients received treatment by centrifugation and the modalities will be compared in a subsequent analysis. Other aspects of the PLEX technique were standardised in the protocol, such as replacement fluids, or were unlikely to influence efficacy, but may have affected safety, such as vascular access. Data has been collected on these factors for further analysis.
Effectiveness of reduced-dose oral glucocorticoids
The factorial design of PEXIVAS permitted examination of a second question, and oral GC dosing was selected because there is uncertainty as to the optimal regimen and GC toxicity contributes to the mortality rate and morbidity of vasculitis. 13 As a fulminant inflammatory disease, initial high-dose GCs are required to gain disease control and either high-dose oral GCs alone or combined oral and i.v. dosing are used. There is no good evidence to guide i.v. dosing, which is recommended in the guidelines, and there are concerns over the safety risks of this route of administration. Furthermore, i.v. GCs are often administered at referring hospitals or before the vasculitis diagnosis has been confirmed. Because PEXIVAS recruited patients from the severe end of the vasculitis spectrum, and most patients were likely to be given i.v. steroids, these were included in the protocol; however, treating physicians could select a total dose of between 1000 and 3000 mg. A subsequent analysis will explore the safety risks and efficacy benefits of the differences in i.v. dose. The randomisation in PEXIVAS focused on the speed of reduction of oral GCs commenced immediately after the i.v. dosing; all patients received the same dose for the first week and then dosing was reduced faster in the reduced-dose group to achieve a difference in cumulative exposure of around 60% by 6 months. Although open label, adherence to the oral GC regimen was good, with physicians and patients noting how helpful it was to have the dosing regimen determined in advance for the whole of the first year. The primary end point was the total number of death and ESRD events over the course of the trial, performed as a per-protocol analysis to avoid falsely inflating a non-inferiority result. No differences were seen between the reduced-dose GCs and standard-dose GCs groups, with an actual risk difference of 2.3% (95% CI –0.034 to 0.08), meeting the 11% non-inferiority margin that was previously determined.
No differences were seen for the two components of the primary end point (i.e. death or ESR) when examined separately, or in the overall occurrence of SAEs. It was hoped that reducing GC exposure would reduce the frequency of severe infections, known to be the most common cause of death in the first year after diagnosis. Although there were no differences in the infection rates over the duration of the trial, there were fewer severe infections in the first year: 96 patients (27%) experienced 142 infections in the reduced-dose GCs group and 116 patients (33%) experienced 180 infections in the standard-dose GCs group.
Safety
Plasma exchange has not been shown to increase the risk of infection or the frequency of SAEs in previous vasculitis studies. There is the potential for vascular access-related problems, haemorrhage, thrombosis, infection, pneumothorax- and procedure-related toxicity, anaphylaxis, TRALI, hypotension, hypocalcaemia and haemorrhage associated with anticoagulation and the removal of coagulation factors. No differences in SAE rates or serious infection rates were seen between the PLEX and no-PLEX groups, but further analysis of adverse event data is required to detect any differences in the pattern of SAEs seen with PLEX.
Sustained remission and quality of life
A long-term observational study of the MEPEX trial participants reported a reduced vasculitis relapse rate in the PLEX group. 21 A sustained-remission secondary end point was selected for PEXIVAS, requiring no disease activity (i.e. a BVAS/WG of zero) at 6 months without relapse by 12 months. This end point has been used in two previous EUVAS trials. No differences in sustained remission were seen between the PLEX and no-PLEX groups. Because early control of disease activity is associated with ESRD, death and vasculitis relapse risk, the lack of effect of PLEX on sustained remission is consistent with the lack of effect on the primary composite end point.
Changes in quality of life were assessed using the SF-36 and EQ-5D. Both tools demonstrated some improvement in quality of life over time, and this is consistent with previous reports highlighting the low quality of life of vasculitis patients at diagnosis, with improvement after initiation of treatment. However, the size of difference seen in the quality-of-life metrics was small and is unlikely to represent a clinically meaningful benefit. If a reduction in the rate of ESRD with PLEX is real, then a health economic analysis may be more sensitive in demonstrating a benefit than the chosen primary composite end point. A year of ESRD care costs £40,000 and PLEX costs £3500 for seven sessions. Thus, if the number of patients with AAV and renal vasculitis needed to be treated with PLEX to save 1 year of ESRD was fewer than 10, PLEX would have a cost advantage. The shape of the Kaplan–Meier curve for time to ESRD suggests that any benefit for PLEX in the first year is lost over the subsequent years, so a sustained financial benefit is unlikely. Exploratory analyses examining whether or not there is a benefit on other renal parameters, such as GFR at 1 year or time-averaged GFR, and an appropriate cost–benefit analysis are planned.
Effectiveness and safety of different glucocorticoid regimens
No previous study has compared different GC regimens in severe AAV. The introduction of GCs in vasculitis occurred in 1947, and this heralded the modern era of treatments for the disease. A dose of prednisolone of 1 mg/kg/day has been used widely in human and veterinary medicine for severe inflammatory disease without a sound evidence base. Vasculitis trials over the last 30 years have typically started treatment at this dose, but have varied in the rate of reduction, cumulative exposure and time to withdrawal. One small study of vasculitis in elderly people found a lower-dose regimen to be safer and no less effective than a standard-dose regimen. 42 A study of two different GC regimens in lupus nephritis, starting at either 1.0 or 0.5 mg/kg/day, found a halving in the number of SAEs at 6 months with the lower dose, with little difference in efficacy. 43 The two GC regimens in PEXIVAS represented the upper and lower ranges of acceptability for steroid dosing at an investigators’ planning meeting for the trial, and almost a 50% difference in cumulative exposure by 6 months. A value of 11% was chosen for the non-inferiority boundary, and this was easily met for the primary composite of death and ESRD. There was also no difference between GC treatment groups in the secondary clinical efficacy end point of sustained remission. In contrast, there were fewer serious infections in the reduced-dose GC group at 1 year. Further studies will assess the safety benefit of the reduced-dose GC regimen on specific GC-related toxicity in this trial. A GC toxicity index has been designed since PEXIVAS started and would have been a validated approach to assessing toxicity. 44
Severe AAV is a fulminant disease, and all patients received an initial treatment with high-dose i.v. GCs and the same oral dose for the first week. This was necessary to ensure adequate treatment, but would have reduced any differential effects between GC treatment groups. In addition, SAEs are most frequent early in the course of treatment, again reducing the potential difference between the groups.
There was concern that physicians or patients would not adhere to their selected GC regimen because of the severity of the patients’ illness, prior beliefs or desire to avoid toxicity. Any convergence between regimens would dilute the possibility of observing a difference in clinical efficacy or safety. The high compliance rates for both standard-dose and reduced-dose GC regimens represented a success for the trial management, investigators and patients. A GC dosing regimen was generated for each patient at the time of randomisation to cover the first 12 months and e-mailed to the investigator for filing in the patient’s hospital record. Feedback to the management team suggested that the presence of an advance plan was appreciated by both the physicians and patients and may have helped compliance.
High-dose i.v. GCs are almost routinely used for severe AAV in the absence of any randomised evidence, with observational studies suggesting both advantages and risks. The range of dosing, between 1000 and 3000 mg in the first 3 days of the trial, was left to physician discretion and reflected investigators’ practice. Further analysis will explore whether differences in delivered i.v. dosing had any impact on clinical efficacy or safety end points.
Strengths and limitations
With 704 patients, PEXIVAS is, to the best of our knowledge, more than three times larger than any previous trial in AAV; however, recruitment took over 6 years, although it did exceed predicted recruitment rates. There was variation in prior beliefs on the clinical efficacy of PLEX and the optimal GC regimen, but any bias in recruitment should have been balanced by the large number of participating sites and the geographic distribution. However, there is still the potential for the highest recruiting sites to have biased their patient selection, and no records were kept on patients who were eligible but not entered to assess this potential. The majority of patients were of white ethnicity. Because previous studies have identified differences in AAV disease organ involvement and severity between Japanese and European cohorts, and differences are suspected between white and other Asian and black ethnicities, there may have been differences in response to the interventions tested in this study between ethnic groups that could not be detected due to the small number of non-white participants.
The trial was unblinded, and treatment allocation may have influenced physician decisions; however, choice of baseline immunosuppressive was determined before randomisation, GC regimen was determined by the protocol and there was good compliance with both PLEX and GCs. Assessment of vasculitis activity using the BVAS/WG is observer dependent and the lack of blinding may have influenced decisions on remission and relapse. However, these occurred at 6 and 12 months, which was a considerable time after the delivery of PLEX. The end points of death and ESRD are robust, although decisions on starting and stopping dialysis (the ESRD definition required a 12-week period of dialysis) are physician dependent. Further analysis of the effects of the intervention on BVAS/WG, including time to BVAS/WG zero and time to relapse, are planned.
Data return, as judged by completed forms for the different assessments at each time point, was good. The patient dropout rate was low despite the long course of the study and the most commonly report reasons for dropout were relocation, distance from the trial centre and the development of ESRD.
A pragmatic approach was taken to the PLEX procedure (which varied between centres), the choice of background immunosuppressive and choice of all medication after 12 months. Systematic bias in these areas may have influenced the results, but we have no evidence that this occurred and the choice of immunosuppressive was balanced in the randomisation process. At the time of the trial design, the routine immunosuppressive was CYC. Rituximab was licensed for AAV in 2011 in the USA and in 2013 in Europe and is slowly becoming the standard of care. Only a minority of patients underwent rituximab induction, but the response to PLEX appeared to be the same across the three immunosuppressive categories.
Other methodological issues
PEXIVAS, a trial of rare disease, demonstrates how a relatively large number of patients can be recruited, provided there is international collaboration and a long recruitment period. Two international networks, EUVAS in Europe and VCRC in North America, existed before PEXIVAS and provided the bulk of the recruiting centres. There was also an existing collaboration between EUVAS and the International Vasculitis Committee of Japan’s Ministry of Health Labour and Welfare, which prepared the ground for Japanese collaboration. PEXIVAS contributed to the formation of the Canadian Vasculitis Network (CANVAS) and to collaborative working in Australia and New Zealand. The existence of centres with research experience in AAV was important for obtaining consensus in the protocol design and use of the trial tools, including BVAS/WG and CDA.
PEXIVAS funding commenced as parallel applications to the Medical Research Council (UK) and the Food and Drug Administration (Silver Spring, MD, USA). Success with both agencies led to applications to the Canadian Institute of Health Research (CIHR), the National Health Medical Research Council (NHMRC; Canberra, ACT, Australia), Programme Hospitalier de Recherche Clinique (Ministry of Health, France) and commercial support from PLEX machine manufacturers Terumo BCT, Inc. (Lakewood, CO, USA), Asahi Kasei Medical Co., Ltd (Tokyo, Japan), and Fresenius (Bad Homburg, Germany). This model of rare disease research funding evolved empirically, but may be useful for research in other diseases.
Implications for health care
Anti-neutrophil cytoplasm antibody-associated vasculitis remains a severe disease and, although outcomes are improving, the rates of death and ESRD are high. PEXIVAS has highlighted the rate of SAEs (almost 1200 events in 704 patients); these represent a major risk and cause of suffering for patients, as well as a major cost burden for health-care systems. A better understanding of the nature and causes of SAEs is required to improve these rates. The data for PLEX will be included in a future meta-analysis before a final conclusion on the relative merits of PLEX can be made and a translation to practice guidelines can occur. Further exploratory analyses may more precisely define any differences between the PLEX and no-PLEX groups, for example in the early improvements in renal function and in those with severe lung haemorrhage, but it is likely that PLEX use will be more restricted than it is currently. The most recent international treatment recommendations statement for the treatment of AAV was made in 2016, by a joint working group between the European League Against Rheumatism (EULAR) and the European Renal Association (ERA), when PLEX was recommended for severe disease and this will need to be amended in any further update. PLEX is expensive, with equipment, disposables, plasma substitute and staff costs associated with its use, and precise costs vary between £500 and £2000 per session (i.e. between £3500 and > £14,000 for seven sessions). There could be cost savings from a reduced requirement for PLEX and associated blood products, which are in periodic short supply and have the potential for transmission of chronic viral infection and prions. In addition, patients are often transferred to regional centres for PLEX, and this may be no longer necessary. Patients could be spared central venous access procedures and the inconvenience of PLEX and in-hospital stays may be shortened.
The success of the reduced-dose GC regimen may permit a specific recommendation regarding how much GC should be used and when the dose could be reduced. This may reduce the number of serious infections, which are a major driver of hospital admission and health costs for vasculitis patients. It could also simplify the work of physicians (who may not need to design their own GC regimens), reduce variation between centres and minimise GC toxicity.
Future research implications
A research plan has been developed based on the PEXIVAS data set and associated biomarker collection. Topics for further studies fall into three areas:
-
Further study of the interventions –
-
a meta-analysis of the efficacy of PLEX in delaying ESRD by combining the PEXIVAS data set with those of previous trials
-
detailed post hoc analysis of the PEXIVAS data set to assess any benefit of PLEX on ESRD risk at 1 year for different baseline subgroups and for early mortality for those with lung haemorrhage
-
further reduction of GC exposure by using different background immunosuppression (e.g. CYC/rituximab combination) or newer therapeutics (e.g. complement inhibition with avacopan)
-
cost-effectiveness analyses for the two interventions
-
-
Clinical epidemiology studies –
-
comparisons between baseline immune suppressives, rituximab and CYC
-
co-morbidity risks and prognostic factors (e.g. thromboembolism, infection)
-
relapse analysis and risk factors
-
-
Biomarker studies –
-
impact of PLEX on ANCA levels
-
identification of prognostic biomarkers for PLEX efficacy
-
histological analysis of renal biopsies from PEXIVAS and the development of prognostic tools
-
A committee has been convened to supervise the management of the further studies using PEXIVAS data or samples.
Acknowledgements
We would like to thank the team at Birmingham Clinical Trials Unit (BCTU) for their data management and statistical support: Elizabeth A Brettell, Paul Crump, Annika Feilbach, Catherine Hewitt, Nick Hilken, Andrew Howman, Terry Hughes, Natalie Ives, Hugh Jarrett, Samir Mehta, Rebecca Record, Gemma Ryan and Chaka Sidile.
Keith Wheatley at the Cancer Research UK Clinical Trials Unit at the University of Birmingham made a major contribution to the design of the trial.
We would also like to thank the members of the Trial Steering Committee for their advice and support for the project: Professor Andrew Rees (Medical University of Vienna, Austria), Professor David Scott (Norfolk and Norwich University Hospital, UK) and Professor Paul Roderick (University of Southampton, UK).
Our thanks also go to the DMC, comprising Professor Martin Landray (University of Oxford, UK), Professor Richard Watts (University of East Anglia, UK) and Dr Jonathan Emberson (University of Oxford).
We express our gratitude to the referring physicians, PEXIVAS investigators and research teams at each site for their support of study recruitment and data collection.
| Centres | Investigators |
|---|---|
| Aarhus University Hospital, Denmark | Dr Per Ivarsen, Dr Henrik Birn, Dr Jon Gregersen and Dr Johan Povlsen |
| Aberdeen Royal Infirmary, UK | Dr Dana Kidder, Dr Lars-Peter Erwig, Dr Neil Basu and Dr Nicholas Fluck |
| Addenbrooke’s Hospital, UK | Professor David Jayne, Dr Rachel Jones, Dr Rona Smith, Dr Lisa Willcocks, Dr Seerapani Gopaluni, Dr Sapna Trivedi and Dr Mark McClure |
| Auckland Hospital, New Zealand | Dr Michael Collins, Dr Liz Curry and Dr John Collins |
| Austin Hospital, Australia | Dr Kathy Paizis, Dr Natasha Cook and Dr Peter Mount |
| Azienda Ospedaliera Spedali Civili di Brescia, Italy | Dr Gina Gregorini, Dr Guido Jeannin and Dr Stefano Possenti |
| Azienda Ospedaliero Universitaria di Parma, Italy | Dr Augusto Vaglio and Professor Carlo Buzio |
| Boston University School Of Medicine, USA | Dr Paul Monach and Dr Karen Quillen |
| Canberra Hospital, Australia | Dr Giles Walters and Dr Darren Roberts |
| Cedars-Sinai Medical Center, USA | Dr Michael Weisman, Dr Daniel Wallace, Dr Lindsy Forbess and Dr Swamy Venuturupalli |
| Centre Hospitalier de Boulogne, France | Dr Pierre Bataille and Dr Rafik Mesbah |
| Centre Hospitalier de la Région d’Annecy, France | Dr Isabelle Rey |
| Centre Hospitalier de Mulhouse, France | Dr François Chantrel |
| Centre Hospitalier de Valenciennes, France | Dr Thomas Quémeneur and Dr Phillipe Vanhille |
| Centre Hospitalier Universitaire de Grenoble, France | Dr Pierre-Louis Carron |
| Centre Hospitalier Regional Universitaire de Brest–Hôpital La Cavale Blanche (Néphrologie), France | Dr Catherine Hanrotel-Saliou and Dr Morgane Gosselin |
| Centre Hospitalier Regional Universitaire de Brest–Hôpital La Cavale Blanche (Médecine Interne), France | Dr Aurélien Delluc and Dr Claire de Moreuil |
| Centre Hospitalier Universitaire de Strasbourg – Médicine Interne, France | Professor Emmanuel Andres and Dr Rachel Cottett |
| Centre Hospitalier Universitaire de Toulouse–Hôtel Dieu Saint Jacques, France | Professor Dominique Chauveau, Dr Joelle Guitard, Dr Antoine Huart and Dr David Ribes |
| Centre Hospitalier Universitaire de Bretonneau, Tours, France | Dr Philippe Gatault and Dr Bernadette Pilorge |
| Cleveland Clinic, USA | Dr Carol Langford, Dr Rula Hajj-Ali, Dr Anna Koo and Dr Gary Hoffman |
| Hôpital de Colmar – Néphrologie, France | Dr Alexandre Klein, Dr Camille Becmeur, Dr Sandrine Muller and Dr Valérie Betz |
| Concord Repatriation General Hospital, Australia | Dr Meg Jardine, Dr Mona Razavian, Dr Sandhya Limaye and Dr Martin Gallagher |
| Dunedin Hospital, New Zealand | Dr John Schollum and Professor Rob Walker |
| Flinders Medical Centre, Australia | Dr Caroline Milton, Dr George Passaris, Dr Jordan Li, Dr Rajiv Juneja, Professor Jonathan Gleadle and Professor Jeffrey Barbara |
| Freeman Hospital, UK | Dr Jim Lordan, Professor Sheerin, Dr Alison Brown and Dr Laura Anne Baines |
| Fremantle Hospital, Australia | Dr Abu Abraham, Dr Brian Jayhan Siva, Dr George Chin, Dr Hermant Kulkarni, Dr Sajan Thomas, Dr Jagadish Jamboti and Dr Paolo Ferrari |
| Fundacio Puigvert, Spain | Dr Jose Ballarin |
| General Faculty Hospital, Czechia | Professor Vladimír Tesař, Dr Zdenka Hruskova and Dr Zdenka Chocova |
| Gold Coast Hospital, Australia | Dr Jagadeesh Kurtkoti and Dr Clare Owens |
| Hammersmith Hospital, UK | Professor Charles D Pusey, Dr Anisha Tanna, Dr Stephen McAdoo, Professor Jeremy Levy and Dr Megan Griffith |
| Herlev University Hospital, Denmark | Dr Elizabeth Krarup |
| Holstebro Hospital and University of Aarhus, Denmark | Dr Ingrid M Thomsen, Professor Erling B Pedersen and Dr Jesper Nørgaard Bech |
| Hôpital Claude Huriez – Centre Hospitalier Regional Universitaire de Lille, France | Dr Céline Lebas and Dr Arnaud Lionet |
| Hôpital Cochin, Paris, France | Dr Xavier Puéchal and Professor Luc Mouthon |
| Hôpital de la Conception, Marseille, France | Professor Noémie Jourde-Chiche |
| Hôpital Européen Georges-Pompidou, Paris, France | Professor Alexandre Karras |
| Hôpital Saint-Luc, Montréal, Canada | Dr Rita Suri and Dr Soumeya Brachemi |
| Hôpital Site Sainte Blandine, Metz, France | Dr François Maurier |
| Instituto Nacional de Enfermedades Respiratorias Ismael Cosio Vilegas, Mexico | Dr Luis Felipe Flores-Suárez |
| Jagiellonian University Medical College, Poland | Dr Wojciech Szczeklik, Dr Agnieszka Padjas, Dr Anna Wludarczyk, Dr Jacek Gorka, Dr Jan Sznajd and Dr Milosz Jankowski |
| John Hunter Hospital, Australia | Dr Eswari Vilayur and Dr Alastair Gillies |
| Karolinska Institute, Sweden | Professor Annette Bruchfeld, Professor Peter Barany and Dr Mats Efvergren |
| Kent and Canterbury Hospital, UK | Dr Timothy Doulton, Dr Bernhard Klebe, Dr Timothy Lewis-Morris and Dr Richard Kingston |
| Kitano Hospital, Japan | Dr Eri Muso, Dr Tatsuo Tsukamoto, Dr Tomomi Endo, Dr Hiroko Kakita, Dr Hiroyuki Suzuki, Dr Takaya Handa, Dr Youngna Kang and Dr Yuki Ariyasu |
| Kyoto University Hospital, Japan | Dr Shuichiro Endo, Dr Tatsuo Tsukamoto and Dr Toshiko Ito-Ihara |
| Leicester General Hospital, UK | Dr Reem Al-Jayyousi, Dr Graham Warwick, Dr James Burton, Dr Jonathon Barratt, Dr Peter Topham, Dr Richard Baines and Professor Nigel Brunskill |
| Linkoping University Hospital, Sweden | Professor Marten Segelmark and Dr Per Eriksson |
| London Health Sciences Centre, Canada | Dr William Clark |
| Centre Hospitalier Lyon Sud (Médecine Interne), France | Professor Jean-Christophe Lega |
| Manchester Royal Infirmary, UK | Dr Sandip Mitra, Dr Michael Venning, Dr Patrick Hamilton, Dr Mumtaz Patel and Dr Nina Brown |
| Mayo Clinic, USA | Dr Ulrich Specks, Dr Jeffrey L Winters, Dr Fernando Fevenza, Dr Steven Ytterberg, Dr Misbah Baqir, Dr Karina Keogh, Dr Kenneth Warrington, Dr Rodrigo Cartin-Ceba and Dr Tobias Peikert |
| Monash Medical Centre, Australia | Dr Jess Ryan, Dr Fiona Brown, Dr Shaun Summers, Professor Kevan Polkinghorne, Professor John Kanellis, Professor Peter Kerr, Dr Jo Ghali, Professor Peter Kerr, Dr Jo Ghali, Professor John Kanellis and Dr Sharon Ford |
| Mount Sinai Hospital, Canada | Dr Simon Carette and Dr Christian Pagnoux |
| Nambour General Hospital, Australia | Dr Euan Noble, Dr Carolyn Clark, Dr Kumar Mahadevan, Dr Nicholas Gray, Dr Peter Hollett and Dr Victoria Campbell |
| North Shore Hospital, New Zealand | Dr Janak De Zoysa and Dr Keith Colvine |
| Nuffield Orthopaedic Centre/Churchill Hospital, UK | Dr Raashid Luqmani, Dr Edward Sharple and Dr Jo Robson |
| Prince of Wales Hospital, Australia | Dr Grant Luxton, Dr James Mackie, Dr Jonathon Erlich, Dr Mangalee Fernando, Professor Bruce Pussell and Professor Zoltan Endre |
| Princess Alexandra Hospital, Australia | Dr David Mudge, Dr Carmel Hawley, Dr Carolyn Van Eps, Dr Nicole Isbel, Dr Scott Campbell and Professor David Johnson |
| Queen Alexandra Hospital, UK | Dr Judith Stevens |
| Queen Elizabeth Hospital Birmingham, UK | Professor Lorraine Harper, Dr Benjamin Rhodes, Dr Dimitrios Chanouzas, Dr Matthew Morgan and Dr Peter Hewins |
| Rigshospitalet University Hospital, Denmark | Dr Wladimir Szpirt and Dr Martin Egfjord |
| Royal Adelaide Hospital, Australia | Dr Chen Au Peh |
| Royal Berkshire Hospital, UK | Dr Oliver Flossmann, Dr Nitin Bhandary and Dr Bassam Alchi |
| Royal Brisbane and Women’s Hospital, Australia | Dr Dwarakanathan Ranganathan, Dr Adrian Kark, Dr Glen Kennedy, Dr Robert Fassett, Dr Helen Healy, Dr George John and Dr Vincent D’intini |
| Royal Devon and Exeter Hospital, UK | Dr Lucy Smyth, Dr Richard D’Souza and Dr Richard Haigh |
| Royal Free Hospital, UK | Professor Alan Salama, Professor Mark Little and Dr Aine Burns |
| Royal Hobart Hospital, Australia | Dr Lisa Jeffs, Dr Matthew Jose, Dr Richard Yu, Dr Geoffrey Kirkland and Dr Steven Yew |
| Royal London Hospital, UK | Dr Ravindra Rajakariar, Dr Stanley Linsun Fan and Professor Magdi Yaqoob |
| Royal Sussex County Hospital, UK | Dr Neil Iggo, Dr Kolitha Basnayake and Dr Edward Kingdon |
| Royal Preston/Sharoe Green Hospital, UK | Dr Ajay Dhaygude |
| Skane University Hospital, Sweden | Dr Daina Selga, Dr Kerstin Westman, Dr Caroline Heijl and Dr Sophie Ohlsson |
| St George’s Hospital London, UK | Professor David Oliveira, Dr Daniel Jones and Dr Iain A M MacPhee |
| St. Joseph’s Healthcare Hamilton, Canada | Dr Michael Walsh and Dr Catherine Clase |
| St Michael’s Hospital, Canada | Dr Ron Wald |
| St Olav’s Hospital, Norway | Dr Knut Aasarod |
| St Paul’s Hospital, Canada | Dr Michael Copland and Professor Adeera Levin |
| St Vincent’s Hospital, Australia | Dr Robyn Langham, Dr Sharon Ford and Dr Veena Roberts |
| Teikyo University Hospital, Japan | Dr Shunya Uchida, Dr Hajime Kono, Dr Suzuki Kazuo, Dr Yoshihide Fujigaki, Dr Hirotoshi Kikuchi, Dr Toshihiro Nanki, Dr Kurumi Asako, Dr Hideki Kato and Dr Akiko Okamoto |
| The Geelong Hospital, Australia | Dr Michael Desmond, Dr Katherine McDougal and Dr Christine Somerville |
| The Ottawa Hospital, Canada | Dr Todd Fairhead |
| The Royal Melbourne Hospital, Australia | Dr Peter Hughes, Dr Kathy Nicholls, Dr Solomon Cohney and Dr Joanna Ghali |
| Tokyo Metropolitan Geriatric Hospital, Japan | Dr Yoshitomo Hamano |
| University Hospitals Leuven, Belgium | Professor Daniel Blockmans, Dr Liesbet Henckaerts and Dr Ben Sprangers |
| University Hospital North Norway, Norway | Dr Wenche Koldingsnes, Dr Marit Solbu and Dr Trude Jannecke Bruun |
| University Hospitals Coventry & Warwickshire, UK | Dr Andrew Short and Dr Sunil Daga |
| University Of Alberta Hospital, Canada | Professor Neesh Pannu, Professor Muhammad Uwais Qarni and Professor Syed Habib |
| University of Calgary, Canada | Professor Louis-Philippe Girard |
| University of Miyazaki Hospital, Japan | Dr Shouichi Fujimoto, Dr Yuji Sato and Dr Masao Kikuchi |
| University of North Carolina, USA | Dr Patrick H Nachman, Dr Amy Mottl, Dr Vimal Derebail, Dr JulieAnne McGregor and Dr Randy Detwiler |
| University of Pennsylvania, USA | Professor Peter A Merkel, Dr Rennie Rhee, Dr Antoine Sreih and Dr Nicole Aqui |
| University of Pittsburgh Medical Centre, USA | Dr Larry W Moreland, Dr Kathleen Maksimowicz-McKinnon, Dr Joseph Kiss, Dr Kimberly P Liang and Dr Niveditha Mohan |
| Waikato Hospital, New Zealand | Dr Peter Sizeland, Dr Vicki Quincey, Dr Eddie Tan, Dr Gerald Waters, Dr Kannaiyan Rabindranath and Dr Rakesh Pandey |
| Washington University School of Medicine, USA | Dr Tingting Li and Dr Richard Brasington |
| Western Infirmary, UK | Dr Bruce Mackinnon, Dr Colin Geddes and Professor Alan G Jardine |
We are also grateful to a number of people at Cambridge University Hospitals NHS Foundation Trust (Cambridge, UK) for their support of the study: Stephen Kelleher, Dr Sabine Klager and Professor John Bradley.
We are grateful to all of the study participants and their carers who generously gave their time to support the study.
This study was also funded by the Food and Drug Administration/National Institutes of Health (USA): FDA R01 FD003516; the National Institutes of Health (USA) – National Institute of Arthritis and Musculoskeletal and Skin Diseases: U54 AR0573319; National Health and Medical Research Council (Australia): 626939; National Health and Medical Research Council (Australia): 1086192; Canadian Institutes of Health Research (Canada): 211079; Programme Hospitalier de Recherche Clinique (Ministry of Health, France): AOM11142; Research Committee on Intractable Vasculitides, the Ministry of Health, Labour and Welfare of Japan; and The Japan Agency for Medical Research and Development (AMED): The Strategic Study Group to Establish the Evidence for Intractable Vasculitis Guideline (Japan). TerumoBCT (USA), Fresenius Australia, Gambro Australia and Asahi Kasei Medical Co., Ltd (Japan), provided replacement plasma exchange disposables.
Contributions of authors
David Jayne (https://orcid.org/0000-0002-1712-0637) was one of the joint chief investigators and had overall responsibility for the trial.
Michael Walsh (https://orcid.org/0000-0001-8292-2014) was one of the joint chief investigators and had overall responsibility for the trial.
Peter A Merkel (https://orcid.org/0000-0001-9284-7345) was one of the joint chief investigators and had overall responsibility for the trial.
Chen Au Peh (https://orcid.org/0000-0002-9712-0396) was a national representative.
Wladimir Szpirt (https://orcid.org/0000-0002-6570-6077) was a national representative.
Xavier Puéchal (https://orcid.org/0000-0003-3573-9203) was a national representative.
Shouichi Fujimoto (https://orcid.org/0000-0003-2854-8169) was a national representative.
Carmel Hawley (https://orcid.org/0000-0002-1392-5649) was a national representative.
Nader Khalidi (https://orcid.org/0000-0002-8270-2617) was a national representative.
Rachel Jones contributed significantly to the recruitment of trial participants.
Oliver Flossmann (https://orcid.org/0000-0001-5976-3609) contributed significantly to the recruitment of trial participants.
Ron Wald (https://orcid.org/0000-0003-4411-8169) contributed significantly to the recruitment of trial participants.
Louis Girard (https://orcid.org/0000-0002-9044-008X) contributed significantly to the recruitment of trial participants.
Adeera Levin (https://orcid.org/0000-0003-2438-8401) contributed significantly to the recruitment of trial participants.
Gina Gregorini (https://orcid.org/0000-0002-8404-3862) contributed significantly to the recruitment of trial participants and was a national representative.
Lorraine Harper (https://orcid.org/0000-0003-1343-9234) contributed significantly to the recruitment of trial participants.
William Clark (https://orcid.org/0000-0003-3294-739X) contributed significantly to the recruitment of trial participants.
Christian Pagnoux (https://orcid.org/0000-0001-6287-9549) contributed significantly to the recruitment of trial participants.
Ulrich Specks (https://orcid.org/0000-0002-6559-1206) contributed significantly to the recruitment of trial participants.
Lucy Smyth (https://orcid.org/0000-0001-6814-4877) contributed significantly to the recruitment of trial participants.
Toshiko Ito-Ihara (https://orcid.org/0000-0001-7152-3026) was responsible for all aspects of trial management.
Janak de Zoysa (https://orcid.org/0000-0001-7528-9230) was a national representative.
Biljana Brezina (https://orcid.org/0000-0001-5060-5604) was responsible for all aspects of trial management.
Andrea Mazzetti (https://orcid.org/0000-0002-8889-092X) was responsible for all aspects of trial management.
Carol A McAlear (https://orcid.org/0000-0002-0016-6866) was responsible for all aspects of trial management.
Donna Reidlinger (https://orcid.org/0000-0002-3365-9923) was responsible for all aspects of trial management.
Samir Mehta (https://orcid.org/0000-0001-7096-513X) was the trial statistician, and undertook both the interim and final analyses.
Natalie Ives (https://orcid.org/0000-0002-1664-7541) provided senior clinical trials unit oversight of the data management and statistical analyses of the trial.
Elizabeth A Brettell (https://orcid.org/0000-0002-0669-3085) was responsible for overseeing the data management of the trial in BCTU.
Hugh Jarrett (https://orcid.org/0000-0001-7374-3700) was responsible for overseeing the data management of the trial in BCTU.
Keith Wheatley (https://orcid.org/0000-0003-0792-580X) contributed to the design of the study.
Elizabeth Broadhurst (https://orcid.org/0000-0001-8464-4353) was responsible for all aspects of trial management.
Alina Casian (https://orcid.org/0000-0001-5822-593X) contributed significantly to the recruitment of trial participants, the study design and set-up, and trial management.
Charles D Pusey (https://orcid.org/0000-0002-1424-3906) contributed significantly to the recruitment of trial participants, the study design and set-up, and trial management.
Andrea Vaulks was responsible for all aspects of trial management.
Publications
Walsh M, Merkel PA, Peh CA, Szpirt W, Guillevin L, Pusey CD, et al. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol for a randomized controlled trial. Trials 2013;14:73.
Walsh M, Merkel PA, Peh CA, Szpirt W, Puéchal X, Fujimoto S, et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 2020;382:622–31.
Ito-Ihara T, Fujimoto S, Suzuki K, Endo T, Muso E, on behalf of the PEXIVAS-JP Group. Apheresis therapy for ANCA-associated vasculitis – plasma exchange and glucocorticoids in anti-neutrophil cytoplasm antibody associated vasculitis: a randomized controlled trial (PEXIVAS). Japanese J Apheresis 2015;34:120–5.
Data-sharing statement
The full database will be placed on a public server hosted by the National Institutes of Health (Bethseda, MD, USA). All queries should be submitted to the corresponding author.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- Jayne DRW, Merkel PA, Walsh M. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis: an international randomized controlled trial 2009. https://fundingawards.nihr.ac.uk/award/08/56/04.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition?. Semin Arthritis Rheum 1995;25:28-34. https://doi.org/10.1016/S0049-0172(95)80015-8.
- Frohnert PP, Sheps SG. Long-term follow-up study of periarteritis nodosa. Am J Med 1967;43:8-14. https://doi.org/10.1016/0002-9343(67)90144-1.
- Rota S, Rambaldi A, Gaspari F, Noris M, Daina E, Benigni A, et al. Methylprednisolone dosage effects on peripheral lymphocyte subpopulations and eicosanoid synthesis. Kidney Int 1992;42:981-90. https://doi.org/10.1038/ki.1992.377.
- Irakam A, Miskolci V, Vancurova I, Davidson D. Dose-related inhibition of proinflammatory cytokine release from neutrophils of the newborn by dexamethasone, betamethasone, and hydrocortisone. Biol Neonate 2002;82:89-95. https://doi.org/10.1159/000063094.
- Saag KG, Koehnke R, Caldwell JR, Brasington R, Burmeister LF, Zimmerman B, et al. Low dose long-term corticosteroid therapy in rheumatoid arthritis: an analysis of serious adverse events. Am J Med 1994;96:115-23. https://doi.org/10.1016/0002-9343(94)90131-7.
- Wolfe F, Caplan L, Michaud K. Treatment for rheumatoid arthritis and the risk of hospitalization for pneumonia: associations with prednisone, disease-modifying antirheumatic drugs, and anti-tumor necrosis factor therapy. Arthritis Rheum 2006;54:628-34. https://doi.org/10.1002/art.21568.
- Booth AD, Almond MK, Burns A, Ellis P, Gaskin G, Neild GH, et al. Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis 2003;41:776-84. https://doi.org/10.1016/S0272-6386(03)00025-8.
- Koldingsnes W, Nossent H. Epidemiology of Wegener’s granulomatosis in northern Norway. Arthritis Rheum 2000;43:2481-7. https://doi.org/10.1002/1529-0131(200011)43:11<2481::AID-ANR15>3.0.CO;2-6.
- De Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150:670-80. https://doi.org/10.7326/0003-4819-150-10-200905190-00004.
- Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniené J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349:36-44. https://doi.org/10.1056/NEJMoa020286.
- De Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52:2461-9. https://doi.org/10.1002/art.21142.
- Walsh M, Merkel PA, Peh CA, Szpirt W, Guillevin L, Pusey CD, et al. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol for a randomized controlled trial. Trials 2013;14. https://doi.org/10.1186/1745-6215-14-73.
- Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007;18:2180-8. https://doi.org/10.1681/ASN.2007010090.
- Lockwood CM, Pinching AJ, Sweny P, Rees AJ, Pussell B, Uff J, et al. Plasma-exchange and immunosuppression in the treatment of fulminating immune-complex crescentic nephritis. Lancet 1977;1:63-7. https://doi.org/10.1016/S0140-6736(77)91079-0.
- Glöckner WM, Dienst C, Kindler J, Sieberth HG. Plasma exchange in rapidly progressive glomerulonephritis (author’s transl). Dtsch Med Wochenschr 1981;106:1616-20. https://doi.org/10.1055/s-2008-1070565.
- Pusey CD, Rees AJ, Evans DJ, Peters DK, Lockwood CM. Plasma exchange in focal necrotizing glomerulonephritis without anti-GBM antibodies. Kidney Int 1991;40:757-63. https://doi.org/10.1038/ki.1991.272.
- Cole E, Cattran D, Magil A, Greenwood C, Churchill D, Sutton D, et al. A prospective randomized trial of plasma exchange as additive therapy in idiopathic crescentic glomerulonephritis: the Canadian Apheresis Study Group. Am J Kidney Dis 1992;20:261-9. https://doi.org/10.1016/S0272-6386(12)80699-8.
- de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, Jayne DR, Gaskin G, Rasmussen N, et al. Clinical and histologic determinants of renal outcome in ANCA-associated vasculitis: a prospective analysis of 100 patients with severe renal involvement. J Am Soc Nephrol 2006;17:2264-74. https://doi.org/10.1681/ASN.2005080870.
- Yamagata K, Hirayama K, Mase K, Yamaguchi N, Kobayashi M, Takahashi H, et al. Apheresis for MPO-ANCA-associated RPGN-indications and efficacy: lessons learned from Japan nationwide survey of RPGN. J Clin Apher 2005;20:244-51. https://doi.org/10.1002/jca.20035.
- Walsh M, Catapano F, Szpirt W, Thorlund K, Bruchfeld A, Guillevin L, et al. Plasma exchange for renal vasculitis and idiopathic rapidly progressive glomerulonephritis: a meta-analysis. Am J Kidney Dis 2011;57:566-74. https://doi.org/10.1053/j.ajkd.2010.10.049.
- Pickering G, Bywaters EGL, Danielli JF, Gell PG, Kellgren JH, Long DA, et al. Treatment of polyarteritis nodosa with cortisone: results after three years: report to the Medical Research Council by the Collagen Diseases and Hypersensitivity Panel. Br Med J 1960;1:1399-400. https://doi.org/10.1136/bmj.1.5183.1399.
- Jayne D, Rasmussen N. Twenty-five years of European Union collaboration in ANCA-associated vasculitis research. Nephrol Dial Transplant 2015;30:i1-7. https://doi.org/10.1093/ndt/gfv060.
- Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group . Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med 2005;352:351-61. https://doi.org/10.1056/NEJMoa041884.
- Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363:221-32. https://doi.org/10.1056/NEJMoa0909905.
- Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol 2017;28:2756-67. https://doi.org/10.1681/ASN.2016111179.
- Stone JH, Hoffman GS, Merkel PA, Min YI, Uhlfelder ML, Hellmann DB, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score: International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum 2001;44:912-20. https://doi.org/10.1002/1529-0131(200104)44:4<912::AID-ANR148>3.0.CO;2-5.
- Seo P, Min YI, Holbrook JT, Hoffman GS, Merkel PA, Spiera R, et al. Damage caused by Wegener’s granulomatosis and its treatment: prospective data from the Wegener’s Granulomatosis Etanercept Trial (WGET). Arthritis Rheum 2005;52:2168-78. https://doi.org/10.1002/art.21117.
- Jayne DRW, Merkel PA, Schall TJ, Bekker P. ADVOCATE Study Group . Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med 2021;384:599-60. https://doi.org/10.1056/NEJMoa2023386.
- Xiao H, Hu P, Falk RJ, Jennette C. Overview of the pathogenesis of ANCA-associated vasculitis. Kidney Dis 2016;1:205-5. https://doi.org/10.1159/000442323.
- Piaggio G, Elbourne DR, Altman DG, Pocock SJ, Evans SJ. CONSORT Group . Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement. JAMA 2006;295:1152-60.
- Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis 2012;71:955-60. https://doi.org/10.1136/annrheumdis-2011-200477.
- Walsh M, Casian A, Flossmann O, Westman K, Höglund P, Pusey C, et al. Long-term follow-up of patients with severe ANCA-associated vasculitis comparing plasma exchange to intravenous methylprednisolone treatment is unclear. Kidney Int 2013;84:397-402. https://doi.org/10.1038/ki.2013.131.
- Hilhorst M, Wilde B, van Paassen P, Winkens B, van Breda Vriesman P, Cohen Tervaert JW. Limburg Renal Registry . Improved outcome in anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis: a 30-year follow-up study. Nephrol Dial Transplant 2013;28:373-9. https://doi.org/10.1093/ndt/gfs428.
- Szpirt WM, Heaf JG, Petersen J. Plasma exchange for induction and cyclosporine A for maintenance of remission in Wegener’s granulomatosis – a clinical randomized controlled trial. Nephrol Dial Transplant 2011;26:206-13. https://doi.org/10.1093/ndt/gfq360.
- Cartin-Ceba R, Diaz-Caballero L, Al-Qadi MO, Tryfon S, Fervenza FC, Ytterberg SR, et al. Diffuse alveolar hemorrhage secondary to antineutrophil cytoplasmic antibody-associated vasculitis: predictors of respiratory failure and clinical outcomes. Arthritis Rheumatol 2016;68:1467-76. https://doi.org/10.1002/art.39562.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002;39:42-7. https://doi.org/10.1053/ajkd.2002.29876.
- Hruskova Z, Casian AL, Konopasek P, Svobodova B, Frausova D, Lanska V, et al. Long-term outcome of severe alveolar haemorrhage in ANCA-associated vasculitis: a retrospective cohort study. Scand J Rheumatol 2013;42:211-14. https://doi.org/10.3109/03009742.2012.754939.
- Casian A, Jayne D. Management of alveolar hemorrhage in lung vasculitides. Semin Respir Crit Care Med 2011;32:335-45. https://doi.org/10.1055/s-0031-1279830.
- Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002;110:955-63. https://doi.org/10.1172/JCI15918.
- Esnault VL, Soleimani B, Keogan MT, Brownlee AA, Jayne DR, Lockwood CM. Association of IgM with IgG ANCA in patients presenting with pulmonary hemorrhage. Kidney Int 1992;41:1304-10. https://doi.org/10.1038/ki.1992.194.
- Pagnoux C, Quéméneur T, Ninet J, Diot E, Kyndt X, de Wazières B, et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol 2015;67:1117-27. https://doi.org/10.1002/art.39011.
- Zeher M, Doria A, Lan J, Aroca G, Jayne D, Boletis I, et al. Efficacy and safety of enteric-coated mycophenolate sodium in combination with two glucocorticoid regimens for the treatment of active lupus nephritis. Lupus 2011;20:1484-93. https://doi.org/10.1177/0961203311418269.
- Miloslavsky EM, Naden RP, Bijlsma JW, Brogan PA, Brown ES, Brunetta P, et al. Development of a Glucocorticoid Toxicity Index (GTI) using multicriteria decision analysis. Ann Rheum Dis 2017;76:543-6. https://doi.org/10.1136/annrheumdis-2016-210002.
List of abbreviations
- AAV
- anti-neutrophil cytoplasm antibody-associated vasculitis
- ANCA
- anti-neutrophil cytoplasm antibody
- BCTU
- Birmingham Clinical Trials Unit
- BVAS/WG
- Birmingham Vasculitis Activity Score/Wegener’s Granulomatosis version
- CDA
- Combined Damage Assessment Index
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CYC
- cyclophosphamide
- DMC
- Data Monitoring Committee
- eGFR
- estimated glomerular filtration rate
- EQ-5D
- EuroQoL-5 Dimensions
- ESRD
- end-stage renal disease
- EUVAS
- European Vasculitis Study Group
- GC
- glucocorticoid
- GFR
- glomerular filtration rate
- GPA
- granulomatosis with polyangiitis
- HR
- hazard ratio
- IgG
- immunoglobulin G
- IgM
- immunoglobulin M
- IRR
- incidence rate ratio
- ITT
- intention to treat
- i.v.
- intravenous
- MEPEX
- Methylprednisolone versus Plasma Exchange
- MPA
- microscopic polyangiitis
- MPO
- myeloperoxidase
- PEXIVAS
- Plasma Exchange In VASculitis
- PLEX
- plasma exchange
- PR3
- proteinase 3
- RR
- relative risk
- SAE
- serious adverse event
- SF-36
- Short Form questionnaire-36 items
- TRALI
- transfusion-associated acute lung injury
- VCRC
- Vasculitis Clinical Research Consortium