

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 15/35/03. The contractual start date was in June 2016. The draft report began editorial review in September 2021 and was accepted for publication in March 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2022 Tesfaye et al. This work was produced by Tesfaye et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2022 Tesfaye et al.
Chapter 1 Introduction
Parts of this report have been reproduced with permission from Tesfaye et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Parts of this chapter have been reproduced from the published OPTION-DM (Optimal Pathway for TreatIng neurOpathic paiN in Diabetes Mellitus) protocol. 2 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text. 3
Scientific background
There are currently 3.9 million people in the UK with a diagnosis of diabetes and, if the numbers continue to increase at the current rate, it is expected that this will increase to 5.3 million people by 2025. 4 Diabetic peripheral neuropathic pain (DPNP) is a serious complication affecting up to 20–26% of these patients. 5,6 With the prevalence of diabetes set to increase by epidemic proportions over the next decade, DPNP will pose a major treatment challenge. 7,8
Diabetic peripheral neuropathic pain causes burning, deep aching and ‘electric shock’-like lancinating (also described as ‘stabbing or knife-like’) pains; contact pain, often with day-time clothes and bedclothes (allodynia); pain on walking, often described as ‘walking barefoot on marbles’ or ‘walking barefoot on hot sand’; sensations of heat or cold in the feet; a persistent achy feeling in the feet and cramp-like sensations in the legs. 8 With advanced disease, the pain can extend above the feet and may involve the whole of the legs. When this is the case, then there is often upper limb involvement also. Moderate to severe unremitting lower limb pain is present in over 70% of sufferers6,9 and can cause insomnia, poor quality of life (QoL), unemployment and depression. 10–13
The mainstay of treatment for DPNP is pharmacotherapy. The National Institute for Health and Care Excellence (NICE) clinical guideline 17314 recommends a choice of amitriptyline, duloxetine, pregabalin or gabapentin as initial treatment. All are licensed treatments for DPNP, except amitriptyline, which has been used off-licence for more than 25 years. There is moderate evidence for the efficacy of each drug based on Cochrane reviews15–18 and meta-analyses,19–21 but the best we can hope for with any monotherapy is 50% pain relief in 50% of patients. 14 This is often accompanied by side effects (dry mouth, constipation, sedation, dizziness, falls, nausea, oedema, etc.) in around 10–20% of patients, depending on dose. NICE recommends combination treatment if initial treatment is not effective (the majority). 14 However, as NICE points out, recommendations are not based on robust evidence because (1) there are few well-designed head-to-head studies comparing the first-line drugs and their combinations, (2) most studies were flawed with inadequate power, inappropriate end points or short duration of follow-up, and (3) many randomised controlled trials (RCTs) lacked appropriate health-related quality of life (HRQoL) measures, including functionality, and failed to measure the impact of drug-related adverse effects on health economics and QoL. 14 A RCT is, therefore, needed to address these deficiencies.
Rationale for research
Recent Cochrane reviews,15–18 meta-analyses,19–21 consensus guidelines22–24 and NICE clinical guidance 17314 support the choice of amitriptyline (25–75 mg/day), duloxetine (60–120 mg/day) and the α2δ agonists pregabalin (300–600 mg/day) and gabapentin (0.9–3.6 g/day) as first-line agents for DPNP. However, these recommendations are not based on solid evidence.
Comparator studies
Two small randomised double-blind crossover short-duration (5 weeks’ follow-up) studies compared amitriptyline with pregabalin (n = 51)25 and amitriptyline with duloxetine (n = 58)26 in DPNP. The studies were underpowered to detect any differences in pain relief between the drugs. Another underpowered, and short (4 weeks), RCT compared amitriptyline (n = 27), duloxetine (n = 28) and pregabalin (n = 28),27 and found no differences between the groups. The lack of head-to-head studies led to an indirect comparison of the efficacy and tolerability of duloxetine with pregabalin, using placebo as a common comparator, but this comparison found no difference in 24-hour pain severity between the two. 28
Combination studies
Low-dose combination therapy with gabapentin and morphine was more effective than higher doses of either,29 although, curiously, there was no difference between placebo and gabapentin. 30 Finally, the COMBO-DN (COmbination vs Monotherapy of pregaBalin and dulOxetine in Diabetic Neuropathy) study,31 which, to the best of our knowledge, is the largest combination study in DPNP (n = 804), assessed whether or not combining standard doses of duloxetine (60 mg/day) and pregabalin (300 mg/day) was superior to maximum doses of either. The COMBO-DN study31 also compared head to head the standard doses of duloxetine and pregabalin and found no difference in the change in 24-hour average pain or number of adverse events (AEs) between standard-dose combination therapy and high-dose monotherapy. 31 Although the standard dose of duloxetine was superior to pregabalin, there was equivalent efficacy with pregabalin at higher doses. 31
Published economic evaluations
To date, no trial has provided conclusive evidence regarding the cost-effectiveness of amitriptyline, duloxetine and pregabalin for DPNP. Wu et al. 32 conducted a cost–utility analysis of duloxetine compared with usual care as part of an open-label study extension and concluded that duloxetine was a dominant treatment (i.e. more effective and less costly). However, methodological issues limit the generalisability of this conclusion. Beard et al. 33 developed a short-term decision tree to estimate alternative treatment sequences that include duloxetine. A standard treatment sequence was defined as amitriptyline, gabapentin and then opioid-related treatment. Duloxetine was evaluated as a first-, second-, third- or fourth-line therapy. First-line use of duloxetine was both the most effective and most cost-effective treatment strategy. O’Connor et al. 34 compared the costs and quality-adjusted life-years (QALYs) of first-line desipramine, duloxetine, gabapentin and pregabalin, and concluded that desipramine and duloxetine may be more cost-effective than gabapentin or pregabalin for first-line treatment of DPNP. In 2012, de Salas Cansado et al. 35 conducted an economic evaluation of pregabalin compared with usual care in the management of community-treated patients with refractory painful diabetic peripheral neuropathy in Spain. de Salas Cansado et al. 35 compared costs and QALYs from a Spanish NHS and societal perspective and concluded that pregabalin may be cost-effective. The limited published evidence highlights the need for a definitive evaluation of the costs and health benefits of alternative treatment sequences for DPNP. This evidence would inform NHS guidance and commissioning and ensure an efficient use of limited health resources.
In summary, there is a lack of head-to-head studies of current drugs and their combinations, highlighting the need for carefully designed RCTs, involving patients recruited from both primary and secondary care, to identify the most cost-effective and best-tolerated treatment pathway for DPNP.
Intervention
The OPTION-DM trial was a randomised crossover trial of treatment pathways to evaluate the superiority of at least one pathway [i.e. amitriptyline supplemented with pregabalin (A-P), duloxetine supplemented with pregabalin (D-P) and pregabalin supplemented with amitriptyline (P-A)] in reducing the 7-day average 24-hour pain in patients with DPNP.
Each treatment pathway consisted of two periods (i.e. 6 weeks’ monotherapy followed by 10 weeks’ combination therapy).
Why exclude gabapentin?
The rationale for not studying two α2δ agonists (i.e. pregabalin and gabapentin) is that:
-
The evidence for gabapentin is derived from only one reasonable-quality RCT with a 4-week titration and a 4-week treatment phase36 (vs. seven RCTs for pregabalin and evidence supported by meta-analysis19).
-
Gabapentin is a thrice-daily drug.
-
In contrast to pregabalin, the pharmacokinetics of gabapentin are not linear, and a long titration period of up to 2 months23 is necessary to avoid toxicity.
Why examine treatment pathways?
Although a head-to-head RCT of individual drugs and a separate RCT of combination therapy could be designed, in our opinion an examination of a treatment pathway as a whole is the most efficient and applicable to current UK clinical practice. This is because most patients are started on monotherapy and will require a second agent added in combination within a few months. Only a minority of patients will either have massive benefit from monotherapy [i.e. 24-hour pain scores of < 3 points on a Numeric Rating Scale (NRS)] and will not need another agent or will not tolerate monotherapy (or monotherapy will be completely ineffective) and will be switched to another agent. Therefore, the OPTION-DM trial, which examined the whole treatment pathway, captured more clinically relevant outcomes than artificially designed head-to-head monotherapy or combination studies. Hence, the outcomes of this study will be readily generalisable to current UK clinical practice.
Which treatment pathways?
The three treatment pathways studied in the OPTION-DM trial were (1) A-P, (2) P-A and (3) D-P.
We did not examine the pathway of pregabalin supplemented by duloxetine because of the COMBO-DN study findings. 31 In the COMBO-DN study, there was no difference in pain reduction if pregabalin was added to duloxetine, or vice versa. 31 However, duloxetine was superior to pregabalin as an initial treatment, is a once daily preparation and is also the cheaper option in the UK. There is, therefore, a good rationale for starting patients on duloxetine and then adding pregabalin in combination. Finally, as both amitriptyline and duloxetine are antidepressants, there was little rationale for combining both.
Efficient design with 16-week treatment pathways
This was an efficiently designed head-to-head crossover RCT,37 with each patient undergoing all pathways. The duration of monotherapy in each pathway was at least 6 weeks, which is an adequate duration to assess treatment effect and whether or not combination therapy is indicated. 23,37 The subsequent 10-week combination therapy in patients with partial benefit from monotherapy is adequate to assess stabilised treatment outcomes. 31
Objectives
The main aims of this study were to determine the most clinically beneficial, cost-effective and tolerated treatment pathway for patients with DPNP.
Efficacy objectives
The efficacy objectives were to evaluate if at least one of the three pathways is superior to the other pathways in improving self-reported pain, as measured by a NRS (the primary outcome), tolerability, QoL and cost-effectiveness over a 16-week treatment period. The secondary efficacy objective was to evaluate if at least one monotherapy is superior to a different monotherapy in improving the same outcomes.
Safety objective
The safety objective was to describe AEs and serious adverse events (SAEs) data (summarised both at patient level and event level) between the different treatment pathways.
Subgroup study objectives
We conducted a subgroup study to investigate if patient phenotypes (demography, type of pain, assessments of mood, sleep, etc.) predict response to treatment.
Chapter 2 Methods
Sections of this chapter have been reproduced from the published OPTION-DM trial protocol. 2 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text. 3
Study design
This was a randomised crossover trial of treatment pathways to evaluate the superiority of at least one pathway (i.e. A-P, D-P and P-A) in reducing the 7-day average 24-hour pain in patients with DPNP. Eligible patients were randomised to one of six treatment sequences, with equal allocation to sequences (1 : 1 : 1 : 1 : 1 : 1). Each sequence examined all three treatment pathways in random order. Each treatment pathway consisted of two phases (i.e. 6 weeks’ monotherapy followed by 10 weeks’ monotherapy or 10 weeks’ combination therapy based on response to treatment).
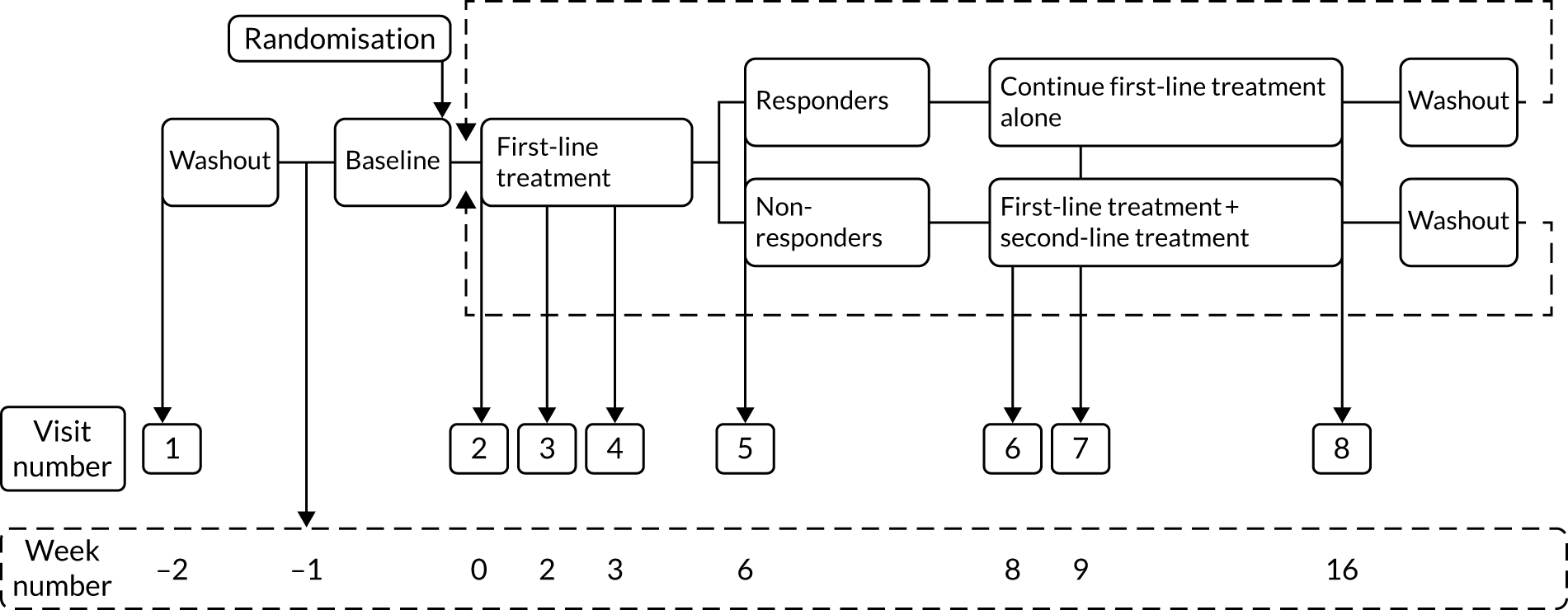
Figure 1 shows a schematic representation of the study schedule. Following the screening, consent and initial washout visits (i.e. weeks –2 to 0), the visits from week 0 to week 16 were repeated until all three treatment pathways were completed. Face-to-face assessments were completed at the week numbers indicated in Figure 1. Weekly telephone calls were carried out between study visits.
FIGURE 1.
Study schedule.
Trial approvals and registration
The trial was approved by the Yorkshire and the Humber – Sheffield Research Ethics Committee (reference 16/YH/0459) on 9 December 2016. The Medicines and Healthcare products Regulatory Agency issued the clinical trial authorisation on 23 June 2017 (reference 21304/0262/001-0001, EudraCT reference 2016-003146-89).
The trial was registered as ISRCTN17545443.
Participant eligibility criteria
Inclusion criteria
Participants were required to meet all of the following inclusion criteria:
-
Age ≥ 18 years.
-
Neuropathic pain affecting feet and/or hands for at least 3 months or taking pain medication for neuropathic pain for at least 3 months.
-
Bilateral distal symmetrical neuropathic pain confirmed by the Douleur Neuropathique 4 (DN4)38 questionnaire at screening visit. The participant was eligible if four or more questions were answered as ‘yes’.
-
Bilateral distal symmetrical polyneuropathy confirmed by a modified Toronto Clinical Neuropathy Score (mTCNS)39 of > 5 points at screening visit.
-
Stable glycaemic control [i.e. glycated haemoglobin (HbA1c) < 108 mmol/mol].
-
A mean total pain intensity of at least 4 points on an 11-point NRS (with 0 being ‘no pain’ and 10 ‘worst pain imaginable’) during 1 week off pain medications (i.e. the baseline period). Patients could be invited to attend the randomisation visit sooner if it was clear that their mean pain score for the week was ≥ 4, that is, as soon as the total sum of the pain scores was ≥ 28 points (e.g. randomisation could take place after 3 days if a patient scored 10 on each of the first 3 days of monitoring). This was to minimise the length of time patients remained off neuropathic pain treatments.
-
Patient is willing and able to comply with all the study requirements and be available for the duration of the study.
-
Patient is willing to discontinue current neuropathic pain-relieving medications.
-
Informed consent form for study participation signed by participant.
Exclusion criteria
Patients were not eligible for the study if they met any of the following exclusion criteria:
-
Non-diabetic symmetrical polyneuropathies.
-
History of alcohol/substance abuse that would, in the opinion of the investigator, impair the patient’s ability to take part in the study.
-
History of severe psychiatric illnesses that would, in the opinion of the investigator, impair the patient’s ability to take part in the study.
-
History of epilepsy.
-
Contraindications to study medications.
-
Pregnancy/breastfeeding or planning pregnancy during the course of the study.
-
Use of prohibited concomitant treatment that could not be discontinued with the exception of prior concomitant and safe use of selective serotonin reuptake inhibitors (SSRIs) with study medication (duloxetine and/or amitriptyline). Note that concomitant use of citalopram was not permitted.
-
Use of a high-dose morphine equivalent (> 100 mg/day).
-
Liver disease [i.e. aspartate aminotransferase (AST)/alanine aminotransferase (ALT) two or more times the upper limit of normal].
-
Significant renal impairment [i.e. an estimated glomerular filtration rate (eGFR) of < 30 ml/minute/1.73 m2].
-
Heart failure (i.e. New York Heart Association ≥ class III).
-
Clinically significant cardiac arrhythmias on 12-lead electrocardiogram, current history of arrhythmia, second- or third-degree heart block or left bundle branch block (patients with right bundle branch block or first-degree heart block may be included following discussion with cardiology team).
-
Patients with a recent myocardial infarction (< 6 months prior to randomisation).
-
Symptomatic postural hypotension that, in the opinion of the investigator, is clinically significant and would be a contraindication to the study medication.
-
Prostatic hypertrophy or urinary retention to an extent that would, in the opinion of the investigators, be a contraindication to the study medication.
-
Patients with other painful medical conditions where the intensity of the pain is significantly more severe than their DPNP (patients were not excluded if the pain was transient in nature).
-
Any suicide risk, as judged by the investigator or as defined by a score of ≥ 2 points on the Suicide Risk Questionnaire.
-
Significant language barriers that are likely to affect the participant’s understanding of the medication schedule or ability to complete outcome questionnaires.
-
Concurrent participation in another clinical trial of an investigational medicinal product (IMP).
-
Major amputations of the lower limbs.
-
Foot ulcers, only if, in the opinion of the local principal investigator (PI), they were likely to have a confounding/detrimental effect on study primary outcome or participation (e.g. localised foot pain from the ulcer site).
Withdrawals
Participants could choose to withdraw from the trial treatment or follow-up at any time. The local research team could also choose to discontinue the study treatment for clinical reasons or if the participant’s condition changed following randomisation so that they met one or more of the exclusion criteria. Outcome data were collected up to the end of the current treatment pathway, if possible. Data already collected up to the point of withdrawal were kept.
Settings and locations where the data were collected
Initially, the trial planned to recruit participants from eight secondary care hospital sites across the UK. Additional sites were added during the trial, and a total of 21 sites were opened to recruitment during the course of the trial.
Potential participants were identified directly through the database and via clinics at participating centres, as well as via participant identification centres, podiatry clinics and general practice mail-outs. Recruitment strategies differed between sites to reflect the local organisational structure at each site.
Participant treatment and outcome data collection was carried out by the recruiting hospital site. All research activity at the site was carried out by hospital employees trained in OPTION-DM trial processes.
Screening, assessment of eligibility and consent
Prior to any study procedures being completed, participants were required to give written informed consent for the study. The participant was given sufficient time to ask questions, consider the study and discuss it with family/friends prior to providing consent, which was taken by medically qualified site investigators.
Eligibility for the study was assessed by the local investigator. Participants were required to stop all existing treatment for neuropathic pain, except paracetamol, if applicable. Treatments were tapered, usually over a period of 3 days followed by a 4-day washout period. Participants then entered the baseline period and the pain scores collected during this period were used to determine eligibility.
Randomisation
Randomisation was completed using the Sheffield Clinical Trials Research Unit (CTRU) online randomisation system (SCRAM). Participants were assigned to one of six sequences (allocation 1 : 1 : 1 : 1 : 1 : 1) based on a predetermined randomisation schedule and stratified by site using permuted blocks of sizes 6 or 12. The trial statistician created the randomisation schedule. Each sequence consisted of three treatment pathways given in random order. The three treatment pathways were:
-
A-P (i.e. first-line amitriptyline, second-line pregabalin)
-
D-P (i.e. first-line duloxetine, second-line pregabalin)
-
P-A (i.e. first-line pregabalin, second-line amitriptyline).
Members of the research teams at participating sites were granted access to the SCRAM system with individual usernames and passwords. These members of staff were responsible for performing the randomisation process once eligibility had been confirmed by the local investigator. After the randomisation was completed, the pharmacy department was informed that a new participant had been randomised. A member of the pharmacy team then accessed the SCRAM system to obtain the unblinded treatment allocation for the participant to allow dispensing. Pharmacy staff were assigned a different level of access to SCRAM to ensure that they were the only members of the local site team with access to the treatment allocation information.
Interventions
Investigational medicinal product details
Study treatment was supplied in capsules with dose levels as follows:
-
amitriptyline – 25-mg capsules
-
amitriptyline – 50-mg capsules
-
duloxetine – 30-mg capsules
-
pregabalin – 75-mg capsules
-
pregabalin – 150-mg capsules
-
matching placebo capsules.
Capsules were supplied in bottles containing nine, 23 or 51 capsules.
Participants were instructed to take medication orally before breakfast and at bedtime. Participants on dose levels 1 or 2 took one tablet in the morning and one tablet in the evening. Participants on dose level 3 took two tablets in the morning and two tablets in the evening. The placebo ensured that the same dosing schedule could be followed for each study drug. For example, a participant on dose level 1 of amitriptyline would take one placebo capsule in the morning and one 25-mg amitriptyline capsule in the evening. A participant on dose level 1 of standard dose pregabalin would take one 75-mg pregabalin capsule in the morning and one 75-mg pregabalin capsule in the evening. This ensured that the medication schedule appeared the same to participants, regardless of which medication they were currently taking.
The total daily dose of each drug was dependent on the dose level prescribed to the participant (Table 1). Participants were provided with clear instructions on the dosing schedule, and this was reinforced with written instructions and a medication diary.
| Dose level | Amitriptyline | Duloxetine | Pregabalin (standarda) | Pregabalin (reducedb) |
|---|---|---|---|---|
| 1 | ||||
| a.m. dose | 1 × placebo | 1 × placebo | 1 × 75 mg | 1 × 75 mg |
| p.m. dose | 1 × 25 mg | 1 × 30 mg | 1 × 75 mg | 1 × placebo |
| 2 | ||||
| a.m. dose | 1 × placebo | 1 × 30 mg | 1 × 150 mg | 1 × 75 mg |
| p.m. dose | 1 × 50 mg | 1 × 30 mg | 1 × 150 mg | 1 × 75 mg |
| 3 | ||||
| a.m. dose | 2 × placebo | 2 × 30 mg | 2 × 150 mg | 2 × 75 mg |
| p.m. dose | 1 × 25 mg &1 × 50 mg | 2 × 30 mg | 2 × 150 mg | 2 × 75 mg |
Dose titration
Participants were titrated to a maximum tolerated dose level on starting each new treatment. The schedule for dose escalation was the same in each pathway (Figure 2).
FIGURE 2.
Dosing and titration schedule for treatment pathways. Each pathway had two treatment phases, each with a 2-week initial titration period towards maximum tolerated dose. Participants continued on maximum tolerated maintenance dose of the drug from the first treatment phase for the duration of the second treatment phase. For patients with an eGFR of ≤ 60 ml/minute, the maximum pregabalin dose was 300 mg/day.
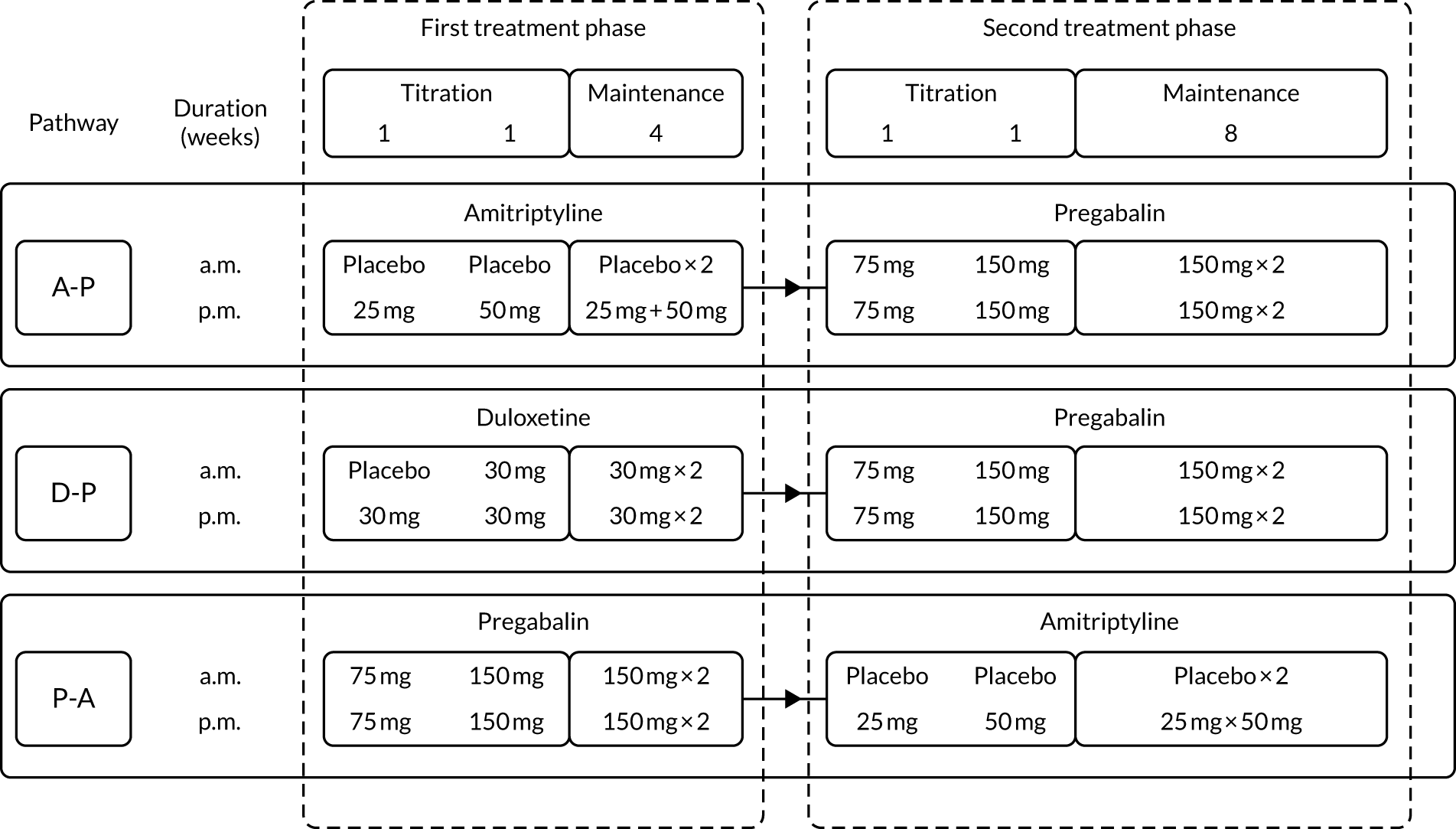
When a new treatment was started, all participants started at dose level 1 and the dose was escalated slowly, one dose level at a time, towards a maximum tolerated dose or maximum permitted dose, whichever was reached first (see Table 1). Dose titration decisions were based on treatment response (i.e. 24-hour pain NRS score), side effect profile and participant preference. Dose titrations were usually made during the first 2 weeks of a new treatment; however, investigators were permitted to make dose changes at any time if deemed necessary. At weekly intervals, the site research nurse evaluated the participant’s response to treatment and AEs and this information was used to guide dose titration.
Treatment response
Adequate pain relief was defined as a 24-hour pain NRS score of ≤ 3 for the purposes of dose titration decisions. Participants who experienced adequate pain relief at dose levels 1 or 2 did not have their dose escalated further.
Adverse events
Participants were asked to report all side effects. The local study team graded the side effects as mild, moderate or severe. The participant was asked to rate whether the side effects were tolerable or intolerable. If a side effect was rated as ‘tolerable’ and was non-severe, dose escalation was continued as indicated by the pain score assessment. However, sites were advised that if a patient was experiencing tolerable non-severe side effects, then it was also acceptable to maintain the current dose for a further week to allow side effects to improve before increasing the dose further. This decision was made by the local site team.
If side effects were severe or were rated as ‘intolerable’, then investigators considered reducing the dose by one dose level or discontinuing the medication based on the overall condition of the participant.
Participant preference
Participant preference was taken into account, where possible, when making dose titration decisions. However, the dose was not increased based on participant preference alone (i.e. the dose was increased only if the participant expressed a preference for an increase in dose and if this was indicated based on treatment response and side effect profile).
Treatment phases
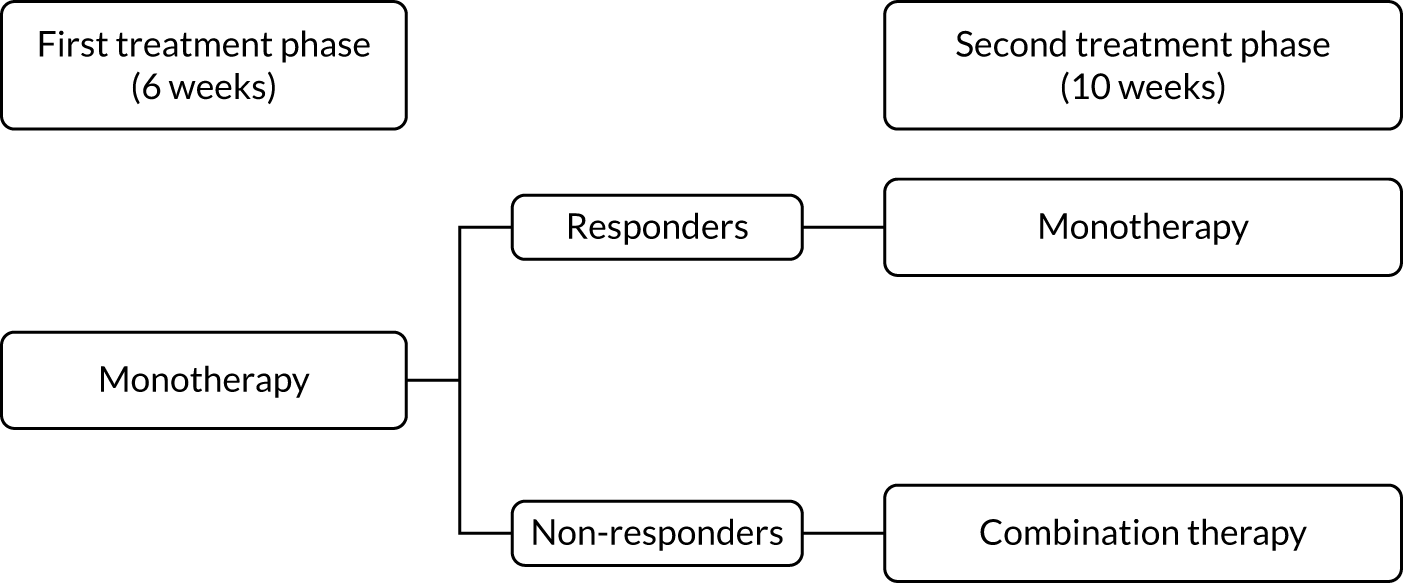
Each treatment pathway was split into two treatment phases, as shown in Figure 3.
FIGURE 3.
Two treatment phases per pathway.
First treatment phase
During the first treatment phase, participants received monotherapy with the first-line treatment in the pathway. This lasted for a total of 6 weeks.
Responder/non-responder assessment
At the end of the first treatment phase, a decision was made either to continue on monotherapy or to start combination therapy with the addition of the second-line treatment in the pathway. This decision was based on the 7-day average pain NRS score during the week preceding the week 6 study visit. Participants were divided into ‘responders’ (with a pain score of ≤ 3 points) and ‘non-responders’ (with a pain score of > 3 points). Responders continued on monotherapy and non-responders commenced combination therapy.
Second treatment phase
The second treatment phase lasted for a total of 10 weeks. Responders continued on first-line treatment as a monotherapy for the remainder of the pathway. Non-responders commenced combination therapy, with the addition of the second-line treatment in the pathway, for 10 weeks.
Taper doses
At the end of a treatment pathway, participants were provided with a taper dose of their current medication. Participants were instructed to take the taper dose for 3 days and then to stop study medication completely for 4 days before commencing the next pathway. The taper dose was one dose level below the maximum tolerated dose, as shown in Table 2.
| Maximum tolerated dose level | Tapered dose level |
|---|---|
| 1 | No taper dose required |
| 2 | 1 |
| 3 | 2 |
Where the patient experienced significant withdrawal side effects, the medication could be tapered down more gradually in accordance with the judgement of the investigator. However, the patient was still required to stop the medication completely for at least 4 days before starting the next pathway.
The first and second treatment phases were repeated until the participant had completed all three treatment pathways.
Permitted changes to the treatment schedule
To make the trial as pragmatic as possible, the following changes were permitted to the treatment schedule as needed:
-
If there was significant intolerance to first-line treatment, participants were permitted to switch to the second-line treatment in the pathway as a monotherapy. This change could be made immediately at any time and without the need to wash out first-line treatment.
-
Non-responders at week 6 who were on dose levels 1 or 2 of first-line treatment were permitted to increase the dose of first-line treatment rather than start combination treatment at the discretion of the local investigator.
-
Second-line treatment could be added as a combination therapy up to week 13 if needed (e.g. if the participant was a responder at week 6, but then, subsequently, became a non-responder).
-
For participants whose pain scores had not reduced at all at week 6, compared with baseline, or if their pain scores had increased first-line treatment was stopped and second-line treatment was started as a monotherapy for the remainder of the pathway.
-
Participants who did not tolerate one treatment pathway could start the next treatment pathway early. In this case, the taper dose was dispensed early and the next pathway started following the appropriate washout period.
Compliance with intervention
Treatment compliance was assessed by the local study team at each study visit via pill counts and this was recorded on a treatment compliance log within the study database. Issues with compliance were discussed with participants and, if needed, participants were re-educated on the study requirements.
Blinding and masking
The study was double-blinded, whereby participants and the local research team were blinded to treatment allocation, with the exception of the site pharmacist who was unblinded. Blinding was maintained with over-encapsulation and matching placebos. As the study drugs have different dosing schedules (e.g. amitriptyline is given once per day, whereas pregabalin is given twice per day), the placebos were used to ensure that the dosing schedule was identical across the three pathways, with dosing twice per day on all treatments. The IMP bottles were supplied with a tear-off label, which the centre’s pharmacist removed prior to dispensing to the participants. Participants and sites were aware of whether monotherapy or combination therapy had been prescribed and of the dose level. Unblinding was considered only in the event of a medical emergency where knowledge of the participant’s treatment allocation would change the clinical management. All participants were unblinded at the end of the study, when the final statistical report was completed.
Data collection and management
Data management was provided by the University of Sheffield CTRU who adhere to their own standard operating procedures relating to all aspects of data management, including data protection and archiving.
Participant confidentiality was respected at all times and the principles of the UK Data Protection Act40 were followed. All participants were assigned a unique study identification number at screening that linked all of the clinical information held about them on the study database. Data were collected on standardised questionnaires and study-specific case report forms (CRFs) and were entered onto the CTRU’s in-house data management system (Prospect). Access to Prospect was controlled by usernames and encrypted passwords, and a comprehensive privilege management feature was used to ensure that users had access to only the minimum number of data required to complete their tasks. This was used to restrict access to personal identifiable data. After data had been entered onto the database, electronic validation rules were applied on a regular basis and discrepancies were tracked and resolved. All entries and corrections were logged, with the person, date and time captured within the electronic audit trail.
Regular site monitoring visits occurred throughout the study and additional visits were undertaken where required. At these visits, the monitor reviewed activity to verify that the data were authentic, accurate and complete. Accurate and reliable data collection was assured by verification and cross-check of the CRFs against investigator’s records (i.e. source document verification). The study monitor contacted and visited sites regularly to inspect CRFs throughout the study to verify adherence to the protocol and the completeness, consistency and accuracy of the data being entered on the CRFs. Monitoring visits also included a pharmacy visit to review processes, documentation and accountability of study drugs. CTRU staff reviewed entered data for possible errors and missing data points. A central review of consent forms was also completed, and sites were requested to post consent forms to CTRU on an ongoing basis. CTRU reviewed pharmacy dispensing logs for some patients centrally.
Study records will be stored for 25 years after the completion of the study before being destroyed.
Outcome measures
The study evaluated the superiority of at least one pathway in reducing the 7-day average 24-hour pain in patients with diabetic neuropathy.
Primary end point
Difference between 7-day average 24-hour pain (evaluated at patient level) among pathways on an 11-point NRS (0 = no pain and 10 = worst pain imaginable), measured during the final follow-up week of the treatment cycle (i.e. week 16). NRS 24-hour average pain is now considered the gold standard for the assessment of neuropathic pain and has been employed in almost all well-designed neuropathic pain studies over the past 10 years. 19,28,37
Secondary end points
Efficacy
-
Difference in 7-day average 24-hour pain (evaluated at patient level) on an 11-point NRS at week 6 among monotherapies.
-
Difference in Short Form questionnaire-36 items (SF-36) physical mean scores (evaluated at patient level) at week 16 among pathways. 41
-
Difference in SF-36 physical mean scores (evaluated at patient level) at week 6 among pathways. 41
-
Difference in SF-36 mental mean scores (evaluated at patient level) at week 16 among pathways. 41
-
Difference in SF-36 mental mean scores (evaluated at patient level) at week 6 among pathways. 41
-
Difference in Hospital Anxiety and Depression Scale (HADS) mean anxiety scores (evaluated at patient level) at week 6 among pathways. 42
-
Difference in HADS mean anxiety scores (evaluated at patient level) at week 16 among pathways. 42
-
Difference in HADS mean depression scores (evaluated at patient level) at week 6 among pathways. 42
-
Difference in HADS mean depression scores (evaluated at patient level) at week 16 among pathways. 42
-
Difference in proportion of patients having treatment success (30%) at week 16 among pathways. Treatment success was defined as a reduction in 30% value at follow-up compared with baseline.
-
Difference in proportion of patients having treatment success (50%) at week 16 among pathways. Treatment success was defined as a reduction in 50% value at follow-up compared with baseline.
-
Difference in Brief Pain Inventory – Modified Short Form (BPI-MSF) measure of pain interference with function total score (evaluated at patient level) at week 6 among pathways. 43
-
Difference in BPI-MSF measure of pain interference with function total score (evaluated at patient level) at week 16 among pathways. 43
-
Difference in Insomnia Severity Index (ISI) total score (evaluated at patient level) at week 6 among pathways. 44
-
Difference in ISI total score (evaluated at patient level) at week 16 among pathways. 44
-
Difference in Patient Global Impression of Change (PGIC) (evaluated at patient level) at week 16 among pathways. 45
-
Difference in proportion of care pathway preferred by participants at week 50.
Cost-effectiveness
-
The EuroQol-5 Dimensions (EQ-5D) is a routinely used generic HRQoL instrument. It is the instrument preferred by NICE for assessing HRQoL, and the newer five-level instrument [i.e. EuroQol-5 Dimensions, five-level version (EQ-5D-5L)] is more sensitive than the original three-level version. 46
-
The Client Service Receipt Inventory (CSRI) is an instrument routinely used to capture health resource use and personal expenses. A modified version of the CSRI, where unnecessary questions were removed to reduce participant burden, was used. 47
Safety
-
The frequency and proportion of patients reporting at least one AE for each of the pathways. In addition, the relationship to intervention (i.e. definite, probable, possible, unlikely, unrelated or not assessable) was reported (frequency and proportion).
-
The frequency and proportion of AEs for each of the pathways.
-
A list of AEs for each of the pathways.
-
The frequency and proportion of patients reporting at least one SAE for each of the pathways. In addition, the following characteristics were summarised (frequency and proportion): intensity (i.e. mild, moderate or severe), relationship (i.e. definite, probable, possible, unlikely, unrelated or not assessable), whether or not a suspected unexpected serious adverse reaction (SUSAR), whether or not resulted in death.
-
Frequencies of SAEs for each of the pathways.
-
A list of SAEs for each of the pathways.
Subgroup
-
Neuropathic Pain Symptom Inventory (NPSI) questionnaire for subgroup analysis relating pain phenotype to treatment response. 48 There is emerging evidence that treatment response may be determined by a patient’s pain phenotype. 49–51 In particular, the following outcomes were evaluated:
-
Difference in ‘burning (superficial) spontaneous pain’ NPSI mean subscores (evaluated at patient level) at week 6 among pathways.
-
Difference in ‘burning (superficial) spontaneous pain’ NPSI mean subscores (evaluated at patient level) at week 16 among pathways.
-
Difference in ‘pressing (deep) spontaneous pain’ NPSI mean subscores (evaluated at patient level) at week 6 among pathways.
-
Difference in ‘pressing (deep) spontaneous pain’ NPSI mean subscores (evaluated at patient level) at week 16 among pathways.
-
Difference in ‘paroxysmal pain’ NPSI mean subscores (evaluated at patient level) at week 6 among pathways.
-
Difference in ‘paroxysmal pain’ NPSI mean subscores (evaluated at patient level) at week 16 among pathways.
-
Difference in ‘evoked pain’ NPSI mean subscores (evaluated at patient level) at week 6 among pathways.
-
Difference in ‘evoked pain’ NPSI mean subscores (evaluated at patient level) at week 16 among pathways.
-
Difference in ‘paresthesia/dysesthesia’ NPSI mean subscores (evaluated at patient level) at week 6 among pathways.
-
Difference in ‘paresthesia/dysesthesia’ NPSI mean subscores (evaluated at patient level) at week 16 among pathways.
-
Difference in NPSI mean total scores (evaluated at patient level) at week 6 among pathways.
-
Difference in NPSI mean total scores (evaluated at patient level) at week 16 among pathways.
-
Patient’s perceived tolerability
-
Difference in tolerability among pathways, evaluated at the patient level on an 11-point NRS at week 6.
-
Difference in tolerability among monotherapies, evaluated at the patient level on an 11-point NRS at week 6
Changes to subgroup analyses and exploratory analyses
The study had intended to characterise pain phenotype by the individual domains of the NPSI; however, a new classification system for pain based on the NPSI was used instead. 52 In addition, the following subgroups were investigated: age, pain score at baseline, anxiety and depression scores at baseline, previous medication and the COVID-19 lockdown restrictions. Additional analyses were performed to compare outcomes among patients on combination therapy against those who remained on monotherapy.
Economic evaluation
A within-trial economic evaluation was completed as part of the study to understand the relative cost-effectiveness of the three treatment pathways.
Data collection tools
A pro forma of each of the forms was available to be used as source documents if needed. These forms were provided to each site electronically, along with a paper copy of each form for reference.
Procedures for assessing efficacy
Numeric Rating Scale 24-hour average pain was assessed via pain diaries that were given to participants at each study visit. Participants were instructed to complete the diaries each morning during the study. Completed diaries were then collected at the subsequent visit. During the weekly telephone calls, the research nurses reminded participants to record their pain scores every day. Pain scores were also collected via daily text messages where participants had given additional consent for this.
Procedures for assessing safety
The following safety assessments were performed to assess safety:
-
Blood tests were performed at week 16 of each pathway.
-
Vital signs were assessed at week 16 of each pathway.
-
AEs were assessed during each study visit or telephone call.
-
Concomitant medications were reviewed during each study visit or telephone call.
Additional procedures for assessing neuropathic pain
The NPSI and BPI-MSF were completed to assess neuropathic pain.
Procedures for assessing quality of life, psychological well-being and health economics
The following questionnaires were completed to assess QoL:
-
ISI
-
HADS
-
SF-36
-
EQ-5D-5L
-
modified CSRI
-
Pain Catastrophizing Scale
-
Suicidal Risk Questionnaire
-
PGIC
-
Tolerability Scale.
Questionnaires could be completed during the visit. Alternatively, questionnaires could be posted to participants in advance of the visit and participants could bring the completed questionnaires when they attended their study visit. In the event that the participant forgot to bring the questionnaires or had not completed them, they were provided with another copy to complete during the visit.
Study-related case report forms
The following additional CRFs were completed by the investigators or delegates (i.e. sub-investigators delegated to consent or collect data) during the study:
-
pre-screening log
-
screening consent
-
demographics
-
medical history
-
mTCNS
-
DN4
-
previous medications
-
weekly contact form
-
pregnancy test
-
confirmation of eligibility
-
randomisation
-
treatment decisions
-
treatment compliance log
-
unblinding
-
unscheduled dose changes
-
pregnancy information
-
protocol non-compliance
-
intervention withdrawal
-
study completion discontinuation.
See Appendix 1 for the CRF completion schedule and Appendix 2 for the questionnaire completion schedule.
Ancillary substudies
Participants were given the opportunity to consent to blood sample collection for future research. This aspect of the study was optional, and participants could take part in the main study without consenting to the blood sample collection. Samples were obtained at the same time as other study blood samples (i.e. week –2 or week 16 of each pathway) and shipped directly to the central labs via Royal Mail (Royal Mail Group plc, London, UK).
Patient and public involvement
Patient and public involvement (PPI) representatives have been involved throughout the study, including involvement in the initial study design, as well as implementation and oversight. The Diabetes PPI Panel at Sheffield Teaching Hospitals NHS Foundation Trust reviewed the study at the proposal stage. The PPI representatives were supportive of the proposal, including the study design, and they contributed to the choice of end points for the study. The panel was later involved in the development of the patient information sheet, consent form and study medication diary, helping to ensure that the study documents were accessible for potential participants. The Trial Steering Committee (TSC) included a PPI representative who provided ongoing input into the oversight of the study. PPI representation on the TSC ensured that the patient perspective was considered throughout the trial, including in decision-making regarding protocol amendments and trial recruitment strategies.
Trial management and oversight
Trial Management Group
The Trial Management Group (TMG) consisted of the chief investigator, collaborators, site investigators, site research nurses and staff from Sheffield CTRU. The TMG was responsible for the day-to-day implementation of the trial.
Data Monitoring and Ethics Committee
A Data Monitoring and Ethics Committee (DMEC) consisted of an independent statistician and two independent clinicians with research expertise. The DMEC reviewed reports provided by Sheffield CTRU to assess the progress of the study, the safety data and the critical end-point data. The DMEC provided feedback to the TSC following each meeting. The chief investigator (or delegate) and members of staff from Sheffield CTRU attended the open sessions of the DMEC meetings as observers. An unblinded statistician from Sheffield CTRU produced the report and attended the closed sessions of the DMEC.
Trial Steering Committee
The TSC consisted of independent clinicians, an independent statistician and a PPI representative. The role of the TSC was to provide supervision of the protocol and statistical analysis plan, to provide advice on and monitor progress of the study, to review information from other sources and to consider the recommendations from the DMEC. The chief investigator (or delegate), a sponsor representative and members of staff from Sheffield CTRU attended the TSC meetings as observers. Sheffield CTRU produced reports for review during the TSC meetings.
Changes to the protocol
All protocol amendments are listed in Appendix 3. A summary of the key changes is provided below.
Eligibility criteria
In substantial amendment 1, the inclusion criteria were updated to clarify that patients must have neuropathic pain affecting both feet and to update the Toronto Clinical Neuropathy Score to the ‘modified’ version. 39 The exclusion criteria were updated to allow investigator judgement to be used when assessing the criteria for alcohol/substance abuse, history of psychiatric illness and prostate hypertrophy or urinary retention. The permitted dose of morphine equivalent was reduced from 120 to 100 mg/day. The exclusion criterion relating to history of ischaemic heart disease was updated to exclude any patient who had suffered a recent myocardial infarction (< 6 months prior to randomisation). New exclusion criteria were added for major amputations of the lower limbs and active diabetic foot ulcers.
Substantial amendment 7 updated the requirements for neuropathic pain to allow the pain to be present in the feet and/or hands. The exclusion criteria were also updated to clarify that only patients with non-diabetic symmetrical polyneuropathies were excluded. Previously, any non-diabetic neuropathy was an exclusion criterion, for example patients with diabetic neuropathy and carpal tunnel syndrome would previously have been excluded from the trial, but this was not the intention. This point was clarified in substantial amendment 7 to ensure that patients were not excluded unnecessarily. The exclusion criterion relating to liver function tests was updated to clarify that only the AST/ALT results were relevant for the eligibility assessment. The exclusion criterion relating to the electrocardiography results was updated to clarify that patients with a current history of arrhythmia were not eligible for the trial.
In substantial amendment 8, the exclusion criteria were updated to allow patients taking concomitant SSRIs to join the study provided that they had prior concomitant and safe use of SSRIs with the study medication (duloxetine and/or amitriptyline). The exclusion criterion relating to active foot ulcers was also updated to allow for investigator discretion. Patients were only excluded if the investigator felt that the ulcer would have a confounding or detrimental effect on the primary outcome or on patient participation.
To minimise the amount of time participants were on no pain medications, substantial amendment 12 allowed participants to be randomised early if the pain scores were high, provided that the mean pain score for the week was > 4 points. The exclusion criterion for heart failure was updated to exclude patients with heart failure class III or above (rather than class II or above). The exclusion criterion for postural hypotension was also updated to allow investigator discretion in the decision. Previously, all patients with a postural drop of > 20 mmHg were excluded.
In substantial amendment 13, the exclusion criteria were updated to clarify that patients taking concomitant citalopram were not eligible for the study. In addition, an update was included to exclude patients with second- or third-degree heart block or left bundle branch block (patients with right bundle branch block or first-degree heart block were permitted to be included following discussion with the cardiology team).
Data collection for the primary end point
At an early meeting, the TSC noted that there were potential issues with recording pain scores using paper diaries, including retrospective completion. The TSC recommended that the trial team consider an alternative method for collecting the primary end-point data. A text message data collection system was included in substantial amendment 5 and implemented in January 2018. This allowed daily text messages to be sent to participants who had provided optional consent for this aspect of the study. The text messages reminded participants to take their study medication and asked them to reply with their pain score, allowing the scores to be captured in real time.
Study treatment
Substantial amendment 1 allowed participants to start the next treatment pathway early if they wanted to withdraw from their current treatment pathway. The requirements for performing a dose review (i.e. to reduce the dose or discontinue a drug) were updated in substantial amendment 2, and it was clarified that only side effects that were severe or intolerable would require a dose review (rather than side effects which were moderate).
In substantial amendment 5, a reduced pregabalin dosing schedule was introduced for participants with an eGFR of 30–59 ml/minute. This ensured that the protocol was in line with British National Formulary (BNF) guidelines for pregabalin dosing. 53 Substantial amendment 5 also allowed participants to start second-line treatment up to week 13 if needed.
Substantial amendment 7 clarified that participant preference could be considered when making dose titration decisions.
In substantial amendment 8, an update was made to allow study medication to be tapered more gradually between pathways in the event of significant withdrawal side effects, at the discretion of the local investigator.
Statistical methods
Sample size
A mean change between groups of 0.5 points was chosen based on the effect size previously reported in a crossover study29 for comparison of two active interventions for neuropathic pain. It was estimated that this would equate to an 8% difference between groups in the proportion of people improving by at least 1 point,54 which is considered a minimally clinically significant change in an individual. 55 By using a within-patient standard deviation (SD) of 1.65,3,9 an alpha of 0.0167 to allow for three comparisons and 90% power, it was calculated that 294 evaluable participants were required. 56
The original plan was to screen 536 patients, in total, for participation in the study. Assuming a 25% dropout rate, the study intended to recruit and randomise 392 participants to ensure that 294 participants completed the study.
However, recruitment for this demanding trial, with multiple study visits and four washout periods, became challenging and difficult to justify, given that most previous similar trials29,31,57 had used a 1 NRS point difference. With approval from the TSC, our PPI Panel and the funder, a decision was made to continue recruitment to a fixed time (July 2019), at which point the trial had recruited 140 participants. Using our original assumptions (i.e. a within-patient SD of 1.65 and alpha of 0.0167), the trial would achieve 90% power to detect a difference of 1 NRS point, assuming at least 74 patients per arm provided outcome data. With a 25% dropout, as originally assumed, the trial would have 95% power to detect a 1-point change and was sufficient to estimate differences in average pain to within a standard error of 0.25 NRS points.
Statistical analysis
General principles
Analyses were limited to randomised and eligible participants who started their pathway. Any participant who withdrew from study in one pathway was excluded from any succeeding pathways. Analyses were undertaken using generalised mixed-effect modelling, with treatment group (i.e. A-P, D-P or P-A) and pathway order (i.e. first, second or third) as fixed-effect covariates and participant as a random intercept. Statistical comparisons used 98.3% confidence intervals (CIs) and a 0.0167 statistical significance level was used for pairwise comparisons. Analyses were undertaken using intention-to-treat principles, which evaluated the policy of pathways rather than adherence to therapies. All analyses were undertaken using Stata® version 16 (StataCorp LP, College Station, TX, USA).
Treatment uptake and response
Treatment uptake was summarised separately for first- and second-line therapies as (1) the proportion of participants stopping treatment prior to week 16 and (2) the dose level being taken at the end of the pathway at week 16 among those on therapy. The uptake of second-line therapy was further categorised as (1) being in combination with first-line therapy or (2) as a switch from first-line monotherapy.
Treatment response at week 6 was defined in relation to first-line monotherapy. A patient was classified as a treatment responder if they remained on first-line monotherapy at the week 6 visit with an average pain score of ≤ 3 points over the previous 7 days. Conversely, non-responders were those who (1) discontinued first-line monotherapy for AE, toxicity or ineffectiveness on or prior to week 6, (2) started second-line treatment (as combination therapy or as treatment switch) on or prior to week 6 or (3) had a NRS pain score > 3 points at week 6.
Treatment response at week 16 was in relation to both therapies. Patients who remained on at least one study medication and reported a 7-day average NRS score of ≤ 3 points were defined as having responded to the pathway, whereas those who discontinued for AE, toxicity or poor effectiveness and/or had a NRS score > 3 points were classed as non-responders.
Numeric Rating Scale pain
Self-reported NRS scores were collected daily for the duration of the pathway, by short message service (SMS) and/or via patient diaries. On days when a participant had provided both SMS and diary data, the NRS was taken from the SMS. A weekly average was calculated only if NRS scores were available on at least 4 out of the 7 days. The time window for the 6-week outcome was –2 weeks to + 1 week (i.e. any consecutive 7-day period ending between 28 and 49 days post commencement), provided that this did not extend to the washout phase or the next pathway. The week 16 data time window was –3 weeks to + 1 week (i.e. any consecutive 7-day period ending between 91 and 119 days post commencement), again, provided that this did not extend to the washout phase or next pathway.
At weeks 6 and 16, NRS scores were analysed using linear mixed-effect modelling. The binary outcomes (i.e. 30% pain reduction from baseline, 50% pain reduction from baseline and NRS score of ≤ 3 points) were analysed by logistic mixed effects regression. Baseline NRS score was the pain score taken during the washout period (i.e. week –1) immediately prior to randomisation and changes from baseline were calculated in reference to this score.
Missing data
The primary analysis was performed using a mixed model on complete-case data, which may be inadequate in situations where differential levels of withdrawal (or differential reasons for withdrawal) occur between groups. To address this, the following three sensitivity analyses were undertaken, using statistical imputation to assess the impact of missing data on pain scores:
-
Last observation carried forward, using the last available weekly NRS data. Although this approach has been widely criticised as oversimplistic,58 it offers a conservative estimate of the treatment effect in the case of conditions that improve with time. 59
-
Multiple imputation,60 which is unbiased under the missing at random assumption if missing data can be predicted by the imputations model’s characteristics.
-
Controlled multiple imputation,61 in which informative withdrawal had more pessimistic values imputed.
Approach 2 used chained predicted mean matching imputation, with 10 nearest neighbours and 100 imputations. Missing data were imputed based on age, sex, baseline total NPSI score, treatment arm, pathway and any previous weekly NRS data available. Trace plots were used to assess convergence and, on the basis of these, 1000 burn-in imputations were used.
Approach 3 assumed that the participants who withdrew from treatment because of toxicity and/or inadequate treatment response would have a worse response than the value imputed by multiple imputation. Specifically, the imputed NRS values yi* created by step 2 were replaced with a pessimistic imputation (yi* + δ), with δ ranging between 0.5 and 2.5. Imputed values were bounded at 10 where applicable.
Subgroups
The NRS responses were analysed in relation to the age, baseline pain, HADS anxiety and depression scores and pain phenotypes, as derived from NPSI scores. 52 Additional post hoc analyses were undertaken to assess whether or not outcomes were temporally associated with the COVID-19 lockdown, which began 3 months before the last patient last visit. Subgroup analyses were undertaken by adding an interaction term to the model and reported as marginal means.
Preferred treatment
After completing all three pathways, participants were asked to choose their preferred treatment. This was reported as a single 3 × 1 contingency table. The hypothesis test of equal proportions was assessed by chi-squared test.
Other efficacy assessments
All other efficacy assessments were undertaken at 6 and 16 weeks after the start of the treatment pathway, which corresponded to the end of monotherapy phase and the end of the treatment pathway.
Harms
Adverse events were recorded at each follow-up visit and categorised prior to unblinding. Any AEs occurring prior to the first treatment pathway were excluded. AEs were presented as the number of patients experiencing each event type and the number of events of each type. Where data allowed, the proportions were compared between arms using a mixed-effect logistic regression approach, as per binary pain outcomes. The following summaries were presented:
-
all AEs
-
all AEs of moderate or severe intensity and related (probably or definitely) to either treatment
-
all SAEs.
Additional post hoc analysis looked at the number of days affected by each event type and the treatment phase during which the event occurred.
Health economic methods
A within-trial cost–utility analysis was conducted alongside the clinical study. The cost–utility analysis estimated the mean differences in costs, QALYs and the incremental cost-effectiveness ratio (ICER) over 16 weeks for each treatment pathway. The cost–utility analysis was conducted in line with the NICE Guide to the Methods of Technology Appraisal62 and is in line with the Consolidated Health Economic Evaluation Reporting Standards (CHEERS) checklist. 63 The analysis is presented from an NHS and Personal Social Services perspective.
Quality of life and quality-adjusted life-years
Quality-adjusted life-years were measured over the 16-week period using the EQ-5D-5L. 64 The EQ-5D-5L is a preference-based QoL measure that can be used in economic evaluations. The EQ-5D-5L consists of five dimensions (mobility, self-care, usual activities, pain or discomfort and anxiety or depression). For each dimension, responders indicate which of five levels their health is at today, with levels ranging from no problems to unable to do/extreme problems. Responses are then scored on a 0–1 scale on which 0 represents death, 1 represents perfect health and negative values indicate states worse than death.
In the OPTION-DM study, and in line with NICE guidelines,65 preference weights were obtained from van Hout et al. ’s66 mapping study in the main analysis, with Devlin et al. ’s46 preference weights applied in sensitivity analysis.
Participants completed the EQ-5D-5L at baseline, prior to the study commencing, and at 6 and 16 weeks in each treatment pathway. Given that this was a crossover study and participants received all three treatment pathways in turn, for the treatment pathway received second or third in order it was assumed that participants’ EQ-5D scores returned to the baseline value during the washout period between treatment pathways. Area under the curve using the trapezium rule was then used to estimate QALYs, which are presented in years throughout this report.
Resource use
NHS resource use was measured for each participant between baseline and the final follow-up (i.e. before crossover/end of follow-up). Resource use included all medication costs, visits to health services and any social care and community support. Medical costs were taken from the study medication records. In addition, other NHS resources used were self-reported by participants using the widely used and validated CSRI questionnaire. 47 Unnecessary questions in the CSRI were removed to reduce the burden for participants; however, questions relating to personal costs incurred and time off work (where relevant) were retained for sensitivity analysis.
Details of unit costs for hospital visits, general practitioner (GP) visits, and social and other health-care services are listed in Table 3 (further details are available from the authors by request). Unit costs for laboratory tests were obtained from the national cost collection for the NHS67 and unit costs for medications are taken from the BNF. 53 For unit costs to be applied to the same year, medication costs were deflated back to 2018/19 prices using the inflation rates provided in the Personal Social Services Research Unit (section 15.2). 76
| Resource | Source | Unit cost (£) |
|---|---|---|
| A&E visit | The National Cost Collection for the NHS (2018/19):67 index sheet unit cost for accident and emergency | 166.00 |
| Hospital admissions | The National Cost Collection for the NHS (2018/19):67 index sheet unit costs averaged for elective and non-elective inpatients | 3477.36 |
| Outpatient visits | The National Cost Collection for the NHS (2018/19):67 total outpatient attendance sheet | 127.00 |
| GP home/surgery visit | Curtis and Burns (section 10.5)68 | 39.00 |
| GP telephone call | Curtis and Burns (section 10.5)68 | 15.52 |
| Practice nurse | Curtis and Burns (section 10.2)68 | 42.00 |
| Practice nurse telephone call | Curtis and Burns (section 10.5)68 | 7.80 |
| Prescription costs | Curtis and Burns (section 10.4)68 | 1.30 |
| Home help | Curtis and Burns (section 11.5)68 | 28.00 |
| Social worker | Curtis and Burns (section 11.1)68 | 51.00 |
| Community pain management | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 116.00 |
| Physiotherapy | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 55.00 |
| Occupational therapy | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 66.00 |
| Podiatry NHS | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 51.00 |
| Podiatry private | Averaged across The Podiatry Clinic,69 A&A Podiatrists70 and the Footcare Centre71 | 54.50 |
| Psychology | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 79.00 |
| Diabetic clinic | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 195.00 |
| Psychiatrist | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 203.00 |
| Counsellor | Agenda for Change 2018/19 (mid-point band 6)72 | 17.37 |
| Eye clinic | The National Cost Collection for the NHS (2018/19):67 non-CL WF01A | 88.00 |
| Vascular surgery | The National Cost Collection for the NHS (2018/19):67 day case | 66.00 |
| Aromatherapy | Averaged across Escape Holistic Therapies,73 Holly’s Holistics74 and Natural at Heart75 | 50.00 |
Treatment costs
Treatment costs consisted of the costs of medication (i.e. amitriptyline, duloxetine and pregabalin), the cost of the clinic visit (face to face or via telephone) and the cost of laboratory tests. Table 4 lists the treatment costs and the source of unit costs. Treatment medication was costed as it would be delivered within the NHS (rather than during a research study) so that results are presented from an NHS perspective. For example, the smallest pack of amitriptyline 25-mg tablets contains 28 tablets, and so the medicine cost at the week 1 visit was £0.94. The frequency of study medication visits were assumed to mirror what would take place in an NHS setting and these were either face to face or via telephone. Laboratory tests were assumed to take place once at the beginning of each treatment pathway and were costed accordingly.
| Treatment | Unit cost source | Unit cost (£) |
|---|---|---|
| Amitriptyline 25 mg: 28 tablets | BNF53 | 0.92 |
| Amitriptyline 50 mg: 28 tablets | BNF53 | 1.68 |
| Duloxetine 30 mg: 28 tablets | BNF53 | 1.63 |
| Pregabalin 75 mg: 56 capsules | BNF53 | 2.12 |
| Pregabalin 150 mg: 56 capsules | BNF53 | 2.94 |
| Diabetic outpatient clinic: face to face | The National Cost Collection for the NHS (2018/19):67 CL WF01A | 145.00 |
| Diabetic outpatient clinic: telephone | The National Cost Collection for the NHS (2018/19):67 CL non-face to face WF01A | 86.00 |
| Laboratory tests (i.e. liver function, blood count, HbA1c, creatinine, eGFR, urea, electrolytes) | The National Cost Collection for the NHS (2018/19):67 pathology services | 328.00 |
As this was a crossover study, the time period in which the participants received the different treatment pathways could affect resource use and QoL. Therefore, linear regression analysis was performed to establish whether or not there was a difference in costs depending on the time period the participant received the treatment pathway. A separate regression model was fitted to costs and QALYs per treatment pathway (i.e. A-P, D-P and P-A). Confidence intervals around the coefficients for differences in costs between those receiving a treatment pathway first and either second or third in order were calculated using 5000 bootstrap simulations. A p-value of < 0.05 was used to establish statistical significance of an ordering affect. If statistical significance was established, then a sensitivity analysis would be carried out to allow for the time period in which participants received the treatment pathway.
Costs are reported using the 2018/19 time frame.
Cost-effectiveness analysis
Results are presented as a within-trial analysis, using a pairwise ICER as cost per QALY gained. It is possible to present this analysis because, within the crossover trial, participants received all three treatments. A non-pairwise (conventional) ICER as cost per QALY gained is also presented. Three cost-effectiveness comparisons are carried out: (1) A-P compared with D-P, (2) A-P compared with P-A and (3) D-P compared with P-A. Results are presented on the cost-effectiveness plane and on cost-effectiveness acceptability curves. No discounting was applied, as the follow-up period was < 1 year.
A total of 5000 simulations were used to obtain 95% CIs using bootstrapping.
All analysis was undertaken using Stata.
Sensitivity analysis
To allow for uncertainty, the following sensitivity analyses were undertaken:
-
Devlin et al. ’s46 algorithm for EQ-5D-5L utility values was used as an alternative to van Hout et al. ’s66 algorithm for utilities.
-
Analysis was undertaken with a wider societal perspective for costs. Personal costs and time off work are included, as reported by participants using the CSRI questionnaire. Participants provided details of any out-of-pockets costs related to employing extra help, transport to health-care appointments, modifications to their home and equipment purchased as a result of their condition. Details were also provided on any time away from usual activities for themselves or for friends or relatives as a result of their condition. Participant and friend/relative time was costed at the average UK wage as detailed in the Office for National Statistics annual survey for hours and earnings (2019). 77
-
EQ-5D responses were missing for 27–32% of participants. Therefore, multiple imputation58 was carried out to impute the missing values assuming responses were missing at random. As with the statistical analysis, predicted mean matching with 10 nearest neighbours and 100 imputations were carried out. Missing data were imputed base on age, sex and treatment pathway.
Chapter 3 Results
Participant recruitment
Figures 4 and 5 detail the patient flow through the study by treatment (i.e. A-P, D-P and P-A) and by chronological treatment pathway (first, second and third). Participating sites collected pre-screening data on all patients who the site had been in contact with regarding the study. Between 2 October 2017 and 31 July 2019, 1004 patients were identified as being potentially eligible for the trial across 18 secondary care hospital centres (a further three centres were activated to recruitment, but did not identify any potential participants). Of these patients, 426 were ineligible. The main reasons for ineligibility at this stage were not having neuropathic pain (n = 202), contraindications for study medications (n = 42), having other painful medical conditions (n = 39) and use of prohibited concomitant treatment (n = 28). A further 314 patients were not interested or unable to continue into the study, with the main reasons being that they were unable or unwilling to attend all study visits (n = 146) or were not wanting to come off current treatment (n = 76). Twelve patients did not continue for other reasons.
FIGURE 4.
Patient disposition and study flow chart. a, Switch to second-line monotherapy before week 6; and b, includes switches to second-line monotherapy after week 6.
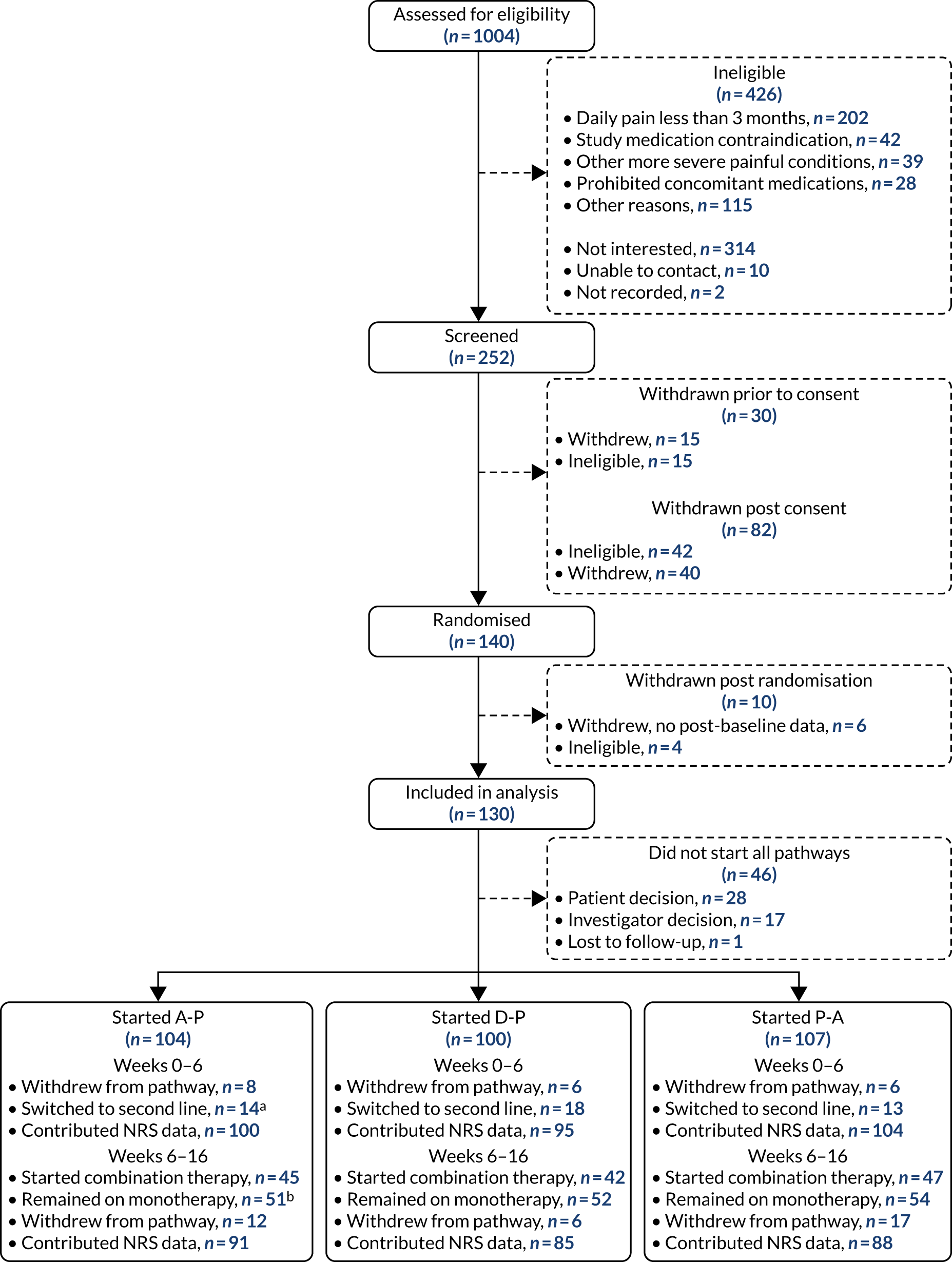
FIGURE 5.
Patient disposition and study flow chart by chronological treatment pathway.
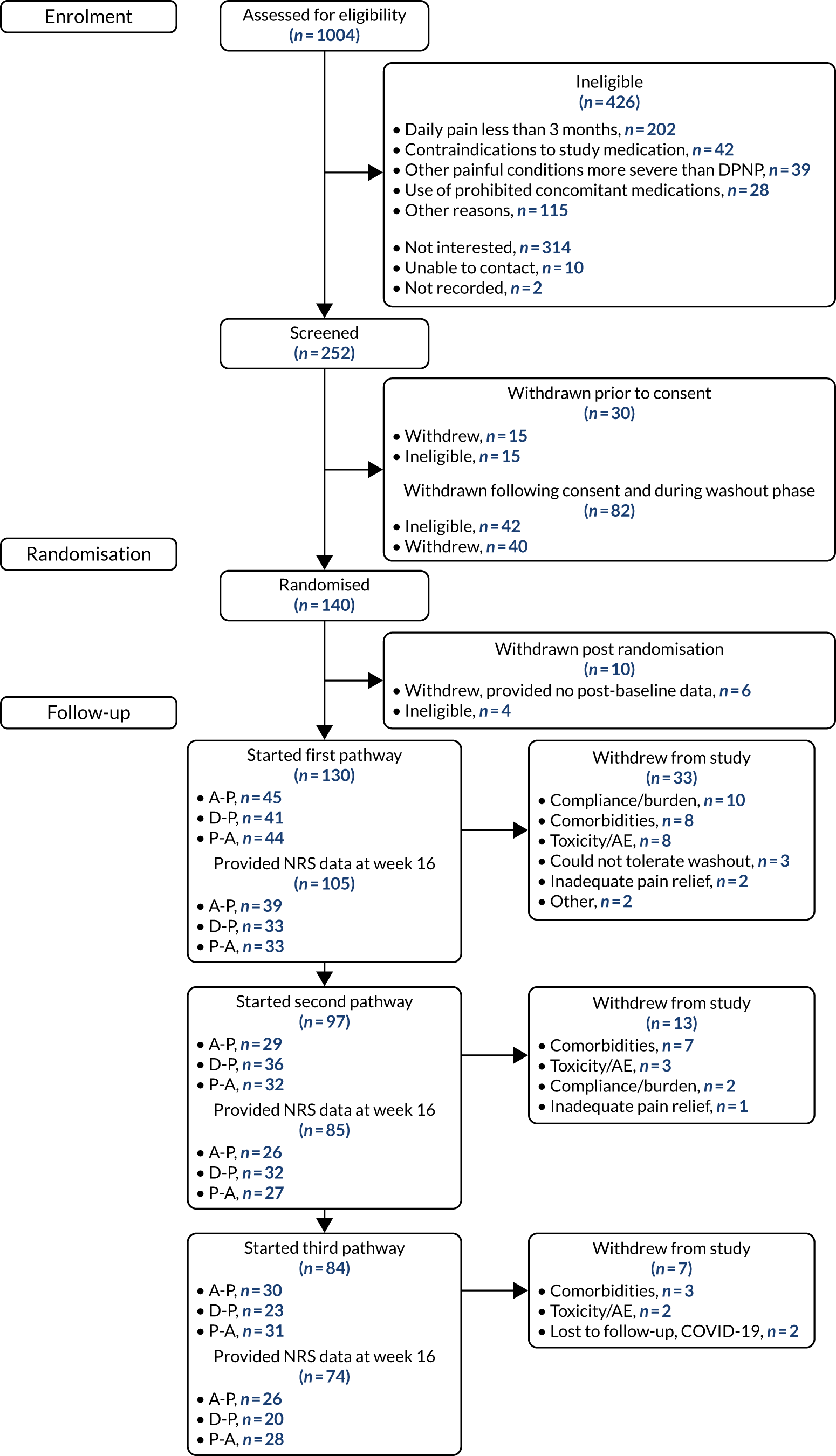
Between 2 October 2017 and 31 July 2019, 252 patients attended the consent visit at week –2 across 17 trial centres, and 222 of these patients provided informed consent. Of those patients who consented, 40 discontinued from the trial before randomisation and a further 42 were ineligible because of unconfirmed or low levels of neuropathic pain (n = 37) or clinically significant arrhythmia (n = 5). One hundred and forty patients proceeded to be randomised at week 0 across 13 sites. Ten patients were excluded following randomisation and within the first treatment pathway. Six patients provided no post-baseline data (three withdrew citing the trial burden, two were lost to follow-up and one developed a significant comorbidity) and four were randomised in error.
A total of 53 patients withdrew from the study before completing all three treatment pathways. The majority (n = 33) of withdrawals came in the first pathway, with a further 13 withdrawals in pathway 2 and seven withdrawals in pathway 3. The numbers of patients starting first, second and third pathways were 130, 97 and 84, respectively, and the numbers of patients contributing 7-day pain scores at week 16 were 105, 85 and 74, respectively.
Recruitment was originally expected to be completed within 12 months by eight trial centres. Owing to slow recruitment rates, the total number of centres was increased to 21 and a number of changes were made to the eligibility criteria and study processes to improve recruitment (see Chapter 2, Changes to the protocol). In addition, assistance was provided to participating centres via regular teleconferences, recruitment packs and one-to-one discussions with the research fellow based at the lead site in Sheffield.
Protocol non-compliances
A total of 146 (major, n = 73; minor, n = 73) protocol non-compliances were reported during the trial and no serious breaches were reported. Five participants were ineligible and were withdrawn from the trial during the first treatment pathway. One participant did not contribute primary outcome data as a result of a good clinical practice non-compliance in the recording of the data at site.
As the study medication was provided in up to eight blinded medication bottles, the dosing schedule for participants was more complicated than in usual care. Thirty-seven cases of participant non-compliance with treatment were reported. These included the participant taking more/less medication than prescribed or taking medication from incorrect bottles.
Fifty-nine non-compliances were reported in relation to trial treatment or procedural issues and this included 22 cases where a patient was prescribed and/or dispensed an incorrect dose of medication. To our knowledge, the errors in treatment doses did not result in any AEs.
Three non-compliances were reported in relation to on-site visits being missed due to COVID-19 restrictions.
Characteristics of trial participants
Trial participants were of similar age to screened patients (median 61.8 years vs. 61.2 years), but fewer females were enrolled (26% vs. 41%).
The full characteristics of the 130 trial participants are presented in Table 5, split according to whether or not the patient completed the three treatment pathways. The majority (82%) of patients had type 2 diabetes, had experienced neuropathic pain for an average of 5 years (median duration 3.4 years, range 4 months to 25 years) and self-rated their pain at 6.6 points out of 10 points (NRS) and 6.1 points out of 10 points (BPI-MSF). Patients more commonly described their pain as relapsing/remitting as opposed to deep or involved (NPSI) and one-third of participants reported having taken each of amitriptyline, pregabalin, duloxetine and gabapentin at some point prior to trial entry.
| Characteristic | Completers (N = 77) | Non-completers (N = 53) | Total (N = 130) |
|---|---|---|---|
| Demographics | |||
| Age (years), mean (SD) | 61.3 (10.9) | 62.5 (11.2) | 61.8 (11.0) |
| Female, n (%) | 22 (29) | 12 (23) | 34 (26) |
| BMI (kg/m2), mean (SD) | 31.7 (6.3) | 31.7 (7.0) | 31.7 (6.6) |
| Diabetes characteristics | |||
| Type 1, n (%) | 12 (16) | 10 (19) | 22 (17) |
| Type 2, n (%) | 63 (82) | 43 (81) | 106 (82) |
| Type missing, n (%) | 2 (3) | 0 | 2 (2) |
| HbA1c (mmol/mol), mean (SD) | 65.4 (13.2) | 68.4 (17.2) | 66.6 (15.0) |
| Duration of diabetes (years), mean (SD) | 14.9 (9.0) | 15.6 (9.7) | 15.1 (9.3) |
| Duration of neuropathic pain (years), mean (SD) | 4.8 (4.1) | 5.0 (4.1) | 4.9 (4.1) |
| Previous medication use, n (%) | |||
| Amitriptyline | 30 (39) | 19 (36) | 49 (38) |
| Pregabalin | 27 (35) | 18 (34) | 45 (35) |
| Duloxetine | 28 (36) | 19 (36) | 47 (36) |
| Gabapentin | 27 (35) | 17 (32) | 44 (34) |
| Any opioid | 27 (35) | 20 (38) | 47 (36) |
| Pain characteristics, mean (SD) | |||
| NRS pain (0–10; higher scores indicate greater pain) | 6.7 (1.5) | 6.5 (1.4) | 6.6 (1.5) |
| BPI-MSF (0–10; higher scores indicate greater pain) | |||
| Pain severity score | 6.1 (1.6) | 6.1 (1.9) | 6.1 (1.7) |
| Pain interference score | 5.8 (2.3) | 6.1 (2.5) | 5.9 (2.4) |
| NPSI (0–10; higher scores indicate greater pain) | |||
| Superficial spontaneous burning pain | 6.0 (2.8) | 6.0 (3.1) | 6.0 (2.9) |
| Deep spontaneous pressing pain | 4.8 (2.8) | 4.7 (2.7) | 4.8 (2.8) |
| Paroxysmal pain | 5.3 (2.9) | 5.8 (2.9) | 5.5 (2.9) |
| Evoked pain | 4.6 (2.5) | 3.9 (2.8) | 4.3 (2.6) |
| Paraesthesia/dysaesthesia | 6.3 (2.4) | 6.4 (3.0) | 6.3 (2.7) |
| NPSI total score (0–100; higher scores indicate greater pain) | 52.6 (18.1) | 52.1 (21.6) | 52.4 (19.5) |
Trial medication usage
The study treatment use is summarised in Table 6. The uptake of first-line therapy was similar for each arm, with around half of patients remaining on the highest dose at the end of each pathway. Likewise, discontinuation overall was similar across the three arms, but more patients discontinued duloxetine because of AEs or toxicity. The proportion of patients starting second-line therapy was also similar, as were the proportions on the highest dose level at week 16. More participants discontinued or switched from duloxetine during first-line therapy and more participants discontinued intervention during the P-A pathway, but neither was statistically significant.
| Treatment use | Pathway | p-value | ||
|---|---|---|---|---|
| A-P | D-P | P-A | ||
| Started treatment pathway, n | 104 | 100 | 107 | |
| First-line therapy | ||||
| Average dose (mg)/day at week 6 | 56 | 76 | 397 | |
| Number (%) on highest dose | 53 (51) | 46 (46) | 59 (55) | |
| Number (%) discontinuing first-line therapy | ||||
| Discontinued at any time (0–16 weeks)a | 29 (18) | 29 (24) | 32 (20) | 0.928 |
| Because of AE/toxicity | 11 (11) | 17 (17) | 5 (5) | 0.031 |
| Because of poor response | 10 (10) | 9 (9) | 14 (13) | 0.518 |
| Because of other reasons | 8 (8) | 3 (3) | 13 (12) | |
| Discontinued while on monotherapyb | 25 (24) | 25 (25) | 23 (21) | 0.797 |
| Because of AE/toxicity | 9 (9) | 14 (14) | 5 (5) | 0.087 |
| Because of poor response | 10 (10) | 9 (9) | 12 (11) | 0.851 |
| Because of other reasons | 6 (6) | 2 (2) | 6 (6) | |
| Second-line therapy | ||||
| Number (%) started | 60 (58) | 61 (61) | 60 (56) | |
| Started as combination therapy | 45 (43) | 42 (42) | 47 (44) | |
| Switched from first line | 15 (14) | 19 (19) | 13 (12) | |
| Average dose (mg)/day at end of study | 347 | 405 | 52 | |
| Number (% of started) on highest dose | 21 (47) | 23 (55) | 2 (47) | |
| Number (% of started) discontinuing second-line therapy | ||||
| Discontinued at any timec | 13 (22) | 7 (11) | 18 (30) | 0.123 |
| Because of AE/toxicity | 4 (7) | 3 (5) | 9 (15) | 0.383 |
| Because of poor response | 6 (10) | 2 (3) | 4 (7) | 0.374 |
| Because of other reasons | 3 (5) | 2 (3) | 5 (8) | |
| Discontinued while on combination therapyd | 7 (12) | 5 (8) | 13 (22) | |
| Because of AE/toxicity | 3 (5) | 2 (3) | 5 (8) | |
| Because of poor response | 2 (3) | 1 (2) | 4 (7) | |
| Because of other reasons | 2 (3) | 2 (3) | 4 (7) | |
| Overall | ||||
| Number (%) discontinuing treatment pathway at any timee | 20 (19) | 12 (12) | 23 (21) | 0.226 |
| Because of AE/toxicity | 6 (6) | 4 (4) | 6 (6) | 0.792 |
| Because of poor response | 4 (4) | 0 | 2 (2) | 0.398 |
| Because of other reasons | 10 (10) | 8 (8) | 15 (14) | |
Numeric Rating Scale pain
None of the pairwise comparisons was statistically significant. At week 16, among participants with outcome data (i.e. complete-case analysis), mean pain in the A-P arm was 0.1 units lower (i.e. better) than in the D-P and P-A arms, but the prespecified clinically important difference of 0.5 units was either excluded from or on the edge of each pairwise CI. For D-P compared with A-P the mean difference was –0.1 (98.3% CI –0.5 to 0.3), for P-A compared with A-P the mean difference was –0.1 (98.3% CI –0.5 to 0.3) and for P-A compared with D-P the mean difference was 0.0 (98.3% CI –0.4 to 0.4) (Table 7). These findings were robust across a range of analyses that assessed missing data under plausible scenarios (Figures 6 and 7), in which the 47 (15%) instances of missing data were imputed by last observation carried forward, multiple imputation or controlled multiple imputation. In all cases, the point effects were within ± 0.1 points.
| Outcome | Baseline (N = 130) | Week 6 (monotherapy phase) | Week 16 (combination therapy phase) | ||||
|---|---|---|---|---|---|---|---|
| A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | ||
| Average weekly pain (NRS; 0–10) | |||||||
| Patients included, n | 130 | 100 | 95 | 104 | 91 | 85 | 88 |
| NRS pain, mean (SD)a | 6.6 (1.5) | 3.8 (2.0) | 3.9 (1.9) | 4.1 (2.1) | 3.3 (1.8) | 3.3 (1.8) | 3.3 (1.8) |
| Change from baseline, mean (SD) | 2.9 (2.0) | 2.8 (2.0) | 2.5 (2.2) | 3.4 (2.1) | 3.5 (2.1) | 3.3 (2.1) | |
| ≥ 30% reduction from baseline, n (%) | 68 (65) | 63 (63) | 60 (56) | 68 (65) | 68 (68) | 68 (64) | |
| ≥ 50% reduction from baseline, n (%) | 42 (40) | 35 (35) | 43 (40) | 50 (48) | 46 (46) | 47 (44) | |
| NRS score of ≤ 3 points, n (%) | 38 (37) | 32 (32) | 36 (34) | 50 (48) | 43 (43) | 50 (47) | |
| Pairwise contrast, mean difference (98.3% CI); p-value | |||||||
| D-P vs. A-P | 0.1 (–0.3 to 0.5); 0.649 | –0.1 (–0.5 to 0.3); 0.613 | |||||
| P-A vs. A-P | 0.3 (–0.1 to 0.8); 0.049 | –0.1 (–0.5 to 0.3); 0.611 | |||||
| P-A vs. D-P | 0.3 (–0.2 to 0.7); 0.137 | 0.0 (–0.4 to 0.4); 0.996 | |||||
| Combined arms | |||||||
| n | 299 | 265 | |||||
| Change from baseline, mean (98.3% CI); p-value | 2.8 (2.2 to 3.0); < 0.001 | 3.4 (2.9 to 3.8); < 0.001b | |||||
| ≥ 50% reduction from baseline, n (%) | 120 (40) | 143 (54) | |||||
| NRS score of ≤ 3 points, n (%) | 106 (35) | 143 (54) | |||||
| Change in NRS from week 6 to week 16 mean (98.3% CI) | |||||||
| Patients on combination therapy | 1.0 (0.6 to 1.3) | ||||||
| Patients on monotherapy | 0.2 (–0.1 to 0.5) | ||||||
| All patients | 0.6 (0.3 to 0.8) | ||||||
FIGURE 6.
Pairwise comparisons of NRS pain score at week 6. MI, multiple imputation. LOCF, Last observation carried forward.
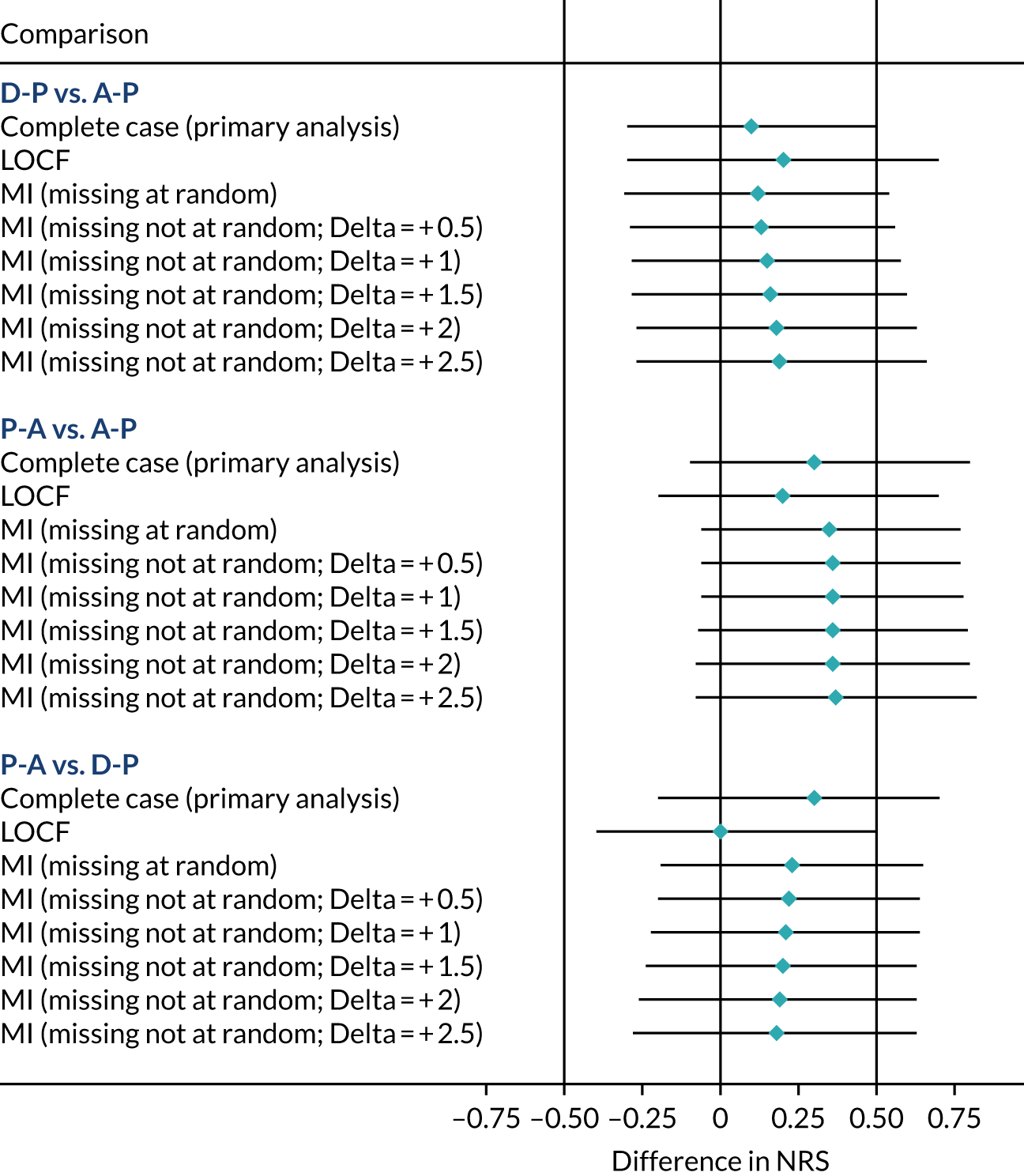
FIGURE 7.
Sensitivity analysis of NRS pain score at week 16. MI, multiple imputation. LOCF, Last observation carried forward.

The differences at week 6 were larger among patients in the P-A arm, having greater average pain than patients in the D-P and A-P arms; however, again, none of these differences was statistically significant. For the complete-case scenario, the difference between P-A and A-P was 0.3 (98.3% CI –0.1 to 0.8; p = 0.0492), with similar findings in analyses of imputed data, reflecting the similar levels of ‘for cause’ missing data in the two arms. Nevertheless, none of the arms differed on average by more than 0.4 points in any scenario and none was statistically significant at the predefined alpha = 0.0167 level. In total, there were 12 (4%) instances of missing data, meaning that the impact of missing data was small.
Response to treatment
Treatment response is summarised in Table 8. As noted above, duloxetine was the least well-tolerated monotherapy and had the lowest response rate (27% vs. 30% for amitriptyline and 34% for pregabalin) at week 6, although, again, this comparison was not statistically significant. The overall response rates at week 16 were 46% (A-P), 43% (D-P) and 46% (P-A).
| Pathway | Number starting | Response status | |||
|---|---|---|---|---|---|
| Week 6 (monotherapy) | n (%) | Week 16 (combination) | n (%) | ||
| A-P | 104 | Respondersa | 31 (30) | Respondersb | 48 (46) |
| Non-respondersc | 69 (66) | Non-responders | 50 (48) | ||
| Stopped first-line therapy | 21 (20) | Stopped treatment pathway for AE/poor response | 11 (11) | ||
| Started second-line therapy | 17 (16) | NRS score > 3 points | 41 (39) | ||
| NRS score > 3 points | 52 (50) | ||||
| Missing | 4 (4) | Missing | 6 (6) | ||
| D-P | 100 | Respondersa | 27 (27) | Respondersb | 43 (43) |
| Non-respondersc | 73 (73) | Non-responders | 52 (52) | ||
| Stopped first-line therapy | 22 (22) | Stopped treatment pathway for AE/poor response | 11 (11) | ||
| Started second-line therapy | 21 (21) | NRS score > 3 points | 42 (42) | ||
| NRS score > 3 points | 57 (57) | ||||
| Missing | 0 | Missing | 5 (5) | ||
| P-A | 107 | Respondersa | 34 (32) | Respondersb | 49 (46) |
| Non-respondersc | 71 (66) | Non-responders | 50 (47) | ||
| Stopped first-line therapy | 19 (18) | Stopped treatment pathway for AE/poor response | 13 (12) | ||
| Started second-line therapy | 16 (15) | NRS score > 3 points | 38 (36) | ||
| NRS score > 3 points | 55 (51) | ||||
| Missing | 2 (2) | Missing | 8 (7) | ||
| Complete case: 7-day average pain, mean (SD) [n] | |||||
| A-P | 3.8 (2.0) [100] | 3.3 (1.8) [91] | |||
| D-P | 3.9 (1.9) [95] | 3.3 (1.8) [85] | |||
| P-A | 4.1 (2.1) [104] | 3.3 (1.8) [88] | |||
Figure 8 shows the trajectory of average self-reported pain by randomised sequence, ranging over the 52 weeks of study (pain scores were not routinely completed for the washout periods and have not been included). Self-rated pain did not return to pre-randomisation levels in the washout periods between pathways 1 and 2 or between pathways 2 and 3. This tempers the conclusions that can be made for the change from baseline outcomes (i.e. 30% or 50% reductions), but the inclusion of period number in the statistical model meant that between-arm comparisons remained valid.
FIGURE 8.
Self-reported NRS pain by time in study.
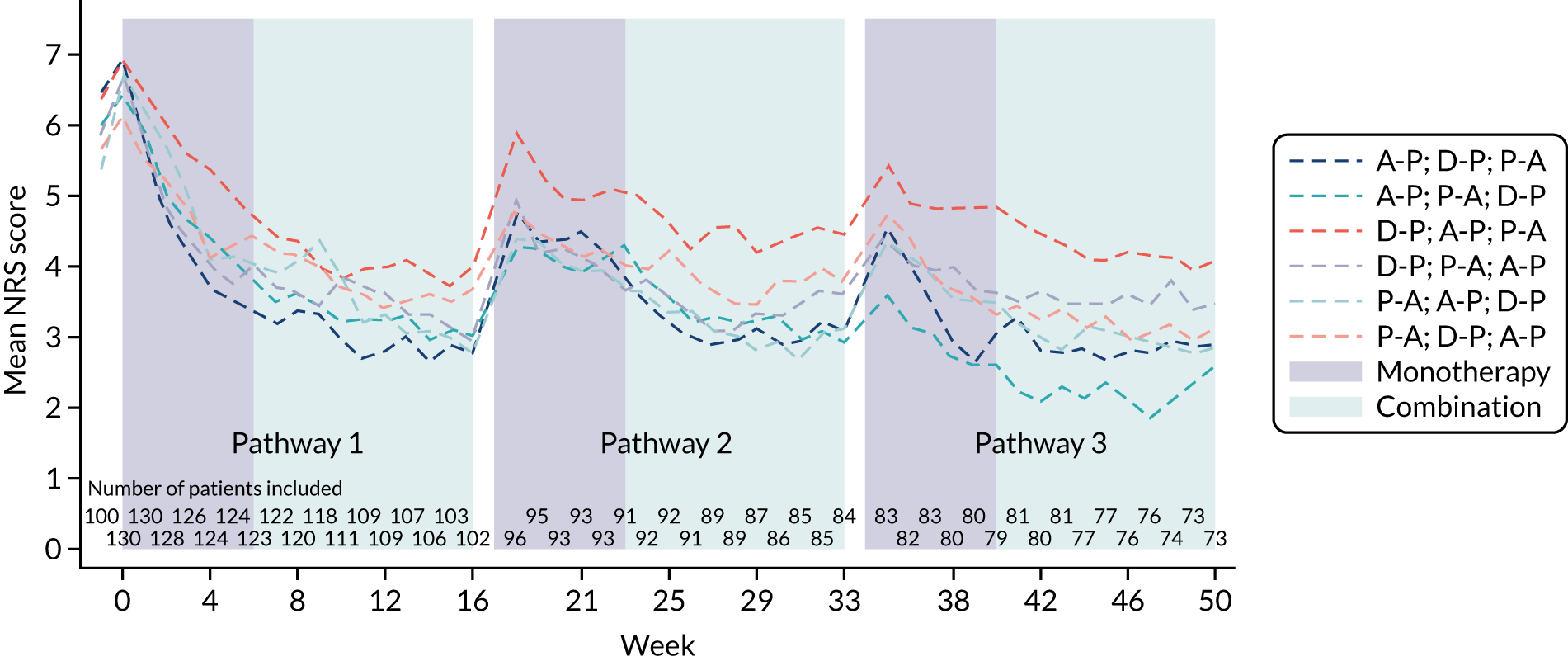
Figure 9 shows the trajectory of average NRS score by time within each pathway. The three curves largely overlap throughout the 16 weeks, suggesting that the arms have equivalent efficacy both in the monotherapy phase (i.e. weeks 0–6) and thereafter. Pain scores continued to decrease after week 6 when combination therapy was offered to participants (mean score: week 16 3.3 vs. week 6 3.9), although qualitatively the scores appeared to plateau around week 12.
FIGURE 9.
Self-reported NRS pain by treatment pathway.
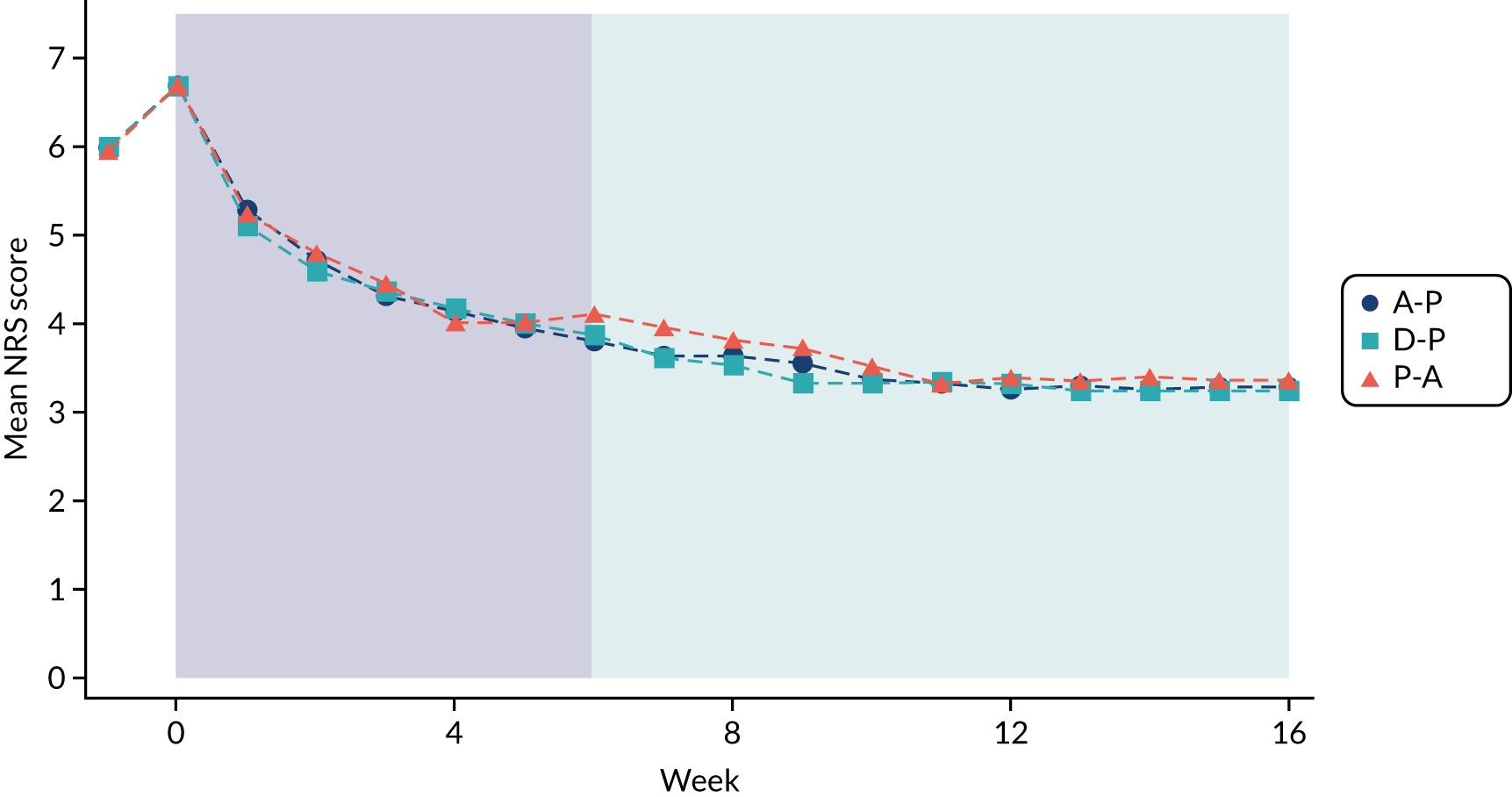
Figure 10 shows the same data as the previous figure, but separately for patients remaining on monotherapy or starting combination therapy on or after week 6. By definition, patients starting combination therapy had higher NRS scores (i.e. worse pain control) at week 6, with average pain levels appearing to plateau in the weeks prior to this. Thereafter, the curves began to converge, with a further mean drop of 1 NRS point among patients on combination therapy (again plateauing over the last weeks of the pathway) compared with 0.2 points among patients on monotherapy (Table 9). This suggests a benefit of combination therapy among patients who had less response to monotherapy alone.
FIGURE 10.
Self-reported NRS pain by use of combination therapy and treatment pathway.
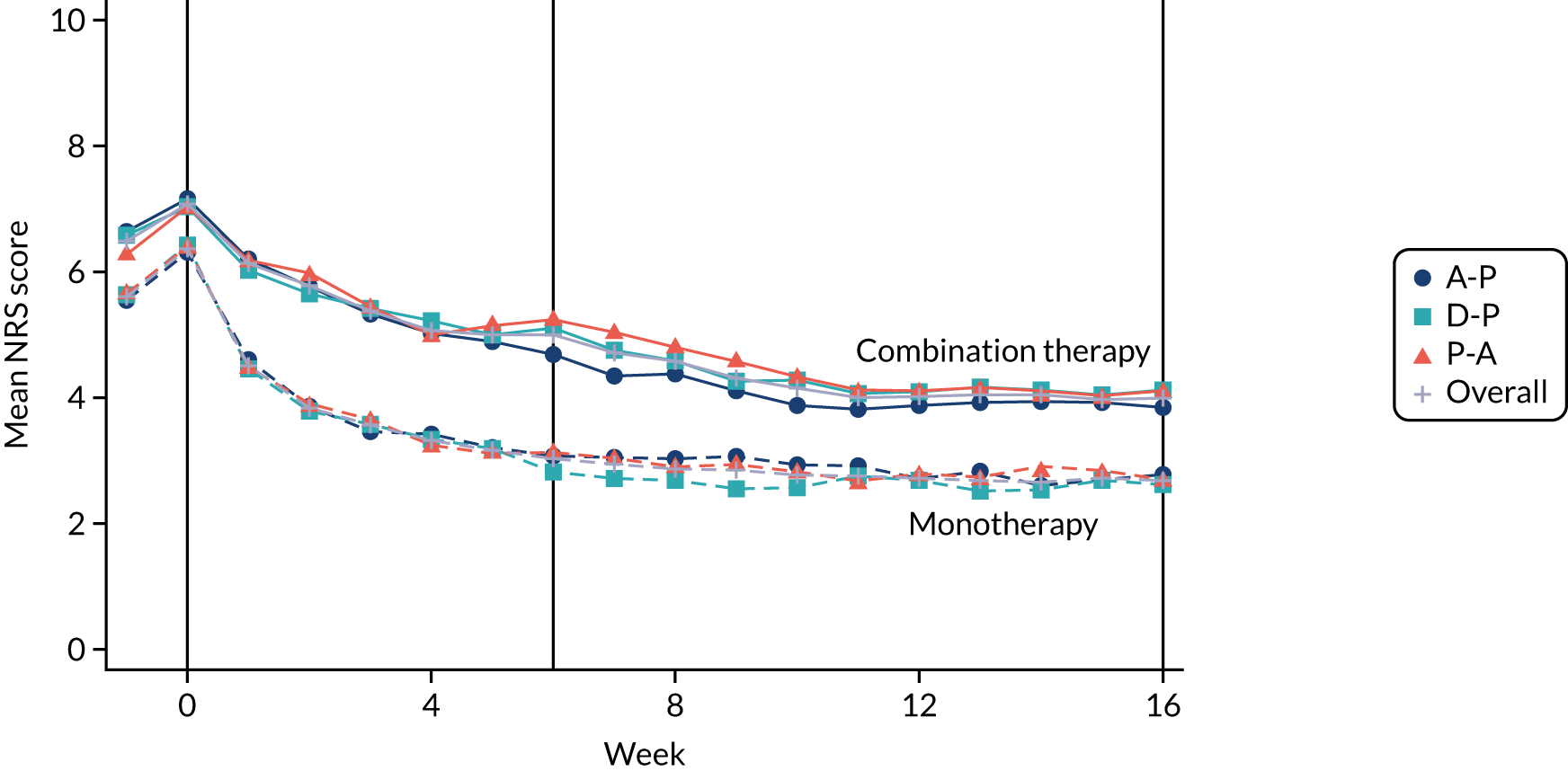
| Outcome | Treatment pathway | All arms | ||
|---|---|---|---|---|
| A-P | D-P | P-A | ||
| Started combination therapy | 0.9 (0.4 to 1.4) | 0.9 (0.4 to 1.5) | 1.1 (0.5 to 1.6) | 1.0 (0.6 to 1.3) |
| Remained on monotherapy | 0.1 (–0.4 to 0.6) | 0.2 (–0.3 to 0.7) | 0.3 (–0.2 to 0.8) | 0.2 (–0.1 to 0.5) |
| Overall change by arm | 0.5 (0.1 to 0.8) | 0.5 (0.2 to 0.9) | 0.7 (0.3 to 1.0) | 0.6 (0.3 to 0.8) |
Subgroup analyses
Further exploratory work looked at whether or not there were subgroups of patients who responded better to some therapies. Figure 11 depicts patient response to monotherapy at week 6 via a Venn diagram. Although most patients responded either to none (n = 37) or all (n = 13) of the therapies, 30 of the 80 patients who provided data had different responses to the treatments.
FIGURE 11.
Response to monotherapy among patients starting all three pathways. Response defined as a NRS score of ≤ 3 points and no informative treatment withdrawal. Four patients provided no data to at least one pathway.
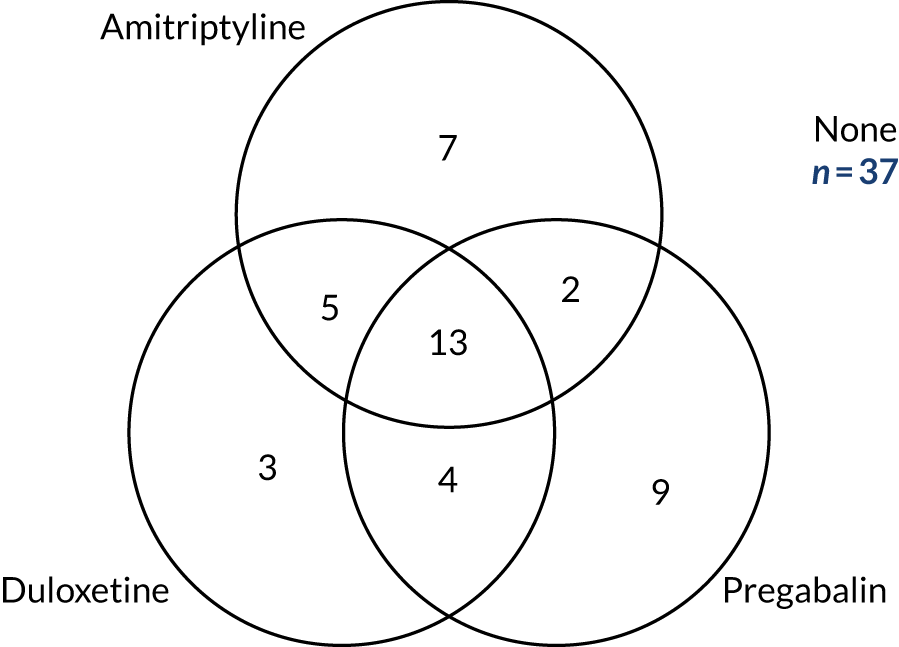
The results of the subgroup analyses are presented in Appendix 4, Figure 16. Pain was, on average, higher among younger (< 60 years) patients than among the older (≥ 60 years) patients, but the three pathways showed similar efficacy within each of the two subgroups (see Figure 16a). A similar theme was apparent for baseline pain score, with week 16 pain being higher among patients with more severe pain (i.e. a NRS score of ≥ 7 points) but no interaction with treatment (see Figure 16b).
The average week 16 NRS scores were similar across the three pain phenotypes (see Figure 16c). There were, however, some interactions among the individual NPSI components, with a suggestion that D-P was less suited to participants with higher NPSI scores.
There was some evidence of an interaction between treatment and baseline mood. The A-P pathway was notably worse at 16 weeks for patients with high levels of anxiety or depression. Patients with moderate anxiety or depression (scores below 15) had similar responses regardless of treatment arm (see Figure 16j and k).
Pain was also investigated in relation to whether or not the patient had previously used the three study drugs (i.e. amitriptyline, duloxetine and pregabalin) or any opioids (see Figure 16l–o). For all four drug options, patients who had previously been prescribed drugs reported higher pain scores than patients who had not, most notably opioids. Nevertheless, the difference between treatment arms appeared unaffected by previous medication use.
Finally, pain responses during the period of the UK lockdown due to COVID-19 (defined as on/after 20 March 2020) were similar to those measured prior to lockdown, meaning that the treatment comparisons were similar both within lockdown and outside lockdown (see Figure 16p and q). There was no apparent change in weekly NRS scores following imposition of lockdown measures, or any other temporal changes.
Tolerability
Tolerability (rated on a 0–10 NRS, with 10 being least tolerable) was similar between arms at weeks 6 and 16. Scores at weeks 6 and 16 were also similar, suggesting no perceived worsening in side effects with the introduction of combination therapy. In fact, self-reported tolerability was slightly better at week 16 than in week 6, albeit not statistically significantly so (Table 10).
| Tolerabilitya | Week 6 | Week 16 | ||||
|---|---|---|---|---|---|---|
| A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | |
| Responses, n | 92 | 86 | 96 | 83 | 84 | 83 |
| Discontinued because of adverse effect or poor response, n (%) | 6 (6) | 2 (2) | 6 (6) | 10 (10) | 4 (4) | 8 (8) |
| Tolerability of unwanted side effects over past 7 days, mean (SD) | 2.2 (2.5) | 2.1 (2.3) | 2.4 (2.9) | 2.3 (2.5) | 2.1 (2.6) | 1.9 (2.5) |
Quality of life
General health (assessed by SF-36), health utility (assessed by EQ-5D-5L) and anxiety/depression (assessed by HADS) are shown in Table 11. Self-reported QoL was similar across the three arms and no consistent patterns were observed among the QoL inventories. Some of the comparisons were statistically significant. After 6 weeks of treatment, duloxetine was associated with worse limitations due to physical health compared with amitriptyline and pregabalin, but was preferable with regard to general health, as assessed via SF-36 (Figure 12) and EQ-5D-5L. All differences were modest in clinical or psychometric terms. No statistically or clinically significant differences were found at 16 weeks.
| Outcome | Week 6 | Week 16 | ||||
|---|---|---|---|---|---|---|
| A-P | D-P | P-A | A-P | D-P | P-A | |
| Starting pathway, n | 104 | 100 | 107 | 104 | 100 | 107 |
| EQ-5D (higher scores indicate better health state) | ||||||
| EQ-5D-5L (crosswalk) | ||||||
| n | 93 | 87 | 99 | 86 | 86 | 86 |
| Mean (SD) | 0.516 (0.266) | 0.540 (0.237) | 0.489 (0.251) | 0.509 (0.253) | 0.511 (0.276) | 0.537 (0.243) |
| Pairwise comparisons, mean difference (98.3% CI); p-value | ||||||
| D-P vs. A-P | 0.029 (–0.023 to 0.081); 0.187 | –0.006 (–0.057 to 0.044); 0.766 | ||||
| P-A vs. A-P | –0.031 (–0.082 to 0.020); 0.149 | 0.009 (–0.041 to 0.059); 0.673 | ||||
| P-A vs. D-P | –0.060 (–0.111 to –0.008); 0.005 | 0.015 (–0.035 to 0.065); 0.468 | ||||
| EQ-5D (thermometer) | ||||||
| n | 92 | 87 | 99 | 86 | 85 | 86 |
| Mean (SD) | 56.5 (22.1) | 55.4 (20.4) | 56.3 (21.7) | 55.7 (22.4) | 57.3 (22.4) | 57.7 (22.4) |
| Pairwise comparisons, mean difference (98.3% CI); p-value | ||||||
| D-P vs. A-P | –0.4 (–5.2 to 4.4); 0.847 | 2.2 (–3.1 to 7.5); 0.316 | ||||
| P-A vs. A-P | –0.8 (–5.5 to 3.9); 0.673 | 1.6 (–3.6 to 6.9); 0.451 | ||||
| P-A vs. D-P | –0.4 (–5.2 to 4.3); 0.821 | –0.6 (–5.8 to 4.7); 0.793 | ||||
| SF-36 (higher scores indicate better QoL) | ||||||
| Physical health component | ||||||
| n | 93 | 87 | 99 | 86 | 86 | 86 |
| Mean (SD) | 25.4 (12.5) | 22.6 (12.6) | 24.5 (13.3) | 23.6 (13.0) | 24.1 (13.8) | 24.1 (13.1) |
| Pairwise comparisons, mean difference (98.3% CI); p-value | ||||||
| D-P vs. A-P | –2.9 (–4.9 to –0.9); < 0.001 | 0.4 (–1.9 to 2.7); 0.711 | ||||
| P-A vs. A-P | –1.4 (–3.4 to 0.5); 0.078 | –0.4 (–2.6 to 1.9); 0.699 | ||||
| P-A vs. D-P | 1.5 (–0.5 to 3.4); 0.067 | –0.7 (–3.0 to 1.5); 0.444 | ||||
| Mental health component | ||||||
| n | 93 | 87 | 99 | 86 | 86 | 86 |
| Mean (SD) | 46.7 (13.0) | 47.8 (11.8) | 47.6 (12.2) | 46.6 (12.8) | 46.3 (11.0) | 47.4 (12.3) |
| Pairwise comparisons, mean difference (98.3% CI); p-value | ||||||
| D-P vs. A-P | 1.3 (–1.0 to 3.6); 0.182 | –0.2 (–2.5 to 2.2); 0.855 | ||||
| P-A vs. A-P | 1.1 (–1.1 to 3.4); 0.229 | 0.8 (–1.5 to 3.1); 0.417 | ||||
| P-A vs. D-P | –0.1 (–2.4 to 2.1); 0.875 | 1.0 (–1.4 to 3.3); 0.317 | ||||
| HADS (higher scores indicate greater symptoms) | ||||||
| Anxiety | ||||||
| n | 93 | 85 | 99 | 86 | 86 | 86 |
| Mean (SD) | 7.5 (5.1) | 7.4 (4.6) | 6.7 (4.4) | 7.7 (5.4) | 7.3 (4.8) | 7.0 (4.6) |
| Pairwise comparisons, mean difference (98.3% CI); p-value | ||||||
| D-P vs. A-P | –0.1 (–0.9 to 0.7); 0.826 | –0.3 (–1.2 to 0.6); 0.449 | ||||
| P-A vs. A-P | –0.5 (–1.3 to 0.3); 0.138 | –0.4 (–1.4 to 0.5); 0.253 | ||||
| P-A vs. D-P | –0.4 (–1.2 to 0.4); 0.213 | –0.1 (–1.1 to 0.8); 0.705 | ||||
| Depression | ||||||
| n | 93 | 85 | 99 | 86 | 86 | 86 |
| Mean (SD) | 7.4 (4.7) | 7.3 (4.4) | 7.0 (4.5) | 7.3 (4.9) | 7.5 (4.5) | 7.2 (4.5) |
| Pairwise comparisons, mean difference (98.3% CI); p-value | ||||||
| D-P vs. A-P | –0.2 (–1.0 to 0.6); 0.489 | –0.1 (–0.9 to 0.6); 0.698 | ||||
| P-A vs. A-P | –0.4 (–1.1 to 0.4); 0.274 | –0.0 (–0.8 to 0.7); 0.903 | ||||
| P-A vs. D-P | –0.1 (–0.9 to 0.7); 0.705 | 0.1 (–0.6 to 0.8); 0.786 | ||||
FIGURE 12.
Short Form questionnaire-36 items domains. Graphs depict the model-based mean and CI for each treatment arm for each of the nine domains at week 16. Higher scores indicate better QoL.
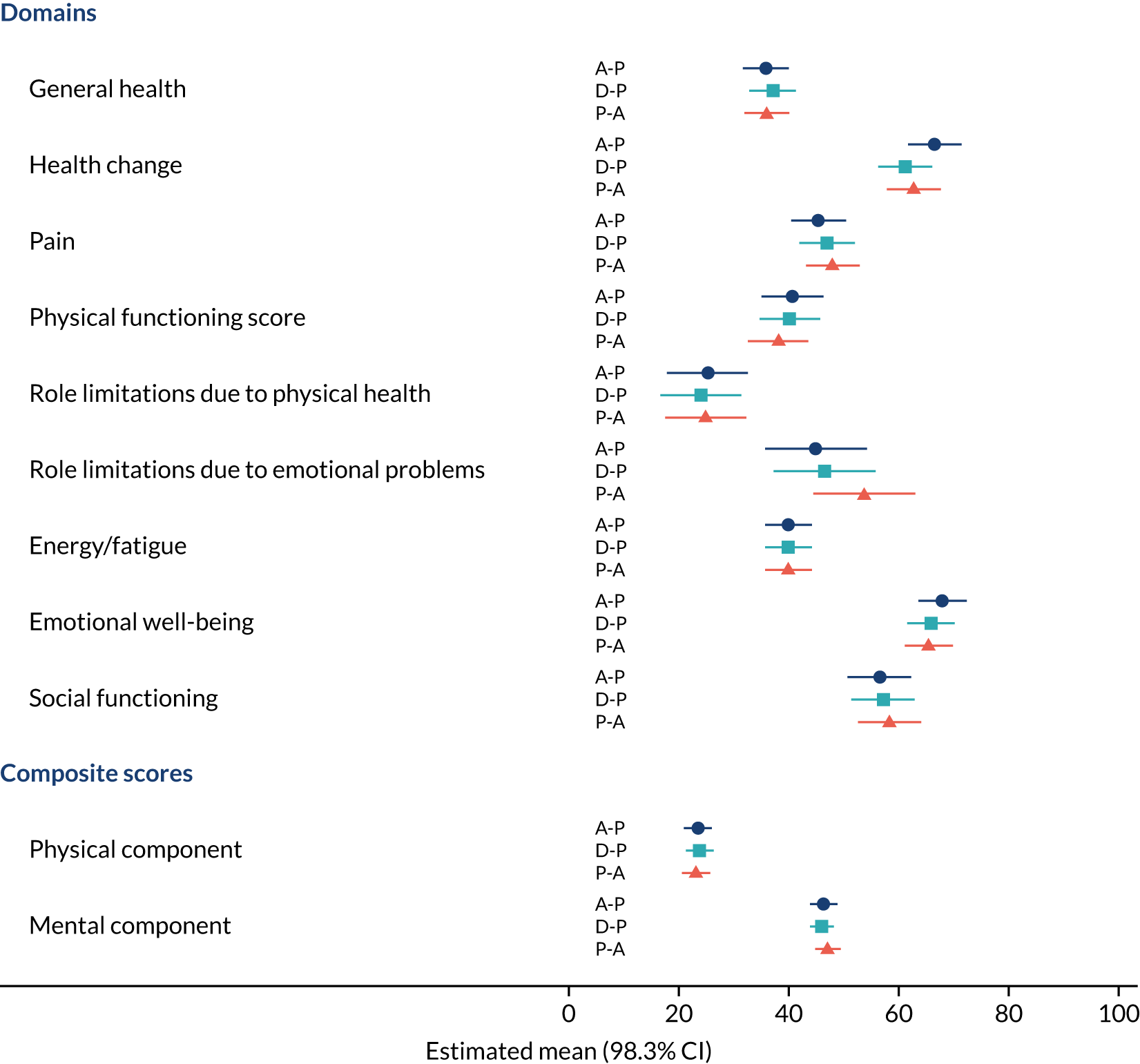
Other pain inventories and insomnia
As with the NRS, the NPSI and BPI-MSF were largely similar across arms at 6 and 16 weeks (Table 12). In general, the D-P arm had the highest pain scores at week 16, with the P-A arm having the lowest, but none of the comparisons was statistically significant. By contrast, week 6 pain scores tended to be higher in the P-A arm, with two BPI-MSF scores (worst pain over 24 hours and overall pain severity) being significantly lower in the A-P treatment pathway.
| Outcome | Baseline (N = 130) | Monotherapy phase (week 6) | Combination treatment phase (week 16) | ||||
|---|---|---|---|---|---|---|---|
| A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | ||
| Patients included, n | 130 | 93 | 87 | 99 | 86 | 86 | 86 |
| NPSI individual components (range 0–10; higher is greater pain), mean (SD) | |||||||
| Superficial spontaneous burning pain | 6.0 (2.9) | 3.4 (2.9) | 3.6 (2.7) | 3.9 (3.0) | 3.2 (3.0) | 3.7 (2.8) | 3.4 (2.8) |
| Deep spontaneous pressing pain | 4.8 (2.8) | 3.0 (2.7) | 2.9 (2.4) | 3.3 (2.9) | 2.9 (2.8) | 3.4 (2.5) | 3.1 (2.8) |
| Paroxysmal pain | 5.5 (2.9) | 3.5 (2.9) | 3.4 (2.5) | 3.6 (2.8) | 3.3 (3.0) | 3.8 (2.9) | 3.6 (2.8) |
| Evoked pain | 4.3 (2.6) | 2.9 (2.5) | 2.7 (2.3) | 2.9 (2.3) | 3.2 (2.7) | 3.0 (2.5) | 3.0 (2.6) |
| Paraesthesia/dysaesthesia | 6.3 (2.7) | 4.2 (2.7) | 4.3 (2.7) | 4.3 (2.8) | 4.1 (3.0) | 4.0 (2.9) | 4.4 (2.9) |
| Total score (range 0–50) | 52.4 (19.5) | 33.7 (21.9) | 33.2 (20.1) | 34.6 (20.9) | 33.8 (24.4) | 35.0 (21.5) | 33.9 (21.9) |
| BPI-MSF (range 0–10; higher is greater pain), mean (SD) | |||||||
| Pain severity score | 6.1 (1.7) | 3.8 (2.0)a | 3.9 (1.7) | 4.3 (2.2) | 3.7 (2.0) | 3.8 (1.9) | 3.5 (2.0) |
| Pain interference score | 5.9 (2.4) | 4.0 (2.6) | 4.2 (2.5) | 4.3 (2.7) | 4.1 (2.7) | 4.0 (2.6) | 3.7 (2.5) |
| Worst pain in last 24 hours | 7.2 (1.8) | 4.3 (2.2)b | 4.5 (2.0) | 5.0 (2.5) | 4.4 (2.3) | 4.5 (2.3) | 4.3 (2.5) |
| Least pain in last 24 hours | 5.0 (2.2) | 3.1 (2.2) | 3.2 (1.9) | 3.5 (2.4) | 3.0 (2.2) | 3.0 (2.0) | 2.8 (2.0) |
| Average pain | 6.1 (1.7) | 4.0 (1.9) | 4.1 (1.6) | 4.4 (2.2) | 3.8 (2.0) | 3.9 (1.9) | 3.7 (2.0) |
| Pain right now | 6.0 (2.2) | 3.5 (2.3) | 3.7 (2.1) | 4.1 (2.6) | 3.5 (2.4) | 3.6 (2.3) | 3.2 (2.3) |
Self-reported insomnia (via the ISI) is summarised in Table 13. Patients rated their insomnia worse in the D-P arm than in the A-P arm, both at week 6 (mean difference = 1.5, 98.3% CI 0.0 to 3.1; p = 0.016) and at week 16 (mean difference = 1.5, 98.3% CI 0.1 to 3.0; p = 0.010).
| Outcome | Baseline (N = 130) | Monotherapy phase (week 6) | Combination treatment phase (week 16) | ||||
|---|---|---|---|---|---|---|---|
| A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | ||
| Patients included, n | 130 | 93 | 87 | 99 | 86 | 86 | 86 |
| ISI total score (0–28; higher scores indicate greater insomnia), mean (SD) | 18.1 (5.9) | 11.8 (7.3)a | 13.8 (6.3) | 12.1 (7.1) | 11.4 (7.3)a | 13.3 (6.8) | 12.1 (6.4) |
Patient impression of change and treatment preference
Finally, participants were asked to complete a global impression of change (using the PGIC) at the end of each pathway and their preferred treatment if they had completed the three pathways (Table 14). The treatments were rated positively among this group, with 44%, 43% and 49% of patients reporting ‘much improved’ or ‘very much improved’ for A-P, D-P and P-A, respectively (Kruskal–Wallis test p = 0.70).
| Outcome | Treatment pathway | ||
|---|---|---|---|
| A-P | D-P | P-A | |
| Starting pathway, n | 104 | 100 | 107 |
| PGIC, n (%) | |||
| Very much improved | 11 (11) | 8 (8) | 12 (12) |
| Much improved | 32 (33) | 33 (35) | 36 (37) |
| Minimally improved | 25 (26) | 26 (27) | 22 (23) |
| No change | 19 (20) | 17 (18) | 16 (16) |
| Minimally worse | 6 (6) | 8 (8) | 10 (10) |
| Much worse | 4 (4) | 3 (3) | 1 (1) |
| Kruskal–Wallis test for difference between groups | p = 0.702 | ||
| Preferred treatment, n (%) | |||
| Stated preference at end of studya | 11 (24) | 15 (33) | 20 (43) |
| Chi-squared test for difference between groups: | p = 0.266 | ||
On reaching the end of the study, participants were asked to rate their preferred treatment of the study. The most popular choice was P-A (43%), followed by D-P (33%) and A-P (24%), although this comparison was not statistically significant (p = 0.266).
Adverse events
Adverse events are summarised by pathway in Table 15. Fatigue was the most commonly reported AE and was similarly prevalent in each pathway. The most notable difference was in the incidence of dry mouth, which was associated with amitriptyline (A-P, 32%; P-A, 17%; D-P, 8%) (p < 0.001). Dizziness was more common in patients in the P-A arm (24%) than in patients in the A-P (12%) and D-P (16%) arms, and nausea was highest (23%) in the D-P arm (vs. 5% in the A-P arm and 7% in the P-A arm; p = 0.001).
| AE category | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | p-value | |||
|---|---|---|---|---|---|---|---|
| Events, n | Patients, n (%) | Events, n | Patients, n (%) | Events, n | Patients, n (%) | ||
| Fatigue | 25 | 21 (20) | 23 | 18 (18) | 25 | 22 (21) | 0.880 |
| Dizziness | 12 | 12 (12) | 17 | 16 (16) | 33 | 26 (24) | 0.036 |
| Dry mouth | 34 | 33 (32) | 8 | 8 (8) | 20 | 18 (17) | < 0.001 |
| Sedation | 22 | 21 (20) | 14 | 11 (11) | 18 | 15 (14) | 0.167 |
| Diarrhoea | 22 | 18 (17) | 17 | 16 (16) | 11 | 9 (8) | 0.122 |
| Fall | 12 | 7 (7) | 17 | 12 (12) | 17 | 10 (9) | 0.880 |
| Oedema | 10 | 9 (9) | 13 | 10 (10) | 18 | 17 (16) | 0.150 |
| Nausea | 5 | 5 (5) | 27 | 23 (23) | 8 | 7 (7) | 0.001 |
| Constipation | 13 | 11 (11) | 15 | 13 (13) | 9 | 8 (7) | 0.469 |
| Headaches | 11 | 9 (9) | 16 | 14 (14) | 10 | 8 (7) | 0.335 |
| Vomiting | 8 | 7 (7) | 12 | 11 (11) | 8 | 8 (7) | 0.513 |
| Excessive sweating | 11 | 9 (9) | 10 | 10 (10) | 6 | 6 (6) | 0.576 |
| Insomnia | 8 | 6 (6) | 9 | 8 (8) | 7 | 7 (7) | 0.902 |
| Abdominal cramping | 6 | 5 (5) | 8 | 6 (6) | 4 | 4 (4) | 0.580 |
| Ataxia | 6 | 4 (4) | 4 | 4 (4) | 8 | 8 (7) | 0.415 |
| Pruritus | 2 | 2 (2) | 9 | 8 (8) | 5 | 5 (5) | 0.170 |
| Weight gain | 3 | 3 (3) | 1 | 1 (1) | 10 | 10 (9) | NC |
| Decreased appetite | 5 | 5 (5) | 5 | 5 (5) | 2 | 2 (2) | 0.401 |
| Inability to concentrate | 5 | 5 (5) | 1 | 1 (1) | 6 | 6 (6) | 0.242 |
| Cardiac ischaemia | 3 | 2 (2) | 3 | 3 (3) | 5 | 5 (5) | NC |
| Hypoglycaemia | 4 | 4 (4) | 3 | 3 (3) | 4 | 4 (4) | NC |
| Blurred vision | 2 | 2 (2) | 1 | 1 (1) | 5 | 5 (5) | NC |
| Low mood | 6 | 6 (6) | 1 | 1 (1) | 1 | 1 (1) | 0.112 |
| Hypotension | 1 | 1 (1) | 5 | 5 (5) | 1 | 1 (1) | NC |
| Restless legs | 2 | 2 (2) | 5 | 5 (5) | 0 | NC | |
| Anxiety | 2 | 2 (2) | 2 | 1 (1) | 2 | 2 (2) | NC |
| Hyperglycaemia | 3 | 2 (2) | 1 | 1 (1) | 2 | 2 (2) | NC |
| Dysarthria | 1 | 1 (1) | 0 | 4 | 4 (4) | NC | |
| Kidney dysfunction | 1 | 1 (1) | 2 | 2 (2) | 2 | 2 (2) | NC |
| Hallucinations | 3 | 1 (1) | 0 | 1 | 1 (1) | NC | |
| Heart failure | 0 | 2 | 2 (2) | 1 | 1 (1) | NC | |
| Liver dysfunction | 1 | 1 (1) | 1 | 1 (1) | 1 | 1 (1) | NC |
| Tachycardia | 2 | 1 (1) | 1 | 1 (1) | 0 | NC | |
| Transient ischaemic attack | 0 | 3 | 3 (3) | 0 | NC | ||
| Increased appetite | 2 | 1 (1) | 0 | 0 | NC | ||
| Urinary retention | 1 | 1 (1) | 1 | 1 (1) | 0 | NC | |
| Seizure | 0 | 1 | 1 (1) | 0 | NC | ||
| Other | 180 | 72 (69) | 181 | 70 (70) | 190 | 77 (72) | |
Table 16 shows AEs further split by the time they occurred (i.e. before or after 6 weeks) and SAEs are tabulated in Table 17. One SUSAR occurred [one participant experienced an atrioventricular heart block (Mobitz II) in the P-A arm 11 weeks into the first treatment pathway and while on amitriptyline, having switched from pregabalin at the week 6 visit].
| AE category | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Events, n | Patients, n (%) | Events, n | Patients, n (%) | Events, n | Patients, n (%) | ||||
| Started < 6 weeks | Started > 6 weeks | Started < 6 weeks | Started > 6 weeks | Started < 6 weeks | Started > 6 weeks | ||||
| Number in study | 104 | 96 | 100 | 94 | 107 | 103 | |||
| Fatigue | 25 | 18 (17) | 5 (5) | 23 | 17 (17) | 4 (4) | 25 | 11 (10) | 13 (13) |
| Dizziness | 12 | 8 (8) | 6 (6) | 17 | 8 (8) | 8 (9) | 33 | 19 (18) | 11 (11) |
| Dry mouth | 34 | 22 (21) | 12 (13) | 8 | 5 (5) | 3 (3) | 20 | 10 (9) | 10 (10) |
| Sedation | 22 | 19 (18) | 2 (2) | 14 | 6 (6) | 6 (6) | 18 | 10 (9) | 7 (7) |
| Diarrhoea | 22 | 8 (8) | 11 (11) | 17 | 10 (10) | 7 (7) | 11 | 6 (6) | 4 (4) |
| Fall | 12 | 3 (3) | 5 (5) | 17 | 6 (6) | 7 (7) | 17 | 5 (5) | 7 (7) |
| Oedema | 10 | 2 (2) | 7 (7) | 13 | 5 (5) | 7 (7) | 18 | 14 (13) | 3 (3) |
| Nausea | 5 | 4 (4) | 1 (1) | 27 | 19 (19) | 6 (6) | 8 | 6 (6) | 2 (2) |
| Constipation | 13 | 9 (9) | 4 (4) | 15 | 8 (8) | 6 (6) | 9 | 5 (5) | 3 (3) |
| Headaches | 11 | 8 (8) | 1 (1) | 16 | 10 (10) | 7 (7) | 10 | 7 (7) | 2 (2) |
| Vomiting | 8 | 5 (5) | 2 (2) | 12 | 9 (9) | 3 (3) | 8 | 1 (1) | 7 (7) |
| Excessive sweating | 11 | 7 (7) | 4 (4) | 10 | 7 (7) | 3 (3) | 6 | 1 (1) | 5 (5) |
| Insomnia | 8 | 3 (3) | 3 (3) | 9 | 7 (7) | 2 (2) | 7 | 3 (3) | 6 (6) |
| Abdominal cramping | 6 | 4 (4) | 2 (2) | 8 | 4 (4) | 2 (2) | 4 | 3 (3) | 1 (1) |
| Ataxia | 6 | 1 (1) | 3 (3) | 4 | 2 (2) | 2 (2) | 8 | 7 (7) | 1 (1) |
| Pruritus | 2 | 1 (1) | 1 (1) | 9 | 5 (5) | 3 (3) | 5 | 3 (3) | 2 (2) |
| Weight gain | 3 | 2 (2) | 1 (1) | 1 | 1 (1) | 0 | 10 | 7 (7) | 3 (3) |
| Decreased appetite | 5 | 4 (4) | 1 (1) | 5 | 5 (5) | 0 | 2 | 0 | 2 (2) |
| Inability to concentrate | 5 | 4 (4) | 1 (1) | 1 | 1 (1) | 0 | 6 | 6 (6) | 0 |
| Low mood | 6 | 5 (5) | 1 (1) | 1 | 1 (1) | 0 | 1 | 1 (1) | 0 |
| Hypotension | 1 | 0 | 1 (1) | 5 | 0 | 5 (5) | 1 | 1 (1) | 0 |
| Restless legs | 2 | 1 (1) | 1 (1) | 5 | 1 (1) | 4 (4) | 0 | 0 | 0 |
| SAE category | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | |||
|---|---|---|---|---|---|---|
| Events, n | Patients, n (%) | Events, n | Patients, n (%) | Events, n | Patients, n (%) | |
| Any SAE | 6 | 4 (4) | 12 | 10 (10) | 13 | 10 (9) |
| Vomiting | 1 | 1 (1) | 1 | 1 (1) | 1 | 1 (1) |
| Cardiac ischaemia | 0 | 0 | 2 | 2 (2) | ||
| Dizziness | 0 | 0 | 2 | 2 (2) | ||
| Headaches | 0 | 2 | 1 (1) | 0 | ||
| Hyperglycaemia | 2 | 1 (1) | 0 | 0 | ||
| Hypoglycaemia | 0 | 1 | 1 (1) | 1 | 1 (1) | |
| Kidney dysfunction | 0 | 0 | 1 | 1 (1) | ||
| Transient ischaemic attack | 0 | 1 | 1 (1) | 0 | ||
| Other | 3 | 3 (3) | 7 | 7 (7) | 6 | 6 (6) |
The remaining tables (Tables 18–20) focus on AEs of moderate or severe intensity that are related to one or both study medications. In total, 97 such events occurred, affecting 19% of patients in the A-P arm, 22% of patients in the D-P arm and 21% of patients in the P-A arm (p = 0.719) (see Table 18). A post hoc analysis looked at days affected by moderate or severe related AEs (see Table 19) while in study follow-up, which for the majority of patients was zero but for the groups as a whole resulted in 84, 69 and 73 days per 1000 days of follow-up in the A-P, D-P and P-A arms, respectively (p = 0.858). Most events started during monotherapy but continued to affect patients beyond week 6, by which point combination therapy may have started. The mean days affected for each category is displayed visually in Figure 13 and in Table 20. The AEs with the longest average person-day impacts were dry mouth in the A-P arm (191 days/1000 days of follow-up) and fatigue in all arms (109 days/1000 days of follow-up).
| AE category | A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | |||
|---|---|---|---|---|---|---|
| Events, n | Patients, n (%) | Events, n | Patients, n (%) | Events, n | Patients, n (%) | |
| Any moderate or severe probably related AEa | 30 | 20 (19) | 35 | 22 (22) | 32 | 22 (21) |
| Fatigue | 11 | 10 (10) | 7 | 7 (7) | 9 | 9 (8) |
| Fall | 7 | 6 (6) | 9 | 5 (5) | 9 | 6 (6) |
| Dizziness | 4 | 4 (4) | 6 | 6 (6) | 12 | 12 (11) |
| Sedation | 7 | 7 (7) | 4 | 4 (4) | 7 | 6 (6) |
| Oedema | 5 | 5 (5) | 6 | 6 (6) | 6 | 6 (6) |
| Vomiting | 3 | 3 (3) | 7 | 7 (7) | 5 | 5 (5) |
| Diarrhoea | 5 | 5 (5) | 7 | 7 (7) | 2 | 2 (2) |
| Insomnia | 3 | 3 (3) | 6 | 6 (6) | 3 | 3 (3) |
| Headaches | 2 | 2 (2) | 8 | 7 (7) | 1 | 1 (1) |
| Nausea | 1 | 1 (1) | 9 | 8 (8) | 1 | 1 (1) |
| Dry mouth | 8 | 8 (8) | 0 | 2 | 2 (2) | |
| Abdominal cramping | 4 | 3 (3) | 4 | 4 (4) | 1 | 1 (1) |
| Ataxia | 4 | 3 (3) | 2 | 2 (2) | 2 | 2 (2) |
| Cardiac ischaemia | 1 | 1 (1) | 2 | 2 (2) | 5 | 5 (5) |
| Excessive sweating | 4 | 4 (4) | 4 | 4 (4) | 0 | |
| Inability to concentrate | 3 | 3 (3) | 1 | 1 (1) | 3 | 3 (3) |
| Constipation | 3 | 3 (3) | 3 | 3 (3) | 0 | |
| Low mood | 5 | 5 (5) | 0 | 1 | 1 (1) | |
| Blurred vision | 1 | 1 (1) | 1 | 1 (1) | 3 | 3 (3) |
| Hypotension | 0 | 4 | 4 (4) | 1 | 1 (1) | |
| Pruritus | 1 | 1 (1) | 2 | 2 (2) | 2 | 2 (2) |
| Hypoglycaemia | 2 | 2 (2) | 1 | 1 (1) | 1 | 1 (1) |
| Kidney dysfunction | 1 | 1 (1) | 1 | 1 (1) | 2 | 2 (2) |
| Anxiety | 1 | 1 (1) | 1 | 1 (1) | 1 | 1 (1) |
| Decreased appetite | 0 | 2 | 2 (2) | 1 | 1 (1) | |
| Liver dysfunction | 1 | 1 (1) | 1 | 1 (1) | 1 | 1 (1) |
| Transient ischaemic attack | 0 | 3 | 3 (3) | 0 | ||
| Dysarthria | 1 | 1 (1) | 0 | 1 | 1 (1) | |
| Heart failure | 0 | 1 | 1 (1) | 1 | 1 (1) | |
| Hyperglycaemia | 2 | 1 (1) | 0 | 0 | ||
| Restless legs | 1 | 1 (1) | 1 | 1 (1) | 0 | |
| Hallucinations | 0 | 0 | 1 | 1 (1) | ||
| Seizure | 0 | 1 | 1 (1) | 0 | ||
| Urinary retention | 1 | 1 (1) | 0 | 0 | ||
| Weight gain | 0 | 0 | 1 | 1 (1) | ||
| Treatment pathway | p-value | |||
|---|---|---|---|---|
| A-P (N = 104) | D-P (N = 100) | P-A (N = 107) | ||
| Total duration | ||||
| Number of patients with AE/number of patients in follow-up | 20/104 | 22/100 | 22/107 | |
| Average weeks affected per person | 1.2 | 1.0 | 1.1 | 0.858 |
| Days affected per 1000 days of follow-up | 84.0 | 68.7 | 72.6 | |
| Pairwise comparisons, IRR (98.3% CI) | ||||
| D-P vs. A-P | 0.70 (0.15 to 3.32) | 0.582 | ||
| P-A vs. A-P | 0.81 (0.18 to 3.75) | 0.747 | ||
| P-A vs. D-P | 1.17 (0.25 to 5.53) | 0.814 | ||
| First-line monotherapy: weeks 1–6 | ||||
| Number of patients with AE/number of patients in follow-up | 14/104 | 11/100 | 11/107 | |
| Average weeks affected per person | 0.4 | 0.4 | 0.2 | 0.742 |
| Days affected per 1000 days of follow-up | 74.6 | 73.8 | 37.4 | |
| Pairwise comparisons, IRR (98.3% CI) | ||||
| D-P vs. A-P | 0.97 (0.12 to 7.62) | 0.971 | ||
| P-A vs. A-P | 0.55 (0.07 to 4.41) | 0.493 | ||
| P-A vs. D-P | 0.57 (0.07 to 4.51) | 0.514 | ||
| Combination therapy | ||||
| Number of patients with AE/number of patients in follow-up | 8/45 | 13/42 | 10/47 | |
| Average weeks affected per person | 1.1 | 0.9 | 0.9 | 0.881 |
| Days affected per 1000 days of follow-up | 283.2 | 206.1 | 244.0 | |
| Pairwise comparisons IRR (98.3% CI) | ||||
| D-P vs. A-P | 0.75 (0.08 to 6.86) | 0.758 | ||
| P-A vs. A-P | 1.19 (0.14 to 10.10) | 0.846 | ||
| P-A vs. D-P | 1.58 (0.18 to 14.07) | 0.617 | ||
| Monotherapy vs. combination therapy | ||||
| Number of days affected/total follow-up | ||||
| Monotherapy | 731/11,998 | |||
| Combination therapy | 894/8480 | |||
| Days affected per 1000 days of follow-up | ||||
| Monotherapy | 60.9 | |||
| Combination therapy | 105.4 | |||
| Combination vs. monotherapy, IRR (98.3% CI) | 1.72 (0.45 to 6.56) | 0.331 | ||
FIGURE 13.
Days impacted by adverse effects.
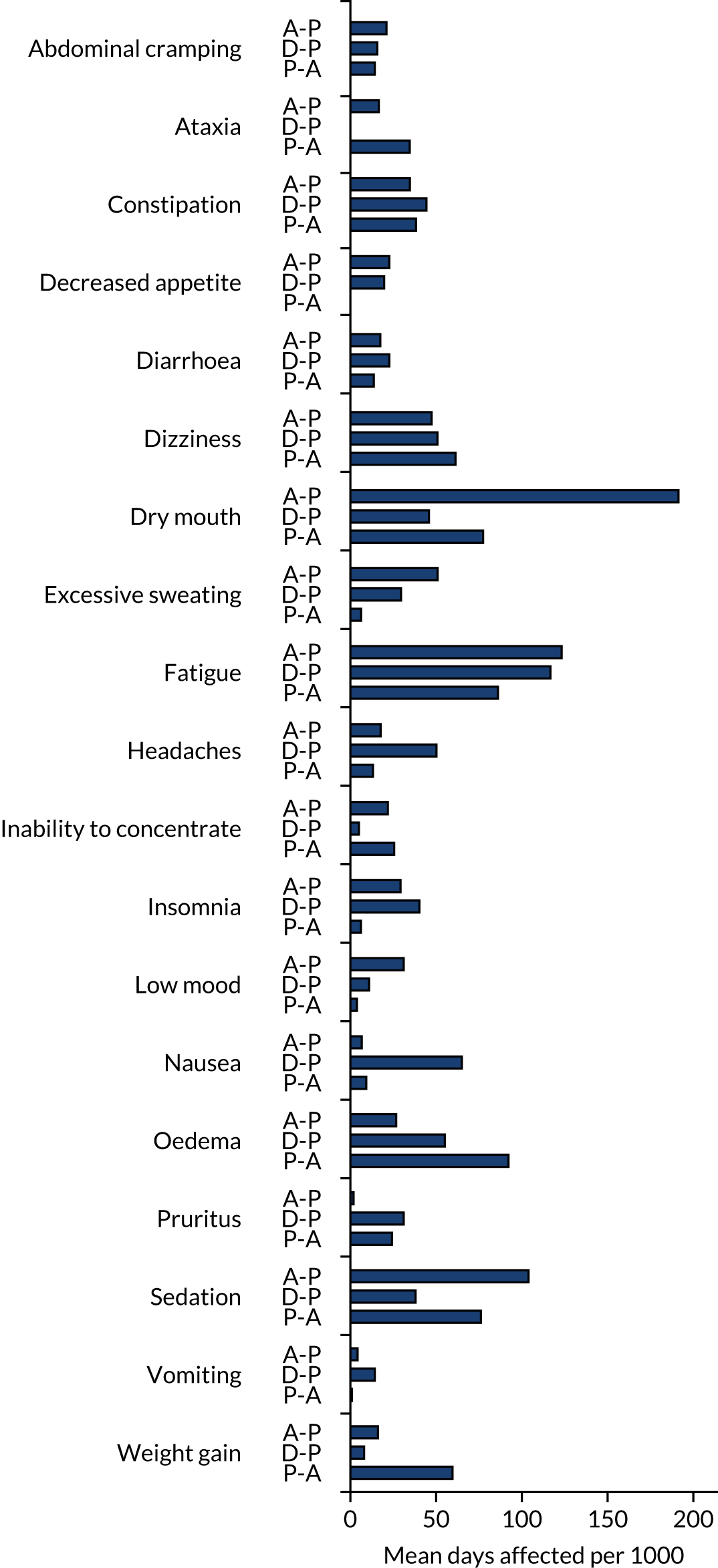
| Days affected by adverse effects | A-P | D-P | P-A | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Overall | Weeks 1–6 | Weeks 7–16 | Overall | Weeks 1–6 | Weeks 7–16 | Overall | Weeks 1–6 | Weeks 7–16 | |
| Patients, n | 104 | 104 | 96 | 100 | 100 | 94 | 107 | 107 | 103 |
| Total person-weeks | 1503.6 | 598.4 | 899.1 | 1483.7 | 582.1 | 897.6 | 1582.0 | 636.9 | 943.4 |
| Abdominal cramping | |||||||||
| Average weeks affected per person | 2.1 | 0.6 | 1.7 | 1.6 | 0.5 | 1.1 | 1.5 | 0.6 | 0.9 |
| Days affected per 1000 days of follow-up | 21.1 | 14.3 | 25.7 | 15.7 | 13.4 | 17.3 | 14.3 | 14.1 | 14.4 |
| Ataxia | |||||||||
| Average weeks affected per person | 1.7 | 0.4 | 1.4 | 0.0 | 0.0 | 0.0 | 3.6 | 1.4 | 2.3 |
| Days affected per 1000 days of follow-up | 16.6 | 10.0 | 21.1 | 0.1 | 0.1 | 0.1 | 34.6 | 32.5 | 36.1 |
| Constipation | |||||||||
| Average weeks affected per person | 3.5 | 1.2 | 2.5 | 4.6 | 1.9 | 2.8 | 4.0 | 0.9 | 3.1 |
| Days affected per 1000 days of follow-up | 34.7 | 29.1 | 38.6 | 44.4 | 47.1 | 42.8 | 38.3 | 22.5 | 49.0 |
| Decreased appetite | |||||||||
| Average weeks affected per person | 2.3 | 0.9 | 1.4 | 2.0 | 1.0 | 1.1 | 0.0 | 0.0 | 0.0 |
| Days affected per 1000 days of follow-up | 22.7 | 23.5 | 22.2 | 19.7 | 23.8 | 17.2 | 0.0 | 0.0 | 0.0 |
| Diarrhoea | |||||||||
| Average weeks affected per person | 1.8 | 0.6 | 1.3 | 2.4 | 0.8 | 1.6 | 1.4 | 1.0 | 0.5 |
| Days affected per 1000 days of follow-up | 17.5 | 14.4 | 19.7 | 22.7 | 19.4 | 24.9 | 13.7 | 23.3 | 7.3 |
| Dizziness | |||||||||
| Average weeks affected per person | 4.8 | 1.2 | 3.8 | 5.3 | 1.1 | 4.4 | 6.3 | 2.9 | 3.6 |
| Days affected per 1000 days of follow-up | 47.4 | 30.8 | 58.7 | 50.8 | 26.0 | 67.2 | 61.3 | 68.5 | 56.5 |
| Dry mouth | |||||||||
| Average weeks affected per person | 19.4 | 5.5 | 14.7 | 4.8 | 1.2 | 3.7 | 8.0 | 1.7 | 6.4 |
| Days affected per 1000 days of follow-up | 191.3 | 137.4 | 228.5 | 45.8 | 30.3 | 56.1 | 77.4 | 41.2 | 102.0 |
| Excessive sweating | |||||||||
| Average weeks affected per person | 5.1 | 2.1 | 3.2 | 3.1 | 1.7 | 1.4 | 0.6 | 0.3 | 0.4 |
| Days affected per 1000 days of follow-up | 50.9 | 52.0 | 50.4 | 29.5 | 41.3 | 22.0 | 6.3 | 6.7 | 6.0 |
| Fatigue | |||||||||
| Average weeks affected per person | 12.5 | 4.5 | 8.5 | 12.1 | 5.0 | 7.4 | 8.9 | 2.1 | 7.0 |
| Days affected per 1000 days of follow-up | 123.2 | 111.4 | 132.0 | 116.7 | 122.1 | 113.8 | 86.1 | 49.9 | 110.6 |
| Headaches | |||||||||
| Average weeks affected per person | 1.8 | 1.1 | 0.7 | 5.2 | 1.4 | 4.0 | 1.4 | 1.0 | 0.3 |
| Days affected per 1000 days of follow-up | 17.7 | 27.1 | 11.5 | 50.2 | 34.7 | 60.4 | 13.2 | 25.1 | 5.2 |
| Inability to concentrate | |||||||||
| Average weeks affected per person | 2.2 | 1.1 | 1.2 | 0.5 | 0.3 | 0.2 | 2.6 | 1.2 | 1.5 |
| Days affected per 1000 days of follow-up | 21.9 | 27.6 | 18.3 | 5.0 | 7.6 | 3.3 | 25.5 | 27.9 | 23.9 |
| Insomnia | |||||||||
| Average weeks affected per person | 3.0 | 0.9 | 2.2 | 4.2 | 1.8 | 2.5 | 0.6 | 0.2 | 0.5 |
| Days affected per 1000 days of follow-up | 29.2 | 22.4 | 33.9 | 40.3 | 43.8 | 38.2 | 6.2 | 4.7 | 7.2 |
| Low mood | |||||||||
| Average weeks affected per person | 3.1 | 1.5 | 1.7 | 1.1 | 0.4 | 0.7 | 0.4 | 0.0 | 0.4 |
| Days affected per 1000 days of follow-up | 31.1 | 37.8 | 26.8 | 10.9 | 10.3 | 11.3 | 3.8 | 0.2 | 6.2 |
| Nausea | |||||||||
| Average weeks affected per person | 0.7 | 0.7 | 0.0 | 6.8 | 3.3 | 3.6 | 1.0 | 0.6 | 0.4 |
| Days affected per 1000 days of follow-up | 6.6 | 16.6 | 0.0 | 65.0 | 81.3 | 54.7 | 9.3 | 14.2 | 5.9 |
| Oedema | |||||||||
| Average weeks affected per person | 2.7 | 0.1 | 2.7 | 5.7 | 0.9 | 5.1 | 9.5 | 2.6 | 7.1 |
| Days affected per 1000 days of follow-up | 26.7 | 3.7 | 42.3 | 55.1 | 21.2 | 77.4 | 92.2 | 61.6 | 113.0 |
| Pruritus | |||||||||
| Average weeks affected per person | 0.2 | 0.2 | 0.0 | 3.2 | 1.2 | 2.1 | 2.5 | 0.6 | 1.9 |
| Days affected per 1000 days of follow-up | 1.9 | 4.3 | 0.4 | 31.1 | 30.3 | 31.7 | 24.3 | 14.5 | 31.0 |
| Sedation | |||||||||
| Average weeks affected per person | 10.5 | 5.0 | 5.9 | 4.0 | 1.0 | 3.1 | 7.9 | 2.3 | 5.7 |
| Days affected per 1000 days of follow-up | 103.9 | 12.7 | 91.5 | 38.0 | 24.3 | 47.1 | 76.1 | 54.5 | 90.8 |
| Vomiting | |||||||||
| Average weeks affected per person | 0.4 | 0.3 | 0.1 | 1.5 | 1.0 | 0.5 | 0.1 | 0.1 | 0.0 |
| Days affected per 1000 days of follow-up | 4.2 | 8.0 | 1.7 | 14.2 | 23.9 | 8.0 | 0.9 | 1.2 | 0.8 |
| Weight gain | |||||||||
| Average weeks affected per person | 1.6 | 0.5 | 1.1 | 0.8 | 0.1 | 0.8 | 6.2 | 1.4 | 4.9 |
| Days affected per 1000 days of follow-up | 16.1 | 13.6 | 17.9 | 8.0 | 2.0 | 11.9 | 59.6 | 33.2 | 77.5 |
Chapter 4 Health economics results
Quality of life and quality-adjusted life-years
Table 21 presents EQ-5D utility values at baseline and at 6 and 16 weeks for the three treatment pathways. QALYs were similar for the three groups and equate to approximately 55 days or just under 2 months over a 16-week period.
| Treatment pathway | Mean EQ-5D values and QALYs (95% bootstrapped CI) | |||
|---|---|---|---|---|
| Baseline | Week 6 | Week 16 | QALYs | |
| A-P (n = 88) | 0.411 (0.352 to 0.466) | 0.515 (0.455 to 0.568) | 0.509 (0.453 to 0.559) | 0.152 (0.136 to 0.167) |
| D-P (n = 88) | 0.408 (0.350 to 0.460) | 0.551 (0.499 to 0.594) | 0.508 (0.448 to 0.562) | 0.157 (0.142 to 0.171) |
| P-A (n = 87) | 0.420 (0.361 to 0.471) | 0.495 (0.438 to 0.545) | 0.545 (0.490 to 0.591) | 0.152 (0.138 to 0.167) |
Table 22 provides a breakdown of EQ-5D-5L responses for each of the five domains of the EQ-5D-5L for participants for whom QALYs were generated. At baseline, no one indicated that they had no pain or discomfort, but this improved at 6 and 16 weeks for all three pathways. Similarly, the proportion of patients with no problems with mobility, usual activities and anxiety or depression also increased over time.
| Time point | EQ-5D-5L item | Response, n (%) | ||||
|---|---|---|---|---|---|---|
| No problems | Slight | Moderate | Severe | Unable to do/extreme | ||
| Baseline (n = 130) | Mobility | 10 (7.7) | 26 (20.0) | 45 (34.6) | 47 (36.2) | 2 (1.5) |
| Self-care | 61 (46.9) | 30 (23.1) | 23 (17.7) | 14 (10.8) | 2 (1.5) | |
| Usual activities | 13 (10.0) | 33 (25.4) | 45 (34.6) | 31 (23.8) | 8 (6.2) | |
| Pain/discomfort | 0 (0.0) | 11 (8.5) | 47 (36.2) | 59 (45.4) | 13 (10.0) | |
| Anxiety/depression | 52 (40.0) | 38 (29.2) | 23 (17.7) | 15 (11.5) | 2 (1.5) | |
| A-P | ||||||
| Week 6 (n = 88) | Mobility | 13 (14.8) | 18 (20.5) | 30 (34.1) | 26 (29.8) | 1 (1.1) |
| Self-care | 43 (48.9) | 20 (22.3) | 17 (19.3) | 7 (8.0) | 1 (1.1) | |
| Usual activities | 14 (15.9) | 28 (31.8) | 25 (28.4) | 18 (20.5) | 3 (3.4) | |
| Pain/discomfort | 3 (3.4) | 19 (21.6) | 46 (52.3) | 18 (20.5) | 2 (2.3) | |
| Anxiety/depression | 39 (44.3) | 20 (22.3) | 18 (20.5) | 6 (6.8) | 5 (5.7) | |
| Week 16 (n = 88) | Mobility | 9 (10.2) | 16 (18.2) | 38 (43.2) | 24 (27.3) | 1 (1.1) |
| Self-care | 43 (48.9) | 20 (22.3) | 21 (24.7) | 3 (3.4) | 1 (1.1) | |
| Usual activities | 17 (19.3) | 20 (22.3) | 33 (37.5) | 16 (18.2) | 2 (2.3) | |
| Pain/discomfort | 4 (4.5) | 16 (18.2) | 38 (43.2) | 28 (31.8) | 2 (2.3) | |
| Anxiety/depression | 42 (47.8) | 23 (26.1) | 13 (14.8) | 9 (10.2) | 1 (1.1) | |
| D-P | ||||||
| Week 6 (n = 91) | Mobility | 14 (15.9) | 17 (19.3) | 36 (40.9) | 20 (22.3) | 1 (1.1) |
| Self-care | 39 (44.3) | 26 (29.5) | 16 (18.2) | 6 (6.8) | 1 (1.1) | |
| Usual activities | 20 (22.3) | 19 (21.6) | 36 (40.9) | 9 (10.2) | 4 (4.5) | |
| Pain/discomfort | 4 (4.5) | 14 (15.9) | 54 (61.4) | 16 (18.2) | 0 (0.0) | |
| Anxiety/depression | 45 (51.1) | 22 (25.0) | 16 (18.2) | 2 (2.3) | 3 (3.4) | |
| Week 16 (n = 88) | Mobility | 20 (22.3) | 11 (12.5) | 29 (30.0) | 27 (30.7) | 1 (1.1) |
| Self-care | 39 (44.3) | 25 (28.4) | 15 (17.0) | 8 (9.1) | 1 (1.1) | |
| Usual activities | 24 (27.3) | 18 (20.5) | 28 (31.8) | 12 (13.6) | 6 (6.8) | |
| Pain/discomfort | 3 (3.4) | 16 (18.2) | 43 (48.9) | 23 (26.1) | 3 (3.4) | |
| Anxiety/depression | 42 (47.7) | 18 (20.5) | 18 (20.5) | 8 (9.1) | 2 (2.3) | |
| P-A | ||||||
| Week 6 (n = 87) | Mobility | 12 (13.8) | 18 (20.7) | 30 (34.5) | 26 (29.9) | 1 (1.1) |
| Self-care | 44 (50.6) | 22 (25.3) | 17 (19.5) | 3 (3.4) | 1 (1.1) | |
| Usual activities | 17 (19.5) | 21 (24.1) | 31 (35.6) | 13 (14.9) | 5 (5.7) | |
| Pain/discomfort | 1 (1.1) | 19 (21.8) | 37 (42.5) | 25 (28.7) | 5 (5.7) | |
| Anxiety/depression | 39 (44.8) | 24 (27.6) | 16 (18.4) | 8 (9.2) | 0 (0.0) | |
| Week 16 (n = 87) | Mobility | 14 (16.1) | 19 (21.8) | 31 (35.6) | 22 (25.3) | 1 (1.1) |
| Self-care | 43 (49.4) | 21 (24.1) | 17 (19.5) | 6 (6.9) | 0 (0.0) | |
| Usual activities | 18 (20.7) | 24 (27.6) | 29 (33.3) | 11 (12.6) | 5 (5.7) | |
| Pain/discomfort | 1 (1.1) | 28 (32.2) | 41 (47.1) | 15 (17.2) | 2 (2.3) | |
| Anxiety/depression | 47 (54.0) | 15 (17.2) | 18 (20.7) | 5 (5.7) | 2 (2.3) | |
Resource use
The most commonly accessed resources were prescription services, podiatry, GPs, practice nurses and outpatient appointments (Table 23). For those attending accident and emergency, patients could attend between one and four times per period. Hospitalisation duration ranged from one to eight nights, and those patients who attended outpatient appointments could attend up to 12 times per period.
| Resource use | A-P | D-P | P-A | |||
|---|---|---|---|---|---|---|
| Week 6 | Week 16 | Week 6 | Week 16 | Week 6 | Week 16 | |
| A&E visit | 0 | 3 | 1 | 2 | 1 | 4 |
| Hospitalisations | 0 | 1 | 1 | 2 | 2 | 2 |
| Outpatient | 21 | 23 | 27 | 22 | 24 | 23 |
| GP surgery visit | 13 | 21 | 11 | 14 | 11 | 15 |
| GP home visit | 1 | 0 | 0 | 1 | 0 | 0 |
| GP telephone call | 7 | 5 | 2 | 5 | 6 | 6 |
| Practice nurse | 8 | 16 | 15 | 15 | 10 | 9 |
| Practice nurse telephone call | 2 | 3 | 5 | 2 | 2 | 8 |
| Prescription | 50 | 59 | 46 | 38 | 45 | 49 |
| Meals on Wheels | 0 | 0 | 0 | 0 | 0 | 0 |
| Home help | 2 | 2 | 3 | 1 | 3 | 1 |
| Social worker | 0 | 0 | 1 | 0 | 0 | 1 |
| Pain management | 0 | 1 | 5 | 1 | 3 | 0 |
| Physiotherapy | 2 | 3 | 5 | 4 | 4 | 4 |
| Occupational therapy | 1 | 1 | 1 | 0 | 1 | 1 |
| Podiatry: NHS | 30 | 34 | 36 | 29 | 36 | 36 |
| Podiatry: private | 3 | 3 | 1 | 1 | 1 | 3 |
| Other: NHSa | 3 | 2 | 4 | 3 | 1 | 3 |
| Other: privatea | 1 | 0 | 0 | 0 | 0 | 0 |
Table 24 presents a summary of resource use costs for each pathway. Costs were similar across the three pathways, with treatment costs being cheaper for A-P (mean £1424) and slightly more expensive for P-A (mean £1448) and D-P (mean £1452). Similarities were also observed for overall costs, with P-A, on average, having the lowest cost (mean £1942), with the average costs for A-P (mean £2012) and D-P (mean £2032) being slightly higher, but similar.
| Treatment cost | Mean treatment cost (£) (95% bootstrapped CI) | ||
|---|---|---|---|
| A-P (n = 88) | D-P (n = 88) | P-A (n = 87) | |
| Treatment medications | 19 (17 to 21) | 33 (29 to 36) | 24 (22 to 26) |
| Treatment visits | 1077 (1031 to 1118) | 1092 (1047 to 1136) | 1096 (1051 to 1140) |
| Treatment totala | 1424 (1376 to 1466) | 1452 (1405 to 1500) | 1448 (1401 to 1493) |
| Concomitant medicationsb | 38 (26 to 58) | 24 (15 to 36) | 33 (24 to 44) |
| Other resource use | 549 (357 to 963) | 555 (377 to 830) | 461 (325 to 701) |
| Total costs | 2012 (1808 to 2421) | 2032 (1852 to 2304) | 1942 (1800 to 2179) |
Order of treatment effect
Regression models were fitted to both costs and QALYs to examine the effect of order of receiving treatment pathways A-P, D-P and P-A. Table 25 presents the results with 95% bootstrap CIs for the order effect compared with receiving treatments in the first time period. The overall effect of order on costs and QALYs was not significant for the three treatment pathways, although it did approach statistical significance for the A-P pathway, with patients receiving A-P in the third time period more likely to have lower costs than patients receiving A-P in the first time period.
| Treatment pathway | Coefficient, mean difference (with 95% bootstrap CIs) | |
|---|---|---|
| Receiving treatment pathway first vs. second | Receiving treatment pathway first vs. third | |
| A-P | ||
| Cost (£) | –470 (–1230 to 19) | –683 (–1456 to –210) |
| QALYs | –0.00009 (–0.0362 to 0.0370) | –0.002 (–0.048 to 0.038) |
| D-P | ||
| Cost (£) | –121 (–570 to 539) | –105 (–555 to 653) |
| QALYs | 0.017 (–0.021 to 0.051) | 0.030 (–0.005 to 0.064) |
| P-A | ||
| Cost (£) | –335 (–799 to 202) | –237 (–681 to 131) |
| QALYs | –0.007 (–0.044 to 0.032) | –0.018 (–0.058 to 0.023) |
Cost-effectiveness analysis
Table 26 presents a summary of the costs and QALYs over 16 weeks for each of the treatment pathways, as well as the pairwise incremental costs, QALYs and cost per QALY gained. Note that participants were eligible to receive all three treatment pathways. However, participants may drop out of the study before receiving all three pathways. Therefore, 67 participants received A-P and D-P, 67 participants received A-P and P-A and 73 participants received D-P and P-A. The incremental QALY gain is small and is uncertain when comparing A-P with D-P and A-P with P-A. However, there is a gain for those on the D-P pathway, equating to approximately 2.5 days when comparing D-P with P-A. The incremental cost gain was not significant for any comparison and ranged between a mean of –£113 for A-P compared with D-P and a mean of £154 for A-P compared with P-A. Figure 14 shows the cost-effectiveness planes for each pathway comparison. On average, D-P is more costly and less effective than A-P, with 57% of points being in the south-west quadrant of the cost-effectiveness plane (mean ICER £7026). A-P and D-P were, on average, more costly but more effective than P-A, with the majority of points being in the north-east quadrant of the cost-effectiveness plane (A-P vs. P-A mean ICER £7441, 79% in north-east quadrant; D-P vs. P-A mean ICER £94,157, 91% points in the north-east quadrant, respectively) (Figure 15). For all comparisons, it is important to note that they are based on a small QALY gain and a small difference in costs between the two treatment pathways.
| Analysis | Cost (£) | QALY |
|---|---|---|
| A-P (n = 88) | 2012 (1808 to 2421) | 0.152 (0.136 to 0.167) |
| D-P (n = 88) | 2032 (1852 to 2304) | 0.157 (0.142 to 0.171) |
| P-A (n = 87) | 1942 (1800 to 2179) | 0.152 (0.138 to 0.167) |
| Pairwise mean incremental | ||
| A-P vs. D-P (n = 67) | –113 (–360 to 102) | –0.002 (–0.011 to 0.007) |
| A-P vs. P-A (n = 67) | 154 (–64 to 510) | 0.006 (–0.002 to 0.014) |
| D-P vs. P-A (n = 73) | 149 (–25 to 366) | 0.007 (0.0002 to 0.015) |
| Pairwise mean ICER (£) | ||
| A-P vs. D-P (n = 67) | 7021 (24,715 to 37,038) | |
| A-P vs. P-A (n = 67) | 7482 (–27,623 to 49,221) | |
| D-P vs. P-A (n = 73) | 94,136 (28,076 to 232,390) | |
FIGURE 14.
Incremental cost-effectiveness plane. (a) A-P vs. D-P; (b) A-P vs. P-A; and (c) D-P vs. P-A.
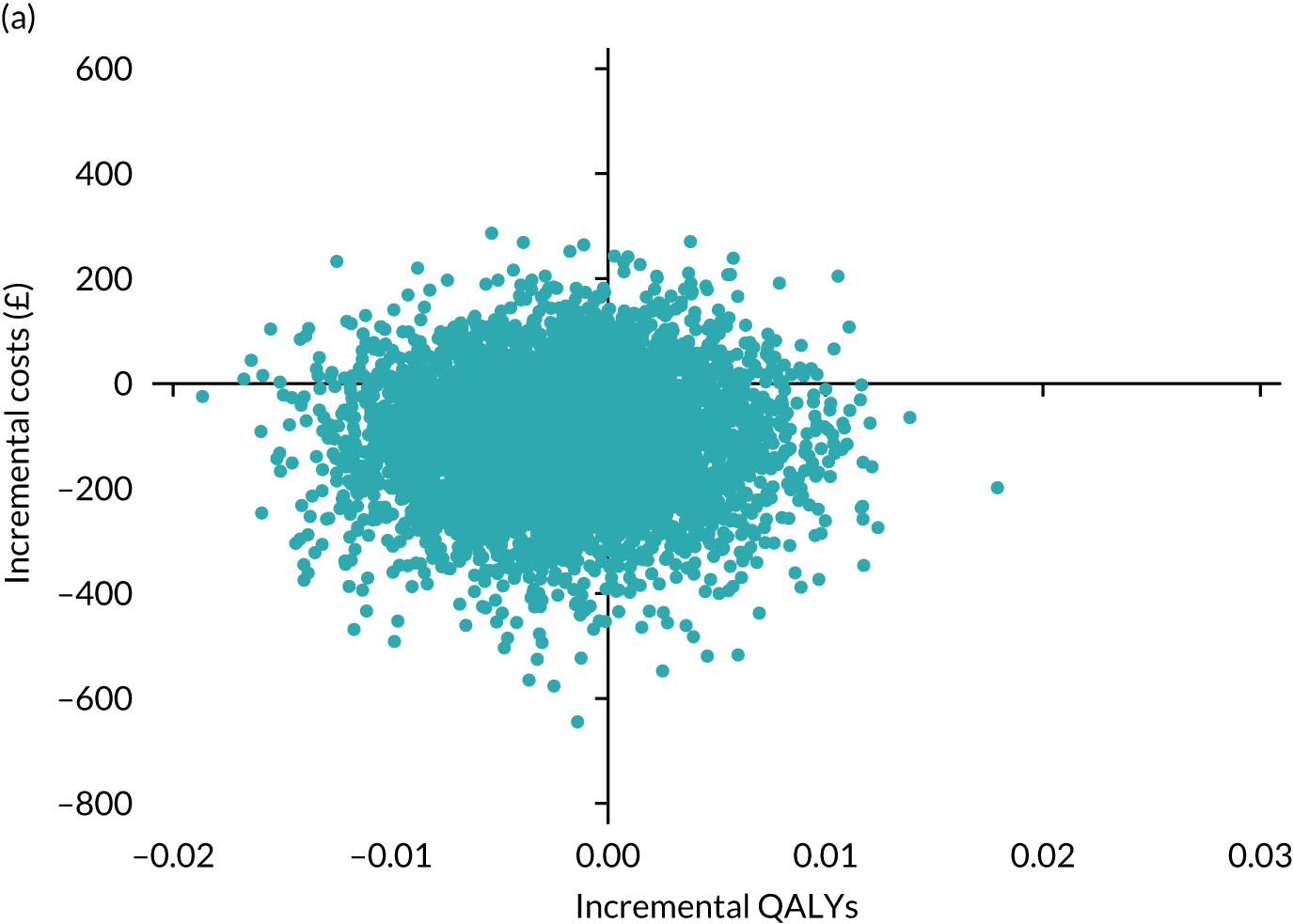
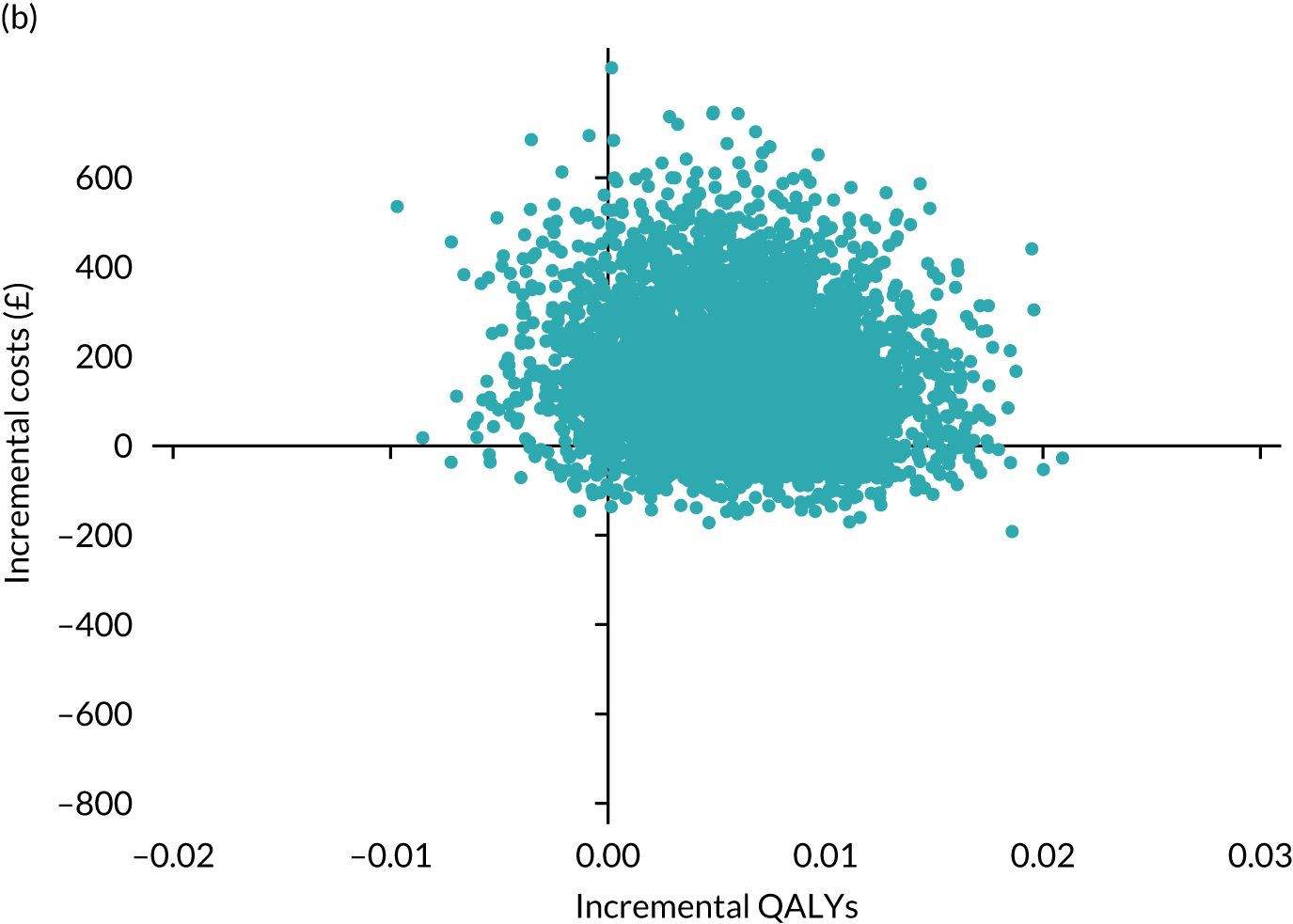
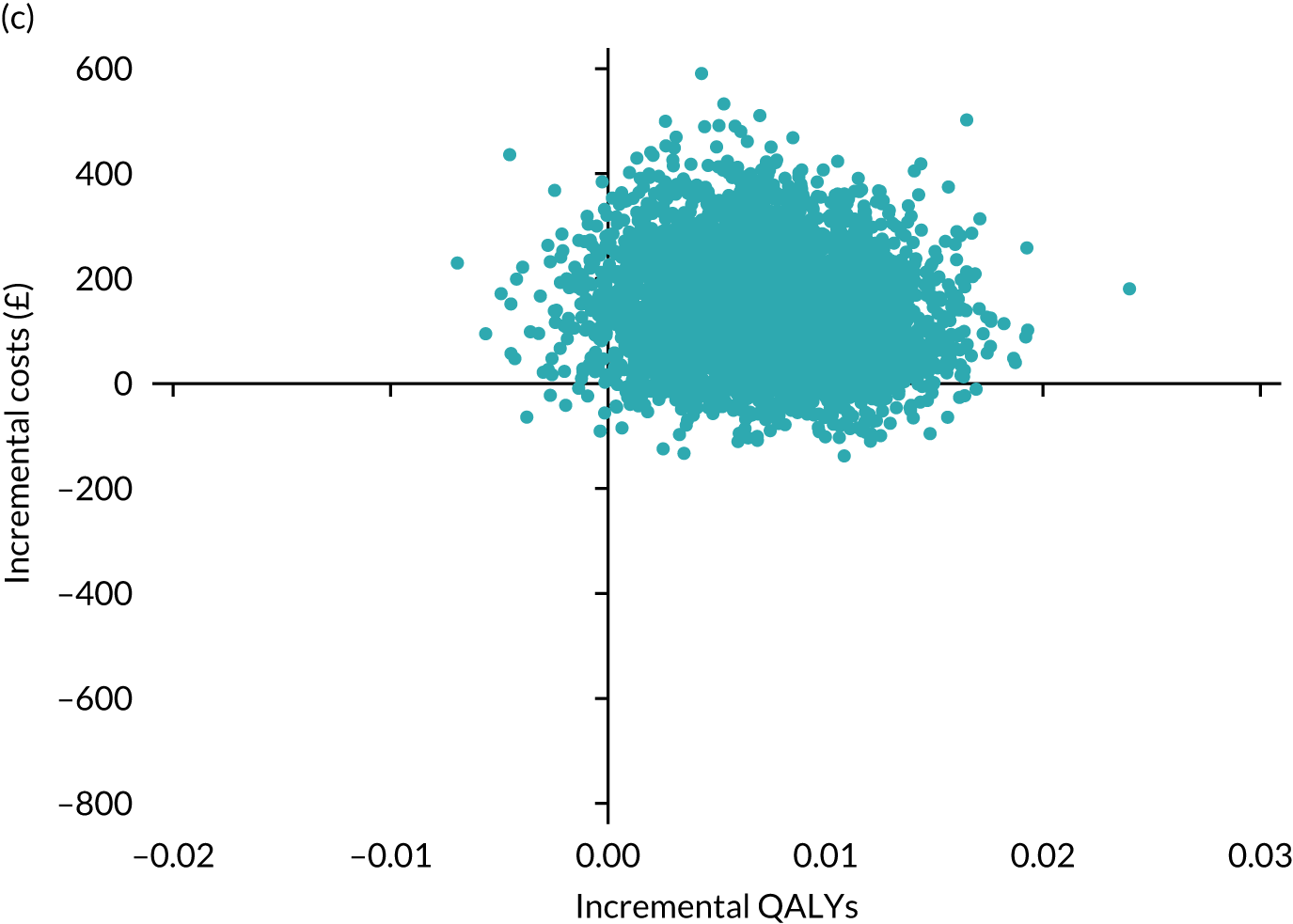
FIGURE 15.
Cost-effectiveness acceptability curve. (a) A-P vs. D-P; (b) A-P vs. P-A; and (c) D-P vs. P-A.
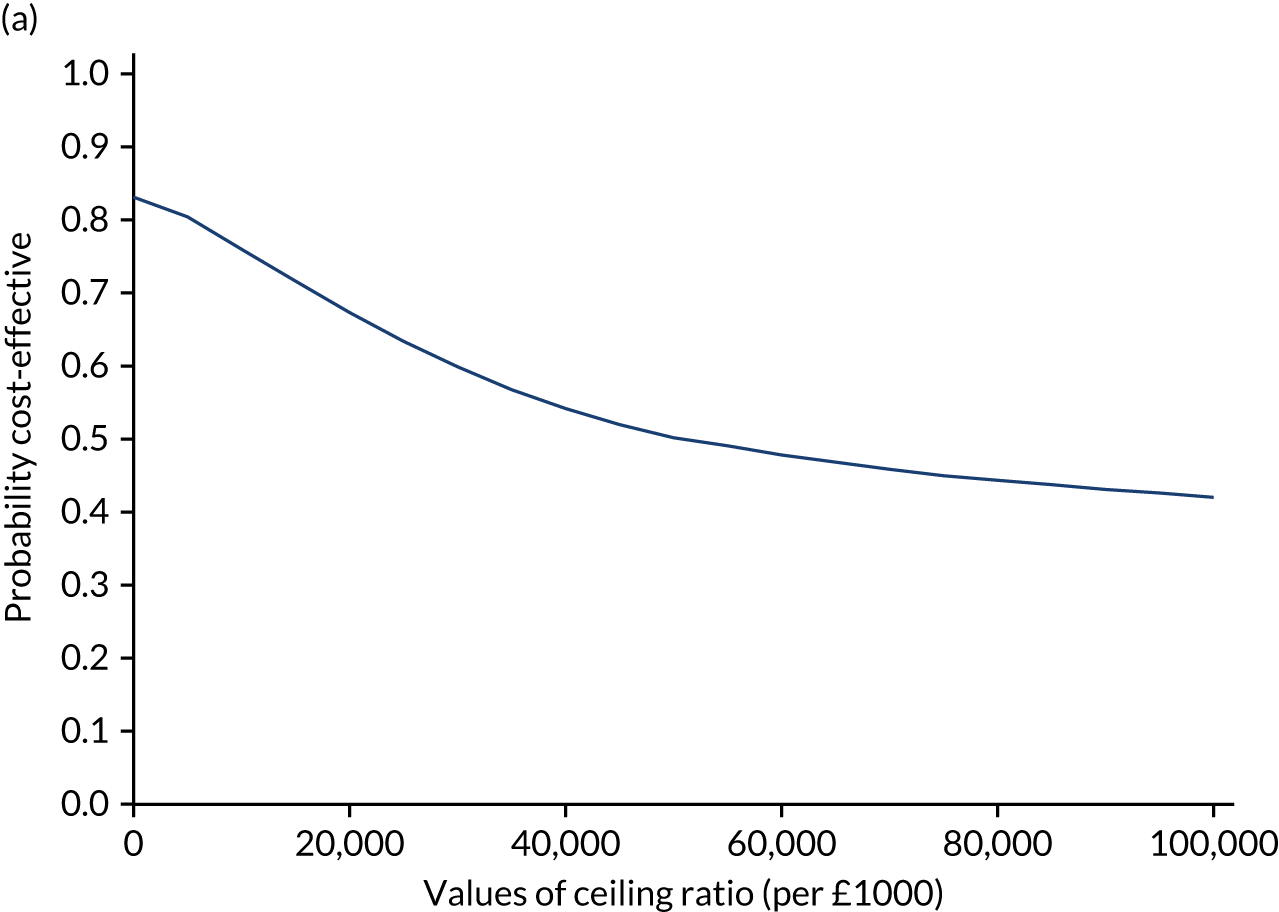
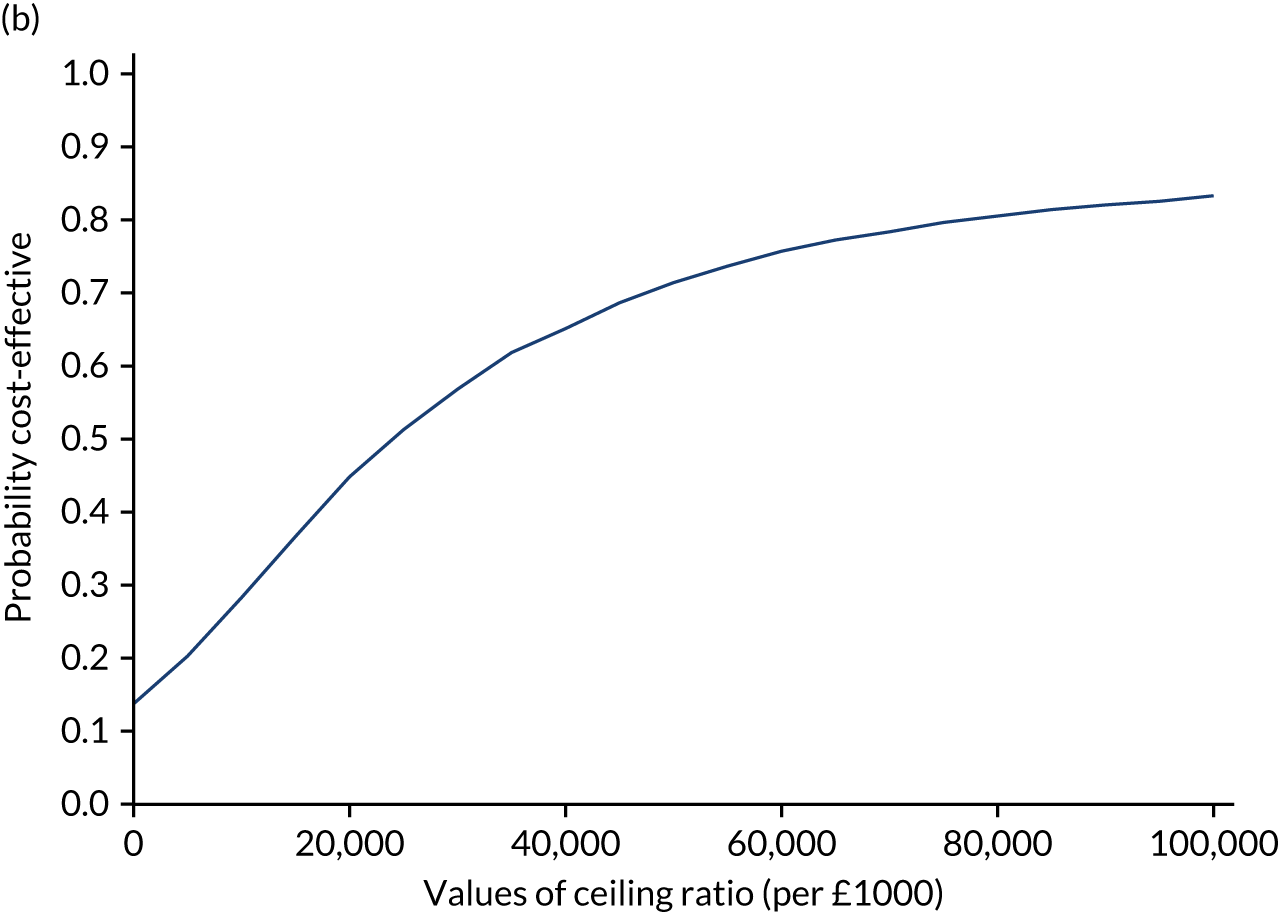
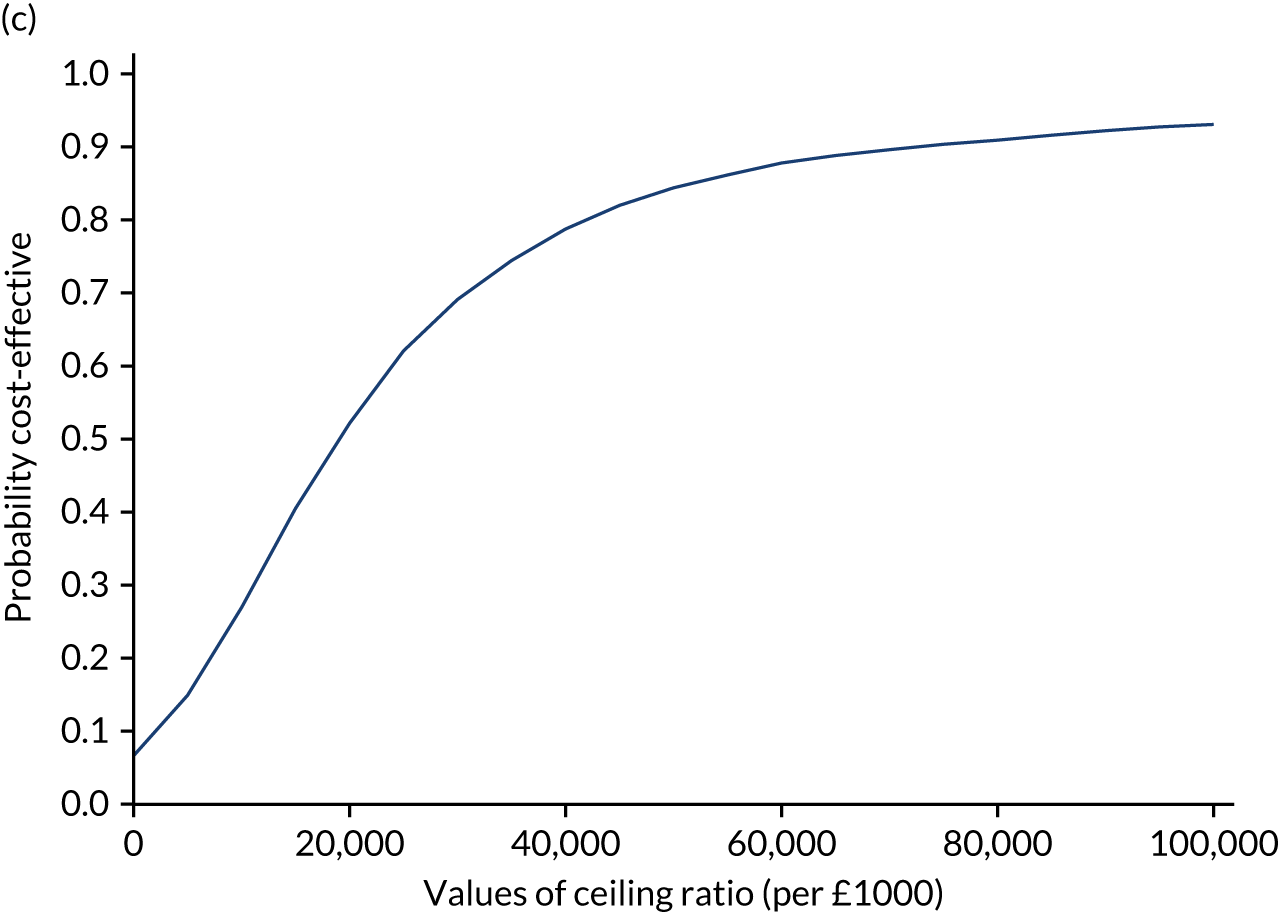
Sensitivity analysis
Non-pairwise comparisons of the three treatment pathways gave ICERs of £4000 for A-P compared with D-P (with D-P on average being more costly but more effective), –£700,000 for A-P compared with P-A (with P-A being less costly and more effective) and £18,000 for D-P compared with P-A (with P-A being less costly and less effective). The incremental QALY difference is very small and uncertain, which is reflected in the difference in the ICERs between pairwise and non-pairwise comparisons.
EuroQol-5 Dimensions, five-level version, Devlin algorithm
Table 27 presents EQ-5D utility values at baseline and at 6 and 16 weeks for the three treatment pathways for the crosswalk and Devlin algorithm. Utilities and QALYs were higher for the Devlin algorithm than the crosswalk, as observed elsewhere,78 but were similar across the three groups for both methods.
| Analysis | Treatment pathway | Mean EQ-5D values and QALYs (95% bootstrapped CI) | |||
|---|---|---|---|---|---|
| Baseline | Week 6 | Week 16 | QALYs | ||
| Crosswalk | A-P | 0.411 (0.352 to 0.466) | 0.515 (0.455 to 0.568) | 0.509 (0.453 to 0.559) | 0.152 (0.136 to 0.167) |
| D-P | 0.408 (0.350 to 0.460) | 0.551 (0.499 to 0.594) | 0.508 (0.448 to 0.562) | 0.157 (0.142 to 0.171) | |
| P-A | 0.420 (0.361 to 0.471) | 0.495 (0.438 to 0.545) | 0.545 (0.490 to 0.591) | 0.152 (0.138 to 0.167) | |
| Devlin algorithm | A-P | 0.506 (0.445 to 0.564) | 0.601 (0.537 to 0.655) | 0.595 (0.535 to 0.648) | 0.179 (0.162 to 0.194) |
| D-P | 0.507 (0.446 to 0.561) | 0.653 (0.600 to 0.698) | 0.599 (0.534 to 0.657) | 0.187 (0.171 to 0.201) | |
| P-A | 0.511 (0.449 to 0.566) | 0.592 (0.529 to 0.645) | 0.642 (0.581 to 0.691) | 0.183 (0.166 to 0.197) | |
The D-P and P-A pathways become less cost-effective, compared with A-P, when the Devlin algorithm is used to estimate the EQ-5D-5L (A-P vs. D-P mean ICER –£37,784 and A-P vs. P-A mean ICER £31,909), with the ICER for D-P compared with P-A being lower (mean ICER £13,097). However, there remains uncertainty in the cost-effectiveness of all pathways.
Including costs borne by participants and their carers
Participants were asked about additional costs, including costs relating to help, transport, changes to the home, specialist equipment, time off work, loss of pay and help from friends and relatives over the past 8 weeks (Table 28). The largest costs were those where adaptions to the home were needed, although this affected only two participants, and costs incurred from caring support from friends and relatives, which were needed by 40–50% of participants.
| Cost | A-P | D-P | P-A | |||
|---|---|---|---|---|---|---|
| Week 6 | Week 16 | Week 6 | Week 16 | Week 6 | Week 16 | |
| Employed extra help | 5; 142 (71 to 212) | 4; 160 (160 to 160) | 6; 180 (0 to 453) | 5; 198 (40 to 356) | 5; 359 (0 to 810) | 3; 60 (0 to 277) |
| Transport to health-care appointments | 7; 22 (5 to 39) | 6; 32 (0 to 74) | 7; 10 (0 to 26) | 5; 17 (0 to 34) | 4; 152 (0 to 2032) | 0; N/A |
| Transport to pain clinics | 0; N/A | 5; 20 (0, 45) | 2; 30 (N/A) | 2; 20 (N/A) | 2; 20 (N/A) | 1; 0 (N/A) |
| Adaption to the home | 2; 1013 (N/A) | 2; 615 (N/A) | 2; 1690 (N/A) | 0; N/A | 1; 3000 (N/A) | 0; N/A |
| Specialist equipment | 1; 40 (N/A) | 1; 280 (N/A) | 1; 600 (N/A) | 1; 150 (N/A) | 2; 840 (N/A) | 2; 325 (N/A) |
| Other costsa | 2; 172 (N/A) | 2; 150 (N/A) | 0; N/A | 1; 10 (N/A) | 2; 80 (N/A) | 1; 80 (N/A) |
| Employment, n (%) | ||||||
| Currently employed | 24 (25.3) | 20 (22.0) | 26 (28.6) | 24 (26.7) | 26 (26.5) | 23 (25.6) |
| Full time | 15 (62.5) | 15 (75.0) | 18 (69.0) | 16 (66.7) | 17 (65.4) | 17 (73.9) |
| Part time | 8 | 5 | 7 | 7 | 8 | 6 |
| Missing | 1 | 0 | 1 | 1 | 1 | 0 |
| Time off in the last 8 weeks | 2 | 0 | 3 | 1 | 3 | 3 |
| Sick leave | 1 | 0 | 1 | 0 | 1 | 2 |
| Paid holiday | 1 | 0 | 2 | 1 | 1 | 0 |
| Unpaid leave | 0 | 0 | 1 | 1 | 0 | 0 |
| Made up timeb | 0 | 0 | 2 | 0 | 0 | 0 |
| Other | 1 | 0 | 0 | 0 | 1 | 1 |
| Total cost (£) of lost earnings (95% CI) | 3.82 (0 to 1140) | 0.00 | 19.71 (0 to 47.80) | 11.96 (0 to 35.71) | 73.27 (0 to 209.22) | 112.83 (0 to 278.01) |
| Help needed from a friend or relative, n (%) | ||||||
| Help from friend or relative | 42 (44.2) | 39 (43.3) | 45 (51.1) | 36 (40.5) | 47 (47.5) | 38 (42.2) |
| With personal care | 16 | 15 | 17 | 16 | 19 | 17 |
| With childcare | 2 | 3 | 2 | 1 | 1 | 3 |
| With housework | 36 | 25 | 33 | 28 | 30 | 27 |
| With transport | 25 | 20 | 20 | 19 | 29 | 22 |
| With general support | 32 | 29 | 38 | 28 | 33 | 31 |
| With other | 4 | 2 | 1 | 5 | 2 | 1 |
| Friend/relative stayed off work to help | 2 | 1 | 2 | 2 | 1 | 0 |
| Total cost (£) of time lost for friend/relative (95% CI) | 3418 (369 to 6467) | 1840 (790 to 2890) | 1439 (822 to 2055) | 1289 (578 to 2000) | 1820 (978 to 2662) | 3385 (670 to 6100) |
| Total costs (£) to participants (95% CI) | 3456 (406 to 6505) | 1860 (808 to 2913) | 1512 (883 to 2142) | 1315 (601 to 2029) | 1962 (1107 to 2818) | 3508 (778 to 6238) |
Over the three treatment pathways, the mean costs borne by participants are more than double the other costs reported for the CSRI (see Table 28) and these were £1207 (95% bootstrap CI £758 to £2376) for A-P, £1740 (95% bootstrap CI £1077 to £3309) for D-P and £2138 (95% bootstrap CI £881 to £6836) for P-A. Overall costs per pathway also increased and these were £2670 (95% bootstrap CI £2220 to £3840) for A-P, £3216 (95% bootstrap CI £2561 to £4807) for D-P and £3619 (95% bootstrap CI £2358 to £8322) for P-A (Table 29). The inclusion of costs to participants and their carers did change the mean ICERs, although the results remained uncertain. D-P was more expensive and less effective than A-P (mean ICER –£19,268). P-A was more costly and more effective than both A-P (mean ICER £21,405) and D-P (mean ICER £229,855), respectively.
| Analysis | Incremental costs (£) (95% bootstrap CI) | Incremental QALYs (95% bootstrap CI) | ICER (£) |
|---|---|---|---|
| Main analysis | |||
| A-P vs. D-P (n = 67) | –113 (–360 to 102) | –0.002 (–0.011 to 0.007) | 7021 (–24,715 to 37,038) |
| A-P vs. P-A (n = 67) | 154 (–64 to 510) | 0.006 (–0.002 to 0.014) | 7482 (–27,623 to 49,211) |
| D-P vs. P-A (n = 73) | 140 (–25 to 366) | 0.007 (0.0002 to 0.015) | 94,136 (28,082 to 232,450) |
| Sensitivity analysis: Devlin algorithm | |||
| A-P vs. D-P (n = 67) | –113 (–360 to 102) | –0.004 (–0.014 to 0.006) | –37,784 (–167,464 to 23,057) |
| A-P vs. P-A (n = 67) | 154 (–64 to 510) | 0.005 (–0.004 to 0.013) | 31,909 (6018 to 71,866) |
| D-P vs. P-A (n = 73) | 140 (–25 to 366) | 0.008 (0.0009 to 0.015) | 13,097 (–23,614 to 48,170) |
| Sensitivity analysis, including costs borne to participant and carers | |||
| A-P vs. D-P (n = 67) | –835 (–3172 to –46) | –0.002 (–0.011 to 0.007) | –19,268 (–113,402 to 48,584) |
| A-P vs. P-A (n = 67) | –968 (–4727 to 1055) | 0.006 (–0.002 to 0.014) | 21,405 (–81,327 to 125,782) |
| D-P vs. P-A (n = 73) | –432 (–3718 to 1650) | 0.007 (0.0002 to 0.015) | 229,855 (–17,691 to 698,208) |
| Sensitivity analysis: multiple imputation of missing EQ-5D responses | |||
| A-P vs. D-P (n = 130) | 17.98 (–214 to 305) | –0.004 (–0.009 to 0.002) | –25,184 (–69,553 to 10,661) |
| A-P vs. P-A (n = 130) | –93.13 (–399 to 196) | 0.004 (–0.0004 to 0.009) | –4615 (–56,525 to 38,410) |
| D-P vs. P-A (n = 130) | –114 (–402 to 139) | 0.008 (0.003 to 0.127) | 56,862 (1701 to 122,604) |
Multiple imputation of missing EuroQol-5 Dimensions responses
Imputing the EQ-5D responses increased the sample size to 130 (i.e. the full sample who participated in the study). On average, this increased the QALYs for A-P (mean QALY 0.157, 95% bootstrap CI 0.145 to 0.168) and D-P (mean QALY 0.160, 95% bootstrap CI 0.149 to 0.171) compared with the observed cases (see Table 26). However, the mean QALY for P-A remained similar to that prior to imputing the data (mean QALY 0.152, 95% bootstrap CI 0.141 to 0.164). As with other sensitivity analysis, imputing missing values did change the mean ICERs, with results remaining uncertain.
Chapter 5 Discussion
Main findings
The OPTION-DM trial showed that all three treatment pathways (i.e. A-P, D-P and P-A) were equally effective in reducing neuropathic pain intensity, significantly reducing the baseline NRS score from a baseline mean of 6.6 (SD 1.5) points to an average of 3.3 (SD 1.8) points at 16 weeks. There were no differences between the treatment pathways, as the effects observed were not different statistically or clinically different between the pathways, with the CIs for the primary end point (i.e. pain at week 16) having the prespecified difference only at their very edge.
As the three treatment options produced similar pain control, it might be important to consider other outcomes when deciding which treatment to recommend; however, no one treatment was uniformly preferable across the end points we assessed.
The P-A pathway led to the fewest discontinuations on monotherapy due to AEs (p = 0.031) and numerically was the most preferred pathway (A-P, 24%; D-P, 33%; P-A, 43%; p = 0.27) at the end of the trial. This is consistent with the American Academy of Neurology recommendations, which give only pregabalin level A evidence, mainly because completion rates in pregabalin trials were more than 80%. 79 The P-A pathway also gave the best pain relief for higher levels of baseline depression (p = 0.011) and both the P-A and D-P pathways provided better pain relief than the A-P pathway for higher levels of baseline anxiety (p = 0.016).
The trial explored a number of secondary end points. The head-to-head trial of maximum tolerated doses of amitriptyline, duloxetine and pregabalin showed similar efficacies for all three monotherapies at the end of 6 weeks, although there were fewer discontinuations due to treatment-emergent AEs with pregabalin. However, only in one-third of patients did monotherapy achieve mild pain (i.e. ‘responders’, with a NRS score of ≤ 3). Similarly, monotherapy achieved 50% pain relief in around 40% of patients. Most patients (‘non-responders’) required combination treatment, which resulted in a 0.6-point further improvement in NRS score and 18% and 14% increases in the achievement of a NRS score of ≤ 3 points and 50% pain relief, respectively. In a crossover trial, Gilron et al. 57 studied 56 patients with neuropathic pain (40 patients with DPNP) treated to maximum tolerated doses of gabapentin, nortriptyline and their combination over 1-month treatment periods. They found that combination treatment was more efficacious than either drug alone. Another crossover trial80 (n = 73), of 5-week treatment periods, compared the combination of imipramine and pregabalin at moderate doses with either treatment on its own in painful polyneuropathy. The study found that combination treatment was more efficacious in relieving neuropathic pain than either drug on its own, but resulted in higher rates of side effects. Despite these results, currently, a number of international bodies do not recommend combination treatment for DPNP because of ‘insufficient evidence’. 81–83 Although the OPTION-DM trial was not designed as a comparison of monotherapy and combination treatment, the data make a compelling case for the recommendation of combination treatment of first-line drugs for DPNP patients with suboptimal response to a monotherapy.
Despite massive variations in the cost and availability of amitriptyline, duloxetine and pregabalin in many countries, it is reassuring that all three drugs are equally efficacious in relieving pain. Furthermore, all monotherapies resulted in a substantial improvement of QoL domains, sleep and measures of mood from baseline. However, amitriptyline was significantly better than duloxetine in improving physical functioning and sleep, and pregabalin was superior to duloxetine in improving role limitation due to physical health. Treatment-emergent AEs were predictable for the monotherapies, although there were no significant differences in their overall frequency and severity. This may, in part, be due to the use of maximum tolerated doses rather than a fixed treatment regime. There were also no significant differences in the frequency of SAEs. However, compared with some previous studies, combination treatment with maximum tolerated doses was well tolerated, with few treatment-emergent AEs, except for larger numbers of patients reporting diarrhoea and dry mouth in the A-P pathway.
Health economics
To the best of our knowledge, this study is the first to present the results of a head-to-head comparison of current medications and their combinations in an economic evaluation over a 16-week period. QALYs were similar over the study time period, with an average difference in costs ranging from –£113 to £154 across the three comparisons. Ninety-five per cent bootstrap CIs around costs and QALYs demonstrated the uncertainty in results. Furthermore, the study showed that greater costs are borne by the patients and their carers rather than the NHS.
Wu et al. 32 examined the cost-effectiveness of duloxetine compared with usual care over a 52-week period. Wu et al. 32 looked at three perspectives and found incremental costs slightly higher than those for our study [i.e. direct (closest possible to NHS) US$1600 (£1194); employment and direct US$2196 (£1639) and societal, direct and employment US$2754 (£2056)]. However, in addition to looking at single rather than combination therapy, the time period is longer in the Wu et al. 32 study and the health-care system is different in the USA and, therefore, caution should be given when comparing these results with those from the OPTION-DM trial.
Two further studies33,34 looked at duloxetine in decision-analytic models. O’Connor et al. 34 presents the cost per QALY of duloxetine compared with desipramine at US$47,700 (or £35,609). This is higher than our average QALY for A-P compared with D-P (£7026) and lower than the average QALY for D-P compared with P-A (£94,157), although it should be noted, again, that this is not a combination therapy study and the time period is shorter and so not comparable. It should also be noted that the O’Connor et al. 34 results were sensitive to obtaining pain relief. Beard et al. 33 also fitted a decision-analytic model and noted a saving of £77,071 for every 1000 patients treated in 2007 or £109,246 per 1000 patients in 2019.
Finally, de Salas-Cansado et al. 35 compared pregabalin with usual care from a Spanish NHS and societal perspective over a 23-week period. The costs per QALY were higher than those for our study [i.e. €5302 (£4426) societal and €14,381 Spanish NHS (£12,005)].
Also worth noting is that the utility values for our sample do appear to be low in comparison with those for the general diabetes population. Table 3 presented mean utility values for the EQ-5D at baseline and at 6 and 16 weeks, and these ranged between 0.408 and 0.551. Collado Mateo et al. 84 present normative values for a Spanish diabetes population by age and sex. The mean EQ-5D values for people aged 60–69 years (the most comparable age band to our population mean of 61.8 years) were 0.860 for males and 0.745 for females, respectively. The authors used the Spanish crosswalk rather than UK crosswalk to obtain utilities for the EQ-5D, but other studies also suggest higher utility values. For example, Wang et al. 85 report a mean utility of 0.720 for patients in Singapore with type 2 diabetes mellitus. Likewise, in a Chinese study, Pan et al. 86 report a mean value of 0.862 for patients with type 2 diabetes mellitus and a mean of 0.815 if the patients also had neuropathy.
Strengths and weaknesses of the research
To the best of our knowledge, this was the first randomised double-blind comparator trial of neuropathic pain treatment pathways. Although head-to-head trials of individual monotherapies and combination treatments could be designed, investigating treatment pathways as a whole was felt most efficient and applicable to current clinical practice. This is because most patients are started on a monotherapy and will require a second agent to be added in combination within a few months. Therefore, the OPTION-DM trial reflected current practice, allowing the outcomes of this study to be readily generalisable. Recruitment from a wide array of sites, including primary and secondary care, further strengthens the generalisability of the study.
The initial prespecified number of participants was not recruited, and this could be taken as a limitation of the study. Early monitoring of the fidelity of recruitment revealed that recruitment for this demanding trial was challenging. Rather than compromise the integrity of the trial, and with the approval of the independent TSC and head of the Health Technology Assessment, the trial continued to a fixed time point when an adequate sample size had been achieved to detect a difference of at least 1 NRS point. The onset of the COVID-19 pandemic further affected follow-up, with some participants having a curtailed follow-up in the final pathway. A strength of the OPTION-DM trial was that, although the trial was curtailed in terms of recruitment, to the best of our knowledge, it is still the longest and largest crossover combination trial. Given the results, and that a 0.5-point NRS difference was at the very edge of plausible difference between the treatment arms, even a significantly larger sample size would probably not have altered study outcomes or conclusions.
The lack of a placebo arm can also be taken as a limitation of the study. However, given the existing wealth of evidence from previous placebo-controlled trials and the advice from our PPI panel, we decided against a placebo control, as including a placebo control would have made the trial longer, more complicated (i.e. a four-way crossover trial) and it would not have been ethically justifiable to leave participants on placebo alone for 17 weeks. The number of dropouts within the 1–2 weeks prior to randomisation in this trial suggest that such a trial is not feasible. The lack of a placebo arm means that we cannot attribute the change from baseline solely to the introduction of treatment, as some improvement may be due to regression to the mean. Nonetheless, previous trials14–22 have demonstrated far smaller changes in participants treated with placebo, and it is reasonable to assume that most of the improvement is due to therapy.
The EQ-5D was captured prior to treatment pathway 1, but not prior to treatment pathways 2 and 3, and was captured for each treatment pathway at 6 and 16 weeks. It may be that measuring HRQoL more frequently would more likely capture any disutilities associated with AEs, and any future study should consider this. The economic analysis reports the within-trial results over 16 weeks, and this is in line with other published economic evaluations,31,32 although future studies could consider modelling the cost-effectiveness over a longer time frame.
There are two further limitations of the economic evaluation. First, the use of a questionnaire to capture resource use means that resource use is self-reported and could be subject to recall issues, thereby potentially underestimating the overall cost of each treatment pathway. The capturing of resource use information from routine sources could be considered in future studies. Second, the crossover nature of this study lent itself to a pairwise comparison of the alternative treatment pathways. This pairwise comparison was varied in a sensitivity analysis, which, owing to the small difference in costs and QALY, meant that the results remained uncertain.
Evidence in the context of other existing research
The OPTION-DM trial was an efficiently designed head-to-head crossover trial,2 with each patient undergoing all three pathways over 50 weeks, making it, to the best of our knowledge, the longest blinded neuropathic pain trial. The durations of monotherapy and combination treatment were long enough to assess full treatment effects. 37 Previous combination trials mainly used fixed-dose titration regimens regardless of treatment response. 31,80 This does not reflect clinical practice and often resulted in high dropout rates. 31 The present trial employed a flexible dosing regimen to achieve maximum tolerated doses, based on individual responses. Moreover, all previous DPNP multiperiod crossover trials were smaller. 25–27 In addition, other studies included neuropathic pain conditions other than DPNP. 29,57,80 Finally, most previous trials did not use combinations of current first-line drugs. 29,57,80 The OPTION-DM trial tried to obtain robust data from current first-line drugs, as, based on the trajectory of new drug developments for DPNP over the past 25 years, the emergence and use of revolutionary new drugs that are considerably more efficacious than current ones seemed unlikely over the foreseeable future.
Implications for practice and policy
The OPTION-DM trial has demonstrated that all three treatment pathways and monotherapies are equally efficacious. Although most international painful diabetic neuropathy management guidelines, including the one by NICE (CG96, updated in 2021),82 place the monotherapies as first-line agents, based on indirect comparisons of the treatments’ efficacy, this is, to the best of our knowledge, the first time that an adequately designed head-to-head comparator trial has been undertaken, and it confirmed that the treatments are of equivalent efficacy. The OPTION-DM trial will hopefully reduce the ambiguity of indirect comparisons that, for example, led the French panel of experts to recommend pregabalin as a second-line agent. 87
Combination treatment is currently not recommended by international guidelines because of insufficient evidence. 81–83 NICE guidance,82 which was updated in 2021, recommends switching to a different treatment rather than combination treatment. In this trial, only one-third of patients saw a reduction in pain to ‘mild’ as quantified as a NRS score of ≤ 3 points (‘responders’). Similarly, monotherapy achieved 50% pain relief in around 40% of patients. Many patients (‘non-responders’) required combination treatment that resulted in a further mean improvement of 1 point on the NRS and an additional 18% and 14% increase in the achievement of NRS score of ≤ 3 points and 50% pain relief, respectively. Although the OPTION-DM trial was not designed as a comparison of monotherapy and combination treatment, the data make a compelling case for the recommendation of combination treatment of first-line drugs for DPNP patients with suboptimal response to a monotherapy, particularly as combination treatment was very well tolerated.
In clinical practice, patients are sometimes prescribed very high doses of amitriptyline (up to 150 mg/day) and, therefore, it is no great wonder that so many patients experience unwanted side effects. The OPTION-DM trial has shown that titration to 60 mg per day can be effective. The OPTION-DM trial also found that the mean dose per day (% on maximum dose) of amitriptyline, duloxetine and pregabalin at week 6 was 60 mg (59%), 82 mg (53%) and 396 mg (56%), respectively. For patients on combination treatments A-P, D-P and P-A the mean dose per day (% on maximum dose) of add-on drug at week 16 was 365 mg (47%), 407 mg (55%) and 55 mg (47%), respectively. These data show that only around half of patients will tolerate maximum doses of these drugs, and this is in the context of a clinical trial that excluded patients with major comorbidities. A key message from the OPTION-DM trial is that drugs need to be titrated gradually, over at least 2 weeks and, in some patients (for example the elderly and those with comorbidities) even more slowly, factoring in the side effect profile and the severity of side effects, as well as efficacy.
The data from OPTION-DM trial are powerful. The OPTION-DM trial was, to the best of our knowledge, the longest blinded neuropathic pain trial to date. Unlike previous combination treatment crossover trials,37 the durations of monotherapy and combination treatments were long enough to assess full treatment effects. Moreover, previous combination trials31,80 mainly used fixed-dose titration regimens regardless of treatment response, which does not reflect clinical practice and resulted in high dropout rates. The OPTION-DM trial employed a flexible dosing regimen to achieve maximum tolerated doses, based on individual responses. Moreover, all previous DPNP multiperiod crossover trials25–27 had smaller sample size and other studies29,57,80 included neuropathic pain conditions other than DPNP. In addition, most previous trials did not use combinations of current first-line drugs. Therefore, the robustness of OPTION-DM trial data will probably influence clinical practice, guidelines and commissioners.
Implications for health care
The current NICE guideline (CG173)82 for the management of painful DPNP states:
Offer a choice of amitriptyline, duloxetine, gabapentin or pregabalin as initial treatment for neuropathic pain.
If initial treatment is not effective or is not tolerated, offer one of the remaining 3 drugs, and consider switching again if the second and third drugs tried are not effective or not tolerated.
The OPTION-DM trial was a pragmatic trial reflecting current best-treatment practice and examined three treatment pathways for painful diabetic neuropathy. In each treatment pathway, patients were initially commenced on monotherapy followed by combination therapy. The results showed significant improvements in pain severity in all three treatment pathways. In addition, the results showed that combination treatment is not only safe but also necessary for a significant proportion of patients to achieve meaningful pain relief (i.e. a NRS pain score of ≤ 3 points).
The most recent NICE CG173 guideline82 recommends that patients switch to a second or third agent if initial/subsequent monotherapy is ineffective. In practice, this often results in poor patient satisfaction and disillusionment, as well as failure to achieve adequate pain relief due to treatment inertia. The results of the OPTION-DM trial suggest that combination treatment may be advocated if inadequate pain relief is achieved with monotherapy. To facilitate this, future pharmacotherapy guidelines for painful diabetic neuropathy should be rationalised into treatment pathways, as the majority of patients end up having combination treatments. Our cost-effectiveness analysis shows that there are no differences between the treatment pathways assessed. Further post hoc analyses are being planned to examine if certain subgroups of patients (e.g. the elderly, patients with mood disorders or clinical pain phenotypes) respond better to a particular treatment pathway.
Recommendations for future research
Implementation
There remains a clinical and economic need to improve outcomes in patients with painful diabetic neuropathy. Based on the trajectory of new drug developments for painful diabetic neuropathy over the past 25 years, the emergence and use of new drugs that are more efficacious than current ones seems unlikely in the next decade. In the OPTION-DM trial, around 40% of patients achieved significant improvement in pain (i.e. 30% improvement in NRS or a NRS score of ≤ 3 points) based on simple pragmatic guidance to treatment escalation. There is, however, a wide variation in treatment outcomes across England. We need to explore the differences in clinical practice and patient behaviour that underlie these differences, as well as promote the outcomes of the OPTION-DM trial.
Mechanistic approach
In all treatment pathways, there was a wide variation in NRS pain intensity scores, with some patients showing marked improvement and others showing minimal or no improvement. Further research is needed to explain why some patients do so well, whereas others do not. This research would involve detailed clinical phenotyping of patients using recent innovations in magnetic resonance neuroimaging, quantitative sensory assessment and genotyping. This research might uncover underlying pain mechanisms that inform treatment responses in individual patients.
Holistic approach
It is notable that we found a significant relationship between emotional distress (e.g. anxiety and depression) and improvement in pain scores in two of the three treatment pathways assessed. The precise role of psychosocial factors in explaining pain severity, distress and treatment response are not well understood. Most studies have investigated psychosocial and biomedical variables independently and the differing contributions of these variables on treatment outcomes are poorly specified.
Pharmacological and non-pharmacological approaches
A significant proportion (around 50%) of patients are not helped by any treatment pathway and this raises the question of ‘what next?’. NICE recommends tramadol only if rescue therapy is needed, but often patients are commenced and remain on long-term opioid therapy. The safe use of opioids and other pharmacological [e.g. high-dose (8%) capsaicin patch or intravenous lidocaine therapy] and non-pharmacological (e.g. spinal cord stimulation) treatments for painful diabetic neuropathy should be explored further.
Chapter 6 Conclusions
The results of the OPTION-DM trial showed that all three treatment pathways had equal pain control, in terms of both statistical and clinical significance. The P-A pathway led to fewer monotherapy discontinuations due to treatment-emergent AEs and was also most preferred by participants at the end of the trial, suggesting that this may be the safest choice as a first line of therapy. Importantly, although the trial was not primarily designed to examine the benefit of combination treatment when there is suboptimal response to a monotherapy, this pragmatic trial demonstrated that combination treatment, where needed, was well tolerated and can lead to further reduction in pain following the introduction of second-line therapy.
Acknowledgements
We are especially grateful to the patients with DPNP who participated in this trial. We would like to thank the members of the TSC and DMEC for their support and guidance. We are grateful to the PIs and their teams for their hard work delivering this complex trial.
The OPTION-DM group
Solomon Tesfaye was the chief investigator.
Dinesh Selvarajah was the co-chief investigator.
The following were co-applicants on the grant, contributed to the study design, were members of the TMG and contributed to data interpretation: David Bennett, Oscar Bortolami, Didier Bouhassira, Cindy Cooper, Rajiv Gandhi, Ravikanth Gouni, Michelle Horspool, Martin Johnson, Edward Jude, Steven A Julious, Satyan Rajbhandari, Gerry Rayman, Prashanth Vas and David White.
The following were site PIs and members of the TMG: Syed Haris Ahmed, Uazman Alam, David Bennett, Augustin Brookes, Marion Devers, Frances Game, Ravikanth Gouni, Christian Hariman, Pallavi Hegde, Edward Jude, Deirdre Maguire, Rory McCrimmon, Claire McDougall, David Price, Satyan Rajbhandari, Gerry Rayman, Abd Tahrani, Solomon Tesfaye, Vasileios Tsatlidis and Prashanth Vas.
The following contributed to participant recruitment, treatment delivery and data collection at sites: Quratul-ain Altaf, Samantha Anderson, Maureen Armstrong, Helen Atikins, Olabisi Awogbemila, Gayna Babington, Sarah Beck, Safia Begum, Elizabeth Bell, Dina Bell, Srikanth Bellary, Lyndsay Bibb, Sharron Birch, Hannah Bond, Elaine Brinkworth, Margaret Broome, Jamie Burbage, Charlotte Busby, Caroline Calver, Christine Cassidy, Tanith Changuion, Carolyn Chee, HoYee Cheung, Kamalakkannan Chokkalingham, Wahid Chugtai, Martyn Clark, Lynda Connor, Amanda Cook, Shirley Cooper, Martina Coulding, Joanna Cox, Helen Cross, Jacqueline Currie, Claire Davies, Caroline Davies, Rebecca Denyer, Laura Dinning, Julie Edwards, Branwen Ellison-Handley, Sarah Finbow, Annette Fleet, Kamini Gandhi, Laura Gardiner, Karyna Gibbons, Amanda Gillespie, Louisa Green, Peter Hammond, Gail Hann, Kate Harrington, Melanie Hayman, Stephanie Horler, Liew Huiling, Wanda Ingham, Ahmed Iqbal, Andrew Irwin, Khalid Jadoon, Victor Jardin, Helen Jenner, Jane Jiao, Katherine Jones, Frank Joseph, Joanne Kadziola, Maqsood Khan, Rosemary Kirk, Alistair Lamb, Rebecca Lenagh, Jonathan Lim, Jackie Lindsay, Linda Macliver, Ellen Malcolm, Barbara Martin, Shamiso Masuka, Alexandra McCarrick, Jacqueline McCormick, Carly McDonald, Nicky McRobert, Aye Min, Ahmad Moolla, Nadim Musah, Alison Musgrove, Sunil Nair, Kilimangalam Narayanan, Abida Nazir, Kelly Nicholson, Francis O’Rourke, Pedro Okoh, Matilde Pascal, Helen Permain, Shirley Peta Navein, Kristina Posadas, James Pratt, Tejpal Purewal, Taslima Rabbi, Sutapa Ray, Rustam Rea, Elsa Redfearn, Gillian Reekie, tephanie Ridgeway, Josephine Rosier, Daniel Rowlandson, Efisia Sais, Lorelei Salazar, Sunita Sandhu, Chandbi Sange, Vivienne Savage, Heather Savill, Caroline Scott, Susan Seal, Ruta Segamogaite, Dinesh Selvarajah, Ajmal Shakoor, Sanjeev Sharma, Gordon Sloan, Dawn Spick, Mara Sprengel, Lisa Stewart, Wendy Stoker, Sharon Storton, Kyla Story, Negar Tadayon, Garry Tan, Andreas Themistocleous, Sue Thomson, Viv Thornton-Jones, Nicola Tinker, Victoria Tobin, Kim Turner, Victoria Turner, Tom Vale, Joanne Vere, Janaka Vithanage, Lisa Wadeson, Taryn Ward, Rachel Whitham, Helen Wild, Andrew Williams, Annie Williamson, Jayne Willson, Ann Wilson, Charlene Wong, Noah Yogo and James Young.
The following contributed to pharmacy and IMP management at sites: Wendy Abbott, Fiona Ashworth, Bhavna Badiana, Liz Bedford, Michelle Beecroft, Sarah Bik, Nicola Blain, Sathan Boonyaprapa, Rachel Bower, Helen Bowler, Louise Brannon, Zara Brown, Shona Carson, Tom Carter, Katie Carter, Stuart Chandler, Vicky Chu, Irene Coburn, Amy Cox, Fionnuala Cunningham, Laura Cureton, Tori Donaldson, James Donworth, Samantha Eccles, Sharon Evans, Jordi Evans, Joanna Flanagan, Emma Galloway, Linda Gemmell, Dilbagh Gill, Marie Goldsworthy, Carlos Gonzalez, Alfonso Gonzalez-Blas, Joanne Gordon, Senait Haile, Clair Hammond, Benson Ho, Sheila Hodgson, Alison Hogan, Sam Holloway, Louise Hough, Matthew Howlett, Cath Hughes, Tracy Jackson, Sam Jackson, Carol Jaques, Dom Jimenez, Natalie Johnston, Paul Jones, Kelly Kauldhar, Laura Kirk, Paige Kissane, Caroline Loan, Jennifer Lynch, Helen Lyon, Jill Lyons, Greg Marchant, Steven Marshall, Peter Mason, Heather Michael, Laura Miller, Gaynor Notcheva, Samuel Nyabam, Matthew Quinn, Andrew Reid, Rosalind Roberts, Fiona Ross, Pam Rushworth, Nita Shah, Victoria Shakespeare, Tania Slater, David Sproates, Anita Stevenson, Kashmira Subramanian, Benham Tabrizi, Rebecca Tangney, Paul Taylor, Nichole Thompson, Christopher Waterhouse, Sue Wiles, Andrew Wilkinson and Elaine Wilson.
Erica Wallis was the sponsor representative and provided support on ethics and governance issues.
Rachel Glover, Jennifer Petrie, Katie Sutherland and Lizzie Swaby provided central trial management and monitoring.
Mike Bradburn provided the statistical support.
Tracey Young provided the health economics support.
Amanda Loban, Emily Turton and Simon Waterhouse provided central data management.
Zoe Furniss and Jess Welburn provided central administration.
Trial Steering Committee
Andrew Rice, independent chairperson (Professor of Pain Research, Imperial College London).
Ralf Baron (Professor of Pain Research, University Medical Centre Schleswig-Holstein).
Sarah Brown (Principal Statistician, University of Leeds).
Arthur Durrant (patient representative).
Nanna Finnerup (Professor of Pain Medicine, Aarhus University).
Roger Knaggs (Associate Professor in Clinical Pharmacy Practice, University of Nottingham).
Jane Lewis (Senior Lecturer in Podiatry, Cardiff School of Sport and Healthcare Science).
Data Monitoring and Ethics Committee
Troels Jensen, independent chairperson (Professor of Neurology and of Experimental and Clinical Pain Research, Aarhus University).
Zoe Hoare (Principal Statistician, Bangor University).
Martin Rutter (Professor of Cardiometabolic Medicine, University of Manchester).
Contributions of authors
Solomon Tesfaye (https://orcid.org/0000-0003-1190-1472) (Consultant Endocrinologist and Honorary Professor of Diabetic Medicine) produced the first draft of the report, was a member of the TMG, contributed to the delivery of the trial, was an applicant on the Health Technology Assessment grant, contributed to the study design and was an investigator at a participating site.
Gordon Sloan (https://orcid.org/0000-0001-6164-2662) (Clinical Research Fellow) produced the first draft of the report, was a member of the TMG and contributed to the delivery of the trial.
Jennifer Petrie (https://orcid.org/0000-0001-6150-6724) (Trial Manager) produced the first draft of the report, was a member of the TMG and contributed to the delivery of the trial.
David White (https://orcid.org/0000-0003-2871-7946) (Senior Research Fellow/Portfolio Lead) produced the first draft of the report, was a member of the TMG, contributed to the delivery of the trial, was an applicant on the Health Technology Assessment grant and contributed to the study design.
Mike Bradburn (https://orcid.org/0000-0002-3783-9761) (Senior Statistician) produced the first draft of the report, was a member of the TMG and contributed to the delivery of the trial.
Tracey Young (https://orcid.org/0000-0001-8467-0471) (Health Economist) produced the first draft of the report, was a member of the TMG and contributed to the delivery of the trial.
Satyan Rajbhandari (https://orcid.org/0000-0002-3995-5336) (Consultant Physician and Diabetologist) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Sanjeev Sharma (https://orcid.org/0000-0001-6386-2052) (Consultant, Endocrinology and Diabetes) was an investigator at a participating site and was a member of the TMG.
Gerry Rayman (https://orcid.org/0000-0003-3331-7015) (Consultant Physician, Diabetes and Endocrinology) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Ravikanth Gouni (https://orcid.org/0000-0002-5852-470X) (Consultant, Diabetes and Endocrinology) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Uazman Alam (https://orcid.org/0000-0002-3190-1122) (Senior Clinical Lecturer and Honorary Consultant Physician, Cardiovascular and Metabolic Medicine) was an investigator at a participating site and was a member of the TMG.
Steven A Julious (https://orcid.org/0000-0002-9917-7636) (Professor of Medical Statistics) was an applicant on the Health Technology Assessment grant, contributed to the study design and was a member of the TMG.
Cindy Cooper (https://orcid.org/0000-0002-2995-5447) (Professor of Health Services Research and Clinical Trials) was an applicant on the Health Technology Assessment grant, contributed to the study design and was a member of the TMG.
Amanda Loban (https://orcid.org/0000-0001-7089-1580) (Lead Data Manager) was a member of the TMG and provided central data management.
Katie Sutherland (https://orcid.org/0000-0002-5162-8119) (Research Assistant) was a member of the TMG and contributed to the delivery of the trial.
Rachel Glover (https://orcid.org/0000-0002-2460-3354) (Trial Manager) was a member of the TMG and contributed to the delivery of the trial.
Simon Waterhouse (https://orcid.org/0000-0002-6303-9610) (Data Manager) was a member of the TMG and provided central data management.
Emily Turton (https://orcid.org/0000-0001-5763-9604) (Data Manager) was a member of the TMG and provided central data management.
Michelle Horspool (https://orcid.org/0000-0002-3069-6091) (Research Development Manager) was an applicant on the Health Technology Assessment grant, contributed to the study design and was a member of the TMG.
Rajiv Gandhi (https://orcid.org/0000-0002-2008-2301) (Consultant Diabetologist) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Deirdre Maguire (https://orcid.org/0000-0001-6730-0997) (Consultant in Diabetes and Endocrinology) was an investigator at a participating site and was a member of the TMG.
Edward Jude (https://orcid.org/0000-0002-3186-4122) (Consultant Physician and Honorary Reader in Medicine) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Syed Haris Ahmed (https://orcid.org/0000-0002-7815-2907) (Consultant Physician in Diabetes, Endocrinology and Metabolic Medicine) was an investigator at a participating site and was a member of the TMG.
Prashanth Vas (https://orcid.org/0000-0001-7448-2995) (Consultant Physician) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Christian Hariman (https://orcid.org/0000-0001-6426-5807) (Consultant Physician, Diabetes and Endocrinology) was an investigator at a participating site and was a member of the TMG.
Claire McDougall (https://orcid.org/0000-0003-0138-5267) (Consultant Diabetologist) was an investigator at a participating site and was a member of the TMG.
Marion Devers (https://orcid.org/0000-0003-2605-4093) (Consultant Physician, Diabetes and Endocrinology) was an investigator at a participating site and was a member of the TMG.
Vasileios Tsatlidis (https://orcid.org/0000-0001-9413-5130) (Consultant, Endocrinology and Diabetes) was an investigator at a participating site and was a member of the TMG.
Martin Johnson (https://orcid.org/0000-0002-8398-5436) (Senior Medical Director, hVIVO) was an applicant on the Health Technology Assessment grant, contributed to the study design and was a member of the TMG.
Didier Bouhassira (https://orcid.org/0000-0001-6446-8719) (Director of Research, Inserm) was an applicant on the Health Technology Assessment grant, contributed to the study design and was a member of the TMG.
David L Bennett (https://orcid.org/0000-0002-7996-2696) (Professor of Neurology and Neurobiology) was an applicant on the Health Technology Assessment grant, contributed to the study design, was a member of the TMG and was an investigator at a participating site.
Dinesh Selvarajah (https://orcid.org/0000-0001-7426-1105) (Senior Clinical Lecturer in Diabetes and Honorary Consultant Physician) produced the first draft of the report, was a member of the TMG, contributed to the delivery of the trial, was an applicant on the Health Technology Assessment grant, contributed to the study design and was an investigator at a participating site.
Publications
Selvarajah D, Petrie J, White D, Julious S, Bortolami O, Cooper C, et al. Multicentre, double-blind, crossover trial to identify the Optimal Pathway for TreatIng neurOpathic paiN in Diabetes Mellitus (OPTION-DM): study protocol for a randomised controlled trial. Trials 2018;19:578. https://doi.org/10.1186/s13063-018-2959
Tesfaye S, Sloan G, Petrie J, White D, Bradburn M, Julious S, et al. Comparison of amitriptyline supplemented with pregabalin, pregabalin supplemented with amitriptyline, and duloxetine supplemented with pregabalin for the treatment of diabetic peripheral neuropathic pain (OPTION-DM): a multicentre, double-blind, randomised crossover trial. Lancet 2022;400:680–90.
Data-sharing statement
Requests for patient-level data and statistical code should be made to the corresponding author and will be considered on a case-by-case basis following the principles for sharing patient-level data. 88
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- Tesfaye S, Sloan G, Petrie J, White D, Bradburn M, Julious S, et al. Comparison of amitriptyline supplemented with pregabalin, pregabalin supplemented with amitriptyline, and duloxetine supplemented with pregabalin for the treatment of diabetic peripheral neuropathic pain (OPTION-DM): a multicentre, double-blind, randomised crossover trial. Lancet 2022;400:680-90. https://doi.org/10.1016/S0140-6736(22)01472-6.
- Selvarajah D, Petrie J, White D, Julious S, Bortolami O, Cooper C, et al. Multicentre, double-blind, crossover trial to identify the Optimal Pathway for TreatIng neurOpathic paiN in Diabetes Mellitus (OPTION-DM): study protocol for a randomised controlled trial. Trials 2018;19. https://doi.org/10.1186/s13063-018-2959-y.
- Creative Commons . Attribution 4.0 International (CC BY 4.0) n.d. https://creativecommons.org/licenses/by/4.0/ (accessed 9 December 2021).
- Diabetes UK . Number of People With Diabetes Reaches 4.8 Million 2020. www.diabetes.org.uk/about_us/news/diabetes-prevalence-2019 (accessed 9 December 2021).
- Abbott CA, Malik RA, van Ross ER, Kulkarni J, Boulton AJ. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 2011;34:2220-4. https://doi.org/10.2337/dc11-1108.
- Davies M, Brophy S, Williams R, Taylor A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care 2006;29:1518-22. https://doi.org/10.2337/dc05-2228.
- Veves A, Backonja M, Malik RA. Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med 2008;9:660-74. https://doi.org/10.1111/j.1526-4637.2007.00347.x.
- Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005;28:956-62. https://doi.org/10.2337/diacare.28.4.956.
- Galer BS, Gianas A, Jensen MP. Painful diabetic polyneuropathy: epidemiology, pain description, and quality of life. Diabetes Res Clin Pract 2000;47:123-8. https://doi.org/10.1016/S0168-8227(99)00112-6.
- Zelman DC, Brandenburg NA, Gore M. Sleep impairment in patients with painful diabetic peripheral neuropathy. Clin J Pain 2006;22:681-5. https://doi.org/10.1097/01.ajp.0000210910.49923.09.
- Gore M, Brandenburg NA, Dukes E, Hoffman DL, Tai KS, Stacey B. Pain severity in diabetic peripheral neuropathy is associated with patient functioning, symptom levels of anxiety and depression, and sleep. J Pain Symptom Manage 2005;30:374-85. https://doi.org/10.1016/j.jpainsymman.2005.04.009.
- Gore M, Brandenburg NA, Hoffman DL, Tai KS, Stacey B. Burden of illness in painful diabetic peripheral neuropathy: the patients’ perspectives. J Pain 2006;7:892-900. https://doi.org/10.1016/j.jpain.2006.04.013.
- Tölle T, Xu X, Sadosky AB. Painful diabetic neuropathy: a cross-sectional survey of health state impairment and treatment patterns. J Diabetes Complications 2006;20:26-33. https://doi.org/10.1016/j.jdiacomp.2005.09.007.
- National Institute for Health and Care Excellence (NICE) . Neuropathic Pain in Adults: Pharmacological Management in Non-Specialist Settings. Clinical Guideline [CG173] 2013.
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev 2015;7. https://doi.org/10.1002/14651858.CD008242.pub3.
- Lunn M, Hughes R, Wiffen P. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia (review). Cochrane Database Syst Rev 2014;3. https://doi.org/10.1002/14651858.CD007115.pub3.
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst Rev 2009;3. https://doi.org/10.1002/14651858.CD007076.pub2.
- Moore R, Wiffen P, Derry S, Mcquay H. Gabapentin for chronic neuropathic pain and fibromyalgia in adults (review). Cochrane Database Syst Rev 2011;3. https://doi.org/10.1002/14651858.CD007938.pub2.
- Freeman R, Durso-Decruz E, Emir B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes Care 2008;31:1448-54. https://doi.org/10.2337/dc07-2105.
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurol 2008;8. https://doi.org/10.1186/1471-2377-8-29.
- Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ 2007;335. https://doi.org/10.1136/bmj.39213.565972.AE.
- Tesfaye S, Vileikyte L, Rayman G, Sindrup SH, Perkins BA, Baconja M, et al. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metab Res Rev 2011;27:629-38. https://doi.org/10.1002/dmrr.1225.
- Dworkin RH, O’Connor AB, Audette J, Baron R, Gourlay GK, Haanpää ML, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc 2010;85:3-14. https://doi.org/10.4065/mcp.2009.0649.
- Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision [published online ahead of print]. Eur J Neurol 2010. https://doi.org/10.1111/j.1468-1331.2010.02999.x.
- Bansal D, Bhansali A, Hota D, Chakrabarti A, Dutta P. Amitriptyline vs. pregabalin in painful diabetic neuropathy: a randomized double blind clinical trial. Diabet Med 2009;26:1019-26. https://doi.org/10.1111/j.1464-5491.2009.02806.x.
- Kaur H, Hota D, Bhansali A, Dutta P, Bansal D, Chakrabarti A. A comparative evaluation of amitriptyline and duloxetine in painful diabetic neuropathy: a randomized, double-blind, cross-over clinical trial. Diabetes Care 2011;34:818-22. https://doi.org/10.2337/dc10-1793.
- Boyle J, Eriksson ME, Gribble L, Gouni R, Johnsen S, Coppini DV, et al. Randomized, placebo-controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: impact on pain, polysomnographic sleep, daytime functioning, and quality of life. Diabetes Care 2012;35:2451-8. https://doi.org/10.2337/dc12-0656.
- Quilici S, Chancellor J, Löthgren M, Simon D, Said G, Le TK, et al. Meta-analysis of duloxetine vs. pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic pain. BMC Neurol 2009;9. https://doi.org/10.1186/1471-2377-9-6.
- Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med 2005;352:1324-34. https://doi.org/10.1056/NEJMoa042580.
- Tesfaye S, Selvarajah D. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med 2005;352:2650-1. https://doi.org/10.1056/NEJM200506233522520.
- Tesfaye S, Wilhelm S, Lledo A, Schacht A, Tölle T, Bouhassira D, et al. Duloxetine and pregabalin: high-dose monotherapy or their combination? The ‘COMBO-DN study’ – a multinational, randomized, double-blind, parallel-group study in patients with diabetic peripheral neuropathic pain. Pain 2013;154:2616-25. https://doi.org/10.1016/j.pain.2013.05.043.
- Wu EQ, Birnbaum HG, Mareva MN, Le TK, Robinson RL, Rosen A, et al. Cost-effectiveness of duloxetine versus routine treatment for U.S. patients with diabetic peripheral neuropathic pain. J Pain 2006;7:399-407. https://doi.org/10.1016/j.jpain.2006.01.443.
- Beard SM, McCrink L, Le TK, Garcia-Cebrian A, Monz B, Malik RA. Cost effectiveness of duloxetine in the treatment of diabetic peripheral neuropathic pain in the UK. Curr Med Res Opin 2008;24:385-99. https://doi.org/10.1185/030079908x253852.
- O’Connor A, Noyes K, Holloway R. A cost–utility comparison of four first-line medications in painful diabetic neuropathy. PharmacoEconomics 2008;26:1045-64.
- de Salas-Cansado M, Pérez C, Saldaña MT, Navarro A, González-Gómez FJ, Ruiz L, et al. An economic evaluation of pregabalin versus usual care in the management of community-treated patients with refractory painful diabetic peripheral neuropathy in primary care settings. Prim Care Diabetes 2012;6:303-12. https://doi.org/10.1016/j.pcd.2012.03.001.
- Backonja M, Beydoun A, Edwards KR, Schwartz SL, Fonseca V, Hes M, et al. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial. JAMA 1998;280:1831-6. https://doi.org/10.1001/jama.280.21.1831.
- Dworkin RH, Turk DC, Peirce-Sandner S, Burke LB, Farrar JT, Gilron I, et al. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. Pain 2012;153:1148-58. https://doi.org/10.1016/j.pain.2012.03.003.
- Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 2005;114:29-36. https://doi.org/10.1016/j.pain.2004.12.010.
- Bril V, Tomioka S, Buchanan RA, Perkins BA. mTCNS Study Group . Reliability and validity of the modified Toronto Clinical Neuropathy Score in diabetic sensorimotor polyneuropathy. Diabet Med 2009;26:240-6. https://doi.org/10.1111/j.1464-5491.2009.02667.x.
- Great Britain . Data Protection Act 2018 2018.
- Ware JE, Gandek B. Overview of the SF-36 Health Survey and the International Quality of Life Assessment (IQOLA) Project. J Clin Epidemiol 1998;51:903-12. https://doi.org/10.1016/S0895-4356(98)00081-X.
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361-70. https://doi.org/10.1111/j.1600-0447.1983.tb09716.x.
- Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singap 1994;23:129-38.
- Bastien CH, Valliéres A, Morin CM. Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep Med 2001;2:297-30. https://doi.org/10.1016/S1389-9457(00)00065-4.
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare; 1976.
- Devlin N, Shah K, Feng Y, Mulhern B, Van Hout B. Valuing health-related quality of life: an EQ-5D-5L value set for England. Health Econ 2018;27:7-22. https://doi.org/10.1002/hec.3564.
- Beecham J, Knapp M, Thornicroft J. Measuring Mental Health Needs. Cambridge: Cambridge University Press; 2001.
- Bouhassira D, Attal N, Fermanian J, Alchaar H, Gautron M, Masquelier E, et al. Development and validation of the Neuropathic Pain Symptom Inventory. Pain 2004;108:248-57. https://doi.org/10.1016/j.pain.2003.12.024.
- Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. Pain 2014;155:2263-73. https://doi.org/10.1016/j.pain.2014.08.014.
- Bouhassira D, Wilhelm S, Schacht A, Perrot S, Kosek E, Cruccu G, et al. Neuropathic pain phenotyping as a predictor of treatment response in painful diabetic neuropathy: data from the randomized, double-blind, COMBO-DN study. Pain 2014;155:2171-9. https://doi.org/10.1016/j.pain.2014.08.020.
- Marchettini P, Wilhelm S, Petto H, Tesfaye S, Tölle T, Bouhassira D, et al. Are there different predictors of analgesic response between antidepressants and anticonvulsants in painful diabetic neuropathy?. Eur J Pain 2016;20:472-82. https://doi.org/10.1002/ejp.763.
- Bouhassira D, Branders S, Attal N, Fernandes AM, Demolle D, Barbour J, et al. Stratification of patients based on the Neuropathic Pain Symptom Inventory: development and validation of a new algorithm. Pain 2021;162:1038-46. https://doi.org/10.1097/j.pain.0000000000002130.
- Joint Formulary Committee . British National Formulary n.d. www.medicinescomplete.com/mc/bnf/current/PHP5799-degarelix.htm (accessed 9 December 2021).
- Julious SA, Walters SJ. Estimating effect sizes for health-related quality of life outcomes. Stat Methods Med Res 2014;23:430-9. https://doi.org/10.1177/0962280213476379.
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain 2008;9:105-21. https://doi.org/10.1016/j.jpain.2007.09.005.
- Julious S. A tutorial in biostatistics: sample sizes for clinical trials with normal data. Stat Med 2004;23:1921-86. https://doi.org/10.1002/sim.1783.
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009;374:1252-61. https://doi.org/10.1016/S0140-6736(09)61081-3.
- Kenward MG, Molenberghs G. Last observation carried forward: a crystal ball?. J Biopharm Stat 2009;19:872-88. https://doi.org/10.1080/10543400903105406.
- European Medicines Agency . Guideline on Missing Data in Confirmatory Clinical Trials 2010. www.ema.europa.eu/en/documents/scientific-guideline/guideline-missing-data-confirmatory-clinical-trials_en.pdf (accessed 6 August 2021).
- Little RRD. Statistical Analysis with Missing Data. Chichester: Wiley; 2019.
- Cro S, Morris TP, Kenward MG, Carpenter JR. Sensitivity analysis for clinical trials with missing continuous outcome data using controlled multiple imputation: a practical guide. Stat Med 2020;39:2815-42. https://doi.org/10.1002/sim.8569.
- National Institute for Health and Care Excellence . Guide to the Methods of Technology Appraisal 2018. www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-technology-appraisals/technology-appraisal-processes-guide-apr-2018.pdf (accessed 9 December 2021).
- Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. Cost Eff Resour Alloc 2013;11. https://doi.org/10.1186/1478-7547-11-6.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- National Institute for Health and Care Excellence . Position Statement on Use of the EQ-5D5L Value Set for England (Updated October 2019) n.d. www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/technology-appraisal-guidance/eq-5d-5l (accessed 17 August 2021).
- van Hout B, Janssen MF, Feng YS, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15. https://doi.org/10.1016/j.jval.2012.02.008.
- NHS . National Cost Collection for the NHS 2018 19 n.d. www.england.nhs.uk/national-cost-collection/ (accessed 9 December).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2019. Canterbury: PSSRU, University of Kent; 2019.
- The Podiatry Clinic . Podiatry Clinic Tariff n.d. www.thepodiatry-clinic.co.uk/prices/ (accessed 16 May 2022).
- A&A Podiatrists . Podiatry Service Prices n.d. www.painfreefeet.co.uk/servicespodiatry/chiropody-cost/ (accessed 16 May 2022).
- The Footcare Centre . Treatments and Prices n.d. www.thefootcarecentre.co.uk/treatments-and-prices/ (accessed 16 May 2022).
- NHS Employers . NHS TCS 2018 (AfC) n.d. www.nhsemployers.org/sites/default/files/media/NHS-terms-and-conditions-pay-poster-201819_0.pdf (accessed 20 December 2021).
- Escape Holistic Therapies . Price List n.d. www.escapeholistictherapies.co.uk/price-list/ (accessed 16 May 2022).
- Holly’s Holistics . Home Massage and Aromatherapy Treatments Price Guide n.d. https://hollysholistics.co.uk/price-guide/ (accessed 16 May 2022).
- Natural at Heart . Treatments and Prices n.d. www.naturalatheart.co.uk/price-list.html (accessed 16 May 2022).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2020. Canterbury: PSSRU, University of Kent; 2020.
- Office for National Statistics . Earnings and Hours Worked, All Employees: ASHE Table 1 2020. www.ons.gov.uk/employmentandlabourmarket/peopleinwork/earningsandworkinghours/datasets/allemployeesashetable1 (accessed 2 July 2021).
- Mulhern B, Feng Y, Shah K, Janssen MF, Herdman M, van Hout B, et al. Comparing the UK EQ-5D-3L and English EQ-5D-5L value sets. PharmacoEconomics 2018;36:699-713. https://doi.org/10.1007/s40273-018-0628-3.
- Bril V, England J, Franklin GM, Backonja M, Cohen J, Del Toro D, et al. Evidence-based guideline: treatment of painful diabetic neuropathy. Neurology 2011;76:1758-65. https://doi.org/10.1212/WNL.0b013e3182166ebe.
- Holbech JV, Bach FW, Finnerup NB, Brøsen K, Jensen TS, Sindrup SH. Imipramine and pregabalin combination for painful polyneuropathy: a randomized controlled trial. Pain 2015;156:958-66. https://doi.org/10.1097/j.pain.0000000000000143.
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162-73. https://doi.org/10.1016/S1474-4422(14)70251-0.
- National Institute for Health and Care Excellence . Neuropathic Pain in Adults: Pharmacological Management in Non-Specialist Settings. Clinical Guideline [CG173] 2020. www.nice.org.uk/guidance/cg173 (accessed 9 December 2021).
- Chaparro LE, Wiffen PJ, Moore RA, Gilron I. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev 2012;7. https://doi.org/10.1002/14651858.CD008943.pub2.
- Collado Mateo D, García Gordillo MA, Olivares PR, Adsuar JC. Normative values of EQ-5D-5L for diabetes patients from Spain. Nutr Hosp 2015;32:1595-602. https://doi.org/10.3305/nh.2015.32.4.9605.
- Wang P, Luo N, Tai ES, Thumboo J. The EQ-5D-5L is more discriminative than the EQ-5D-3L in patients with diabetes in Singapore. Value Health Reg Issues 2016;9:57-62. https://doi.org/10.1016/j.vhri.2015.11.003.
- Pan CW, Sun HP, Wang X, Ma Q, Xu Y, Luo N, et al. The EQ-5D-5L index score is more discriminative than the EQ-5D-3L index score in diabetes patients. Qual Life Res 2015;24:1767-74. https://doi.org/10.1007/s11136-014-0902-6.
- Moisset X, Bouhassira D, Avez Couturier J, Alchaar H, Conradi S, Delmotte MH, et al. Pharmacological and non-pharmacological treatments for neuropathic pain: systematic review and French recommendations. Rev Neurol 2020;176:325-52. https://doi.org/10.1016/j.neurol.2020.01.361.
- Smith CT, Hopkins C, Sydes M, Woolfall K, Clarke M, Murray G, et al. Good practice principles for sharing individual participant data from publicly funded clinical trials. Trials 2015;16. https://doi.org/10.1186/1745-6215-16-S2-O1.
Appendix 1 Case report form completion schedule
| Form | Pre-screening | Week | Repeat through each pathway: week | Week 17 | Unscheduled forms | Study discontinuation | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| –2 | –1 | 0 (randomisation) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | |||||
| Pre-screening log | ✗ | ||||||||||||||||||||||
| Screening consent | ✗ | ||||||||||||||||||||||
| Demographics | ✗ | ||||||||||||||||||||||
| Medical history | ✗ | ||||||||||||||||||||||
| Vital signs | ✗ | ✗ | |||||||||||||||||||||
| mTCNS | ✗ | ||||||||||||||||||||||
| DN4 | ✗ | ||||||||||||||||||||||
| Laboratory tests | ✗ | ✗ | |||||||||||||||||||||
| Previous medications | ✗ | ||||||||||||||||||||||
| Weekly contact form | ✗ | ✗ | ✗ | ✗a | ✗a | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||||||
| Pregnancy testb | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||||||||||||
| Confirmation of eligibility | ✗ | ||||||||||||||||||||||
| Randomisation | ✗ | ||||||||||||||||||||||
| Treatment decisions | ✗ | ✗ | ✗ | ✗ | ✗c | ✗c | ✗c | ||||||||||||||||
| Treatment compliance log | ✗ | ✗ | ✗ | ✗c | ✗ | ✗ | ✗ | ✗ | |||||||||||||||
| Concomitant medications | ✗ | ||||||||||||||||||||||
| AEs | ✗ | ||||||||||||||||||||||
| Unblinding | ✗ | ||||||||||||||||||||||
| Unscheduled dose changes | ✗ | ||||||||||||||||||||||
| Pregnancy information | ✗ | ||||||||||||||||||||||
| Protocol non-compliance | ✗ | ||||||||||||||||||||||
| Intervention withdrawal | ✗ | ✗ | |||||||||||||||||||||
| Study completion discontinuation | ✗d | ✗d | |||||||||||||||||||||
Appendix 2 Questionnaire completion schedule
| Form/event | Week | Repeat through each pathway: week | Study discontinuation | ||
|---|---|---|---|---|---|
| –2 | 0 (randomisation) | 6 | 16 | ||
| Assessment of suicidal riska | ✗ | ||||
| Pain diaryb | ✗ | ||||
| BPI-MSF | ✗ | ✗ | ✗ | ✗ | |
| ISI | ✗ | ✗ | ✗ | ✗ | |
| NPSI | ✗ | ✗ | ✗ | ✗ | |
| HADS | ✗ | ✗ | ✗ | ✗ | |
| SF-36 | ✗ | ✗ | ✗ | ✗ | |
| EQ-5D-5L | ✗ | ✗ | ✗ | ✗ | |
| CSRI | ✗ | ✗ | ✗ | ✗ | |
| Pain Catastrophizing Scale | ✗ | ||||
| Tolerability Scale | ✗ | ✗ | ✗ | ✗ | |
| Study medicine and pain diaryc | ✗ | ✗ | ✗ | ✗ | |
| PGIC | ✗ | ✗ | |||
Appendix 3 Summary of protocol amendments
| Amendment number | Summary of amendment |
|---|---|
| Substantial amendment 1, protocol version 3.0, approved 24 March 2017 (REC only) | Patient-perceived tolerability end points added. Scoring system for SF-36 updated and HADS end points updated. Inclusion criteria clarified:
|
| Substantial amendment 2, protocol version 4.0, approved 26 May 2017 (REC only) | Clarified that study treatment dose review is required for only severe or intolerable side effects. Updated information on emergency unblinding out of hours to allow for variation across centres. Other minor corrections and clarification throughout the protocol. New site added (Chester) |
| Substantial amendment 3, protocol version 5.0, approved 24 July 2017 | Added serum creatinine to the list of safety blood tests. Added pregnancy test to list of assessments at randomisation and then at weeks 3, 6, 9 and 16 of each pathway for women of childbearing potential. Clarified process for emergency unblinding during office hours. Clarified methods of contraception acceptable within the trial. Removed existing site (Poole) |
| Substantial amendment 4, approved 18 October 2017 (REC only) | Added new sites (Bournemouth, Liverpool and Harrogate) |
| Substantial amendment 5, protocol version 6.0, approved 28 November 2017 | Added a reduced pregabalin dosing schedule for participants with a eGFR of 30–59 ml/minute. Clarified that if a participant withdraws from the study, then any blood samples already collected would be kept unless the participant requests otherwise. Clarified the pre-screening procedures and data to be collected at the screening and randomisation visits. Added the option for participants to consent to provide daily pain scores via text message. Clarified that second-line treatment could be started up to week 13, if needed. Updated the unblinding process for safety reporting. Clarified that the blood sample for future research could be taken at the same time as any other study blood sample. Clarified the requirements for source data and the process for validating data within the Prospect database. Other minor corrections and clarifications throughout protocol |
| Substantial amendment 6, approved 15 January 2018 (REC only) | Added new sites (Aintree, Queen Elizabeth University Hospital, Edinburgh Royal Infirmary, Hairmyres Hospital, Monklands Hospital, Bradford Teaching Hospitals and New Cross Hospital) |
| Substantial amendment 7, protocol version 7.0, approved 13 March 2018 | Clarified that assessments at weeks 1 and 7 can be completed over the telephone or via a face-to-face visit. Added details regarding the recruitment process in Scotland. Updated inclusion criteria to clarify that neuropathic pain may be present in the hands. Updated exclusion criteria:
|
| Non-substantial amendment 1, approved 4 April 2018 | Study medicine and pain diary updated to allow participants to start treatment on the day of randomisation or the following morning. Minor corrections made to the OPTION-DM trial leaflet and the GP letter |
| Substantial amendment 8, protocol v9.0, approved 11 May 2018 | Updated exclusion criteria:
|
| Substantial amendment 9, approved 21 June 2018 | Updated reference safety information for amitriptyline and duloxetine |
| Substantial amendment 10, approved 24 July 2018 | Added new sites (Derby, Gateshead and Morriston). Changed PI to Marion Devers at Monklands Hospital |
| Non-substantial amendment 2, approved 12 October 2018 | Updated recruitment end date to February 2019 |
| Substantial amendment 11, approved 1 November 2018 | Added new sites (St Georges and Croydon) |
| Substantial amendment 12, protocol version 10.0, approved 8 January 2019 | Updated inclusion criteria:
|
| Non-substantial amendment 3, approved 13 March 2019 | Introduced the ‘end of study participant information sheet’. Updated recruitment end date to April 2019. Invitation letters updated to clarify that potential participants may be contacted by telephone. Minor changes made to the ‘study medicine and pain diary’ and the ‘participant information sheet’ |
| Substantial amendment 13, protocol version 11.0, approved 3 June 2019 | Amended predicted sample size. Updated recruitment end date to July 2019. Updated exclusion criteria:
|
| Substantial amendment 14, approved 26 September 2019 | Genetic analysis substudy protocol submitted for approval |
| Substantial amendment 15, approved 22 September 2020 | End-of-trial definition updated to allow sample analyses to be completed. The end of trial is now defined as the completion of all sample analyses |
Appendix 4 Subgroup analyses
FIGURE 16.
Numeric Rating Scale pain at week 16 by subgroup. (a) NRS by age subgroup (values are least square means ± 98.3% CI); (b) NRS by pain at baseline (values are least square means ± 98.3% CI); (c) NRS by NPSI pain phenotype – overall classification52 (values are least square means ± 98.3% CI); (d) NRS by baseline NPSI paraesthesia/dysaesthesia pain score (values are derived from lowess plot of response in relation to baseline NPSI score); (e) NRS by baseline NPSI evoked pain score (values are derived from lowess plot of response in relation to baseline NPSI score); (f) NRS by baseline NPSI paroxysmal pain score (values are derived from lowess plot of response in relation to baseline NPSI score); (g) NRS by baseline NPSI deep spontaneous/pressing pain score (values are derived from lowess plot of response in relation to baseline NPSI score); (h) NRS by baseline NPSI superficial spontaneous/burning pain score (values are derived from lowess plot of response in relation to baseline NPSI score); (i) NRS by NPSI pain phenotype – total pain score (values are derived from lowess plot of response in relation to baseline NPSI score); (j) NRS by HADS anxiety (values are derived from lowess plot of response in relation to baseline HADS score); (k) NRS by HADS depression (values are derived from lowess plot of response in relation to baseline HADS score); (l) NRS by previous use of amitriptyline (values are least square means ± 98.3% CI); (m) NRS by previous use of duloxetine (values are least square means ± 98.3% CI); (n) NRS by previous use of pregabalin (values are least square means ± 98.3% CI); (o) NRS by previous use of opioids (values are least square means ± 98.3% CI); (p) longitudinal NRS by time [values are derived from lowess plot of the residuals for response in a longitudinal model in which the covariates are treatment, period and week of pathway (0–16); residual standardised for treatment pathway, pathway week and treatment group]; and (q) NRS by pre-/post-lockdown (values are least square means ± 98.3% CI).
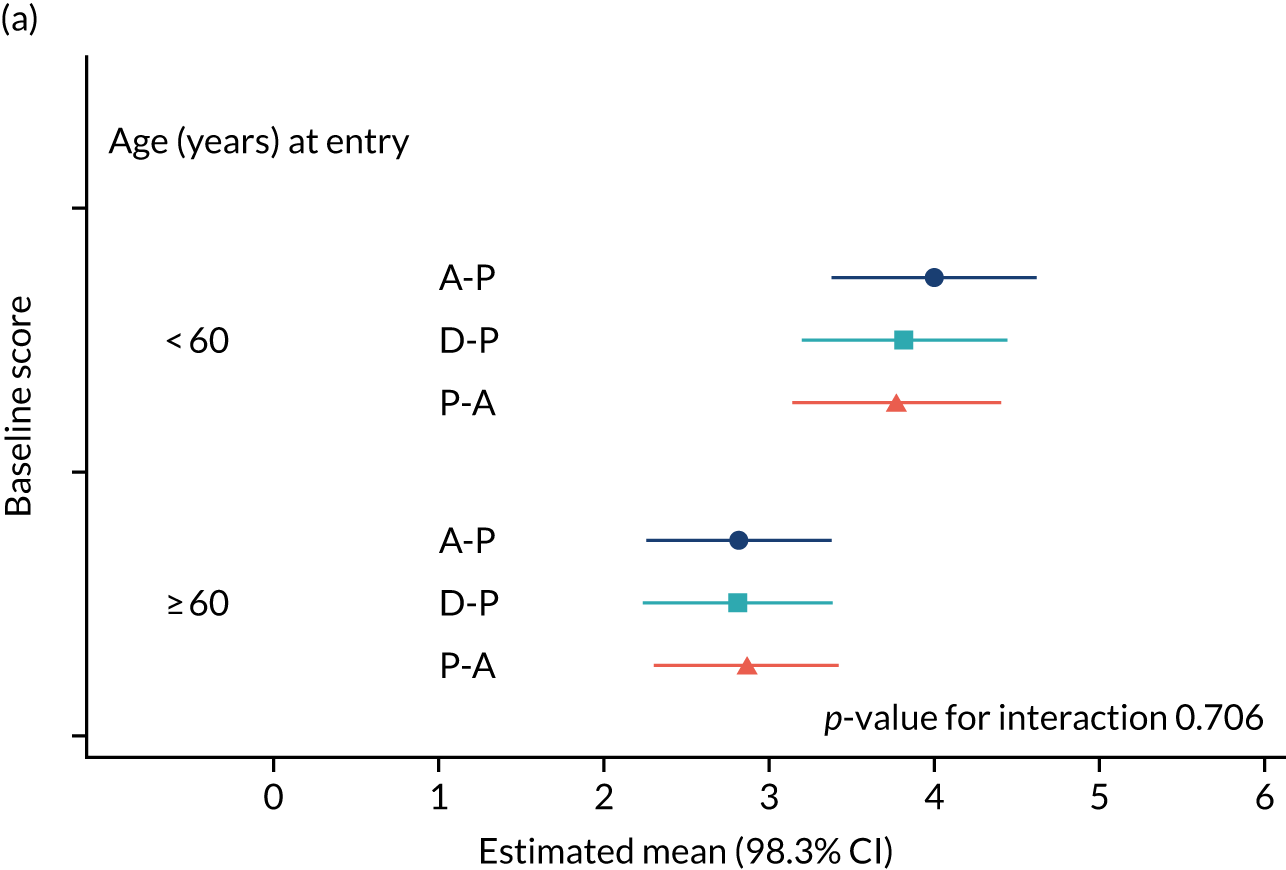

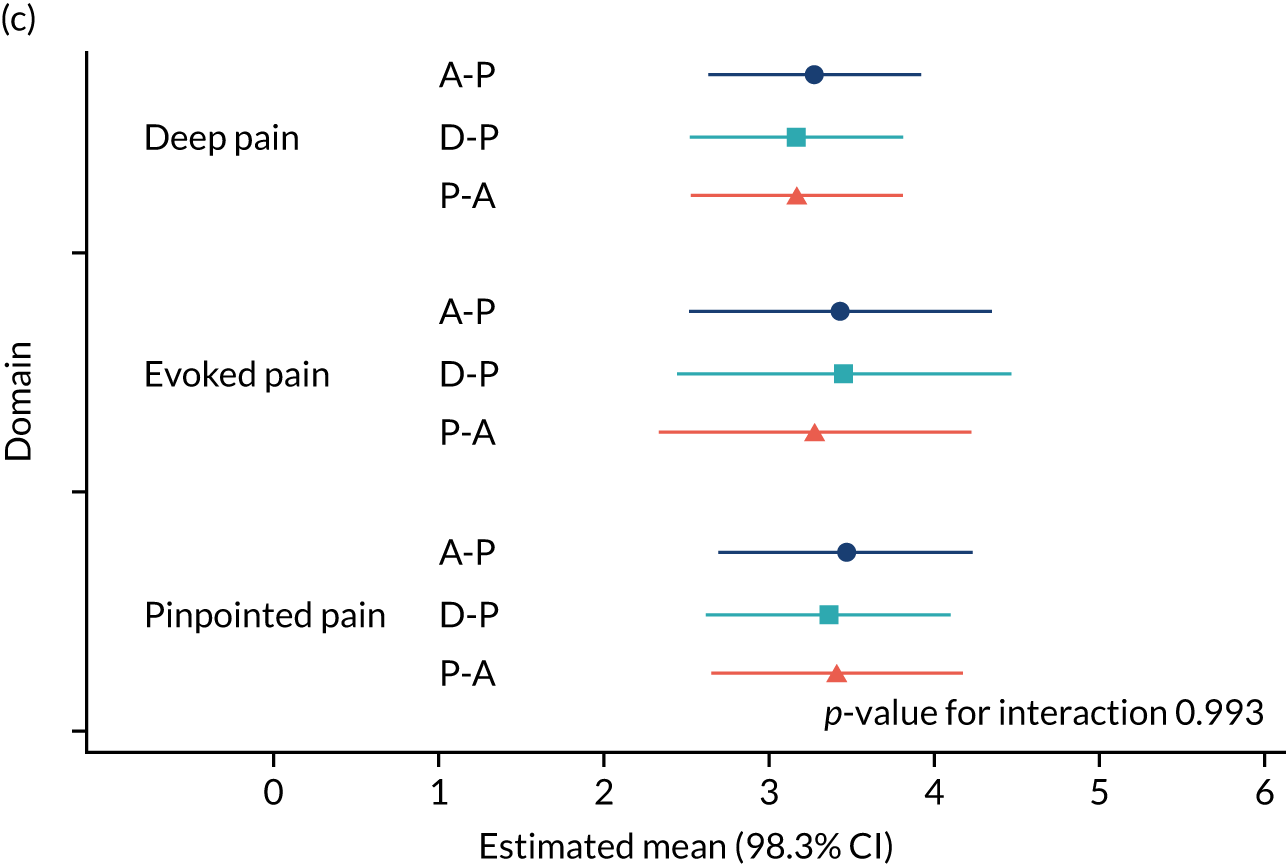
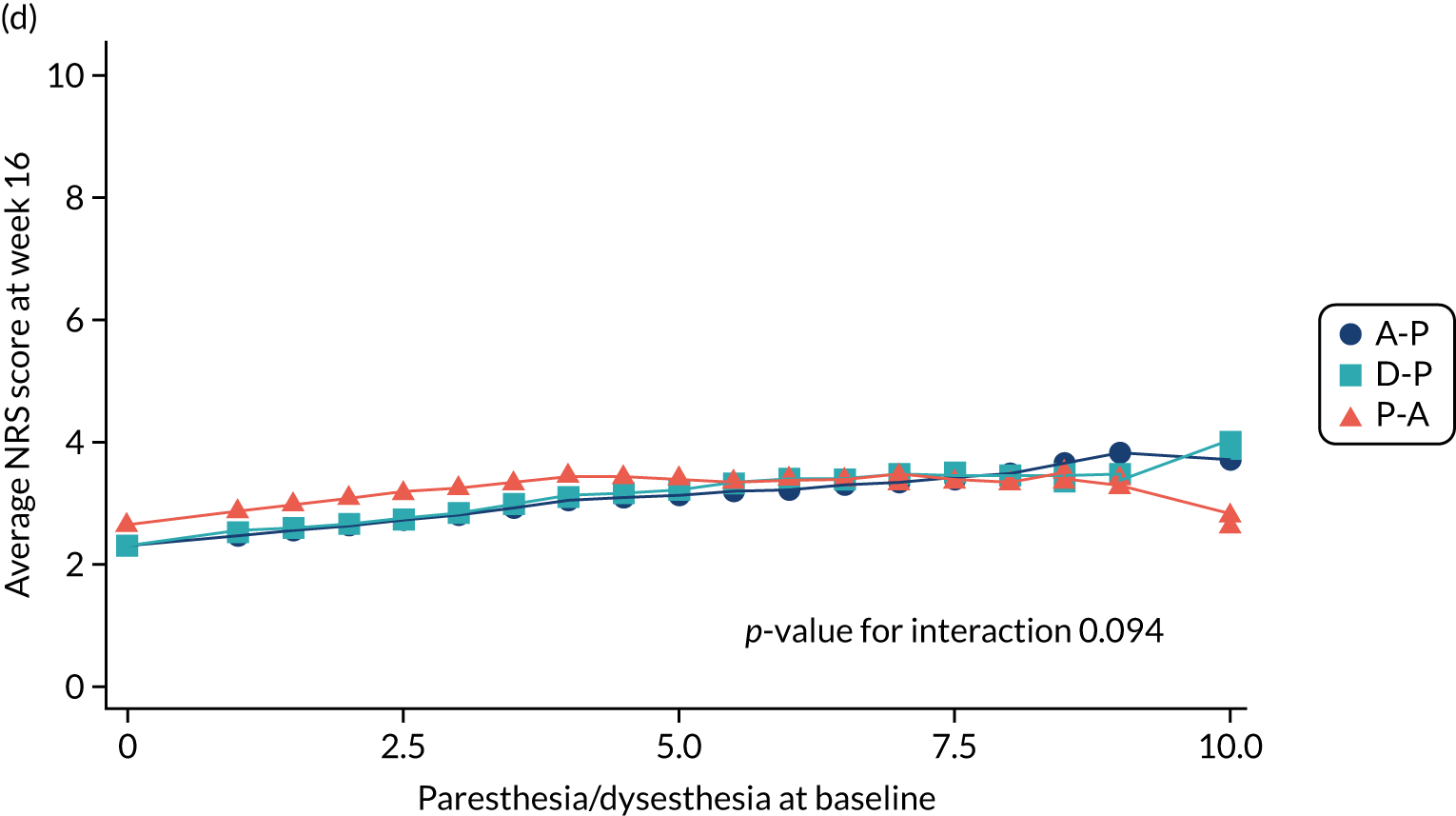
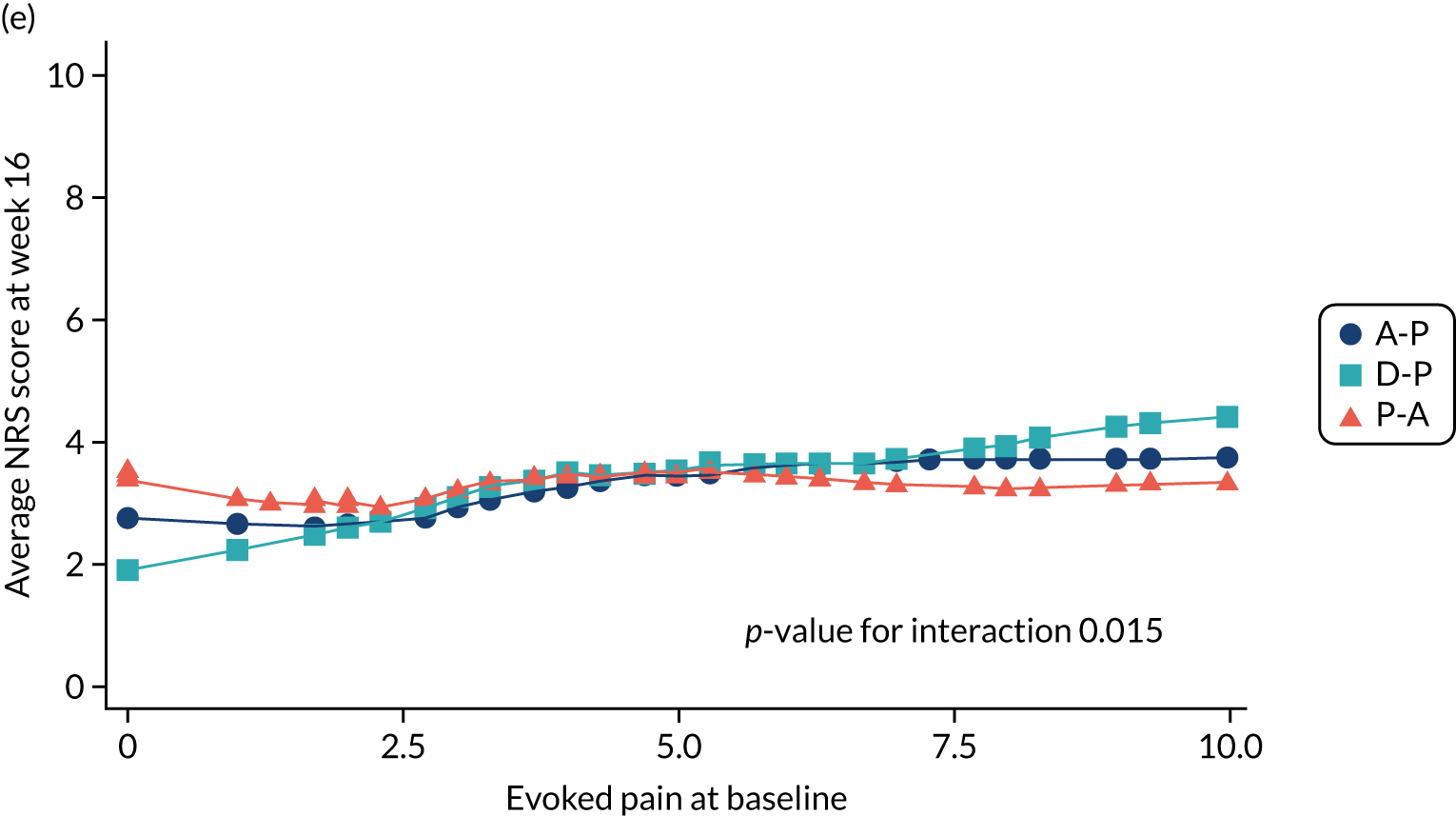


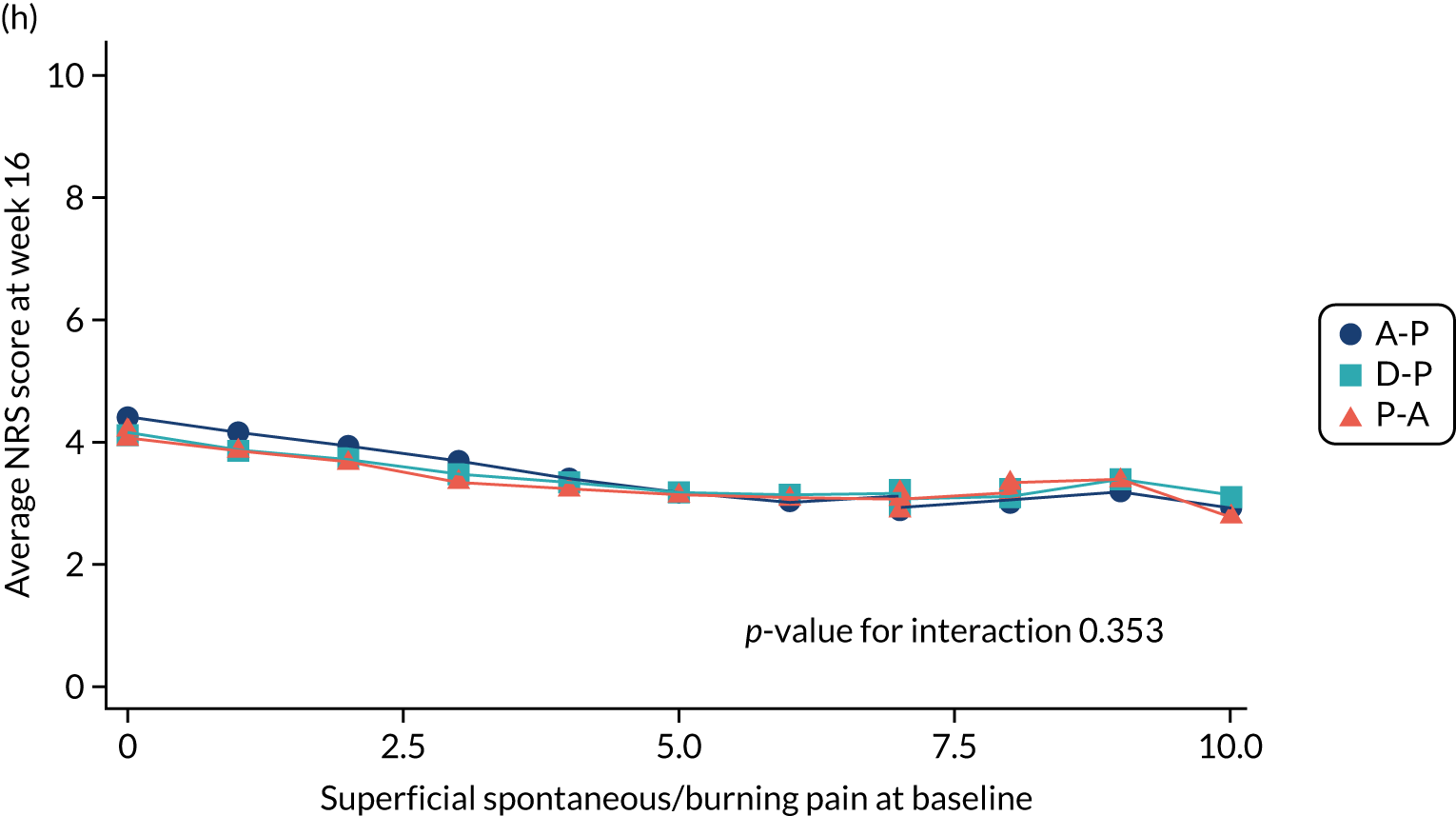
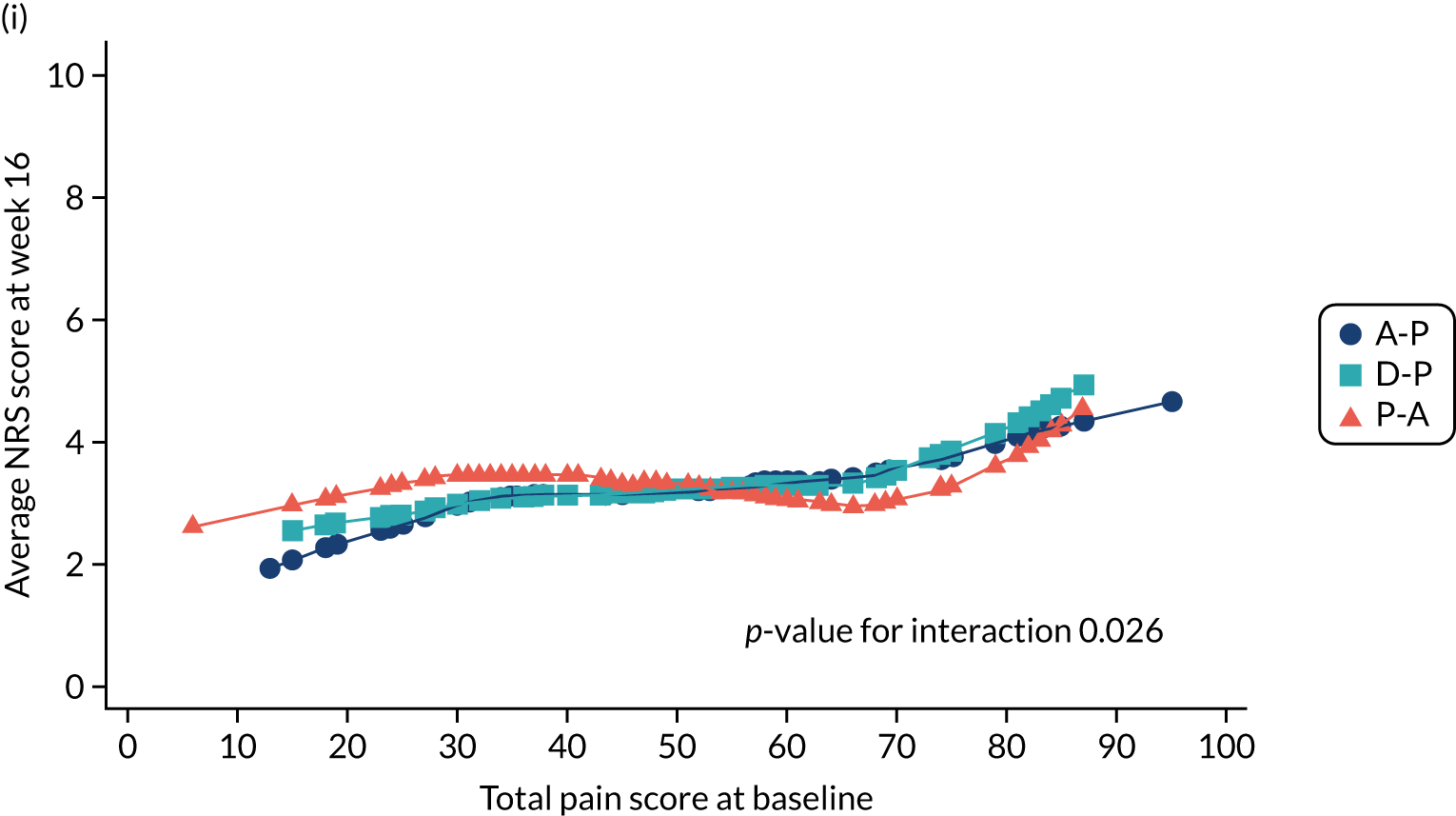
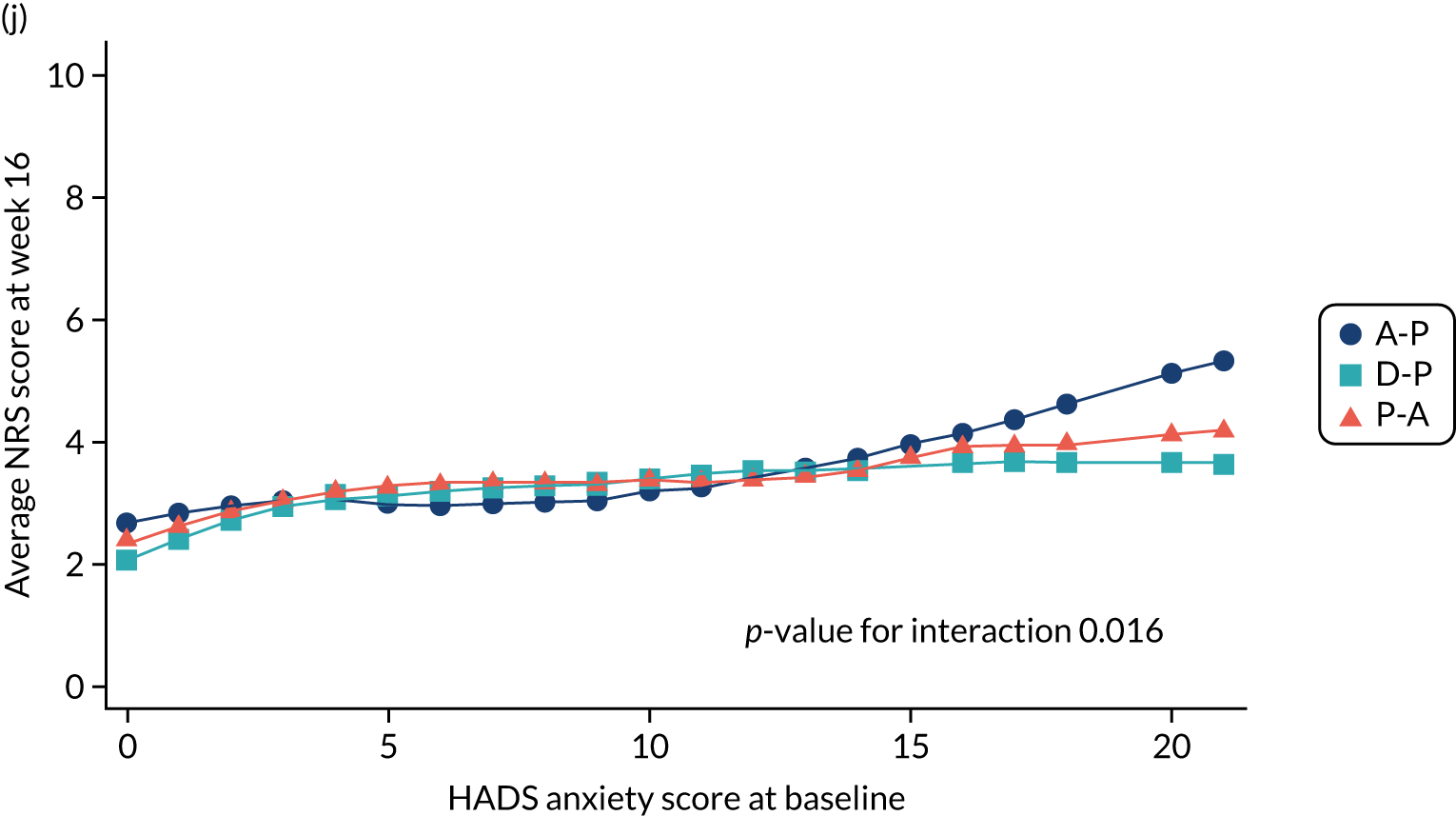
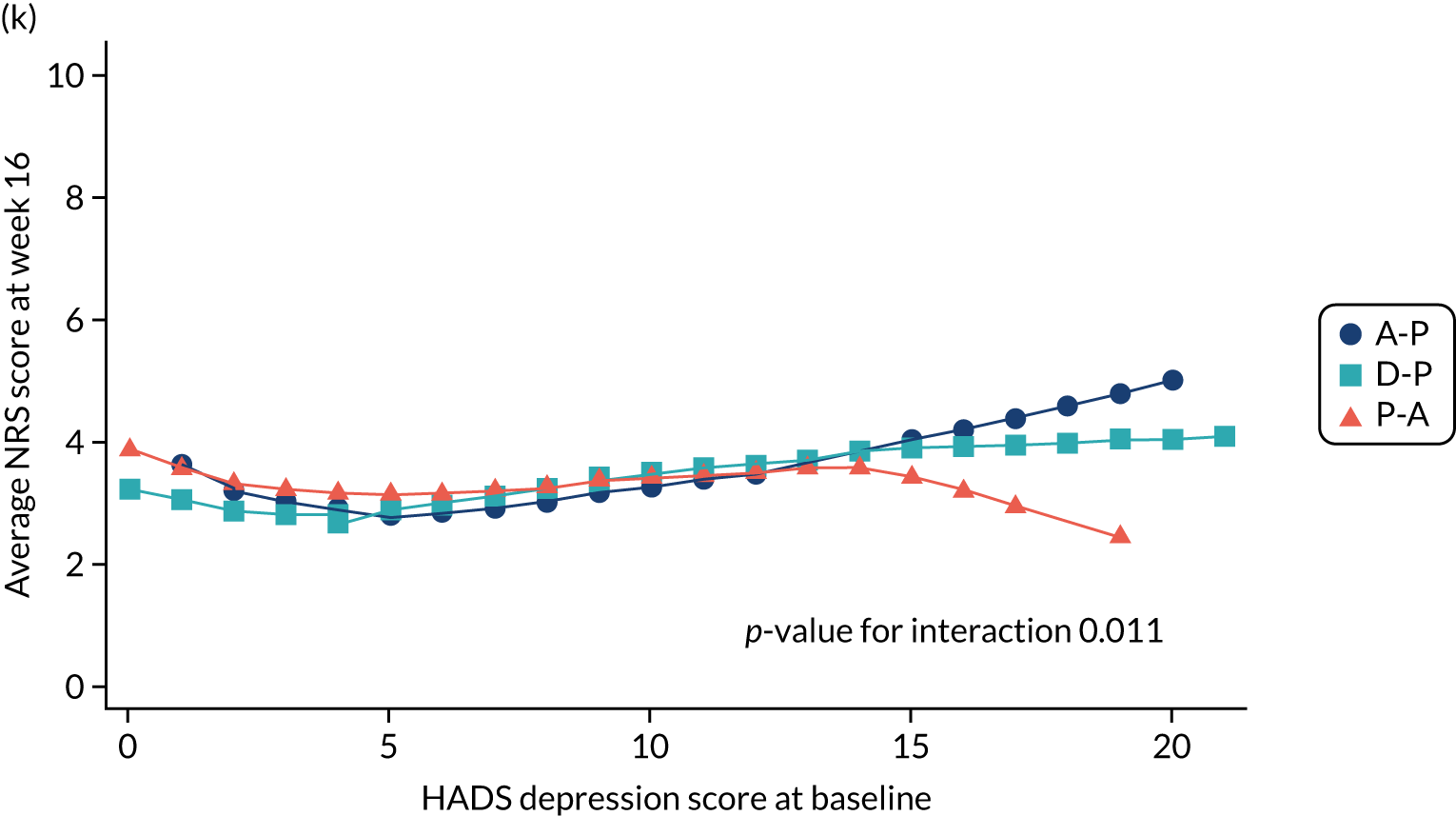

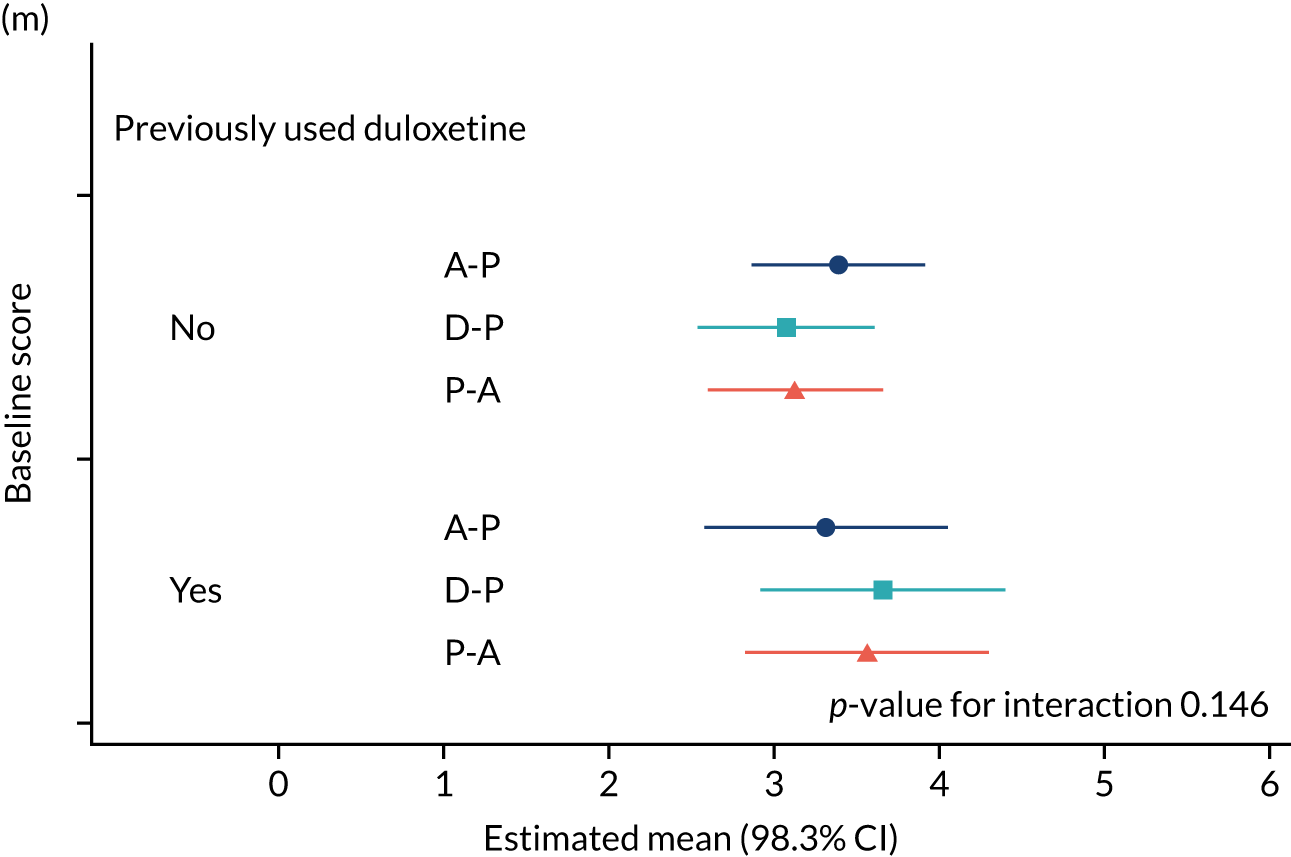
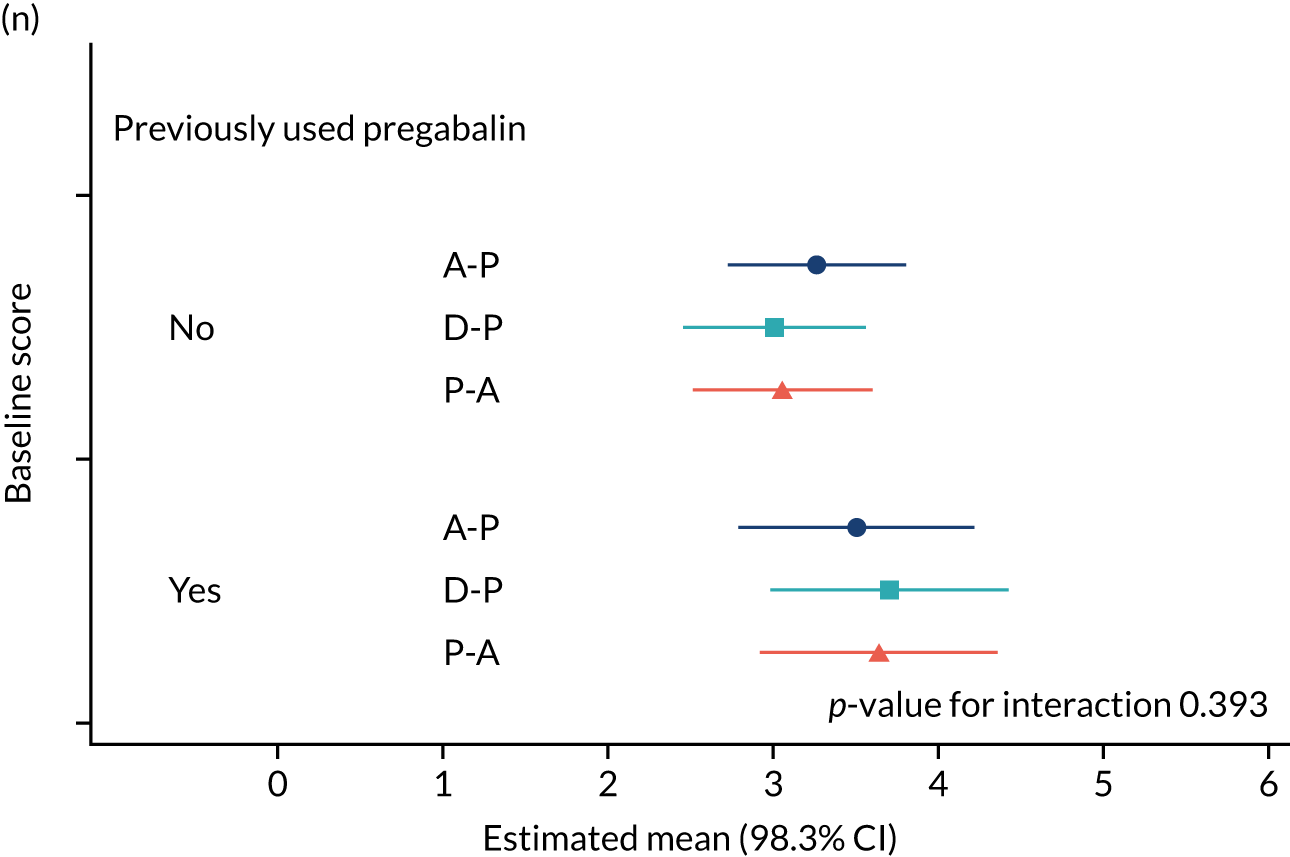

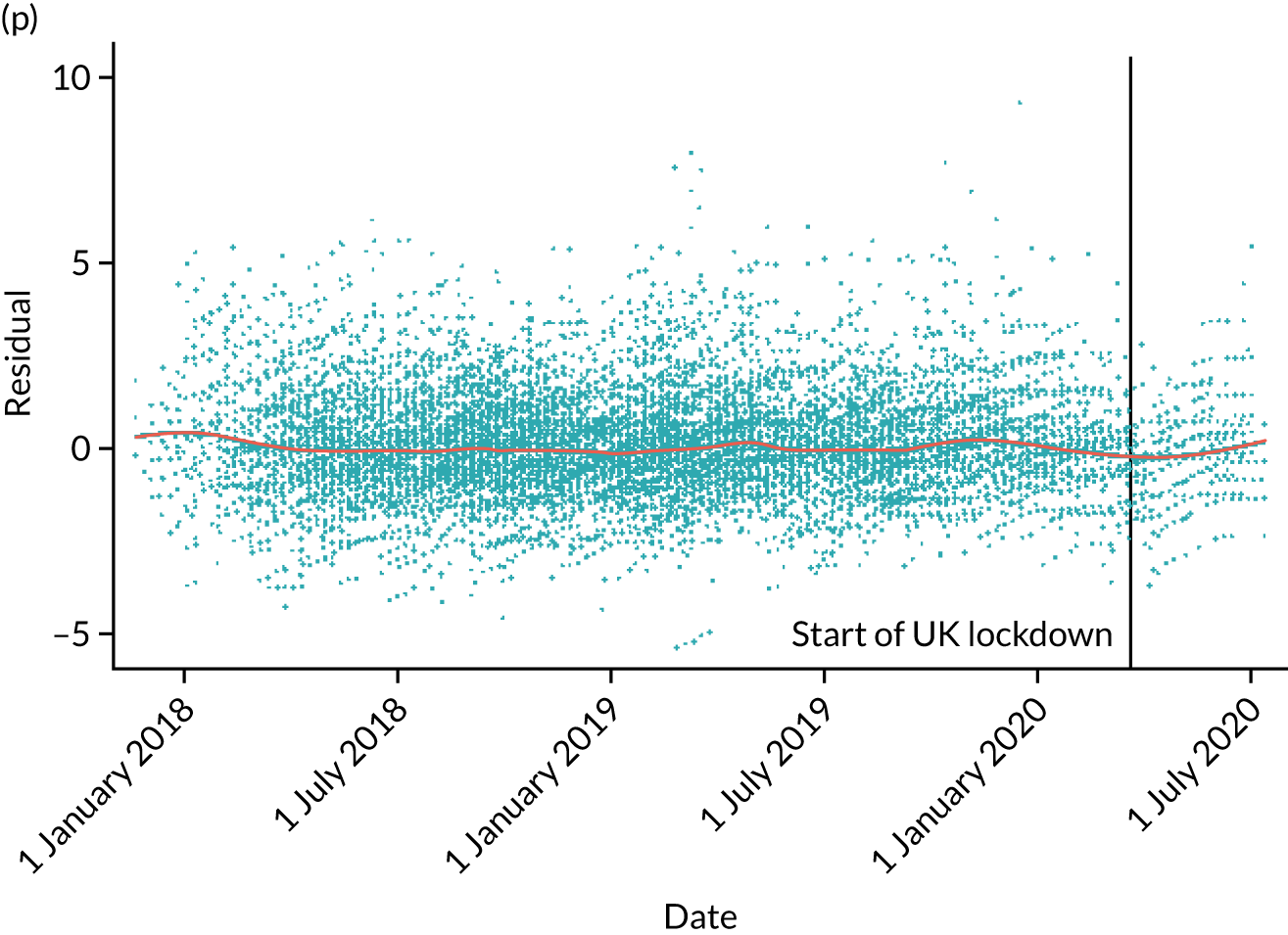
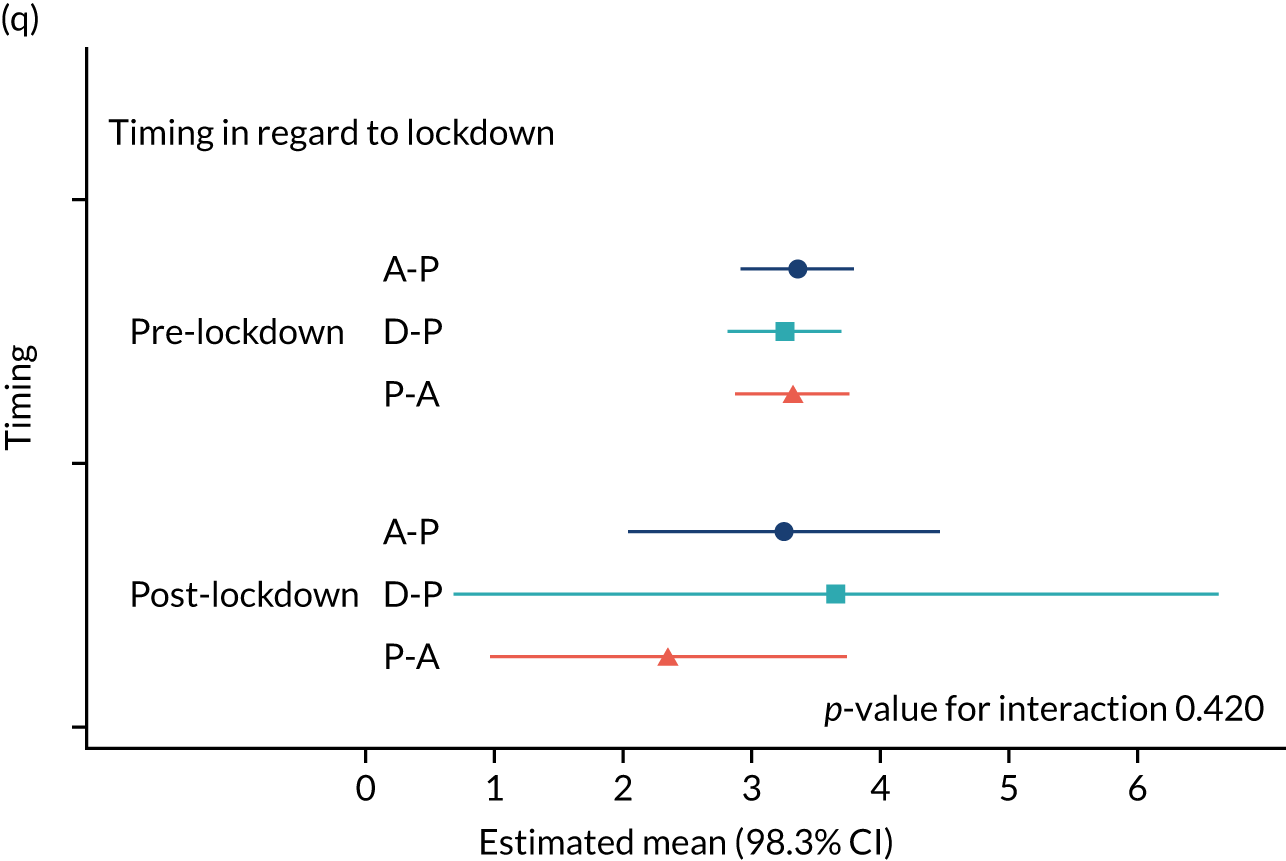
Appendix 5 Concomitant medications
| Medication | Treatment pathway | ||
|---|---|---|---|
| A-P | D-P | P-A | |
| Insulin | 10 | 16 | 13 |
| Canagliflozin | 2 | 0 | 1 |
| Dapagliflozin | 3 | 5 | 0 |
| Dulaglutide (Trulicit) | 1 | 5 | 3 |
| Empagliflozin | 6 | 4 | 4 |
| Exenatide (Bydureon) | 0 | 1 | 0 |
| Gliclazide | 12 | 10 | 13 |
| Linagliptin | 2 | 2 | 3 |
| Liraglutide | 5 | 3 | 4 |
| Metformin | 27 | 29 | 25 |
| Pioglitazone | 1 | 3 | 1 |
| Saxagliptin | 0 | 1 | 0 |
| Sitagliptin | 2 | 0 | 6 |
| Amlodipine | 13 | 11 | 8 |
| Atenolol | 3 | 4 | 1 |
| Bendroflumethiazide | 2 | 5 | 4 |
| Furosemide | 3 | 2 | 3 |
| Lansoprazole | 7 | 9 | 8 |
| Omeprazole | 8 | 7 | 9 |
| Ramipril | 14 | 14 | 9 |
| Simvastatin | 13 | 11 | 7 |
| Total | 134 | 142 | 122 |
List of abbreviations
- AE
- adverse event
- ALT
- alanine aminotransferase
- A-P
- amitriptyline supplemented with pregabalin
- AST
- aspartate aminotransferase
- BNF
- British National Formulary
- BPI-MSF
- Brief Pain Inventory – Modified Short Form
- CI
- confidence interval
- CRF
- case report form
- CSRI
- Client Service Receipt Inventory
- CTRU
- Clinical Trials Research Unit
- DMEC
- Data Monitoring and Ethics Committee
- DN4
- Douleur Neuropathique 4
- D-P
- duloxetine supplemented with pregabalin
- DPNP
- diabetic peripheral neuropathic pain
- eGFR
- estimated glomerular filtration rate
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- GP
- general practitioner
- HADS
- Hospital Anxiety and Depression Scale
- HbA1c
- glycated haemoglobin
- HRQoL
- health-related quality of life
- ICER
- incremental cost-effectiveness ratio
- IMP
- investigational medicinal product
- ISI
- Insomnia Severity Index
- mTCNS
- modified Toronto Clinical Neuropathy Score
- NICE
- National Institute for Health and Care Excellence
- NPSI
- Neuropathic Pain Symptom Inventory
- NRS
- Numeric Rating Scale
- OPTION-DM
- Optimal Pathway for TreatIng neurOpathic paiN in Diabetes Mellitus
- P-A
- pregabalin supplemented with amitriptyline
- PGIC
- Patient Global Impression of Change
- PI
- principal investigator
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SCRAM
- Sheffield Clinical Trials Research Unit online randomisation system
- SD
- standard deviation
- SF-36
- Short Form questionnaire-36 items
- SMS
- short message service
- SSRI
- selective serotonin reuptake inhibitor
- SUSAR
- suspected unexpected serious adverse reaction
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee