
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 09/102/01. The contractual start date was in August 2009. The draft report began editorial review in August 2011 and was accepted for publication in October 2011. As the funder, by devising a commissioning brief, the HTA programme specified the research question and study design. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Perel et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2012 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Traumatic brain injury (TBI), which can be defined as an alteration in brain function or other evidence of brain pathology caused by an external force, is a leading cause of death and disability worldwide. 1 It is estimated that each year more than 1.5 million people die and about 10 million people are hospitalised following a TBI. 2 TBI is commonly accompanied by intracranial bleeding. Its frequency varies according to TBI severity. In the CRASH-1 (Corticosteroid Randomisation after Significant Head Injury) trial, which included 10,008 patients with mild, moderate and severe TBI, 67% of participants had computerised tomography (CT) scan evidence of intracranial bleeding. 3
Regardless of location, patients with large haematomas have a substantially higher mortality rate than patients with small haematomas. We conducted an analysis of 13,962 patients with TBI from the Trauma Audit & Research Network (TARN) and found that large intracranial bleeds were associated with an increased risk of mortality compared with small ones. The odds ratios (ORs) for mortality for large subdural, parenchymal and epidural bleeds, in comparison with small bleeds, were 3.41 [95% confidence interval (CI) 2.68 to 4.33], 3.47 (95% CI 2.26 to 5.33) and 2.86 (95% CI 1.86 to 4.38), respectively. 4
In about half of TBI patients with intracranial bleeding, the haematomas enlarge after hospital admission. Studies involving repeated CT scanning have found that intracranial bleeds can develop or expand in the 24 hours after injury. Oertel et al. 5 studied a group of patients in whom two CT scans were obtained within 24 hours of injury to determine the prevalence of progressive intracranial haemorrhage. Among patients who had their first CT scan within 2 hours of injury, 49% had radiological evidence of progressive haemorrhage. Yadav et al. 6 conducted repeat CT scanning of TBI patients at hospital admission and 24 hours later and found that 16% of 262 parenchymal haematomas and contusions increased in size in the first 24 hours. Similarly, Sullivan et al. 7 found that traumatic epidural haemorrhages enlarged in 23% of 160 TBI patients treated non-operatively. The mean enlargement was 7 mm, and the mean time to enlargement was 8 hours from injury and 5.3 hours from CT diagnosis. More recently, Narayan et al. 8 reported a study in which they included patients with TBI and parenchymal intracranial bleeding confirmed by CT scan of ≥ 2 ml. They repeated the CT scan at 24 and 72 hours and found that in 51% of the included patients the lesions expanded.
In summary, traumatic intracranial bleeding appears to be a common complication after TBI, it is associated with a worse prognosis, larger bleeds have worse outcomes and the bleeding appears to continue after hospital admission. These observations raise the possibility that an intervention administered in the first hours after the injury may prevent the enlargement of intracranial bleeding and therefore might improve patients’ outcomes.
In the haemostatic process, coagulation occurs rapidly at the site of a damaged vessel, building a tight net of fibrin, while at the same time the fibrinolytic system removes the fibrin deposits that could cause permanent vascular occlusion once vascular repair has taken place. 9 The coagulation and fibrinolytic system are believed to be in a state of dynamic balance that maintains an intact vascular system. Tranexamic acid (TXA) is a potent antifibrinolytic agent that exerts its effect by blocking lysine binding sites on plasminogen molecules and has the potential to enhance the effectiveness of the patient’s own haemostatic mechanisms. Consequently, clot breakdown (fibrinolysis) is inhibited and excessive or recurrent bleeding is reduced. TXA is commonly used in surgery to reduce blood loss. A systematic review of randomised controlled trials of TXA in elective surgery showed that it reduces the need for transfusion by one-third, reduces donor exposure by one unit and halves the need for further surgery to control bleeding. 10
It is biologically plausible that TXA reduces haemorrhage growth in TBI patients. About one-third of TBI patients have coagulopathy at hospital admission. 11 TBI patients with coagulopathy are more likely to have progressive haematomas and are more likely to die. 11 Increased fibrinolysis, as indicated by low levels of fibrinogen and high levels of fibrinogen degradation products and d-dimers, is a common feature of the coagulopathy seen in TBI patients, raising the possibility that an antifibrinolytic agent might reduce intracranial bleeding. 12 To date, there have been no randomised controlled trials of TXA in TBI. 13
The possibility that TXA might reduce intracranial bleeding has, however, been evaluated in spontaneous subarachnoid haemorrhage (sSH). A systematic review of randomised controlled trials of TXA administration in patients with sSH found that TXA reduced the rate of rebleeding by approximately 40%. 14 However, because of an increase in cerebral ischaemia there was no overall clinical benefit. This finding has resulted in scepticism about its potential for benefit in TBI. 15 However, the effect of TXA from the sSH trials might not be directly generalisable to TBI patients. The characteristics and risk of ischaemia of patients with sSH are different from those of patients with TBI. Moreover, the duration of TXA treatment in the sSH trials was up to 6 weeks and this may account for the increase in cerebral ischaemia reported. It is possible that a shorter treatment might have prevented rebleeding while avoiding the risk of ischaemia.
The potential benefit of a simple and affordable intervention such as TXA for TBI patients could have important public health implications. The CRASH-2 (Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage) trial16 represented a unique opportunity to evaluate the effect of TXA in TBI patients, in order to reassess the existing belief of harm, and to generate a plausible prior of benefit, which could be informative to design future studies. The CRASH-2 trial recruited 20,211 trauma patients with significant (extracranial) haemorrhage. 16 Although TBI was not an inclusion criterion, it was expected that a significant proportion of patients with multiple trauma would also have TBI. However, to keep data collection to a minimum, and to ensure recruitment to detect the main outcome (overall mortality), CT scan data were not routinely collected. Nevertheless, the CRASH-2 trial represented a unique opportunity to nest an exploratory study collecting CT scan data to evaluate the effect of TXA on imaging outcomes in TBI patients. Collection of evidence to inform future studies of TXA in this population, and specifically addressing the existing safety concerns about the increase of cerebral ischaemia in patients with TBI who receive TXA, also formed part of this study. 13
The Intracranial Bleeding Study (CRASH-2 IBS) was a prospective randomised controlled trial nested within the CRASH-2 trial to quantify the effect of an early short course of TXA on intracranial haemorrhage and new focal cerebral ischaemic lesions in patients with TBI.
Chapter 2 Methods
Trial design
The CRASH-2 IBS is a double-blind randomised placebo-controlled trial nested in a cohort of CRASH-2 trial participants of the effects of TXA on intracranial bleeding and focal ischaemic brain lesions in adult trauma patients with significant haemorrhage and TBI (the trial protocol can be found in Appendix 1).
Participants
The CRASH-2 IBS was conducted in a subset of CRASH-2 trial participants. Eligible patients fulfil CRASH-2 trial inclusion criteria ‘Adult trauma patients with significant haemorrhage (systolic blood pressure < 90 mmHg or heart rate > 110 beats per min, or both), or who were considered to be at risk of significant haemorrhage, and who were within 8 h of injury, were eligible for the trial. Entry was governed by the uncertainty principle’17 and have a TBI [Glasgow Coma Scale (GCS) score of ≤ 14 and a CT scan compatible with TBI]. Pregnant women and patients for whom a second CT scan was not possible were excluded. Consent procedures at participating hospitals were established by local regulation and the appropriate ethics committees. Informed consent was obtained from patients if physical and mental capacity allowed. If patients could not give consent, proxy consent was obtained from a relative or representative. If a proxy was unavailable then, if permitted by local regulation, consent was deferred or waived. When consent was deferred or given by a proxy, the patient was informed about the trial as soon as possible and consent obtained for use of the data collected if required.
Study settings
Ten hospitals were selected based on level of interest by the principal investigator in the research question, recruitment rate in the CRASH-2 trial and ability of the hospital to collect and send the necessary CT scan data to the trial co-ordinating centre. The selected hospitals were (India) Aditya Neuroscience Centre, Sanjivani Hospital, CARE Hospital, Christian Medical College, Medical Trust Hospital, Jeevan Jyoti Hospital and (Colombia) Hospital Universitario San Vicente de Paul, Hospital Pablo Tobón Uribe, Hospital Universitario San José de Popayán and Fundación Valle del Lili.
Interventions
Patients were randomly allocated to receive either a loading dose of 1 g of TXA infused over 10 minutes followed by an intravenous infusion of 1 g over 8 hours or matching placebo (sodium chloride 0.9%). Each patient was assigned a uniquely numbered treatment pack that contained four ampoules of either TXA 500 mg or placebo, one 100-ml bag of 0.9% sodium chloride (for use with the loading dose), instructions for administration of trial treatment, a syringe and needle and stickers with the trial details and randomisation number (for attaching to infusion bags, data forms and patient medical records). Information leaflets for patients and their representatives, consent forms and data collection forms were available in each box. The stickers, instructions, leaflets and forms were translated into local languages where needed.
Outcomes
Brain imaging outcomes
We obtained two brain CT studies for each participant, the first before randomisation and the second 24–48 hours later. A neuroradiologist (ZM), who was blind to treatment allocation, evaluated the pre-randomisation and 24- to 48-hour scans. She made the readings of the two scans twice (with the second reading blind to the results of the first reading), by central reading of the Digital Imaging and Communications in Medicine (DICOM) image files in Digital Jacket™ (DesAcc, Inc, Chicago, IL, USA) software. She measured the size of four types of intracranial haematomas (intraparenchymal, haemorrhagic contusion, subdural and epidural), subarachnoid haemorrhage, mass effect findings and the overall amount of tissue damage, using validated rating scales based on previous work. The individual ratings and measurements were combined into a rating form developed for the purposes of this study (see Appendix 2). She followed structured guidance to complete the CT scan form. This guidance provided definitions of the overall appearance of the scan, haemorrhagic findings, non-haemorrhagic findings and mass effect findings as detailed in the following section.
Computerised tomography scan guidance
Computerised tomography parameters
Slice thickness, interval, matrix and field of view should be recorded on CT 1 and 2 to allow comparison in case of differences in parameters between CT 1 and 2. For best comparison, parameters on CT 1 and 2 should be identical.
Angulation need not be identical but should allow direct comparison between CT 1 and 2 to be made (subjective assessment, based on planning scan and axial imaging).
Overall appearance of scan
Subjective assessment based on summation of all lesions present:18
-
mild focal lesion (e.g. small contusion in one area of brain only, but rest of brain normal)
-
medium focal lesion (e.g. several contusions in one or two immediately adjacent areas of brain, or small epidural haematoma or small subdural haematoma but rest of brain normal)
-
mild or moderate diffuse injury (several small contusions or haematomas in several non-adjacent areas of brain, but normal appearance elsewhere)
-
massive focal injury (e.g. large epidural haematoma or subdural haematoma or massive contusions or parenchymal haematoma in one area of the brain)
-
massive diffuse injury (severe generalised swelling of the brain or many contusions or haematomas in multiple areas).
Haemorrhagic findings
Parenchymal haematoma
Definition: brain parenchymal blood collection secondary to local loss of vascular integrity. 19
Volume measurement: ABC/2. A representative slice at the centre of the haematoma will be selected. The maximum linear length (A) in cm will be multiplied by the maximum width perpendicular to A (B) and the maximum depth (C) in cm. The depth (C) will be determined by multiplying the number of slices on which the haematoma is visible by the slice thickness listed on the CT scan. To obtain the volume in cm3 the final product will be divided by 2. 20,21
If there is more than one haematoma, list all of them, specify the location and estimate the volume of each individual bleed and give total by adding volumes. Codes for location are: L = left, R = right, T = temporal, F = frontal, P = parietal, O = occipital, BG = basal ganglia, B = brainstem.
Subdural haematoma
Definition: haemorrhagic collection in subdural space. 19
Volume measurement: adaptation of ABC/2 method. A representative slice near the centre of the haematoma will be selected. The linear distance in cm between each corner of the subdural crescent will be used to determine the length (A). The width (B) will be measured as the maximum thickness in cm of haematoma from the inner table of the skull perpendicular to the length. The depth (C) will be determined by multiplying the number of slices on which the haematoma is visible by the slice thickness listed on the CT scan. To obtain the volume in cm3 the final product will be divided by 2. 21 If there is more than one haematoma, list all of them, specify the location and estimate the volume of each individual bleed and give total by adding volumes. Codes for location are: L = left R = right, T = temporal, F = frontal, P = parietal, O = occipital, BG = basal ganglia, B = brainstem.
Also, specify if there is tentorial subdural haematoma (TSH).
Epidural haematoma
Definition: blood collection within potential space between skull inner table and dura mater. 19
Volume measurement: ABC/2, method as above. 22
If there is more than one haematoma, list all of them, specify the location and estimate the volume of each individual bleed and give total by adding volumes. Codes for location are: L = left, R = right, T = temporal, F = frontal, P = parietal, O = occipital, BG = basal ganglia, B = brainstem.
Subarachnoid haemorrhage
Definition: blood within subarachnoid spaces between pia and arachnoid membranes.
Subarachnoid haemorrhage score:23 rates the thickness of subarachnoid haemorrhage within a representative sulcus: subarachnoid haemorrhage thickness ≤ 5 mm, subarachnoid haemorrhage thickness > 5 mm.
Specify whether subarachnoid haemorrhage is basal (B) or convexity (C).
Intraventricular haemorrhage
Definition: blood within ventricular system.
Intraventricular haemorrhage score:24 the amount of intraventricular haemorrhage will be quantified in the lateral, third and fourth ventricles as follows: 0 indicates no blood; 1 – sedimentation (< 25% filled); 2 – moderately filled; 3 – completely filled, leading to an intraventricular haemorrhage score ranging from 0 to 12.
Haemorrhagic contusions
Definition (contusion): injury to brain surfaces involving superficial grey matter. 19
Refinement of definition: injury to superficial parenchyma secondary to trauma against the skull or fixed dural fold, characterised by focal low attenuation with or without oedema. If haemorrhagic, haemorrhage is patchy and relatively ill-defined. The distinction between contusion and parenchymal haemorrhage is blurred because both involve bleeding within the brain tissue; however, an arbitrary cut-off exists that the injury is a contusion if two-thirds or less of the tissue involved is blood, and a haemorrhage otherwise.
Volume measurement: ABC/2, method as above. Measure volume of entire contusion rather than just volume of haemorrhagic component.
If there is more than one contusion, list all of them, specify the location and estimate the volume of each individual bleed and give total by adding volumes. Codes for location are: L = left, R = right, T = temporal, F = frontal, P = parietal, O = occipital, BG = basal ganglia, B = brainstem.
Petechial haemorrhages
Definition: small punctuate haemorrhages measuring ≤ 15 mm in diameter indicative of diffuse axonal injury.
Non-haemorrhagic findings
Acute focal ischaemic lesion
Definition: focal low attenuation in distribution indicating arterial ischaemic cause rather than traumatic contusional injury.
Non-haemorrhagic contusion
Definition: see Haemorrhagic contusions.
Volume measurement: ABC/2, method as above.
If there is more than one contusion, give total by adding volumes.
Mass effect findings
Scoring system based on Wardlaw and Sellar:25
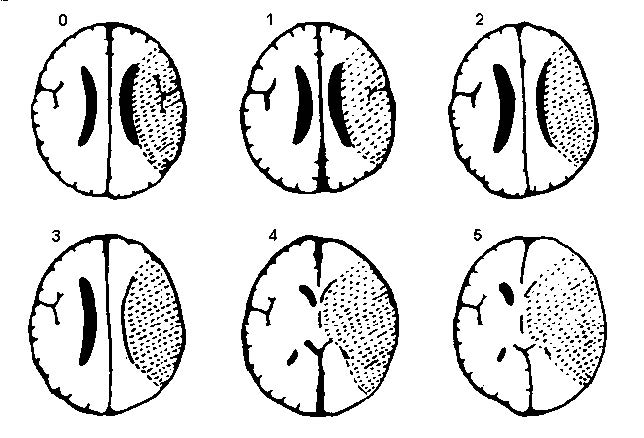
-
0 = no swelling
-
1 = effacement of the sulci overlying the infarct
-
2 = 1 + minor effacement of adjacent lateral ventricle
-
3 = 1 + complete effacement of lateral ventricle
-
4 = 1 + effacement of the lateral and third ventricles
-
5 = 4 + shift of the midline away from the side of the ventricle
-
6 = 5 + effacement of the basal cisterns/uncal herniation.
Measurement of midline shift
FIGURE 1.
Computerised tomography scan image showing midline shift.
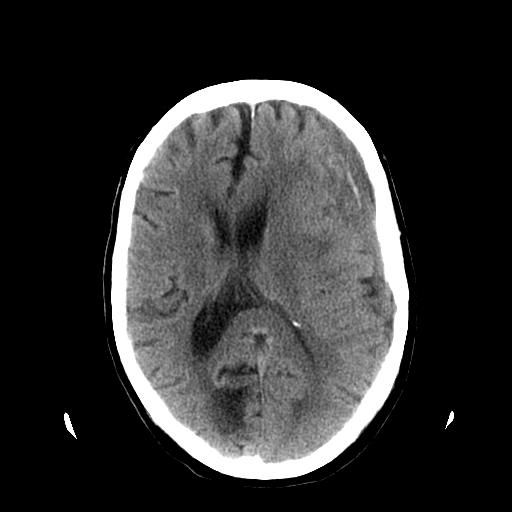
Method of measurement
-
Define the midline: straight line connecting the fixed anterior and posterior margins of the falx cerebri.
-
Degree of midline shift = longest distance to midline structures (interhemispheric fissure, septum pellucidum or third ventricle) measured perpendicular to the midline.
The primary outcome was the adjusted change in total haemorrhage growth from the first to the follow-up scan, defined as the difference in the combined volume (ml) of all intracranial haemorrhagic lesions (intraparenchymal haematoma + haemorrhagic contusion + subdural haematoma + epidural haematoma) from the first to the second scan. The secondary imaging outcomes were (1) the occurrence of significant haemorrhage growth (any haematoma that increased by 25% in relation to its initial volume), (2) new intracranial haemorrhage (apparent on the follow-up brain CT but not on the baseline brain CT), (3) change in subarachnoid haemorrhage grade, (4) the mass effect and (5) the occurrence of new focal cerebral ischaemic lesions (apparent on the follow-up brain CT, but not on the baseline brain CT).
Clinical outcomes
The clinical outcomes were death and the need for neurosurgical intervention. Dependency was measured using the five-point modified Oxford Handicap Scale (mOHS). 26 The scale was dichotomised into ‘dependent’ (fully dependent requiring attention day and night, or dependent but not requiring constant attention) or ‘independent’ (some restriction in lifestyle but independent, minor symptoms or no symptoms). Clinical outcomes were recorded at hospital discharge, 28 days or death, whichever occurred first. The corresponding form is shown in Appendix 3.
Combined outcome
We defined ‘poor outcome’ as a patient who developed one or more of the following during the scheduled follow-up period: significant haemorrhage growth, new intracranial haemorrhage, new focal cerebral ischaemic lesions, the need for neurosurgery or death.
Sample size
Assuming an initial intracranial haematoma volume of 20 ml, an average haemorrhage growth of 7 ml in the control group and a correlation of 0.6 between initial and follow-up volumes, we estimated that a trial with 300 patients would have 80% power (alpha = 0.05) to detect a 35% reduction in haemorrhage growth. A trial with 200 patients would have the same power to detect a 40% reduction. We prespecified in the protocol that, as this study was nested within the main CRASH-2 trial, even if the planned sample size of 300 patients was not achieved, recruitment would stop at the same time as that for the main CRASH-2 trial.
Randomisation
Patients were randomised using a local pack system. Consecutively numbered treatment packs were taken from a box of eight packs (with a random allocation sequence based on a block size of eight). After eligibility had been confirmed and the locally approved consent procedures had been completed, patients were randomised by selecting the lowest numbered treatment pack from a box containing eight numbered packs.
Blinding
Blinding was achieved through the use of matching placebo. TXA and placebo ampoules were indistinguishable. The placebo was manufactured by St Mary’s Pharmaceutical Unit, Cardiff, UK. TXA was manufactured by Pharmacia (Pfizer, UK). The treatment packs were prepared by an independent clinical trial supply company (Bilcare GCS Ltd, Crickhowell, UK). Correct blinding and coding of the ampoules was assured by independent random testing of each batch by high-performance liquid chromatography to confirm the contents. Emergency unblinding of treatment allocation was available by telephoning the Clinical Trials Service Unit at the University of Oxford.
Statistical methods
We assessed the intraobserver reliability of haemorrhage growth measurements on brain imaging using the intraclass correlation coefficient. We assessed the reliability of the measurement of binary outcomes using a kappa statistic. In all subsequent analyses, we used the average measurement of the two readings of continuous variables and considered binary outcomes to be positive when reported as positive on both of the radiologist’s readings of a particular brain image for a patient.
We used a Bayesian statistical approach as it has several advantages in comparison with the traditional frequentist approach for the purposes of our study. A Bayesian approach can be defined as a mathematical method for combining the prior belief that we have about an effect size with the information observed in the actual study we conduct, to produce a ‘posterior’ belief of the effect size. This ‘posterior belief’ is presented in the form of a 95% credibility interval (CrI), that is, there is a 95% probability that the true effect lies within this interval. 27 Furthermore, a Bayesian approach allows us to calculate probabilities for specific effect sizes. For example, the probability of an OR < 1 can be estimated. We used a Bayesian statistical approach because it fitted with the aims of this exploratory study. We were interested in obtaining a reliable ‘degree of belief’ about the effect of TXA in patients with TBI that could justify (or not) a future study in this population. At the time that the study was designed there was no evidence about the effect of TXA on TBI patients. The Bayesian approach allowed us to generate a probability distribution of the effect of TXA that could be used as a prior for future research. Unlike traditional frequentist analysis, in which you can only reject (or not) a null hypothesis, the Bayesian approach allows you to report how probable a specific effect will be (e.g. an OR < 0.8).
Because this was the first randomised controlled trial of TXA in TBI patients, we used non-informative priors for the primary analysis to reflect the lack of previous knowledge in this area. For the difference of continuous variables between groups the prior was a normal distribution mean = 0 and standard deviation (SD) = 100. For the relative risk (RR) the prior was defined in the log scale with a normal distribution ln(RR) ∼ N(0, SD = 1.74). This is equivalent to having a 95% prior belief that the RR will be between 1/30 and 30, centred on 1. We conducted a sensitivity analysis using an enthusiastic prior for significant haemorrhage growth and a sceptical prior for new focal cerebral ischaemic lesions. These priors were provided by the systematic review of TXA in subarachnoid haemorrhage that reported an OR of 0.49 (95% CI 0.37 to 0.65) for rebleeding and an OR of 1.28 (95% CI 0.93 to 1.75) for confirmed cerebral ischaemia. 14
For all the estimated effects we reported a posterior 95% CrI. We also reported the probability of a beneficial effect of TXA for all the outcomes, except for new focal cerebral ischaemic lesions, for which we reported the probability of a harmful effect. We defined the effect as beneficial if the effect size was < 0 (for differences) or < 1 (for ratios). For the sensitivity analysis we also reported the posterior probability of a ‘clinical significant beneficial effect’ (OR < 0.8) for significant haemorrhage growth and the posterior probability of a ‘clinical significant harmful effect’ (OR > 1.25) for new focal cerebral ischaemic lesions.
All analyses were undertaken on an intention-to-treat basis. We used generalised linear mixed models adjusted for baseline variables. The covariates included in the adjustment were GCS score and age because these variables are predictors of poor outcome. 28 In addition, for CT outcomes we adjusted for time from injury to first and second CT scan and for initial bleeding volume. In our analysis of mass effect we adjusted for initial mass effect. Adjusted effects are considered in the primary analysis, but both adjusted and unadjusted effect measures are reported.
We evaluated haemorrhage growth for four types of intracranial haematomas (intraparenchymal, haemorrhagic contusion, subdural and epidural) combined and separately. Haemorrhage growth was analysed using multiple linear regression (analysis of covariance, ANCOVA), the main factor being the treatment group. Outcomes are reported combined and separately for patients who did or did not undergo neurosurgery evacuation between the first and second CT scan. The binary outcomes, significant haemorrhage growth, new intracranial haemorrhage, new focal cerebral ischaemic lesion, mass effect, need for neurosurgery (other than the one indicated based on the pre-randomisation CT scan) and mortality, were analysed using logistic regression. Subarachnoid haemorrhage was measured on an ordinal scale ranging from 0 to 4 points. We compared the distribution of this outcome in the two groups using a non-parametric rank test (Kruskal–Wallis).
Neurosurgery was defined as initial (if decision of evacuation was based on the pre-randomisation CT scan), intermediate (if the evacuation was conducted after the pre-randomisation CT scan but before the follow-up CT scan) and late (if the evacuation was conducted after the follow-up CT scan).
To evaluate the clinical relevance of the primary surrogate outcome selected in this study we also analysed the clinical effect of haemorrhage growth. We conducted a logistic regression analysis with dependency (as defined by mOHS) as the outcome and haemorrhage growth as the main exposure variable, adjusting by the potential confounders initial volume, GCS score, age, time from injury to CT scan and treatment.
Funding
The funder of the study had no role in study design, data collection, data analysis, data interpretation or writing of the report. The Writing Committee had full access to all data in the study and had final responsibility for the decision to submit for publication.
This study is registered as ISRCTN86750102.
Chapter 3 Results
Summary of the trial
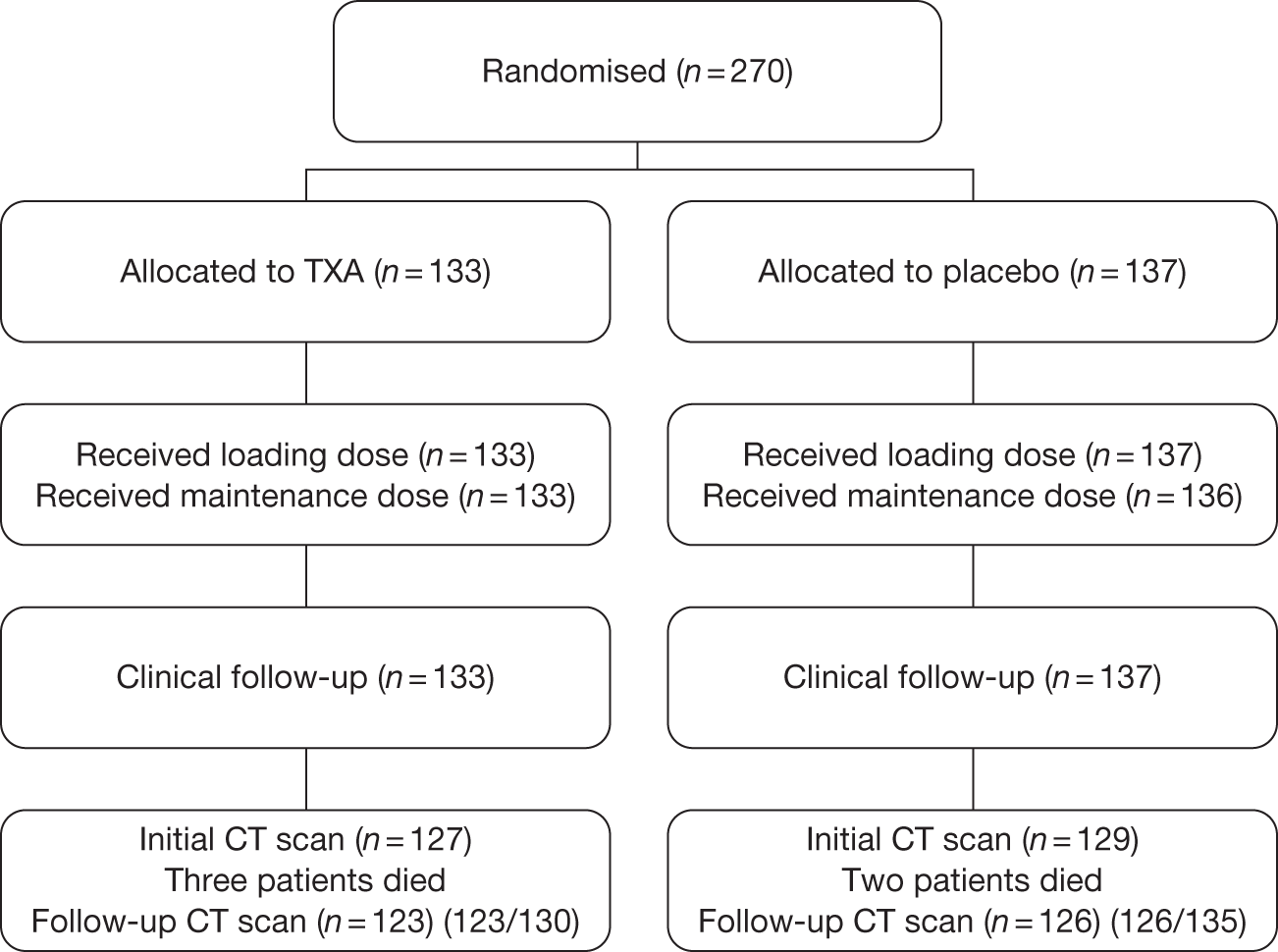
We recruited 270 patients between August 2008 and January 2010, when the main CRASH-2 trial reached its prespecified sample size. All patients (Figure 2) received the loading and maintenance doses except for one placebo-allocated patient who did not receive the maintenance dose. All patients were followed up for clinical outcomes. A total of 256 patients (95%) had an initial CT scan. In 14 patients (six TXA allocated, eight placebo allocated) the initial CT scan was unavailable for reading for technical reasons. Five patients died before the second CT scan (three TXA allocated, two placebo allocated). In terms of protocol deviations, nine (3%) patients were randomised before the initial CT scan (six TXA allocated, three placebo allocated). A total of 31 (11%) patients had a GCS score of 15 at baseline (17 TXA allocated, 14 placebo allocated). In 51 (19%) patients the second CT scan was conducted outside the 24- to 48-hour window (25 TXA allocated, 26 placebo allocated). We included all 270 patients in our analyses.
FIGURE 2.
Summary of the trial.
Baseline characteristics
Baseline characteristics of the included patients and their initial CT scan findings are shown in Tables 1 and 2, respectively. In total, 211 patients (82%) had an intraparenchymal haematoma, haemorrhagic contusion, subdural haematoma or epidural haematoma. A focal cerebral ischaemic lesion was present in three patients (2%) in the placebo group and two (2%) in the TXA group at the initial CT scan. A total of 40 patients (20 TXA allocated, 20 placebo allocated) had an initial neurosurgery evacuation.
| TXA (n = 133) | Placebo (n = 137) | |
|---|---|---|
| Sex | ||
| Male | 111 (83.5%) | 117 (85.4%) |
| Female | 22 (16.5%) | 20 (14.6%) |
| Injury type | ||
| Blunt | 132 (99.2%) | 136 (99.3%) |
| Penetrating | 1 (0.8%) | 1 (0.7%) |
| Age (years), mean (SD) | 36.2 (14.0) | 37.0 (13.7) |
| Hours since injury, mean (SD) | 4.4 (1.8) | 4.2 (1.7) |
| Glasgow Coma Scale score, mean (SD) | 10.5 (3.6) | 10.5 (3.6) |
| Systolic blood pressure (mmHg), mean (SD) | 116.4 (31.2) | 113.5 (29.4) |
| Central capillary refill time (seconds), mean (SD) | 3.4 (1.0) | 3.5 (1.1) |
| Heart rate (beats per minute), mean (SD) | 100.7 (25.7) | 101.6 (23.5) |
| TXA (n = 127), n (%) | Placebo (n = 129), n (%) | Total no. of patients (%) | |
|---|---|---|---|
| CT scan baseline characteristics | |||
| Normal | 4 (3) | 3 (2) | 7 (3) |
| Mild focal injury | 26 (20) | 22 (17) | 48 (19) |
| Medium focal injury | 39 (31) | 41 (32) | 80 (31) |
| Mild/moderate diffuse injury | 23 (18) | 19 (15) | 42 (16) |
| Massive focal (± diffuse) | 17 (13) | 23 (18) | 40 (16) |
| Massive diffuse (± focal) | 18 (14) | 21 (16) | 39 (15) |
| Types of bleeding | |||
| Intracranial haemorrhage (intraparenchymal, haemorrhagic contusion, subdural and epidural) | 106 (83) | 105 (81) | 211 (82) |
| Intraparenchymal haematoma | 9 (7) | 15 (12) | 24 (9) |
| Haemorrhagic contusion | 61 (48) | 66 (51) | 127 (50) |
| Subdural haematoma | 38 (30) | 45 (35) | 83 (32) |
| Epidural haematoma | 38 (30) | 28 (22) | 66 (26) |
| Subarachnoid haemorrhage | 55 (43) | 79 (61) | 134 (52) |
| Mass effect findings | |||
| Ventricular effacement | 45 (35) | 47 (36) | 92 (36) |
| Uncal herniation | 15 (12) | 18 (14) | 33 (13) |
| Cisterns compressed | 12 (9) | 19 (15) | 31 (12) |
| Midline shift | 28 (22) | 29 (22) | 57 (22) |
| Any mass effect | 69 (54) | 78 (60) | 147 (57) |
Imaging outcomes
Intraobserver agreement
The intraobserver agreement between the two CT scan readings by the radiologist was high for all outcomes except new intracranial haemorrhage. The intraclass correlation coefficient for total haemorrhage growth was 0.89 and the corresponding kappas for significant haemorrhage growth, new intracranial haemorrhage, any mass effect and new focal cerebral ischaemic lesions were 0.82, 0.32, 0.81 and 0.89, respectively. Imaging outcomes were available for 249 (99%) of the 251 patients who had an initial CT scan and were alive at 24 hours.
Effect on haemorrhage growth
The mean total haemorrhage growth was 5.9 ml (SD 26.8 ml) and 8.1 ml (SD 29.2 ml) in the TXA and placebo group, respectively. The adjusted analysis showed a reduction in total haemorrhage growth in the arm that received TXA in comparison with placebo (–3.8 ml, 95% CrI –11.5 to 3.9 ml). In patients who had neurosurgical evacuation before the second CT scan, the reduction in total haemorrhage growth in the TXA arm was larger (–12 ml, 95% CrI –45.7 to 15.5 ml). The posterior probability that TXA reduces haemorrhage growth was high for total haematoma and each type separately except for epidural haematoma (Table 3). The change in the subarachnoid haemorrhage scale was –0.11 for TXA-allocated patients and –0.12 for placebo-allocated patients (p = 0.93).
| Total no. of patients | Haemorrhage growth (ml), unadjusted (95% CrI) | Haemorrhage growth (ml), adjusted (95% CrI) | Probability of benefit (%) | |
|---|---|---|---|---|
| All haematomas combined | ||||
| All patients | 206 | –2.12 (–9.82 to 5.56) | –3.79 (–11.45 to 3.88) | 84 |
| Evacuated patients | 46 | –6.160 (–34.603 to 22.284) | –15.120 (–45.744 to 15.504) | 84 |
| Non-evacuated patients | 160 | –1.62 (–7.25 to 4.00) | –2.11 (–7.12 to 2.89) | 80 |
| Intraparenchymal haematoma | ||||
| All patients | 24 | –6.19 (–17.57 to 5.18) | –4.50 (–17.69 – 8.68) | 76 |
| Evacuated patients | 4 | –1.49 (–33.69 to 30.71) | NA | NA |
| Non-evacuated patients | 20 | –7.99 (–20.88 to 4.90) | –9.14 (–24.89 to 6.61) | 89 |
| Haemorrhagic contusions | ||||
| All patients | 124 | –5.69 (–15.15 to 3.77) | –1.57 (–9.08 to 5.92) | 66 |
| Evacuated patients | 31 | –17.52 (–46.87 to 11.82) | –14.34 (–40.78 to 12.09) | 87 |
| Non-evacuated patients | 93 | –0.61 (–8.50 to 7.27) | 2.04 (–3.75 to 7.84) | 24 |
| Subdural haematoma | ||||
| All patients | 78 | 1.50 (–9.85 to 12.86) | –2.80 (–8.68 to 3.06) | 83 |
| Evacuated patients | 29 | 0.52 (–25.60 to 26.66) | –14.37 (–32.89 to 4.14) | 95 |
| Non-evacuated patients | 49 | –0.31 (–3.48 to 2.86) | 0.26 (–1.76 to 2.29) | 39 |
| Epidural haematoma | ||||
| All patients | 66 | 5.68 (–4.88 to 16.24) | 3.86 (–1.85 to 9.58) | 9 |
| Evacuated patients | 21 | 2.70 (–25.02 to 30.43) | –5.24 (–14.21 to 3.73) | 89 |
| Non-evacuated patients | 45 | 2.12 (–1.77 to 6.01) | 2.71 (–1.54 to 6.98) | 10 |
Computerised tomography scan binary outcomes
There were 44 (35.8%) patients with significant haemorrhage growth in the TXA group compared with 56 (44.4%) in the placebo group. In total, 13 (10.6%) patients had new haemorrhage area in the TXA group compared with 20 (15.9%) in the placebo group, and there were 58 (47.2%) patients with signs of mass effect in the TXA group compared with 76 (60.3%) in the placebo group. In Table 4 the unadjusted and adjusted ORs are reported for all of the CT scan binary outcomes. A beneficial effect was highly probable for all of the binary CT scan outcomes considered (range 89–93%).
| CT scan finding | Unadjusted OR (95% CrI) (n = 249) | Adjusted OR (95% CrI) (n = 249) | Probability of benefit (%) |
|---|---|---|---|
| New haemorrhage | 0.64 (0.31 to 1.33) | 0.63 (0.29 to 1.35) | 88 |
| Mass effect | 0.59 (0.36 to 0.98) | 0.55 (0.24 to 1.22) | 93 |
| Significant haemorrhage growth | 0.70 (0.42 to 1.16) | 0.68 (0.41 to 1.13) | 93 |
Sensitivity analysis for significant haemorrhage growth
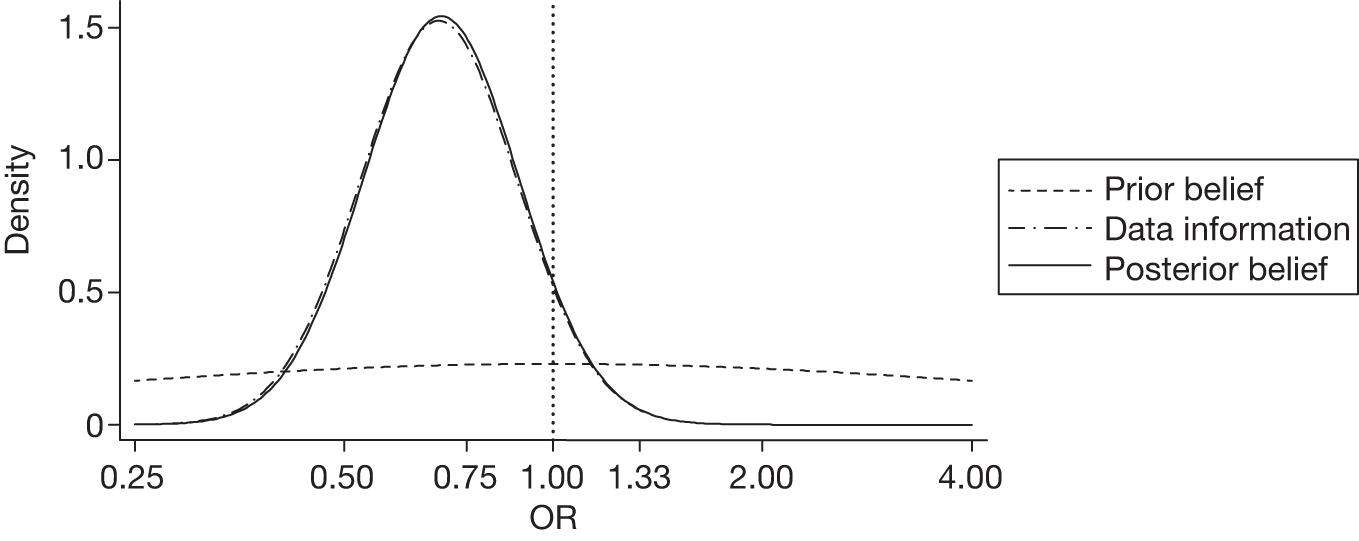
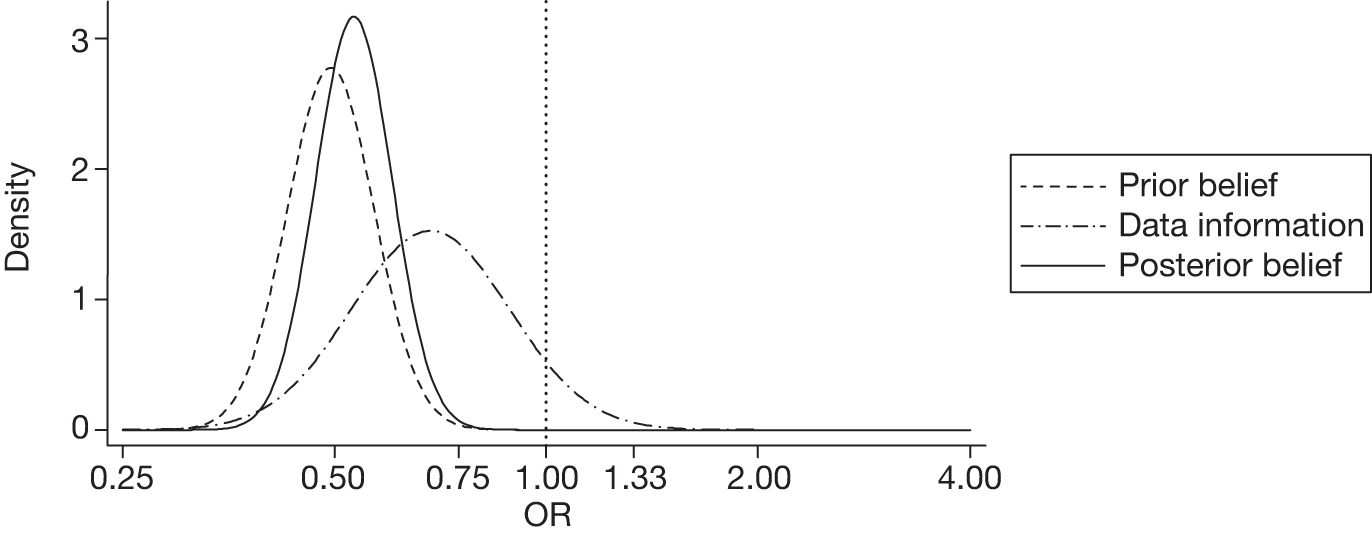
The sensitivity analysis for significant haemorrhage growth with an enthusiastic prior gave an adjusted OR of 0.52 (95% CrI 0.41 to 0.68) with a very high posterior probability (99%) of a clinically significant beneficial effect. Figure 3 displays the probability distributions of the effect of TXA on significant haemorrhage growth, assuming a non-informative prior and an enthusiastic prior.
FIGURE 3.
Probability distributions of the effect of TXA on significant haemorrhage growth using (a) a non-informative prior and (b) an enthusiastic prior.
New focal cerebral ischaemic lesions
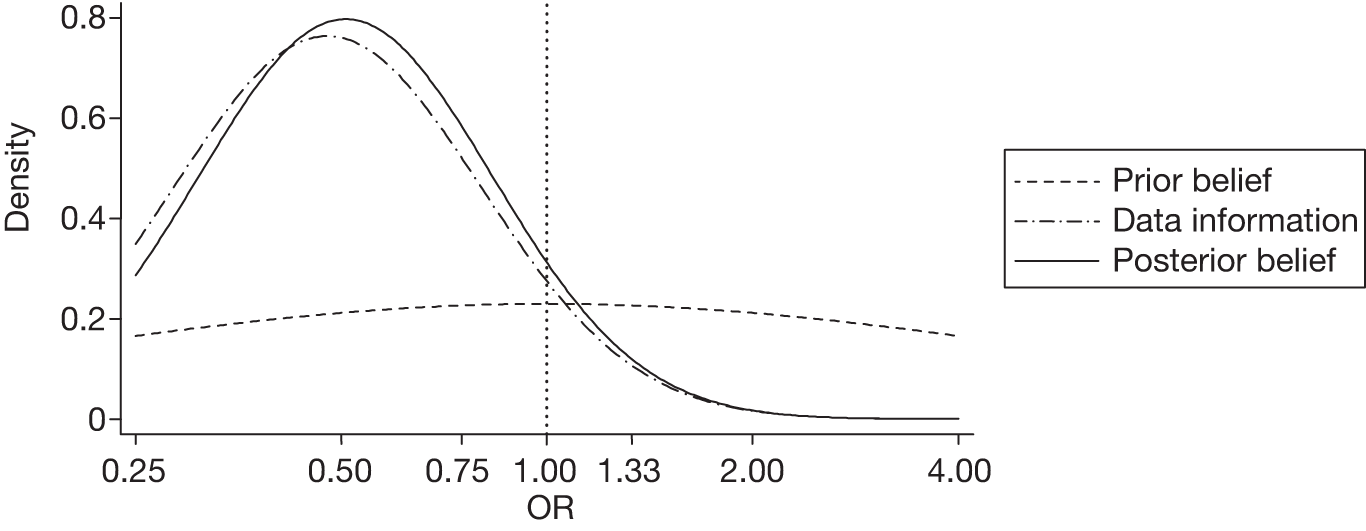
There were six (4.9%) patients with new focal cerebral ischaemic lesions in the TXA group compared with 12 (9.5%) in the placebo group. The unadjusted and adjusted ORs were 0.51 (95% CrI 0.19 to 1.37) and 0.54 (95% CrI 0.20 to 1.46), respectively. The probability of a harmful effect of TXA was 10%. The sensitivity analysis with a sceptical prior gave an adjusted OR of 1.18 (95% CrI 0.87 to 1.60) and a posterior probability of a clinically significant harmful effect of 35%. Figure 4 displays the probability distributions of the effect of TXA on new focal cerebral ischaemic lesions, assuming a non-informative prior and a sceptical prior.
FIGURE 4.
Probability distributions of the effect of TXA on new focal cerebral ischaemic lesions using (a) a non-informative prior and (b) a sceptical prior.
Clinical outcomes
Clinical outcomes were available for all patients (Table 5). A total of 14 (10.5%) patients died in the TXA group compared with 24 (17.5%) in the placebo group. There were 12 (9.0%) deaths in the TXA group due to head injury compared with 20 (14.6%) in the placebo group. Intermediate or late neurosurgical evacuation was required in 20 (15.0%) patients in the TXA group compared with 21 (15.3%) in the placebo group. Except for the need for neurosurgical intervention, there was a high probability of a beneficial effect of TXA on all of the clinical outcomes (Table 5). Finally, a poor outcome was observed in 60 (45.1%) patients in the TXA group and 80 (58.4%) in the placebo group (adjusted OR 0.59, 95% CrI 0.37 to 0.96).
| Unadjusted OR (95% CrI) (n = 270) | Adjusted OR (95% CrI) (n = 270) | Probability of benefit (%) | |
|---|---|---|---|
| Mortality | 0.57 (0.28 to 1.14) | 0.49 (0.22 to 1.06) | 96 |
| Mortality due to head injury | 0.59 (0.28 to 1.25) | 0.54 (0.24 to 1.22) | 93 |
| Need for neurosurgery | 0.98 (0.50 to 1.93) | 0.98 (0.50 to 1.91) | 53 |
Relationship between haemorrhage growth and dependency
We found that an increase of 10 ml in haematoma growth was strongly associated with dependency at hospital discharge (adjusted OR 1.32, 95% CI 1.08 to 1.62).
Adverse events
No emergency unblinding was needed and there were no adverse events regarded as serious, unexpected or suspected to be related to the study treatment.
Chapter 4 Discussion
This was the first randomised controlled study to evaluate the effect of TXA in TBI patients and it found that neither moderate benefits nor moderate harmful effects can be excluded. However, although uncertainty remains, our analyses suggest that TXA administration might improve outcome in TBI patients and provide grounds for evaluating this hypothesis in future research.
If we assume no prior knowledge of the effect of TXA on significant haemorrhage growth with a non-informative prior, the posterior probability of benefit provides reasonable grounds for therapeutic optimism. Using an enthusiastic prior, based on the effect of TXA on intracranial bleeding observed in trials in subarachnoid haemorrhage, it appears highly probable that TXA reduces total haemorrhage growth in TBI.
The reduction in haemorrhage growth observed in this study could be because of several different reasons. There is good evidence that a large proportion of TBI patients become coagulopathic early on after injury. 11 The exact mechanism of this coagulopathy is still disputed. Among the existing hypotheses it has been suggested that the release of thromboplastin after TBI is followed by the activation of the coagulation and fibrinolytic pathways. Pathak et al. 29 have recently reported an increase in activity of tissue thromboplastin among TBI patients. The fact that a larger effect was found in intraparenchymal haematoma, for which release of thromboplastin is more likely, is consistent with this hypothesis. Alternatively, it has been suggested that only TBI patients with hypotension activate the fibrinolytic response. 30 CRASH-2 IBS was conducted among TBI patients with significant (extracranial) haemorrhage and the effect of TXA might be different in patients with isolated TBI. It is possible that in our study population TXA reduced extracranial bleeding, and therefore patients in the TXA group were less hypotensive and less coagulopathic, and through this mechanism TXA reduced haemorrhage growth. However, only 7% of the included patients had systolic blood pressure < 90 mmHg and we did not find evidence of interaction according to blood pressure at admission (data not shown) that would support this hypothesis; however, we acknowledge that our study was not powered to detect this interaction. Additionally, a recent paper has challenged this hypothesis by reporting that coagulopathy is also found in TBI patients without hypotension. 31 Antifibrinolytic agents have been shown to reduce blood loss in surgical patients. 10 Therefore, the larger effect observed in surgical patients is consistent with the evidence of effectiveness of antifibrinolytic agents in elective surgery.
There are several potential explanations for the observed reduction in mortality. It could be because of the reduction in haemorrhage growth, mass effect and focal ischaemic lesions, or alternatively the observed effect on mortality could be related to the survival benefit observed in the main CRASH-2 study for trauma patients and significant (extracranial) bleeding. However, in the main CRASH-2 study the largest and most significant effect was observed in deaths because of bleeding, while in the CRASH-2 IBS 85% of the observed deaths were because of brain injury.
With regard to new focal cerebral ischaemic lesions, we found a reduction in TXA-allocated patients. The posterior probabilities were compatible with either a beneficial or a harmful effect, although a beneficial effect was more likely assuming a non-informative prior. Overall, the incidence of these lesions was low and it is possible that the observed difference between the groups may have arisen by chance alone. On the other hand, it is plausible that, if TXA reduces haemorrhage growth and volume, this will reduce local pressure on arteries and also total intracranial pressure and any consequent ischaemia. Given that TXA has been shown to reduce mortality because of bleeding, it is also possible that TXA-allocated patients may have had a more stable circulation with higher blood pressure and that this may have accounted for the observed reduction in ischaemic lesions. Using a sceptical prior, based on the effect of TXA on ischaemic lesions in subarachnoid haemorrhage trials, a harmful effect of TXA appeared more probable, although the probability of a clinically significant harmful effect remains low.
Our study has several strengths. The randomisation methods ensured that participating clinicians did not have foreknowledge of treatment allocation, baseline clinical factors were well balanced, there was a high follow-up rate and all analyses were performed on an intention-to-treat basis. Although there were baseline differences between TXA- and placebo-allocated patients for some CT scan characteristics, we conducted adjusted analyses that should have accounted for any imbalance. We attempted to minimise measurement error through the use of central CT reading by a neuroradiologist following a strict protocol made up mostly of already validated methods (those that had not been validated were further tested in the present study). We found that intra-rater reliability was high for most imaging measurements.
The Bayesian analysis presented many advantages. We were able to report the 95% CrI, which is the intuitive interpretation that most readers (incorrectly) make of the traditional 95% CI. 25 It also allowed us to explicitly incorporate sceptical prior beliefs about the potential harms of TXA (from prior experience with a different disease, aneurysmal subarachnoid haemorrhage); thus, taking into account some clinicians’ concerns about the potential for harm. Finally, the posterior probability about the effect size can be used very conveniently as a ‘prior’ belief for future trials.
Among the limitations is the fact that our study included only a relatively small sample of the CRASH-2 participants with TBI, and a larger sample size could have provided more precise results. The nature of a large pragmatic trial such as the CRASH-2 trial constrained our ability to collect CT scans in all TBI patients included in the CRASH-2 trial. Nevertheless, the relatively small sample size does not affect the internal validity of this study and, although imprecise, the results should be unbiased. Another important limitation is that for our sensitivity analysis we used information from a systematic review of the effect of TXA in a different clinical condition (aneurysmal subarachnoid haemorrhage). The doses of TXA used in these trials were larger and more prolonged than in our study and so the enthusiastic and sceptical priors could overestimate the reduction in significant haemorrhage growth and the risk of cerebral ischaemic lesions, respectively. A more recent trial of a short course of TXA in aneurysmal subarachnoid haemorrhage has shown that TXA reduces rebleeding without apparently increasing the risk of ischaemia. 32 These data provide support for our findings that a short course of TXA could reduce intracranial haemorrhage after TBI without increasing the risk of cerebral ischaemia. A further limitation is that 19% of the patients had their second CT scan conducted outside the 24- to 48-hour window; however, the distribution was similar in both groups and because we adjusted for time from randomisation to first and second CT scan the results should be unbiased.
Chapter 5 Conclusion
Implications for health care
The CRASH-2 trial has shown reliably that early administration of TXA in trauma patients with, or at risk of, significant bleeding reduces the risk of all-cause mortality. However, many patients with traumatic haemorrhage also have TBI and concerns about the risk of cerebral ischaemia may influence the decision of some doctors to give TXA to these patients. The results presented here provide reassurance about the safety of TXA in bleeding trauma patients with TBI.
Implications for research
Our results also have implications for research. If TXA reduces intracranial bleeding after TBI without increasing the risk of ischaemic lesions, then TXA clearly has the potential to improve patient outcomes. The results of this trial provide a reasonable basis to expect such effects. Although an increased risk of cerebral ischaemia cannot be ruled out, our results provide a sound basis to believe that the benefits of TXA administration could outweigh the risks. If such an inexpensive and widely practicable treatment were found to improve patient outcomes after traumatic intracranial bleeding this would have major implications for clinical care. Future research should be conducted to reliably assess the effectiveness and safety of the early administration of a short course of TXA in patients with isolated TBI.
Acknowledgements
Writing Committee
Pablo Perel (UK) (chairperson), Rustam Al-Shahi Salman (UK), Taemi Kawahara (UK), Zoe Morris (UK), David Prieto-Merino (UK), Ian Roberts (UK), Peter Sandercock (UK), Haleema Shakur (UK) and Joanna Wardlaw (UK).
Collaborators by site
India: Aditya Neuroscience Centre and Sanjivani Hospital: Anil P Lal and Natasha Gohain; CARE Hospital: Pamidimukkala Venkataramana Ramana; Christian Medical College: Yashbir Dewan, Sarvpreet Singh Grewal and Pradipta Tripathy; Medical Trust Hospital: Ramalingam Ramanathan Ravi, Subbiah Raja and Anand Doshi; Jeevan Jyoti Hospital: Prakash Ketan.
Colombia: University of Antioquia, Hospital Universitario San Vicente de Paul: Carlos Morales and Santiago Naranjo; Hospital Pablo Tobón Uribe: Alfredo Constain and Edwin Vazquez Salazar; Hospital Universitario San José de Popayán: Jorge Herrera and Liliana Caicedo; Fundación Valle del Lili: Jorge Mejía-Mantilla and Ana MariaVarela.
UK Co-ordinating Team
London School of Hygiene and Tropical Medicine: Eni Balogun (clinical trials associate), Lin Barnetson (data manager), Collette Barrow (assistant trials administrator), Matthew Berle (trials assistant), Lisa Cook (assistant trials manager), Avni Gadhiya (data assistant), Taemi Kawahara (assistant trials manager), Pablo Perel (clinical lecturer), Maria Ramos (trials administrator), Ian Roberts (clinical co-ordinator), Chris Rubery (data assistant), Haleema Shakur (clinical lecturer) and Cynthia To (data assistant).
University of Edinburgh: Rustam Al-Shahi Salman, Zoe Morris, David Perry, Peter Sandercock and Joanna Wardlaw.
Steering Committee
Ian Franklin (chairperson), Brigitte Chaudhry, Tim Coats, Charles Deakin, Steve Goodacre, BJ Hunt, David Meddings, Richard Peto, Ian Roberts and Peter Sandercock.
Funding
The CRASH-2 study was funded by the UK NIHR Health Technology Assessment programme, Pfizer, the Bupa Foundation and the JP Moulton Charitable Foundation. This substudy was funded by the UK Health Technology Assessment programme. RA-SS was funded by a UK MRC clinician scientist fellowship. JMW was funded by the Scottish Funding Council through the SINAPSE Collaboration (Scottish Imaging Network, A Platform for Scientific Excellence, www.sinapse.ac.uk).
Contributions of authors
Pablo Perel, Rustam Al-Shahi Salman, Taemi Kawahara, Zoe Morris, Ian Roberts, Peter Sandercock, Haleema Shakur, and Joanna Wardlaw contributed to the study design including the study protocol and study data forms. David Prieto Merino provided expertise in statistics and undertook the statistical analysis. Joanna Wardlaw and Zoe Morris contributed with expertise on neuroradiology. Zoe Morris conducted the reading of all the CT scans. Peter Sandercock and Rustam Al-Shahi Salman contributed with expertise in neurology. Pablo Perel, Ian Roberts and Haleema Shakur provided expertise in clinical trials. Taemi Kawahara and Haleema Shakur provided expertise in trial management. Pablo Perel drafted the first version of the monograph and all the authors commented and contributed to the final version.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Menon DK, Schwab K, Wright DW, Maas AI. Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil 2010;91:1637-40.
- Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia 2003;44:2-10.
- CRASH trial Collaborators . Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury – outcomes at 6 months. Lancet 2005;365:1957-9.
- Perel P, Roberts I, Bouamra O, Woodford M, Mooney J, Lecky F. Intracranial bleeding in patients with traumatic brain injury: a prognostic study. BMC Emerg Med 2009;9.
- Oertel M, Kelly DF, McArthur D, Boscardin WJ, Glenn TC, Lee JH, et al. Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J Neurosurg 2002;96:109-16.
- Yadav YR, Basoor A, Jain G, Nelson A. Expanding traumatic intracerebral contusion/hematoma. Neurol India 2006;54:377-81.
- Sullivan TP, Jarvik JG, Cohen WA. Follow-up of conservatively managed epidural hematomas: implications for timing of repeat CT. Am J Neuroradiol 1999;20:107-13.
- Narayan RK, Maas AI, Servadei F, Skolnick BE, Tillinger MN, Marshall LF. Progression of traumatic intracerebral hemorrhage: a prospective observational study. J Neurotrauma 2008;25:629-39.
- Prentice CR. Basis of antifibrinolytic therapy. J Clin Pathol Suppl 1980;14:35-40.
- Henry DA, Carless PA, Moxey AJ, O’Connell D, Stokes BJ, McClelland B, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev 2007;4.
- Harhangi BS, Kompanje EJ, Leebeek FW, Maas AI. Coagulation disorders after traumatic brain injury. Acta Neurochir 2008;150:165-75.
- Bayir A, Kalkan E, Koçak S, Ak A, Cander B, Bodur S. Fibrinolytic markers and neurologic outcome in traumatic brain injury. Neurol India 2006;54:363-5.
- Perel P, Roberts I, Shakur H, Thinkhamrop B, Phuenpathom N, Yutthakasemsunt S. Haemostatic drugs for traumatic brain injury. Cochrane Database Syst Rev 2010;1.
- Roos YB, Rinkel GJ, Vermeulen M, Algra A, van Gijn J. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2003;2.
- Stein SC, Halpern C, Reilly PM. Upsetting the balance. Indian J Med Res 2007;125:186-7.
- CRASH-2 trial collaborators . Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23-32.
- Peto R, Baigent C. Trials: the next 50 years. BMJ 1998;317:1170-1.
- Wardlaw JM, Easton VJ, Statham P. Which CT features help predict outcome after head injury?. J Neurol Neurosurg Psychiatry 2002;72:188-92.
- Osborn A. Diagnostic imaging: brain. Altona, MB: Amirsys; 2004.
- Kwak R, Kadoya S, Suzuki T. Factors affecting the prognosis in thalamic hemorrhage. Stroke 1983;14:493-500.
- Gebel JM, Sila CA, Sloan MA, Granger CB, Weisenberger JP, Green CL, et al. Comparison of the ABC/2 estimation technique to computer-assisted volumetric analysis of intraparenchymal and subdural haematomas complicating the GUSTO-1 trial. Stroke 1998;29:1799-801.
- Petersen OF, Espersen JO. Extradural hematomas: measurement of size by volume summation on CT scanning. Neuroradiology 2004;26:363-7.
- Greene KA, Marciano FF, Johnson BA, Jacobowitz R, Spetzler RF, Harrington TR. Impact of traumatic subarachnoid hemorrhage on outcome in nonpenetrating head injury. Part I: a proposed computerized tomography grading scale. J Neurosurg 1995;83:445-52.
- Hijdra A, Brouwers PJAM, Vermeulen M, van Gijn J. Grading the amount of blood on computed tomograms after subarachnoid hemorrhage. Stroke 1990;21:1156-61.
- Wardlaw JM, Sellar RJ. A simple practical classification of cerebral infarcts on CT and its interobserver reliability. Am J Neuroradiol 1994;15:1933-9.
- Perel P, Edwards P, Shakur H, Roberts I. Use of the Oxford Handicap Scale at hospital discharge to predict Glasgow Outcome Scale at 6 months in patients with traumatic brain injury. BMC Med Res Methodol 2008;8.
- Spiegelhalter DJ, Abrahms KR, Myles JP. Bayesian approaches to clinical trials and health care evaluation. Chichester: John Wiley; 2004.
- Perel P. for the MRC CRASH Collaborators . Predicting outcome after traumatic brain injury: practical prognostic models based on large cohort of international patients. BMJ 2008;336:425-9.
- Pathak A, Dutta S, Marwaha N, Singh D, Varma N, Mathuriya SN. Change in tissue thromboplastin content of brain following trauma. Neurol India 2005;53:178-82.
- Cohen MJ, Brohi K, Ganter MT, Manley GT, Mackersie RC, Pittet JF. Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway. J Trauma 2007;63:1254-61.
- Lustenberger T, Talving P, Kobayashi L, Barmparas G, Inaba K, Lam L, et al. Early coagulopathy after isolated severe traumatic brain injury: relationship with hypoperfusion challenged. J Trauma 2010;69:1410-14.
- Astrup J. [‘Ultra-early’ antifibrinolytic treatment of subarachnoidal bleeding with tranexamic acid. Ugeskr Laeger 2006;168:1107-11.
Appendix 1 CRASH-2 IBS trial protocol

CRASH-2 substudy
The effect of tranexamic acid on intracranial bleeding among CRASH-2 trial participants
1. Background
CRASH-2 is a large randomised controlled trial of the effect on mortality and transfusion requirements of tranexamic acid (TXA) in trauma patients with significant bleeding (www.crash2.Lshtm.ac.uk). Over 9,000 patients have been enrolled so far and recruitment will continue until 20,000 patients have been randomised. Many of the patients included in the CRASH-2 trial have multiple injuries and in about 40% of patients this includes traumatic brain injury (TBI). The CRASH-2 trial will assess as sub-group analyses, the effect of TXA in patients who also have TBI. These analyses will examine the effect of TXA on mortality, on the need for a neurosurgical operation and on neurological impairment, using a modified version of the Oxford Handicap Score (our previous analyses have shown that this score is strongly correlated with outcome on the Glasgow Outcome Scale at six months).
Traumatic intracranial bleeding: TBI is commonly accompanied by intracranial bleeding, which can be epidural, subdural, subarachnoid or parenchymal. Of the 7,814 patients with TBI enrolled in the MRC CRASH trial who had a computerised tomography (CT) scan, 31% had subarachnoid haemorrhage and 40% had an intracranial haematoma. Overall 56% of TBI patients had some type of intracranial bleeding. 1
Prognostic studies show that intracranial bleeding is associated with increased mortality and disability six months after injury. In the MRC CRASH trial, the presence of subarachnoid haemorrhage, petechial haemorrhage or intracranial haematoma were independently associated with poor outcome at 2 weeks and 6 months. 2 Similarly, the IMPACT study found that after controlling for age, Glasgow Coma Score (GCS) motor score and pupil reactions, subarachnoid and subdural haemorrhages more than doubled the odds of poor outcome at six months. 3 The larger the intracranial bleeding, wherever the location, the worse prognosis it is associated with. The Brain Trauma Foundation Guideline for Surgery takes into account the bleeding size to recommend surgical evacuation: 50 cm3 for parenchymal haematoma, 30 cm3 for epidural and 10 mm for subdural haematoma. 5
In patients with TBI, intracranial bleeding can develop or worsen after hospital admission. Studies involving repeated CT scanning have found that intracranial bleeds can develop or expand in the 24 hours after injury. Ortel studied a group of patients in whom two CT scans were obtained within 24 hours of injury to determine the prevalence of progressive intracranial haemorrhage. 4 Among patients who had their first CT scan within 2 hours of injury, 49% had radiological evidence of progressive haemorrhage. Yadav conducted repeat CT scanning of TBI patients at hospital admission and 24 hours later, and found that 16% of 262 parenchymal haematomas and contusions increased in size in the first 24 hours. 6 Similarly, Sullivan et al. found that traumatic epidural haemorrhages enlarged in 23% of 160 TBI patients treated non-operatively. 7 The mean enlargement was 7 mm, and the mean time to enlargement was 8 hours from injury and 5.3 hours from CT diagnosis. Although these studies provide estimates of the occurrence of intracranial bleeding and expansion they all have limitations. All included patients who have an abnormal initial CT scan and there is little information on the proportion of patients that develop new intracranial bleeds in the first 24 hours who have the potential to benefit from early treatment.
Tranexamic acid and intracranial bleeding: Tranexamic acid is commonly used in surgery to reduce blood loss. A systematic review of randomised controlled trials of TXA in elective surgery showed that it reduces the need for transfusion by one third, reduces donor exposure by one unit, and halves the need for further surgery to control bleeding. 8 A systematic review of randomised trials of TXA in patients with aneurismal subarachnoid haemorrhage showed that TXA reduced the rate of rebleeding by approximately 40%, but because of an increase in cerebral ischaemia there was no overall benefit. 9 However, the duration of TXA treatment in these trials was six weeks and it is possible that a shorter treatment might prevent rebleeding whilst avoiding the risk of ischaemia. The systematic review was conducted in 2003 but since then a randomised controlled trial of the early administration of a short course (3 days) of TXA in aneurysmal subarachnoid haemorrhage found that TXA reduced the occurrence of rebleeding from 10.8% to 2.4% with no evidence of increased side effects. 10 Almost all of the effect on rebleeding was observed within the first few hours after hospital admission. Tranexamic acid could also improve outcome after TBI by reducing systemic blood loss. Hypotension is an established risk factor for poor outcome after TBI.
Hypothesis: Early administration of TXA can prevent the occurrence or increase of intracranial bleeding in patients with TBI and significant bleeding.
Aim: The aim of the proposed substudy is to quantify the effect on intracranial bleeding of the early administration of TXA, in CRASH-2 trial participants with traumatic brain injury.
Primary outcome:
-
Increase in volume of intracranial bleeding
Secondary outcomes:
-
Frequency of progressive haematomas
-
Frequency of delayed haematomas
-
New focal ischaemic lesions
Other relevant outcomes including mortality, disability, need of neurosurgical operation and non fatal thromboembolic events are collected in the CRASH-2 trial and will be reported for the TBI subgroup in the main analysis.
2. Study design
Methods
Participating hospitals: Participating hospitals have been selected based on level of interest by the principal investigator in the research question, the recruitment rate in the CRASH-2 trial and the ability of the hospital to collect and send the necessary CT scan data to the trial co-ordinating centre. This substudy will be conducted in the hospitals listed in Appendix 1.
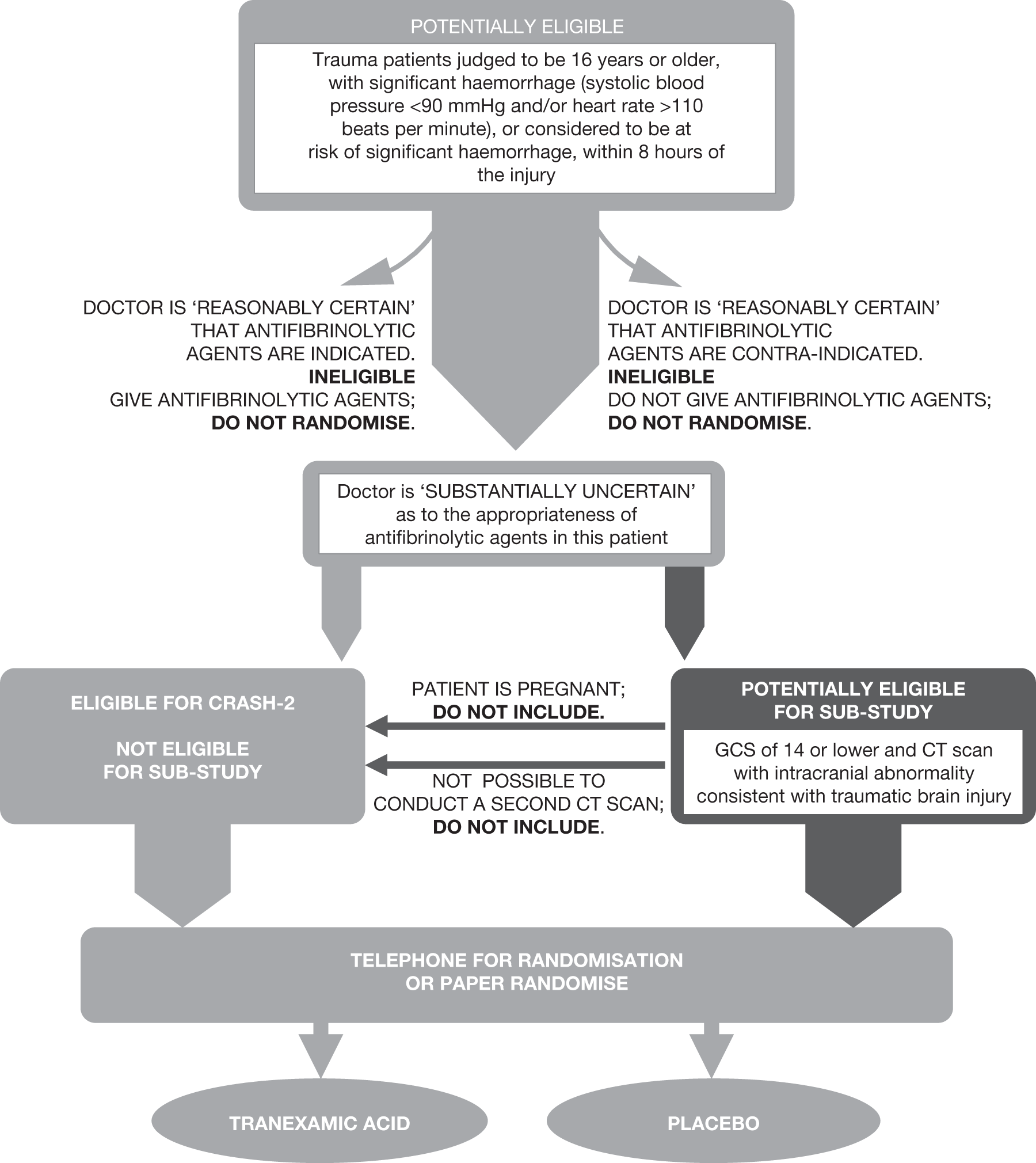
Inclusion criteria: All patients meeting the following criteria will be eligible for inclusion in the substudy:
-
Fulfils the inclusion criteria for the CRASH-2 trial
-
GCS of 14 or less
-
Baseline clinical CT scan shows intracranial abnormality consistent with TBI
-
Non pregnant
Number of patients needed: Assuming a baseline intracranial bleeding volume of 20 ml, an average increase of 7 ml in the control group and a correlation of 0.6 between baseline and follow-up bleeding, we need to recruit 900 patients to have 80% power (with alpha = 0.05) to detect a 20% reduction in the increase of intracranial bleeding volume in the control group. If 400 patients were recruited the trial could detect a 30% reduction in the primary outcome. A sample size of 200 patients could detect a 40% reduction in the primary outcome.
FIGURE 1.
Eligibility.
Procedures: Patients will follow all procedures as per CRASH-2 protocol; Additional procedures for the substudy are as follows:
Patients enrolled in the substudy will be identified by the completion of one form that will collect data on time since injury, time of initial (pre-randomisation) and follow-up CT scans and the file identifiers of the respective CT scan images. This form will also collect information on whether or not a decision to undertake neurosurgery was taken based on the CT scan result (see Appendix 2). This form will be sent to the trial co-ordinating centre in the same way as the CRASH-2 trial data.
CT scan protocol:
CT scan data acquisition: Two CT scans will be obtained for each participant, a clinical pre-randomisation scan and second scan 24–48 hours later. CT scans will be sent to the co-ordinating centre by uploading them onto the CRASH-2 trial server. Scans will be checked by the trial data manager to ensure that they are of the head, from the correct patient, performed on the correct date and of sufficient quality to be read. All study sites will be required to provide documentation as to the standard parameters used for each CT scanner. The specific scan protocol/parameters of the initial CT evaluation will be limited by the emergent nature at the time of admission. However, after a patient is enrolled, the follow-up scan must match the baseline CT scan with regard to section thickness, section spacing (overlap or no gap), matrix, field of view, and scan angulations. Consistency in these parameters across all CT evaluations for a patient allow comparable measurements because of identical spatial resolution. All name identifiers will be removed before loading the scans onto the CT reading system. The CT scans will be allocated to an expert CT scan reader for evaluation who will be blind to the treatment allocation.
CT scan data analysis: In both CT scans we will measure: type of bleeding (subdural, epidural, subarachnoid haemorrhage, parenchymal haematoma), volume of bleeding, ischaemic lesions and indirect signs of intracranial pressure. Volume of intracranial bleeding will be measured using validated methods; further details are described in the statistical analysis plan.
Consent
Because patients included in this substudy will have significant TBI, relatives or legal representatives would be asked to sign the informed consent in line with local legal requirements. To minimise the need for multiple information sheets and consent forms, one form which combines the CRASH-2 and substudy information will be used (Appendix 3).
Randomisation
As per CRASH-2 protocol.
Treatment
As per CRASH-2 protocol.
Serious unexpected suspected adverse events
As per CRASH-2 protocol.
Expected side effects
As per CRASH-2 protocol.
Potential risks associated with this substudy
It is standard care for all patients with a history of TBI and associated clinical signs to have a CT scan. Therefore, the initial scan will form part of standard care. The substudy requires one additional CT Scan to be done 24 to 48 hours after the first, which in many cases is likely to be clinically indicated. The effective radiation dose from a CT scan is about 2 mSv, which is about the amount received from background radiation in eight months.
Unblinding
As per CRASH-2 protocol.
Analysis
Haemorrhage volume from CT scans will be analysed with use of generalised linear mixed models. The baseline bleeding volume and the time from injury to CT will be included as covariates. Because patients who undergo a neurosurgery based on the pre-randomisation CT scan would not have a baseline haematoma they would be excluded of this analysis.
We will express the effect of TXA on the occurrence of secondary endpoints using relative risks and 95% confidence intervals. All analysis will be based on the intention to treat principle. We will conduct subgroup analysis according to type of bleeding. No interim analysis is planned for the sub-study; the analysis will be done at the end of the CRASH-2 Trial. Further details are described in the statistical analysis plan.
Definitions
Progressive haematoma will be defined as a growth of the haematoma larger than 25% from the initial to the follow-up CT scan.
Delayed haematoma will be defined as appearance of an haematoma in the follow-up CT scan where there was not one on the initial scan.
New focal ischaemic lesions will be defined as those ischaemic lesions which appear in the follow-up CT scan but not in the initial one.
3. Organisation
Data monitoring committee
The data monitoring committee members would be the same as CRASH-2.
Standard Operating Procedures: As per CRASH-2 protocol.
Steering committee
The steering committee members would be the same as CRASH-2 plus Professor Peter Sandercock.
Standard Operating Procedures: As per CRASH-2 protocol.
Substudy Protocol Committee
Rustam Al-Shahi Salman (UK), Yashbir Dewan (India), Anil P Lal (India), Carlos Morales (Colombia), Zoe Morris (UK), Pablo Perel (UK), P V Ramana (India), R R Ravi (India), Ian Roberts (UK), Peter Sandercock (UK), Haleema Shakur (UK), Joanna Wardlaw (UK).
Co-ordinating centre responsibilities
As per CRASH-2 protocol.
Publication
The results of the trial will be reported first to trial collaborators. Dissemination of results to patients will take place via the media, trial website (www.crash2@Lshtm.ac.uk) and relevant patient organisations.
Indemnity
As per CRASH-2 protocol.
Financial support
LSHTM.
4. IBS protocol references
- The CRASH Trial Collaborators . Effect of intravenous corticosteroids on death within 14 days in 10,008 adults with clinically significant head injury (MRC CRASH Trial): a randomised placebo-controlled trial. Lancet 2004;364:1321-28.
- MRC CRASH Trial Collaborators . Predicting outcome after traumatic brain injury: practical prognostic models based on large cohort of international patients. BMJ 2008;12.
- Maas AI, Steyerberg EW, Butcher I, Dammers R, Lu J, Marmarou A, et al. Prognostic value of computerized tomography scan characteristics in traumatic brain injury: results from the IMPACT study. J Neurotrauma 2007;24:303-14.
- Oertel M, Kelly DF, McArthur D, Boscardin WJ, Glenn TC, Lee JH, et al. Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J Neurosurg 2002;96:109-16.
- Bullock M, Chesnut, Randall, Ghajar, Jamshid, Gordon, et al. Surgical Management of Traumatic Brain Injury. Neurosurgery 2006;58.
- Yadav YR, Basoor A, Jain G, Nelson A. Expanding traumatic parenchymal contusion/hematoma. Neurol India 2006;54:377-81.
- Sullivan TP, Jarvik JG, Cohen WA. Follow-up of conservatively managed epidural hematomas: implications for timing of repeat CT. AJNR Am J Neuroradiol 1999;20:107-13.
- Henry DA, Moxey AJ, Carless PA, O’Connell D, McClelland B, Henderson KM, et al. The Cochrane Library. Chichester, UK: John Wiley & Sons, Ltd; 2007.
- Roos YB, Rinkel GJE, Vermeulen M, Algra A, van Gijn J. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. The Cochrane Database of Systematic Reviews 2003. URL: 10.1002/14651858.CD001245.
- Astrup J. ‘Ultra-early’ antifibrinolytic treatment of subarachnoidal bleeding with tranexamic acid. Ugeskr Laeger 2006;168:1107-11.
Appendix 1 Hospitals that will participate in the substudy
-
Aditya Neuroscience Centre, Dibrugarh, India
-
Sanjivani Hospital, Dibrugarh, India
-
Care Hospital, Visakhapatnam, India
-
Christian Medical College, Ludhiana, India
-
Medical Trust Hospital, Kochi, India
-
Hospital Universitario San Vicente de Paul, Medellín, Colombia
-
Hospital Universitario San Jose De Popayan, Colombia
-
Hospital Pablo Tobon Uribe, Medellín, Colombia
-
Fundacion Clinica Valle del Lili, Colombia
-
Jeevan Jyoti Hospital & Research Centre, Allahabad, India.
Other hospitals may be added if recruitment falls below predicted.
Appendix 2 Investigator computerised tomography scan form
Appendix 3a Information for relatives and representatives
Appendix 3b Information for patients
Appendix 2 Computerised tomography scan reader forms
Appendix 3 CRASH-2 outcome form
List of abbreviations
- CI
- confidence interval
- CRASH-2
- Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage
- CRASH-2 IBS
- CRASH-2 Intracranial Bleeding Study
- CrI
- credibility interval
- CT
- computerised tomography
- GCS
- Glasgow Coma Scale
- mOHS
- modified Oxford Handicap Scale
- OR
- odds ratio
- RR
- relative risk
- SD
- standard deviation
- sSH
- spontaneous subarachnoid haemorrhage
- TBI
- traumatic brain injury
- TXA
- tranexamic acid
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health