
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 06/303/20. The contractual start date was in April 2007. The draft report began editorial review in January 2012 and was accepted for publication in August 2012. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors' report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen's Printer and Controller of HMSO 2013. This work was produced by Roberts et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Among people aged 5–45 years, trauma is second only to human immunodeficiency virus (HIV) infection or acquired immunodeficiency syndrome (AIDS) as a cause of death. Each year, worldwide, about 3 million people die as a result of trauma, many after reaching hospital. 1 Among trauma patients who do survive to reach hospital, exsanguination is a common cause of death, accounting for up to half of in-hospital trauma deaths. 2 Central nervous system injury and multiorgan failure account for most of the remainder, both of which can be exacerbated by severe bleeding. 3
Mechanisms
The haemostatic system helps to maintain the integrity of the circulatory system after severe vascular injury, whether traumatic or surgical in origin. 4 Major surgery and trauma trigger similar haemostatic responses and the consequent massive blood loss presents an extreme challenge to the coagulation system. Part of the response to surgery and trauma, in any patient, is stimulation of clot breakdown (fibrinolysis) which may become pathological (hyper-fibrinolysis) in some. 4 Antifibrinolytic agents have been shown to reduce blood loss in patients with both normal and exaggerated fibrinolytic responses to surgery, and do so without apparently increasing the risk of postoperative complications; most notably there is no increased risk of venous thromboembolism. 5
Existing knowledge
Tranexamic acid (TXA) is widely used in major surgery to prevent fibrinolysis and reduce surgical blood loss. A systematic review6 of randomised controlled trials of TXA in elective surgical patients identified 53 studies including 3836 participants. TXA reduced the need for blood transfusion by one-third [relative risk (RR) 0.61; 95% confidence interval (CI) 0.54 to 0.70]. TXA reduced the need for blood transfusion in cardiac surgery, orthopaedic surgery, liver surgery and vascular surgery. When the analysis was restricted to those trials that had good-quality allocation concealment, there was again a significant reduction in the need for blood transfusion (RR = 0.60; 95% CI 0.49 to 0.72). Although there was some statistical evidence of heterogeneity in the treatment effect, such that some trials showed a larger effect than others, the effect was consistent in as much as nearly all trials showed a reduction in the need for blood transfusion. There was also a non-significant reduction in the risk of death with TXA (RR = 0.60; 95% CI 0.32 to 1.12). The effect was large but imprecise, reflecting the fact that mortality is relatively rare in elective surgery. Importantly, there was no evidence of any increased risk of thromboembolic events with TXA. Specifically, there was no evidence of any increased risk of myocardial infarction, stroke, deep-vein thrombosis or pulmonary embolism. Taken together, the evidence that TXA reduces bleeding in surgical patients is reasonably strong. This evidence was an important motivation for the conduct of the Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage-2 (CRASH-2) clinical trial.
Tranexamic acid is a member of a class of drugs called antifibrinolytic agents. Other members of this class are aprotinin and aminocaproic acid. At the time that the CRASH-2 trial protocol was being developed there was also evidence that aprotinin reduced blood loss in surgery, again without compelling evidence of an increase in thromboembolic complications. Indeed, because there had been many more clinical trials of aprotinin in elective surgery than of TXA, the estimate of the effect of aprotinin on the need for blood transfusion was even more precise than for TXA (RR = 0.66; 95% CI 0.62 to 0.71). However, a major drawback of aprotinin is that it is considerably more expensive than TXA. Another disadvantage of aprotinin is that unlike TXA, which is a simple synthetic molecule, aprotinin is a bovine product with a consequent risk of allergic reaction and a hypothetical risk of disease transmission. Indeed, some sources recommended that before using aprotinin there should be a test dose to assess the presence or absence of allergic reactions.
At the time the CRASH-2 trial was being designed there was considerable interest in the potential of a drug called activated factor VII (factor VIIa) as a treatment for bleeding. Activated recombinant factor VII [rFVIIa or NovoSeven®(Novo Nordisk Inc., Princeton, NJ, USA)] is a pro-coagulant drug used to treat clotting disorders including haemophilia with inhibitors. It was suggested that it might also be useful in controlling non-haemophiliac bleeding. However, despite the considerable hype at the time, there was little reliable evidence that the benefits of factor VIIa outweighed the harms. One of the earliest reports of rFVIIa use in non-haemophiliac bleeding was published in The Lancet in 1999. 7 This report documented the use of rFVIIa in one Israeli soldier who suffered a thoracic gunshot wound. It reported that surgical attempts to stop bleeding had failed and the patient was near death but that then, ‘in a desperate attempt to control the bleeding’, he was given two doses of rFVIIa. Minutes later the bleeding stopped, allowing surgeons to repair the vessel. Although case reports such as this are a notoriously unreliable basis for treatment decisions, such narratives are remarkably influential. Following this early case report hundreds more were published, which may have influenced practitioners to take similar ‘desperate’ measures in comparable situations. However, the evidence base was weak. The scientific ‘prior’ for TXA as a treatment for bleeding trauma patients was much more compelling and, hence, TXA was selected for use in the CRASH-2 clinical trial.
Hypothesis
The extent to which a treatment effect observed in one clinical setting (elective surgery) might reasonably be generalised to another clinical setting (e.g. trauma) is a scientific judgement based largely on a consideration of whether or not the same mechanism of action is likely to apply in both cases. Of course, the most important criterion for any scientific generalisation is that the treatment effect to be generalised is valid and precise. The Cochrane systematic review6 of TXA in surgical patients had provided evidence of a treatment effect that was apparently valid – in as much as it was obtained from well-concealed randomised controlled trials and reasonably precise. TXA appeared to reduce bleeding in surgical patients and the effect was large. Is it possible, then, that it might also reduce bleeding in trauma patients? In both of these clinical situations patients experience tissue damage and bleed as a result. In both of these clinical situations the coagulation system is involved in achieving haemostasis. Antifibrinolytics such as TXA are believed to work by inhibiting clot breakdown. They work by inhibiting plasmin, an enzyme that causes clot breakdown. Because we judged that the mechanism of action of TXA on blood coagulation after injury was likely to be similar to the effect of TXA in surgery, we considered it possible that TXA might reduce blood loss, the need for transfusion and mortality following trauma. However, prior to the CRASH-2 trial there had been only one small randomised controlled trial of an antifibrinolytic agent in bleeding trauma patients (70 randomised patients: drug vs placebo: 0 vs 3 deaths). 8 As a result, there was insufficient evidence to either support or refute a clinically important treatment effect. Systemic antifibrinolytic agents have been used in the management of eye injuries, where there is some evidence that they reduce the rate of secondary haemorrhage. 9
Need for a trial
A simple and widely practicable treatment that reduces blood loss following trauma might prevent thousands of premature trauma deaths each year and could also reduce exposure to the risks of blood transfusion. Blood is a scarce and expensive resource and major concerns remain about the risk of transfusion-transmitted infection. Trauma is common in parts of the world where the safety of blood transfusion is not assured. A recent study in Uganda estimated that the population-attributable fraction of HIV acquisition as a result of blood transfusion to be around 2%, although some estimates are much higher. 10,11 Only 43% of the 191 World Health Organization (WHO) member states test blood for HIV and hepatitis C and B viruses. Every year unsafe transfusion and injection practices are estimated to account for 8–16 million hepatitis B infections, 2.3–4.7 million hepatitis C infections and 80,000–160,000 HIV infections. 12 A large randomised trial is therefore needed of the use of a simple, inexpensive, widely practicable antifibrinolytic treatment such as TXA, in a wide range of trauma patients who, when they reach hospital, are thought to be at risk of major haemorrhage that could significantly affect their chances of survival.
Dose selection
The systematic review of randomised controlled trials of antifibrinolytic agents in surgery showed that dose regimens of TXA vary widely. 6 Loading doses range from 2.5 mg/kg to 100 mg/kg and maintenance doses from 0.25 mg/kg/hour to 4 mg/kg/hour delivered over time periods of 1–12 hours. Studies examining the impact of different doses of TXA on bleeding and transfusion requirements showed no significant difference between a high dose and a low dose.
Studies in cardiac surgery have shown that a 10 mg/kg initial dose of TXA followed by an infusion of 1 mg/kg/hour produces plasma concentrations sufficient to inhibit fibrinolysis in vitro. 13 The dose–response relationship of TXA was examined by Horrow et al. ,14 who concluded that 10 mg/kg followed by 1 mg/kg/hour decreases bleeding after extracorporeal circulation and that larger doses did not provide any additional haemostatic benefit.
In this emergency situation, administration of a fixed dose would be more practicable, as determining the weight of a patient would be impossible. Therefore, a fixed dose within the dose range which has been shown to inhibit fibrinolysis and provide haemostatic benefit is being used for this trial. The fixed dose chosen would be efficacious for larger patients (> 100 kg) but also safe in smaller patients (< 50 kg), as the estimated dose/kg that the latter group would receive has been applied in other trials without adverse effects. The planned duration of administration allows for the full effect of TXA on the immediate risk of haemorrhage without extending too far into the acute phase response seen after surgery and trauma.
Chapter 2 Methods
Study design
Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage-2 is a large placebo-controlled trial of the effects of early administration of a short course of TXA on death, vascular occlusive events and the receipt of blood transfusion. The trial protocol was peer-reviewed and published on The Lancet website in 2005. 15
Although the effect of TXA on blood transfusion in surgical patients was reasonably large, we felt that it would be unreasonable to expect a large reduction in mortality with TXA in bleeding trauma patients. On the other hand, a modest reduction in all-cause mortality was scientifically plausible. However, to detect such a modest treatment effect the trial would need to be large and would have to include many thousands of randomised trauma patients. It would clearly have to be an international clinical trial, as it would take many years to enrol such a large number of trauma patients in the UK alone. The trial co-ordinating centre at the London School of Hygiene and Tropical Medicine (LSHTM) had experience in conducting large international clinical trials. Indeed, the CRASH-2 trial followed on closely from the Corticosteroid Randomisation After Significant Head Injury (CRASH-1) trial of corticosteroids in head injury. The Medical Research Council (MRC)-funded CRASH-1 trial had enrolled 10,000 patients with significant traumatic brain injury from many countries worldwide. As a result of the CRASH-1 clinical trial, the LSHTM trial co-ordinating centre had established an excellent global network of collaborating trauma hospitals. Over many years of collaboration, LSHTM had developed good working relationships with a large number of trauma doctors around the world. The trial network thus had extensive experience in conducting large-scale clinical trials and it was a critically important human resource when it came to the conduct of the CRASH-2 trial. Planning for the CRASH-2 trial was already under way before the end of the CRASH-1 trial. Indeed, the draft protocol of the CRASH-2 trial was presented for discussion at the closure meeting of the CRASH-1 clinical trial. There was considerable enthusiasm within the trial network for the conduct of the CRASH-2 clinical trial. This was a major asset in ensuring rapid recruitment in the CRASH-2 trial.
The trial was undertaken in 274 hospitals in 40 countries. The main criteria for selecting participating hospitals were that the hospitals provide definitive trauma care for a sufficiently large number of trauma patients; the hospital doctors are substantially uncertain as regards the effect of TXA in the management of bleeding trauma patients; and the hospitals have the necessary research infrastructure to conduct the trial. As an example of the hospitals having the necessary research infrastructure to conduct the trial, it is essential that the trial co-ordinating centre has a reliable means of communication with all the participating hospitals and reliable e-mail and telephone communication with the principal investigator at the participating hospital.
Although some clinicians believe that standardisation of clinical care is a prerequisite for the conduct of multicentre randomised trials, the CRASH-2 investigators did not stipulate how trauma patients at participating hospitals should be managed. Providing that a trial is large enough, randomisation will ensure that the intervention and control groups are identical with regard to both known and unknown confounders. It is of course conceivable that the size of the intervention effect may vary a little depending on the other aspects of care given, but not the direction of the effect. Patients in the future will almost certainly receive different forms of care from those given today and treatments shown to be effective today may be more or less effective in the future, but the direction of the effect is likely to be the same. Rather than standardise care, the CRASH-2 trial investigators believed that it was much more important to make sure that the CRASH-2 trial was large enough to detect reliably moderate but clinically important treatment effects. Indeed, if the trial was large enough, it might have sufficient statistical power to assess the overall treatment effect and examine how the effect varies according to other factors. One of the major obstacles to conducting large clinical trials is that triallists attempt to collect too much information about other interventions. The more information participating doctors are required to collect, the more burdensome the trial becomes and so they tend to recruit fewer patients. In other words, the best way to ensure that patients in the treatment and comparison group have a similar prognosis, apart from the treatment, is not to collect more information on potential confounders but to collect much less and instead randomise a greater number of patients.
Pilot phase
A pilot phase was conducted to test the procedures for patient recruitment and data collection and provide reliable estimates of event rates for the sample size calculation. This was supported by the WHO, the Bupa Foundation and the J P Moulton Charitable Foundation. Two thousand patients were recruited over a 1-year period. The trial procedures were found to work efficiently. Most patients (> 70%) were recruited within 3 hours of their injury, there was 98% data completeness at 28 days, data audits at 20 hospitals confirmed the validity of the trial data, and the predicted event rates used in our sample size calculations were found to be accurate (20% mortality at 28 days and 60% transfusion requirement).
Study patients
The first patient was enrolled in May 2005. The last patient was enrolled in January 2010. Adult trauma patients with significant haemorrhage [systolic blood pressure (BP) < 90 mmHg, heart rate > 110 beats per minute or both] or were considered to be at risk of significant haemorrhage and who were within 8 hours of injury, were eligible for the trial. Patients were included if the responsible doctor was substantially uncertain about whether or not to treat with TXA (i.e. entry was governed by the uncertainty principle). 16 Patients in whom the responsible doctor considered that there was a clear indication for TXA were not randomly assigned. Similarly, patients in whom there was considered to be a clear contraindication to TXA treatment were not randomly assigned. However, when the responsible doctor was substantially uncertain whether or not to treat with this agent, these patients were eligible for randomisation. The use of simple entry criteria and the uncertainty principle, as recommended in the context of large trials, allows participating doctors to use clinical judgement when deciding whether or not to enrol patients into the trial, just as in normal medical practice. This is particularly appropriate in the context of traumatic haemorrhage, where it is necessary to evaluate a range of clinical signs (also taking into account remedial measures such as fluid resuscitation) when establishing the presence or absence of major haemorrhage. For example, patients with haemorrhagic hypovolaemia can maintain a reasonable BP by vasoconstriction and about one-third of patients with traumatic haemorrhage will present with bradycardia. Although the use of clinical judgement is very likely to result in variation in the types of patients entering the trial, this heterogeneity is a scientific strength, not a weakness. If a wide range of patients are randomised (the relevant clinical characteristics will have been carefully recorded), then it may be possible for a large trial such as this one to help determine which (if any) particular types of patient are most likely to benefit from treatment. In clinical trials in trauma care it is particularly important that the inclusion criteria are simple and straightforward. Acute severe trauma is a medical and surgical emergency and the responsible doctor's primary responsibility is to provide urgent clinical care for the bleeding trauma patient. It would be inappropriate to ask doctors in this situation to consider a long list of inclusion and exclusion criteria. Fortunately, there were few absolute contraindications to the administration of TXA and so it was possible to allow a wide range of patients to be enrolled without a long list of exclusions.
It was expected from the outset that a proportion of the patients with significant haemorrhage enrolled in CRASH-2 would also have head injuries. Indeed, because haemorrhagic hypotension is an important risk factor for poor outcome in head injury, if TXA reduces haemorrhage it could improve outcome in these patients. Early TXA administration may also prevent, or limit the extent of, delayed intracranial bleeding, an important treatable cause of secondary damage after head injury. On the other hand, up to one-third of patients with head injury have computerised tomography (CT) evidence of traumatic subarachnoid haemorrhage and randomised controlled trials of TXA in patients with aneurysmal subarachnoid haemorrhage had shown that, although a 6-week course of TXA reduced the rate of rebleeding by approximately 40%, there was no overall clinical benefit because of an increase in cerebral ischaemia. Of course, the duration of TXA treatment in the CRASH-2 trial (8 hours of treatment in the acute phase while the patient is bleeding) was much shorter than the 6 weeks used in aneurysmal subarachnoid haemorrhage, in which case this should be less of a concern. Nevertheless, we elected to collect data on the occurrence of head injury in order to examine the effect of TXA administration in patients with and without head injury. We also conducted a CT scan substudy to examine the effect of TXA administration on neuroradiological and clinical outcomes in trauma patients with significant haemorrhage and traumatic brain injury. These data are reported in a separate Health Technology Assessment journal publication. 17 Data on the effects of TXA in patients with and without head injury were also shown to the Data Monitoring Committee so that the overall effect and the effect within the head injury subgroup could be monitored.
Consent procedures at participating hospitals were established by local regulation and the appropriate ethics committees. Informed consent was obtained from patients if physical and mental capacity allowed. If patients could not give consent, proxy consent was obtained from a relative or representative. If a proxy was unavailable, then, if permitted by local regulation, consent was deferred or waived. When consent was deferred or given by a proxy, the patient was informed about the trial as soon as possible and consent obtained for use of the data collected if needed.
Randomisation and masking
After eligibility had been confirmed and the locally approved consent procedures had been completed, patients were randomly assigned. Randomisation was balanced by centre, with an allocation sequence based on a block size of eight, generated with a computer random number generator. In hospitals in which telephone randomisation was not practicable, we used a local pack system that selected the lowest-numbered treatment pack from a box containing eight numbered packs. Apart from the pack number, the treatment packs were identical. The pack number was recorded on the entry form which was sent to the international trial co-ordinating centre in London, UK. Hospitals with reliable telephone access used the University of Oxford's Clinical Trial Service Unit (CTSU) telephone randomisation service. The randomisation service used a minimisation algorithm balancing for sex, age, time since injury, type of injury (blunt or penetrating), Glasgow Coma Scale (GCS) score, systolic BP, respiratory rate, central capillary refill time and country, taking into account what packs were available at that hospital. Once the treatment pack number was recorded, the patient was included in the trial whether the treatment pack was opened or the allocated treatment started. Both participants and study staff (site investigators and trial co-ordinating centre staff) were masked to treatment allocation.
Tranexamic acid and placebo ampoules were indistinguishable. TXA was manufactured by Pharmacia (Pfizer, Sandwich, UK) and the placebo by St Mary's Pharmaceutical Unit, Cardiff, UK. The treatment packs were prepared by an independent clinical trial supply company (Bilcare, Crickhowell, UK). Correct blinding and coding of ampoules was assured by independent random testing of each batch by high-performance liquid chromatography to confirm the contents. Emergency unblinding was available by telephoning CTSU.
Procedures
Patients were randomly allocated to receive a loading dose of 1 g of TXA infused over 10 minutes, followed by an intravenous (i.v.) infusion of 1 g over 8 hours or matching placebo (0.9% saline). Every patient was assigned a uniquely numbered treatment pack that contained four ampoules of either 500 mg TXA or placebo, one 100-ml bag of 0.9% saline (for use with the loading dose), a syringe and needle, stickers with the trial details and randomisation number (for attaching to infusion bags, data forms and patient medical records), and instructions. Each box contained information leaflets for patients and their representatives, consent forms and data collection forms. The stickers, instructions, leaflets and forms were in local languages.
Outcome measures and prespecified subgroup analyses
The primary outcome was death in hospital within 4 weeks of injury. Cause of death was described by the following categories: bleeding, vascular occlusion (myocardial infarction, stroke and pulmonary embolism), multiorgan failure, head injury and other. Secondary outcomes were vascular occlusive events (myocardial infarction, stroke, pulmonary embolism and deep-vein thrombosis), surgical intervention (neurosurgery, thoracic, abdominal and pelvic surgery), receipt of blood transfusion and units of blood products transfused. Dependency was measured at hospital discharge, or on day 28 if still in hospital, with the five-point Modified Oxford Handicap Scale. The scale was dichotomised into dead, dependent (fully dependent requiring attention day and night or dependent but not needing constant attention) or independent (some restriction in lifestyle but independent, minor symptoms or no symptoms). 18 Data for the use of rFVIIa and for gastrointestinal bleeding as a complication were also collected. Because the expected complications of the trial treatment were collected on the outcome form, only adverse events that were serious, unexpected and suspected to be related to the study treatment were reported separately. Outcomes were recorded if they occurred while the patient was still in hospital for up to 28 days after randomisation. Data were sent to the co-ordinating centre either electronically (by encrypted electronic data forms that could be sent by e-mail or uploaded to a secure server) or by fax, and were entered onto a central database at the trial co-ordinating centre in London, UK. We monitored the quality of the trial data using a combination of centralised statistical data checking and site visits at which patient outcome forms were compared with clinical case notes. 19
We planned to report the effects of treatment on the primary outcome subdivided by four baseline characteristics: (1) estimated hours since injury (< 1, 1–3, 3–8 hours); (2) systolic BP (≤ 75, 76–89, > 89 mmHg); (3) GCS score (severe 3–8, moderate 9–12, mild 13–15); and (4) type of injury (penetrating only or blunt, which included blunt and penetrating).
Statistical analyses
The statistical analysis plan was sent to all ethics committees and regulatory agencies before unblinding. As the risk of death might be around 20%, and even a 2% survival difference (corresponding to an RR of death with TXA of 0.9) would be important, a trial of 20,000 patients was planned, which would then have an 85% chance of achieving a two-sided p-value of < 0.01 and a 95% chance of a two-sided p-value of < 0.05. All analyses were undertaken on an intention-to-treat basis. For each binary outcome we calculated RRs and 95% CIs, and two-sided p-values for statistical significance. The RR gives the number of times more likely (RR > 1) or less likely (RR < 1) an event is to happen in the TXA group than in the placebo group. For analysis of the prespecified subgroups (primary outcome only) we calculated RRs with 99% CIs with two-sided p-values. Heterogeneity in treatment effects across subgroups was assessed with chi-squared tests. We prespecified that, unless there was strong evidence (p < 0.001) against homogeneity of effects, the overall RR would be considered the most reliable guide to the approximate RRs in all subgroups. Means and standard deviations (SDs) were estimated for count outcomes, and we calculated two-sided p-values of the difference in means of logarithms. A complete case analysis, including only cases for which the relevant outcome data were available, was undertaken. There was no imputation for missing data. During the study, unblinded interim analyses were supplied by an independent statistician to the Data Monitoring Committee.
Role of the funding source
The trial was funded by the UK National Institute for Health Research Health Technology Assessment programme, Pfizer, the Bupa Foundation and the J P Moulton Charitable Foundation. The funders of the study had no role in study design, data collection, data analysis, data interpretation or writing of the report. The Writing Committee had full access to all data in the study and had final responsibility for the decision to submit for publication.
Chapter 3 Main results
The first patient was randomised on 19 May 2005 and the follow-up was completed on 9 March 2010. The trial ended when it reached its planned sample size.
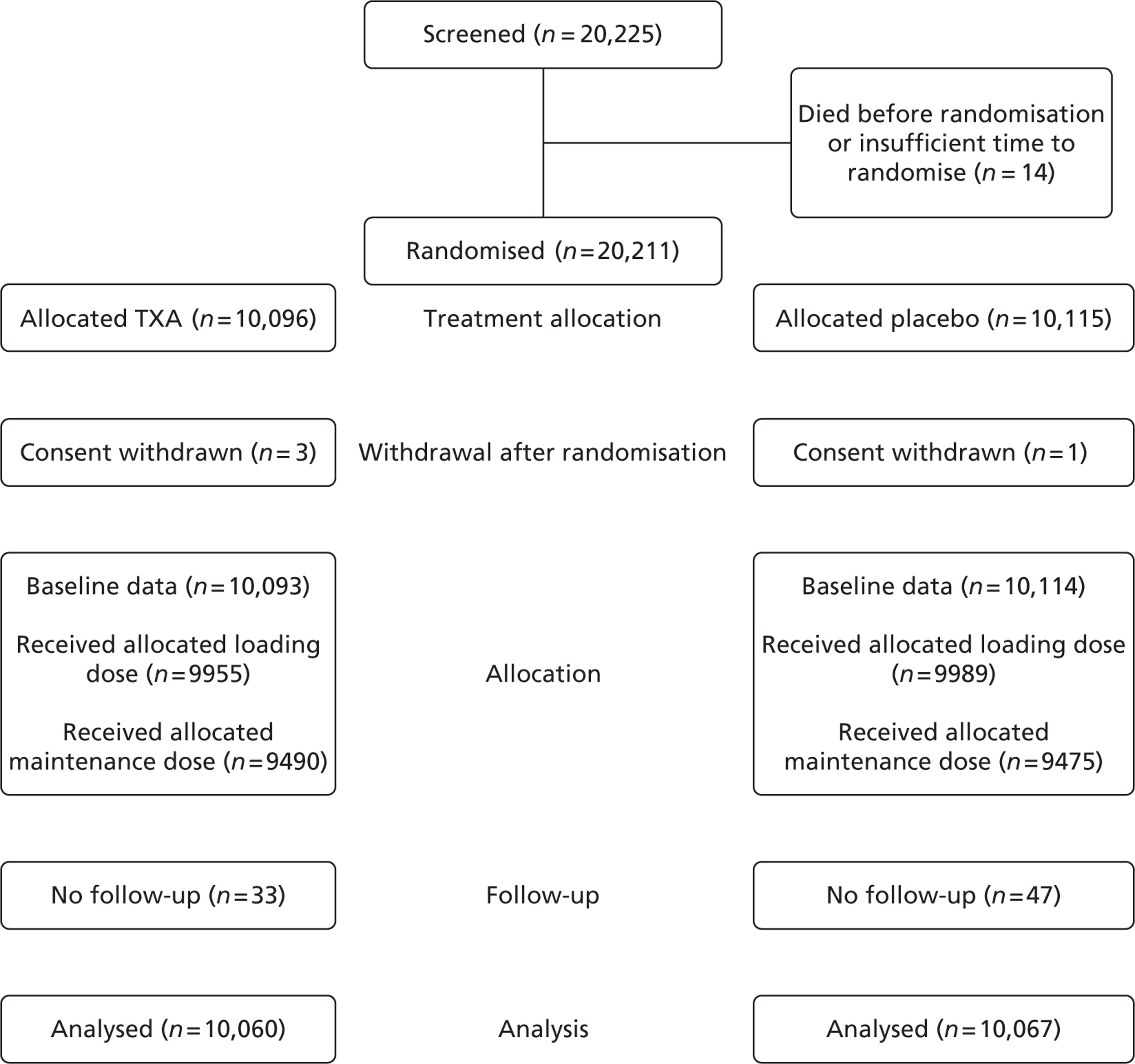
Figure 1 shows the trial profile. A total of 20,211 patients were randomly assigned to TXA or placebo (see Figure 1), of whom 20,116 patients were randomly assigned through the local pack system and 95 patients through telephone randomisation. The data from four patients were removed from the trial because their consent was withdrawn after randomisation. Five patients enrolled in the study were later found to be < 16 years of age. Age was unknown for four patients. Twenty-three patients were enrolled > 8 hours after their injury. Time of injury was not known for 11 patients. Nine patients had haemorrhage from non-traumatic conditions. Three patients were given a pack that differed from that allocated. The planned consent procedures were not fully followed in 34 patients. The relevant ethics committees were informed and approval for use of data was obtained. All the patients, apart from the four in whom consent was withdrawn, were included in the analysis. Because of digit preference (the tendency when reporting figures to round to specific digits), the number of patients in the early time since injury category (< 1 hour) was low and the subgroup estimate was imprecise. We therefore (post hoc) defined the early category as those treated ≤ 1 hour from injury (Figure 2). Treatment groups were balanced with respect to all baseline patient characteristics (Table 1 shows baseline data of patients with follow-up).
FIGURE 1.
Trial profile. No follow-up relates to those patients where there was no information on the primary end point.
| Variable | TXA (n = 10,093) | Placebo (n = 10,114) |
|---|---|---|
| Sex | ||
| Male | 8439 (83.6%) | 8496 (84%) |
| Female | 1654 (16.4%) | 1617 (16%) |
| Not known | 0 | 1 (0.01%) |
| Age (years) | ||
| Mean age (SD) | 34.6 (14.1) | 34.5 (14.4) |
| < 25a | 2783 (27.6%) | 2855 (28.2%) |
| 25–34 | 3012 (29.8%) | 3081 (30.5%) |
| 35–44 | 1975 (19.6%) | 1841 (18.2%) |
| > 44 | 2321 (23.0%) | 2335 (23.1%) |
| Not known | 2 (0.02%) | 2 (0.02%) |
| Time since injury (hours) | ||
| Mean (SD) | 2.8 (2.2) | 2.9 (2.6) |
| ≤ 1 | 3756 (37.2%) | 3722 (36.8%) |
| > 1 to ≤ 3 | 3045 (30.2%) | 3006 (29.7%) |
| > 3b | 3287 (32.6%) | 3380 (33.4%) |
| Not known | 5 (0.05%) | 6 (0.06%) |
| Type of injury | ||
| Bluntc | 6812 (67.5%) | 6843 (67.7%) |
| Penetrating | 3281 (32.5%) | 3271 (32.3%) |
| Systolic BP (mmHg) | ||
| ≤ 75 | 1566 (15.5%) | 1608 (15.9%) |
| 76–89 | 1615 (16.0%) | 1697 (16.8%) |
| > 89 | 6901 (68.4%) | 6791 (67.1%) |
| Not known | 11 (0.11%) | 18 (0.18%) |
| Respiratory rate (per minute) | ||
| < 10 | 160 (1.6%) | 149 (1.5%) |
| 10–29 | 8355 (82.8%) | 8436 (83.4%) |
| > 29 | 1491 (14.8%) | 1429 (14.1%) |
| Not known | 87 (0.86%) | 100 (0.99%) |
| Central capillary refill time (seconds) | ||
| ≤ 2 | 3432 (34.0%) | 3406 (33.7%) |
| 3–4 | 4665 (46.2%) | 4722 (46.7%) |
| > 4 | 1699 (16.8%) | 1672 (16.5%) |
| Not known | 297 (2.9%) | 314 (3.1%) |
| Heart rate (beats per minute) | ||
| < 77 | 875 (8.7%) | 871 (8.6%) |
| 77–91 | 1727 (17.1%) | 1770 (17.5%) |
| 92–107 | 2556 (25.3%) | 2546 (25.2%) |
| > 107 | 4872 (48.3%) | 4853 (48.0%) |
| Not known | 63 (0.62%) | 74 (0.73%) |
| GCS score (total) | ||
| Severe (3–8) | 1799 (17.8%) | 1839 (18.2%) |
| Moderate (9–12) | 1353 (13.4%) | 1351 (13.4%) |
| Mild (13–15) | 6934 (68.7%) | 6908 (68.3%) |
| Not known | 7 (0.07%) | 16 (0.16%) |
| Any protocol violation | 39 (0.4%) | 39 (0.4%) |
Primary outcome data were available for 20,127 (99.6%) randomised patients (10,060 patients allocated to TXA group and 10,067 patients to placebo group), of whom 19,944 (99.1%) patients were known to have completed the loading dose and 18,965 (94.2%) patients completed the 8-hours maintenance dose. A total of 3076 (15.3%) patients died, of whom 1086 (35.3%) died on the day of randomisation (see Figure 2). There were 1063 deaths due to bleeding, of which 637 (59.9%) were on the day of randomisation.
FIGURE 2.
All-cause mortality by subgroup.
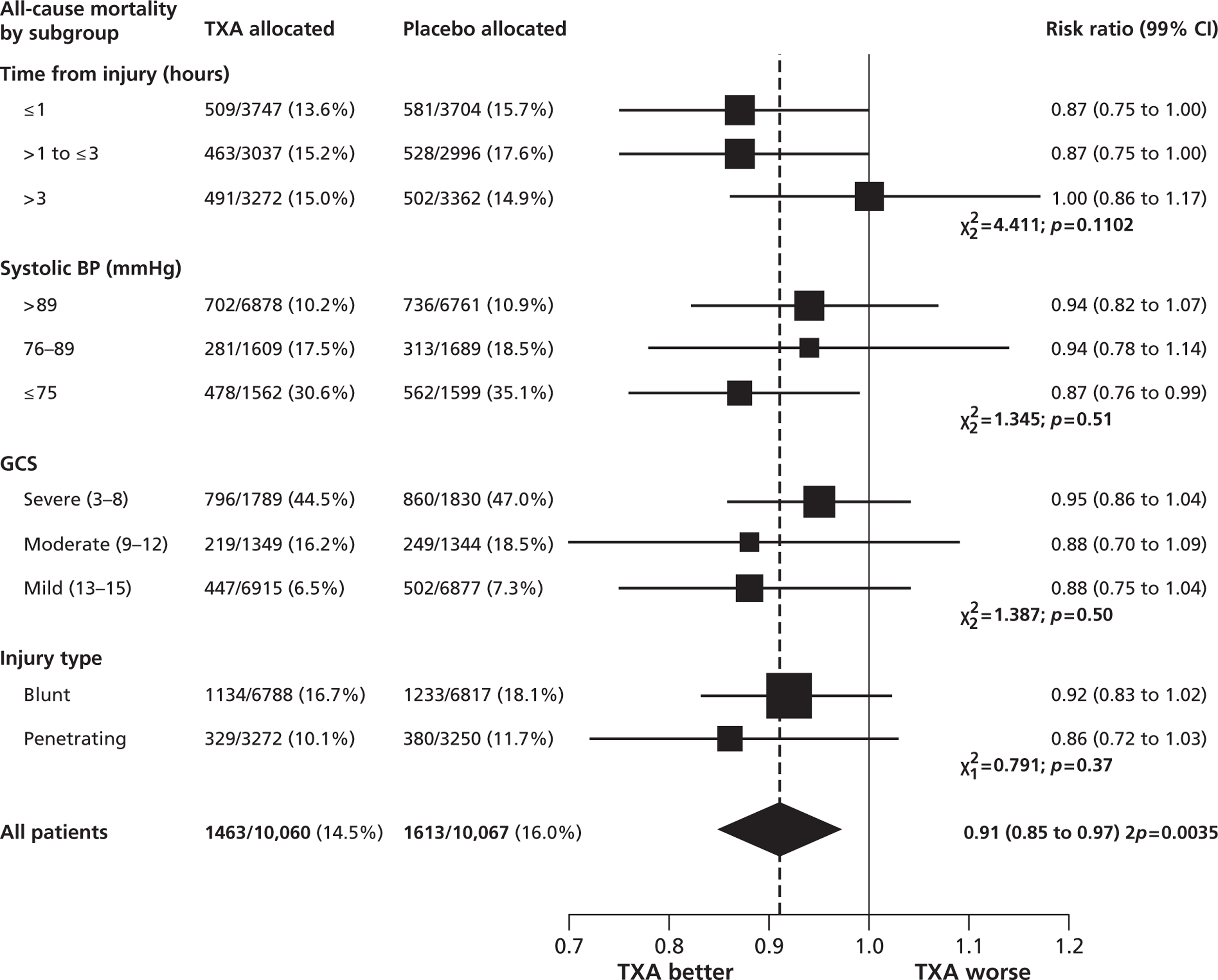
All-cause mortality was significantly reduced with TXA (Table 2). The RR of death with TXA was 0.91 (95% CI 0.85 to 0.97; p = 0.0035). The risk of death due to bleeding was significantly reduced. This effect was also apparent for deaths due to bleeding on the day of randomisation [282 patients (2.8%) died in the TXA group vs 355 (3.5%) in the placebo group: RR 0.80; 95% CI 0.68 to 0.93; p = 0.0036]. There were 33 (0.3%) deaths in the TXA group compared with 48 (0.5%) deaths in the placebo group from vascular occlusion (these included 7 vs 22 deaths from a myocardial infarction, 8 vs 5 deaths from a stroke and 18 vs 21 deaths from pulmonary embolism). Deaths from multiorgan failure, from head injury, or due to other causes did not differ significantly in the TXA group compared with the placebo group.
| Cause of death | TXA (n = 10,060) | Placebo (n = 10,067) | RR (95% CI) | p-value |
|---|---|---|---|---|
| Any cause of death | 1463 (14.5%) | 1613 (16.0%) | 0.91 (0.85 to 0.97) | 0.0035 |
| Bleeding | 489 (4.9%) | 574 (5.7%) | 0.85 (0.76 to 0.96) | 0.0077 |
| Vascular occlusiona | 33 (0.3%) | 48 (0.5%) | 0.69 (0.44 to 1.07) | 0.096 |
| Multiorgan failure | 209 (2.1%) | 233 (2.3%) | 0.90 (0.75 to 1.08) | 0.25 |
| Head injury | 603 (6.0%) | 621 (6.2%) | 0.97 (0.87 to 1.08) | 0.60 |
| Other causes | 129 (1.3%) | 137 (1.4%) | 0.94 (0.74 to 1.20) | 0.63 |
Vascular occlusive events (fatal or non-fatal) did not differ significantly, with 168 (1.7%) patients having one or more vascular occlusive events (myocardial infarction, stroke, pulmonary embolism or deep-vein thrombosis) in the TXA group compared with 201 (2.0%) patients in the placebo group (Table 3).
| TXA (n = 10,060) | Placebo (n = 10,067) | RR (95% Cl) | p-value | |
|---|---|---|---|---|
| Vascular occlusive eventsa | ||||
| Any vascular occlusive event | 168 (1.7%) | 201 (2.0%) | 0.84 (0.68 to 1.02) | 0.084 |
| Myocardial infarction | 35 (0.4%) | 55 (0.5%) | 0.64 (0.42 to 0.97) | 0.035 |
| Stroke | 57 (0.6%) | 66 (0.7%) | 0.86 (0.61 to 1.23) | 0.42 |
| Pulmonary embolism | 72 (0.7%) | 71 (0.7%) | 1.00 (0.73 to 1.40) | 0.93 |
| Deep-vein thrombosis | 40 (0.4%) | 41 (0.4%) | 0.98 (0.63 to 1.51) | 0.91 |
| Management | ||||
| Any surgery | 4814 (47.9%) | 4836 (48.0%) | 1.00 (0.97 to 1.02) | 0.79 |
| Neurosurgery | 1040 (10.3%) | 1059 (10.5%) | 0.98 (0.91 to 1.07) | 0.67 |
| Chest surgery | 1518 (15.1%) | 1525 (15.1%) | 1.00 (0.93 to 1.06) | 0.91 |
| Abdominal surgery | 2487 (24.7%) | 2555 (25.4%) | 0.97 (0.92 to 1.00) | 0.28 |
| Pelvic surgery | 683 (6.8%) | 648 (6.4%) | 1.05 (0.95 to 1.17) | 0.31 |
| Blood product transfused | 5067 (50.4%) | 5160 (51.3%) | 0.98 (0.96 to 1.01) | 0.21 |
| Median (IQR) units of blood product transfusedb | 3 (2–6) | 3 (2–6) | 0.59c | |
| Dependency | ||||
| No symptoms | 1483 (14.7%) | 1334 (13.3%) | 1.09 (1.02 to 1.17) | 0.0086 |
| Minor symptoms | 3054 (30.4%) | 3061 (30.4%) | 0.98 (0.94 to 1.02) | 0.39 |
| Some restriction | 2016 (20.0%) | 2069 (20.6%) | 0.96 (0.91 to 1.01) | 0.13 |
| Dependent (not requiring constant attention) | 1294 (12.9%) | 1273 (12.6%) | 1.00 (0.93 to 1.07) | 0.98 |
| Fully dependent | 696 (9.9%) | 676 (6.7%) | 1.01 (0.92 to 1.12) | 0.79 |
| Alive (disability status not known) | 54 (0.5%) | 41 (0.4%) | ||
| Dead | 1463 (14.5%) | 1613 (16.0%) | 0.91 (0.85 to 0.97) | 0.0035 |
Blood product transfusions were given to 5067 (50.4%) patients allocated to TXA compared with 5160 (51.3%) patients allocated to placebo (see Table 3). Those patients allocated to TXA and transfused received a mean of 6.06 (SD 9.98) blood units compared with a mean of 6.29 (SD 10.31) blood units for those allocated to the placebo. A total of 4814 (47.9%) patients in the TXA group received one or more surgical intervention (neurosurgery, chest surgery, abdominal surgery or pelvic surgery) compared with 4836 (48.0%) patients in the placebo group. Only 17 patients received treatment with rFVIIa (13 in the TXA group vs 4 in the placebo group). One hundred and thirty-two patients in each group had gastrointestinal bleeding (p = 0.99).
Of patients allocated to TXA, 3453 (34.3%) were classified as dead or dependent at discharge or on day 28 if still in hospital compared with 3562 (35.4%) of those allocated to placebo (RR 0.97; 95% CI 0.93 to 1.00; p = 0.12). A total of 1483 (14.7%) patients in the TXA group had no symptoms at discharge or on day 28 if still in hospital compared with 1334 (13.3%) patients in the placebo group (see Table 3). A total of 1846 (9.2%) patients were still in hospital at 28 days (958 patients in the TXA group vs 888 patients in the placebo group).
We had prespecified that, unless there was strong evidence (p < 0.001) against homogeneity of effects, the overall RR would be regarded as the most reliable guide as to the approximate RRs in all subgroups. We recorded no such evidence of heterogeneity for any of the prespecified subgroup analyses: systolic BP (heterogeneity p = 0.51); GCS score at randomisation (p = 0.50); type of injury (p = 0.37); or time from injury to randomisation (p = 0.11). No emergency unblinding was needed, and there were no adverse events regarded as serious, unexpected, or suspected to be related to the study treatment.
Chapter 4 Exploratory analyses on death due to bleeding
Introduction
The CRASH-2 trial was motivated by the evidence that TXA reduces bleeding in patients undergoing elective surgery, and the hypothesised mechanism was inhibition of fibrinolysis leading to improved clinical effectiveness of haemostasis. 20 However, as seen in the previous chapter, no significant difference was recorded in transfusion requirements between the TXA and placebo groups, and the CRASH-2 trial did not measure the effect of this drug on fibrinolytic assays. Thus, an alternative hypothesis is that TXA might act by reducing the pro-inflammatory effects of plasmin rather than by improving haemostasis. 21 In order to examine this issue, we conducted the same prespecified subgroup analyses as we did for all cause mortality, but for the outcome that we hypothesised would be most affected by TXA, specifically mortality due to bleeding.
Methods
We examined the effect of the trial treatment on death due to bleeding subdivided by four baseline characteristics: (1) time from injury to treatment (≤ 1, > 1 to ≤ 3 and > 3 hours); (2) severity of haemorrhage as assessed by systolic BP (≤ 75, 76–89 and > 89 mmHg); (3) GCS score (severe 3–8, moderate 9–12 and mild 13–15); and (4) type of injury (penetrating only, blunt plus blunt and penetrating). These were the same subgroup analyses that were reported in the previous chapter, but for the outcome of death due to bleeding rather than for all-cause mortality.
Heterogeneity in treatment effects across subgroups was assessed by a chi-squared test. We had prespecified that, unless there was strong evidence against the null hypothesis of homogeneity of effects (i.e. p < 0.001), the overall RR would be considered the most reliable guide to the approximate RRs in all subgroups. To test the independence of any observed treatment interactions we ran a logistic model including all possible interactions in the four prespecified baseline characteristics and treatment subgroups.
A logistic regression was estimated with death due to bleeding as the dependent variable, with treatment group and time to treatment as explanatory factors. We included an interaction parameter to allow for a proportional change in the odds ratio (OR) as time to treatment increases. ORs and 95% CIs were estimated for different times to treatment. CIs were calculated with a logistic model with time as a continuous term and an interaction term between time and TXA. We also ran a model with an interaction term for time to treatment squared to allow for a non-constant proportional change in the OR.
Results
Of the 3076 deaths from all causes, death due to bleeding accounted for 1063 (35%). The risk of death due to bleeding was significantly reduced with TXA. A total of 489 of 10,060 (4.9%) patients died because of bleeding in the TXA group compared with 574 of 10,067 (5.7%) patients in the placebo group (RR 0.85; 95% CI 0.76 to 0.96; p = 0.0077). We noted no significant effect on the risk of death for all other (non-bleeding) causes combined (Table 4).
| All causes of death | Bleeding death | Non-bleeding death | |
|---|---|---|---|
| RR (95% CI) | 0.91 (0.85 to 0.97); p = 0.0035 | 0.85 (0.76 to 0.96); p = 0.0077 | 0.94 (0.86 to 1.02); p = 0.13 |
| Time to treatment (hours) | |||
| ≤ 1 | 0.87 (0.76 to 0.97) | 0.68 (0.57 to 0.82) | 1.04 (0.89 to 1.21) |
| > 1 to ≤ 3 | 0.87 (0.77 to 0.97) | 0.79 (0.64 to 0.97) | 0.91 (0.78 to 1.05) |
| > 3 | 1.00 (0.90 to 1.13) | 1.44 (1.12 to 1.84) | 0.89 (0.78 to 1.02) |
| Chi-squared test of homogeneity | 4.411 (p = 0.11) | 23.516 (p = 0.0000) | 2.537 (p = 0.28) |
Table 5 shows the baseline characteristics of patients according to time to treatment.
| Characteristic | Time since injury (hours) | ||
|---|---|---|---|
| ≤ 1 (n = 7451) | 1–3 (n = 6033) | > 3 (n = 6634) | |
| Age (years) | |||
| Mean (SD) | 33.4 (13.9) | 35.0 (14.0) | 35.5 (14.8) |
| < 25 | 2283 (30.6%) | 1557 (25.8%) | 1773 (26.7%) |
| 25–34 | 2360 (31.7%) | 1832 (30.4%) | 1882 (28.4%) |
| 35–44 | 1356 (18.2%) | 1177 (19.5%) | 1262 (19.0%) |
| > 44 | 1452 (19.5%) | 1467 (24.3%) | 1716 (25.9%) |
| BP (mmHg) | |||
| ≤ 75 | 1380 (18.5%) | 1012 (16.8%) | 768 (11.6%) |
| 76–89 | 1203 (16.1%) | 1064 (17.6%) | 1029 (15.5%) |
| > 89 | 4857 (65.2%) | 3955 (65.6%) | 4821 (72.7%) |
| Heart rate (beats per minute) | |||
| < 77 | 681 (9.1%) | 450 (7.5%) | 603 (9.1%) |
| 77–91 | 1189 (16.0%) | 971 (16.1%) | 1326 (20.0%) |
| 92–107 | 1888 (25.3%) | 1562 (25.9%) | 1625 (24.5%) |
| > 107 | 3637 (48.8%) | 2990 (49.6%) | 3059 (46.1%) |
| Respiratory rate (per minute) | |||
| < 10 | 149 (2.0%) | 82 (1.4%) | 77 (1.2%) |
| 10–29 | 6144 (82.5%) | 4992 (82.7%) | 5590 (84.3%) |
| > 29 | 1077 (14.5%) | 901 (14.9%) | 923 (13.9%) |
| Capillary refill time (seconds) | |||
| ≤ 2 | 2450 (32.9%) | 2140 (35.5%) | 2217 (33.4%) |
| 3–4 | 3472 (46.6%) | 2773 (46.0%) | 3110 (46.9%) |
| > 4 | 1131 (15.2%) | 963 (16.0%) | 1257 (19.0%) |
| GCS score (total) | |||
| Severe | 1000 (13.4%) | 1124 (18.6%) | 1494 (22.5%) |
| Moderate | 868 (11.7%) | 915 (15.2%) | 909 (13.7%) |
| Mild | 5577 (74.9%) | 3994 (66.2%) | 4214 (63.5%) |
| Continents | |||
| Asia | 1213 (16.3%) | 2475 (41.0%) | 3656 (55.1%) |
| Africa | 2490 (33.4%) | 1437 (23.8%) | 872 (13.1%) |
| Central and South America | 2453 (32.9%) | 1456 (24.1%) | 1355 (20.4%) |
| North America, Europe and Oceania | 1295 (17.4%) | 665 (11.0%) | 751 (11.3%) |
Figure 3 shows the results of the subgroup analyses for death due to bleeding. Time to treatment was unknown in nine participants. Treatment given ≤ 1hour from injury significantly reduced the risk of death due to bleeding [198 out of 3747 patients (5.3%) died in the TXA group vs 286 out of 3704 patients (7.7%) in the placebo group: RR 0.68; 95% CI 0.57 to 0.82; p < 0.0001]. Treatment given between 1 and 3 hours also reduced the risk of death due to bleeding [147 out of 3037 patients (4.8%) died in the TXA group vs 184 out of 2996 patients (6.1%) in the placebo group: RR 0.79; 95% CI 0.64 to 0.97; p = 0.03]. Treatment given > 3 hours after injury significantly increased the risk of death due to bleeding [144 out of 3272 patients (4.4%) died in the TXA group vs 103 out of 3362 patients (3.1%) in the placebo group: RR 1.44; 95% CI 1.12 to 1.84; p = 0.004].
FIGURE 3.
Mortality due to bleeding by subgroups.
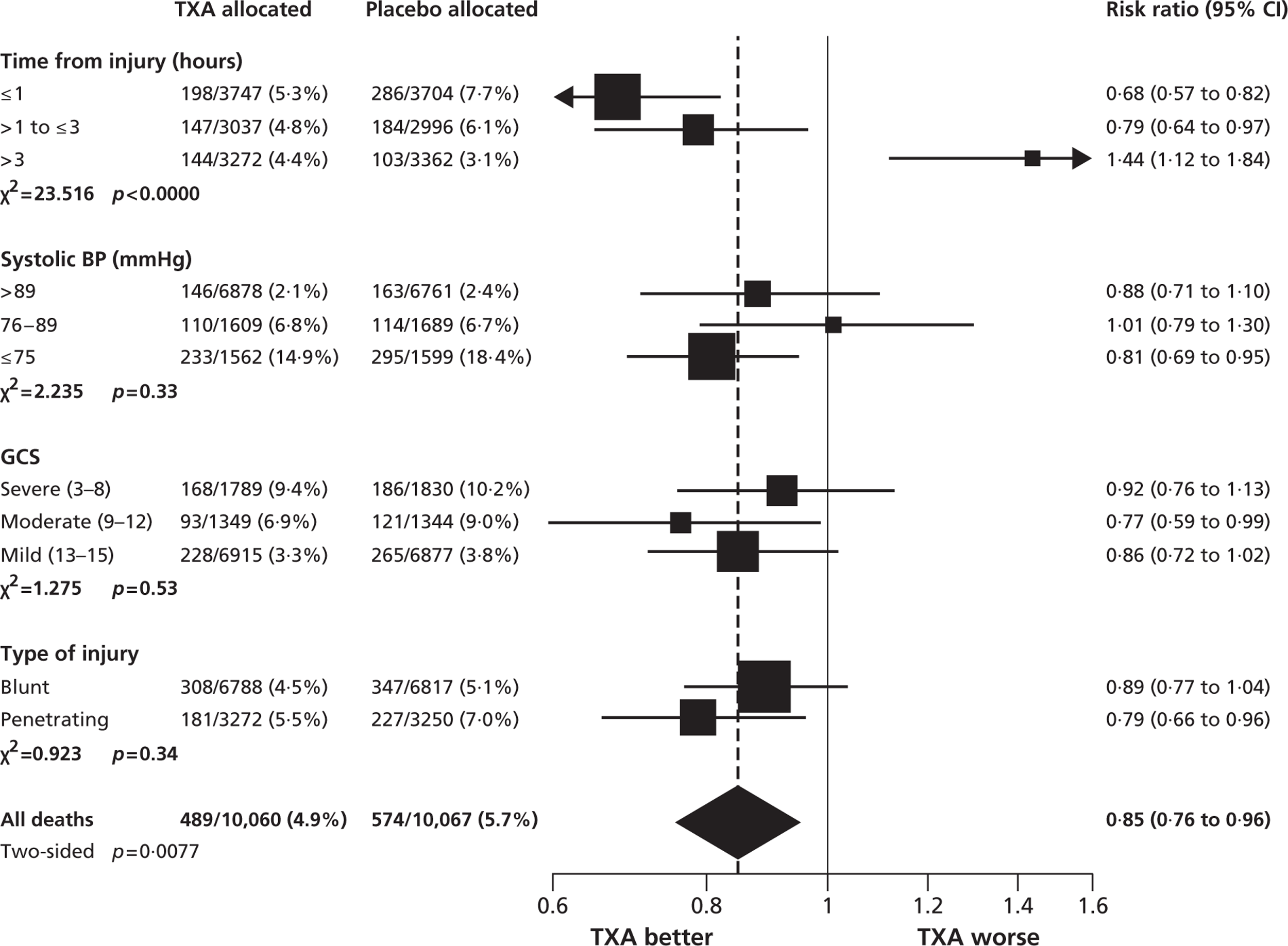
We recorded strong evidence that the effect of TXA on death due to bleeding varied according to time from injury to treatment (p < 0.0001). The evidence for interaction remained strong even after adjustment for interactions between the other prespecified baseline characteristics and treatment (p < 0.0001; data not shown).
The estimated OR of TXA on death due to bleeding when given immediately after injury was 0.61 (95% CI 0.50 to 0.74). We estimated that this OR is multiplied by 1.15 (95% CI 1.08 to 1.23) for every hour that passes since the injury. Figure 4 shows how the OR and 95% CIs vary with time to treatment. The interaction term for time to treatment squared was not significant (OR = 0.99; p = 0.38).
FIGURE 4.
Effect of TXA on death due to bleeding by time to treatment. Shaded area represents 95% CI.
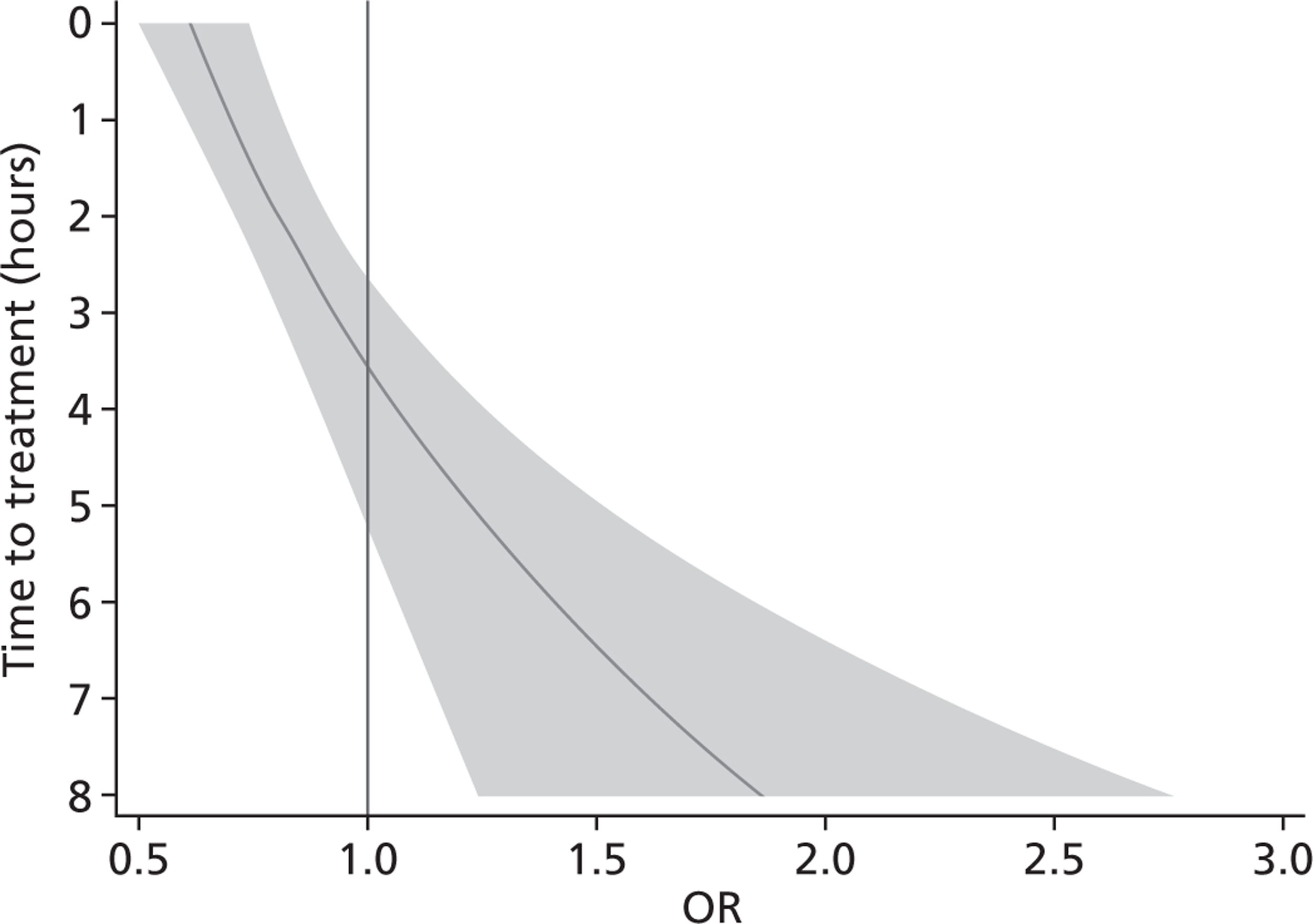
We recorded no evidence of heterogeneity for the subgroup analyses according to systolic BP, GCS score at randomisation or type of injury (see Figure 3). We detected no evidence of heterogeneity in the effect of TXA on the risk of non-bleeding deaths (see Table 4).
Chapter 5 Economic evaluation
Introduction
Although the primary objective of the trial was to assess the clinical effectiveness of TXA, there is a widespread recognition that, should TXA prove to be a clinically effective intervention, information is also required regarding its cost-effectiveness. Given that TXA appears to be clinically effective, its cost-effectiveness might appear obvious given that it is a relatively inexpensive intervention. However, in countries with limited health-care budgets, it is very important to establish the ratio of incremental cost to incremental effect. In the absence of any evidence of heterogeneity in the treatment effect between countries, both the cost of TXA and the effectiveness of TXA [in terms of life-years (LYs) gained] will still depend on the setting of analysis. In low-income countries (LICs) and in middle-income countries (MICs) life expectancies are shorter than in high-income countries (HICs). Consequently, any gain in LYs associated with TXA will tend to be lower than in HICs. The incremental cost of a policy of giving TXA will also tend to be higher in HICs than in both MICs and LICs (see Appendix 6).
The objective of the economic evaluation is to assess the cost-effectiveness of TXA for the treatment of significant haemorrhage following trauma in three different health-care settings: Tanzania [LIC with gross domestic product (GDP) per capita $509], India (MIC with GDP per capita $1134) and the UK (HIC with GDP per capita $35,165). 22
Methods
The cost-effectiveness of TXA was measured in terms of the additional costs per LY gained. A lifetime Markov model was developed in order to estimate the LYs gained as a result of the intervention. In each cycle, assumed to last 1 year, patients are either alive or dead. LYs gained were discounted at an annual rate of 3.5% in accordance with the National Institute for Health and Clinical Excellence recommendation [and broadly consistent with the 3% rate used for disability-adjusted life-years (DALYs)]. The number of LYs saved for each death averted by TXA depends on the age and sex of the patients. In the CRASH-2 trial, consistent with the statistics reported by Norton et al. ,23 the majority of the trauma victims were aged between 16 and 35 years, and only a small proportion were female. In order to estimate the incremental LYs gained by TXA, five age groups were identified, each containing about one-fifth of the trial participants: 16–20 years, 21–25 years, 26–34 years, 35–50 years and > 50 years. The mean age of trial participants in each age group was calculated and the model was then run for cohorts with these starting ages: 18 years, 22 years, 30 years, 42 years and 75 years old. The overall number of LYs saved in each country was then estimated as a weighted average of the results for the five age groups using the age distribution for trial participants in countries of the relevant income group (low, middle or high). All analyses were carried out using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA) and Stata 11 (StataCorp LP, College Station, TX, USA).
Since the CRASH-2 trial recorded data up to 28 days or death, a parametric survival function was fitted to extrapolate mortality experience over the 12 months following injury. Four different parametric survival functions (Weibull, Gompertz, log-logistic and log-normal) were fitted to data from the placebo arm and the best-fitting model was selected using the Akaike information criterion (AIC). Cox–Snell residuals were plotted as a confirmatory test. 24 Both the AIC test and the Cox–Snell residuals suggest that the Gompertz parametric model fits the data best. The AIC scores were 10,088 (Weibull), 10,065 (log-logistic), 9,954 (log-normal) and 9,425 (Gompertz). The four functions and the Cox–Snell plots are reported in Appendix 6.
The Gompertz survival function,24 which has been extensively used in medical research to model mortality data, has the following cumulative hazard rate:
where γ determines if the hazard function increases with time (if γ is positive) or decreases (if γ is negative). If γ is equal to zero, the hazard function follows an exponential model. t is the time frame over which the cumulative probability of dying is estimated.
In the analyses, three covariates were explored: age, sex and GDP. When covariates are considered in the analysis, λ is given by the following equation:24
Using CRASH-2 data the cumulative hazard rate is:
As expected, γ (–0.20; 95% CI –0.21 to –0.18) is negative, implying that after trauma the hazard rate decreases over time. The probability of dying increases with age (age coefficient = 0.020; 95% CI 0.016 to 0.024), whereas sex was not found to be influential for the hazard rate (–0.06; 95% CI –0.22 to 0.11). A GDP per capita was assigned to each country in the trial according to the latest World Bank estimates and two binary variables, x2 and x3, were constructed to estimate whether or not the likelihood of death changes according to the GDP. 22x2 took the value 1 for LICs and 0 otherwise. Similarly, x3 took the value of 1 for MICs and 0 otherwise. GDP coefficients (β2 = −0.31; β3 = −0.61) were found to be highly significant. Thus, the function for λ is:
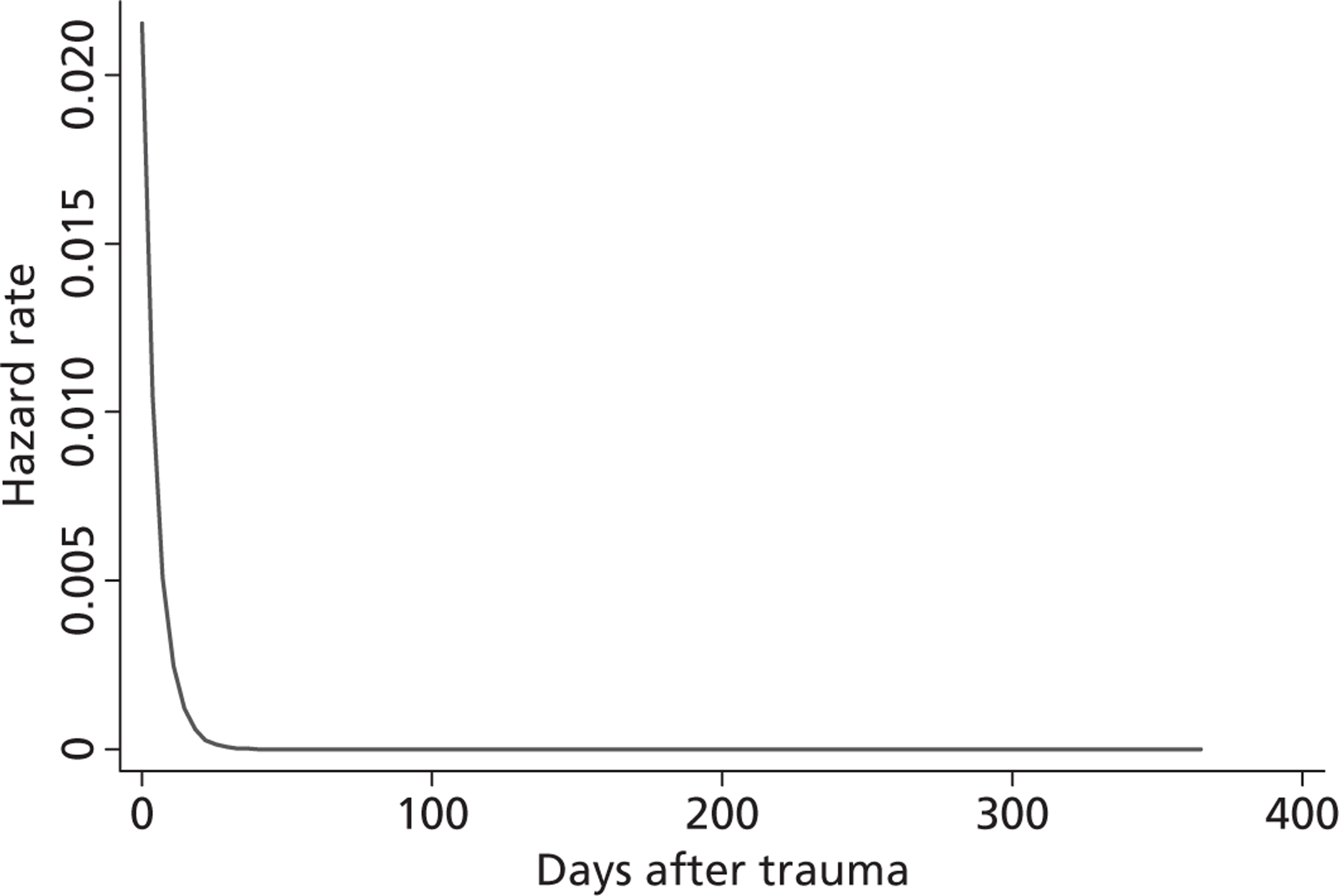
Figure 5 shows the hazard rate function during the first year after trauma observed in the placebo group. The hazard rate decreases almost to zero in the first 40 days after hospital admission and remains constant for the rest of the year. This finding is consistent with previous studies which suggest that the majority of trauma-related deaths occur within a few weeks after injury. The figure also validates the model assumption that, after 1 year, the baseline risk of death is the same observed in the general population. 25
FIGURE 5.
Gompertz regression. Hazard rate function during the first year after trauma observed in the placebo group.
Risk of death during the first year following trauma in the TXA group was estimated by multiplying the cumulative hazard for the placebo group by the RR reduction in all-cause mortality estimated by the CRASH-2 trial (RR = 0.87; 95% CI 0.81 to 0.95). Beyond 12 months, the risk of death is assumed to be equal whether or not the patient received TXA, and is set equal to the risk of death for the relevant age–sex group in the general population.
The evaluation was conducted from a health services' perspective. 26 Resource use in the TXA and placebo arms was compared. As a consequence, two cost items were considered in the present study: (1) the cost of administering TXA to bleeding trauma patients; and (2) the incremental cost of non-intensive care hospital stay. There was no evidence from the trial of any other differences in resource use, such as time spent in intensive care, the frequency of transfusion or the volumes transfused. Costs have been converted from national currencies into international dollars ($) using purchasing power parities (Organisation for Economic Co-operation and Development and Penn World Table). 27 Where necessary, the US Consumer Price Index was used to inflate prices. 28 All costs are expressed in 2008–9 values. Costs were not discounted, as the costs associated with giving TXA occur within the year following trauma.
One-way sensitivity analysis was undertaken in order to assess the impact of uncertainty regarding the input parameters [RR of death with TXA, cost of TXA, cost of additional non-intensive care unit (ICU) stay and cost per non-ICU day] and the choice of cumulative hazard function. The incremental cost per LY saved for TXA compared with no TXA was estimated using the 95% confidence limits for the RR of death with TXA observed in the trial (0.81 and 0.95). 29 In order to evaluate the effect of variation in the price of TXA on the incremental cost-effectiveness ratio (ICER), the price was assumed to vary between $2.57/g (from Casati et al. 30) and $45.67/g (from Eaton31). One-way sensitivity analysis was also conducted on the increase in non-ICU hospital stay following TXA administration, again using the confidence limits from the trial (0.007 and 0.08). For the cost of a non-ICU day, the lowest estimates, for both Tanzania and India, were the cost per day in a primary health centre ($9.84 and $18.75, respectively) whereas the highest estimates ($18.69 and $35.41) were obtained from tertiary hospitals. 32,33 For the UK, the lower and the upper estimates of cost per non-ICU day ($90–784) were taken from UK reference costs. 34
A further deterministic sensitivity analysis was performed to investigate how the results change if different parametric distributions are adopted (Weibull, log-normal and log-logistic) when estimating survival beyond 28 days.
In the probabilistic sensitivity analysis, uncertainty in the data was captured by fitting probability distributions to each parameter (see Appendix 6 for details). Beta and log-normal distributions were used for binomial data and RR parameters, respectively. For costs a ‘right-tailed’ γ distribution was adopted. Finally, to account for correlation among the parameters of the Gompertz survival function a Cholesky decomposition of the covariance matrix was performed. For each country, 1000 second-order Monte Carlo simulations of the expected incremental cost, LYs saved and net benefits of TXA were produced using the following formula:
where willingness to pay (WTP) is the WTP per LY saved, ΔE is the incremental number of LYs saved and ΔC is the incremental cost of the TXA. Cost-effectiveness acceptability curves (CEACs) were drawn for each of the three countries to show the probability that TXA is cost-effective for WTP values from $0 to $250 per LY saved.
Economic evaluation results
Cost of administering tranexamic acid
Resource use and unit cost associated with TXA administration are reported in Table 6. There are no recent studies reporting the national cost of TXA per gram in Tanzania and India. In the main analysis, the global cost of TXA, $5.70/g, was obtained from the British National Formulary and assumed to be the same for all the three countries considered. 35 The average cost per hour of a nurse was obtained through personal communications with local hospitals. 36–38 In the UK, it was retrieved from the publication Unit Costs of Health and Social Care. 39,40 The cost of a syringe was retrieved from Dziekan et al.'s41 study, which estimated the average unit cost of syringes and needle set in the different regions of the world. i.v. infusion and saline bag prices were obtained from the British National Formulary35 and converted into international dollars ($). As TXA can be stored at room temperature (15–30°C), storage and distribution costs per intervention are negligible. The overall cost of administering TXA per patient is estimated to be $17.48, $19.55 and $30.83 in Tanzania, India and the UK, respectively. Owing to the low cost of health-care personnel, in both MICs and LICs, the drug cost constitutes almost 70% of the overall intervention cost.
| Resource use and unit cost | Tanzania | India | UK |
|---|---|---|---|
| Resource use | |||
| TXA dosage (g) | 2 | 2 | 2 |
| 100-ml saline bag | 1 | 1 | 1 |
| 500-ml saline bag | 1 | 1 | 1 |
| Minutes spent by nurse preparing and administering TXA | 21 | 21 | 21 |
| Average number of non-ICU days | 10 | 7 | 8 |
| Unit cost ($) | |||
| TXA per gram | 5.70 | 5.70 | 5.70 |
| Nurse per hour | 2.37 | 8.08 | 38 |
| Syringes and needles | 0.18 | 0.19 | 0.23 |
| i.v. administration set | 4.35 | 4.35 | 4.35 |
| Non-ICU hospital cost per day | 13 | 28 | 429 |
Cost of non-intensive care unit hospital stay
The CRASH-2 trial data indicated that receipt of TXA is associated with a slight increase in the number of days spent in non-ICU hospital facilities (mean difference = 0.04; 95% CI 0.007 to 0.08; p = 0.0095). The estimated unit cost per day in non-ICU facilities for Tanzania and India were obtained from WHO CHOsing Interventions that are Cost Effective (CHOICE)32,33 and uplifted to 2008–9 values. For the UK, the estimated cost comes from the Reference Costs. 34
Base-case analysis
Table 7 presents the base-case cost-effectiveness results. As shown, the TXA strategy is always cost increasing because of the administration cost and the longer non-ICU hospital stay. As expected, the UK shows the highest incremental cost, at $48,002, because health-care services are more expensive. The incremental cost of TXA is considerably lower in Tanzania and India ($18,025 and $20,670, respectively). Over a lifetime horizon, TXA would save 372, 315 and 755 LYs per 1000 patients in Tanzania, India and the UK, respectively. Each of these estimates takes into account the age distribution of trauma patients in the three countries.
| Item | Tanzania | India | UK |
|---|---|---|---|
| Non-ICU hospital stay ($)a | |||
| TXA | 135,183 | 213,435 | 3,272,416 |
| No TXA | 134,641 | 212,315 | 3,255,244 |
| TXA administration cost ($)a | |||
| TXA | 17,483 | 19,550 | 30,830 |
| Overall incremental costa | 18,025 | 20,670 | 48,002 |
| LYs gained discounteda | |||
| TXA | 13,079 | 18,176 | 24,162 |
| No TXA | 12,707 | 17,861 | 23,407 |
| Incremental LY saveda | 372 | 315 | 755 |
| Incremental cost per LY saved ($) | 48 | 66 | 64 |
Where life expectancy is higher, as in the UK, TXA will potentially save a greater number of LYs. Nevertheless, the number of LYs saved in Tanzania is higher than in India because Tanzanian patients are more likely to die in the first year following trauma. The incremental cost per LY saved of TXA is $48, $66 and $64 for Tanzania, India and the UK, respectively (see Table 7 and Appendix 6).
One-way sensitivity analysis
One-way sensitivity analyses were conducted on various model parameters. As shown in Table 8, the incremental cost per LY saved of TXA is influenced by the clinical effectiveness of the intervention. If TXA was associated with a 5% reduction in the probability of death (higher bound of the 95% CI), the incremental cost of TXA would be $126 in Tanzania, $170 in India and $168 in the UK.
| Sensitivity analysis | Cost per LY gained ($) | ||
|---|---|---|---|
| Tanzania | India | UK | |
| Base case | 48 | 66 | 64 |
| RR of death with TXA vs non-TXA | |||
| 0.81 | 33 | 45 | 43 |
| 0.95 | 126 | 170 | 168 |
| TXA drug cost | |||
| $2.57 | 8 | 12 | 26 |
| $45.60 | 124 | 148 | 86 |
| Additional non-ICU hospital stay for TXA patients | |||
| 0.007 days | 47 | 63 | 45 |
| 0.080 days | 50 | 69 | 86 |
| Cost of non-ICU hospital stay (per day) | |||
| Low unit cost | 47 | 64 | 46 |
| High unit cost | 49 | 67 | 82 |
| Parametric distribution used | |||
| Weibull | 31 | 37 | 34 |
| Log-normal | 35 | 46 | 39 |
| Log-logistic | 25 | 42 | 32 |
If the price of TXA is as low as $2.57, as suggested by Casati et al.,30 the incremental cost per LY saved would be $8 (Tanzania), $12 (India) and $26 (UK). Assuming a TXA price of $45.60 the TXA incremental cost per LY saved would be $124, $148, $86 in Tanzania, India and the UK, respectively. Assuming a longer incremental non-ICU hospital stay in Tanzania and India (mean difference range of 0.007–0.08 days) has little impact on the results. In UK, where the cost per non-ICU day is high, variations in the incremental non-ICU length of stay have a greater impact on the cost-effectiveness of TXA.
The graphs of the cumulative hazard rates, by age quintile, extrapolated using different parametric functions are shown in Appendix 6 for LICs, MICs and HICs separately. As observed, during the first 30 days, all four of the parametric functions produce similar shapes of the cumulative hazard. After 30 days, the extrapolated Gompertz cumulative hazard rate levels off to a constant value, whereas the cumulative hazard rates extrapolated using other parametric survival functions keep increasing for the entire time frame of analysis. As a result, use of these other parametric distributions would generate higher cumulative baseline hazards at 1 year and consequently lower ICERs. For example, the incremental cost per LY saved is $35, $46 and $39 in Tanzania, India and the UK, respectively, if a log-normal parametric function were to be adopted.
Probabilistic sensitivity analysis
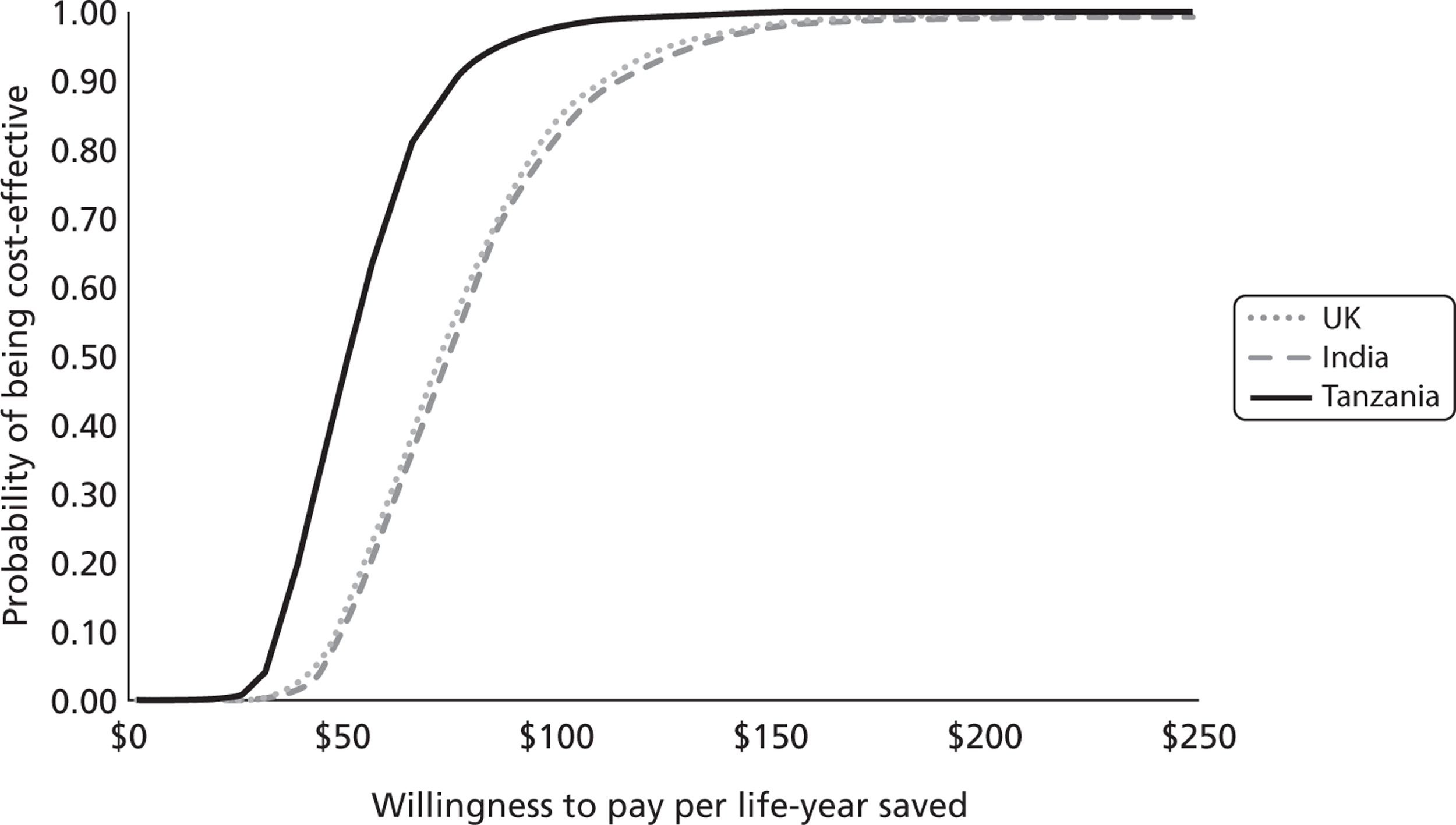
Figure 6 presents the CEACs for Tanzania, India and the UK. The CEACs are similar for India and the UK, whereas Tanzania is more likely to be cost-effective even for lower WTP values because of the lower incremental cost of TXA. For example, when WTP is $100 per LY saved, about 80% of simulations produce a positive net benefit in India and in the UK, whereas in Tanzania the corresponding probability is close to 100%.
FIGURE 6.
Willingness to pay per LY saved.
Chapter 6 Discussion
The results show that the early administration of TXA to trauma patients with, or at risk of, significant bleeding reduces the risk of death from haemorrhage, with no apparent increase in fatal or non-fatal vascular occlusive events. All-cause mortality was significantly reduced with TXA.
The trial inclusion criteria were clinical and did not depend on the results of laboratory tests. Patients were enrolled if they were judged to have ongoing significant haemorrhage, as evidenced by hypotension or tachycardia, or if they were considered to be at risk of significant haemorrhage (e.g. patients with compensated haemorrhage and stable vital signs, or those in whom bleeding might have stopped but in whom bleeding might recommence following volume resuscitation). The use of clinical inclusion criteria is appropriate in the context of traumatic bleeding, in which a range of clinical signs need to be assessed when establishing the presence or absence of haemorrhage, while taking into account remedial measures such as fluid resuscitation. The clinical inclusion criteria, and the large numbers of patients studied in a range of different health-care settings, help these results to be generalised widely.
Our study had strengths and limitations. The randomisation methods ensured that participating clinicians did not have foreknowledge of treatment allocation. Although few participating hospitals used the telephone randomisation system, which included a minimisation algorithm, baseline prognostic factors were well balanced. In many low- and middle-income hospitals, 24-hour access to outside telephone lines is difficult to obtain and the next pack system was found to be more convenient. All analyses were on an intention-to-treat basis and, because almost all randomised patients were followed up, there was no need to use imputation methods for missing data. 42 The primary end point was all-cause mortality, and the observed reduction in mortality with TXA was both statistically significant and clinically important. The diagnosis of traumatic haemorrhage can be difficult, and some of the included patients might not have been bleeding at the time of randomisation. This misdiagnosis would have reduced the power of the trial to show an effect of TXA on mortality from bleeding. Nevertheless, we recorded a significant reduction in death due to bleeding.
Although we recorded no increased risk of non-fatal vascular occlusive events with TXA, the precision of the estimates was low and we cannot exclude the possibility of some increase in risk. In the context of outcome assessment in clinical trials, estimates of the RR are unbiased even when the sensitivity of diagnosis is imperfect, provided that there are few false-positives (high specificity). 43 Therefore, we sought high specificity in the diagnosis of non-fatal vascular occlusive events and stipulated that occlusive events should be recorded only when there was clear clinical evidence. As a result, we might have under-reported the frequency of these events. However, our estimates of the RR of non-fatal occlusive events should be unbiased. 43
We did not find any substantial reduction in the receipt of a blood transfusion or the amount of blood transfused in trauma patients. This finding could be an indication of the difficulty of accurate estimation of blood loss in trauma patients when assessing the need for transfusion. Another possible explanation is that, after the loading dose, TXA was infused over 8 hours, whereas decisions about transfusion are made soon after admission. Finally, fewer deaths occurred in patients allocated to the TXA group than to the placebo group, and the patients who survived as a result of TXA administration would have had a greater opportunity to receive a blood transfusion (competing risks).
The TXA loading dose was given within 8 hours of injury, followed by a maintenance infusion over 8 hours. We chose the early administration of a short course of TXA because most deaths from bleeding occur on the day of the injury and we postulated that the drug would act by reducing bleeding. Generally, after the first day, the risk of death from haemorrhage is reduced but the risk of vascular occlusive events might remain. We therefore selected a regimen that would allow for the effect of TXA on the early risk of haemorrhage without extending into the period when the risk of vascular occlusive events might be increased by this treatment. The absence of any increase in vascular occlusion with TXA, whether fatal or non-fatal, provides reassurance that this regimen is safe.
The dose of TXA used in this trial was based on studies of this drug in surgical patients in which loading doses range from 2.5 mg/kg to 100 mg/kg, and maintenance doses from 0.25 mg/kg/hour to 4 mg/kg/hour, delivered over 1–12 hours. 20 Findings from studies of the effect of different doses of TXA on blood loss and blood transfusion showed no significant difference between high and low doses. Studies in cardiac surgery have noted that a 10 mg/kg-loading dose of TXA followed by an infusion of 1 mg/kg/hour produces plasma concentrations sufficient to inhibit fibrinolysis, and that a larger dose does not provide any additional haemostatic benefit. 13,14 In emergency situations, the administration of a fixed dose is practicable since determining the weight of a seriously injured patient can be difficult. We therefore selected a fixed dose within the range shown to inhibit fibrinolysis and provide haemostatic benefit that would be efficacious for larger patients (> 100 kg) but also safe in smaller patients (< 50 kg), to the extent that the dose per kg that smaller patients would receive has been used in surgical trials without adverse effects. The possibility that a higher dose of TXA would have a greater treatment effect remains open to debate and warrants further study.
Our exploratory analyses show clearly that the effect of TXA on death due to bleeding depends importantly on the time between injury and onset of treatment. Early treatment seems to be much more effective than late treatment. These results also raise the possibility that late treatment with TXA might increase the risk of death due to bleeding, although there was no evidence of any increase in all-cause mortality in patients treated after 3 hours. This finding might indicate that patients treated with TXA beyond 3 hours who died from bleeding might otherwise have died from some other non-bleeding cause (competing risks). If late administration does cause harm, this finding would be important, as many bleeding trauma patients in LICs and MICs have long prehospital times. Indeed, about one-third of trauma patients in the CRASH-2 trial were treated > 3 hours after the injury.
In clinical trials, a treatment is rarely found to be beneficial in one subgroup but harmful in another (qualitative interaction), and some methodologists recommend that qualitative interactions should generally be disbelieved. 44 The results of our exploratory analysis of the effect of TXA on death due to bleeding do, however, satisfy most of the criteria against which the credibility of subgroup results should be judged:44 time from injury was measured at baseline; the hypothesis that early treatment with TXA might be more clinically effective was prespecified in the trial protocol; the interaction suggests a very low likelihood that chance explains the findings; the interaction remained significant after controlling for the non-significant interactions between treatment and the other prespecified baseline prognostic factors; the subgroup effect is large; and a biological rationale supports the interaction. Although this clinical trial was not powered to examine subgroup effects, the interaction recorded is large and highly significant. 46
Nevertheless, we prespecified in our trial protocol that the main subgroup analyses would be undertaken for all-cause mortality and not for mortality due to bleeding. Even though we postulated that TXA would act by reducing bleeding, we focused on all-cause mortality because overall survival is most important to patients. However, in view of the significant reduction in all-cause mortality, most of which was attributable to the effect of TXA on death due to bleeding, and the biological rationale that this drug would act by improving haemostasis, our analyses, although not prespecified, would seem justified.
Acute severe trauma is associated with increased fibrinolysis that contributes to an early coagulopathy and increased mortality. 47,48 Fibrinolysis can be assessed by measurement of fibrin degradation products, which include small protein fragments called D-dimers. Brohi et al. 47 showed that D-dimer concentrations are raised in trauma patients at the time of hospital admission (median prehospital time 28 minutes), with the highest concentrations measured in the most severely injured patients. 47 Similar results were recorded in a 2009 study from Japan that measured fibrin degradation product and D-dimers in 314 severe trauma patients. 49 If this early increased fibrinolysis exacerbates bleeding and increases the risk of death, then we might expect that an antifibrinolytic drug such as TXA would be most effective in this period.
Although we had expected that early treatment with TXA might be most effective, the apparent increase in the risk of death due to bleeding in patients treated > 3 hours after the injury is unexpected and cannot readily be explained. It could be a chance finding and there might be no real biological effect. However, patients in the late phase of trauma can develop thrombotic disseminated intravascular coagulation and antifibrinolytics could be contraindicated in this period. 49,50 Although disseminated intravascular coagulation is characterised by fibrin formation and coagulation, the rapid consumption of coagulation proteins can lead to their exhaustion, resulting in uncontrolled bleeding. The need to avoid giving an antifibrinolytic in this late phase was why we restricted trial inclusion to patients who were within 8 hours of injury. The possibility that the change to a prothrombotic state might occur sooner than was previously expected is open to debate and needs further research. We should also bear in mind that patients who arrive at hospital many hours after injury are likely to differ from those who arrive early. For example, there could be an increased prevalence of hypothermia and acidosis. These or other differences could explain the decreased efficacy of TXA administration when it is given late.
The results of the exploratory analyses nevertheless strongly endorse the importance of early administration of TXA in bleeding trauma patients and suggest that trauma systems should be configured to facilitate this recommendation. In patients presenting late (several hours after injury), the clinician should be cautious and make an assessment of the individual benefits and risks of this treatment, as the drug is likely to be much less effective and possibly even harmful. To the extent that our subgroup analyses are consistent with the results of studies showing an early increased fibrinolytic coagulopathy, they support the hypothesis that TXA acts through the inhibition of fibrinolysis with improved haemostasis.
Can the result of the CRASH-2 trial be generalised?
There is widespread misunderstanding about the scientific basis for generalisation of the result of medical research. Some clinicians believe that the results of medical research can be generalised only to populations that are similar to those that were studied. This leads to a misplaced emphasis on the extent to which the study population is representative of the target population, to which the results are to be generalised. So, for example, when considering whether or not the results of the CRASH-2 trial, in which a large proportion of the randomised patients were recruited from MICs, can be applied to patients in the NHS, they consider whether or not the clinical care patients received in the trial was similar to that of patients in the NHS. As it is reasonably easy to imagine at least some differences, it is tempting to conclude that the results cannot be generalised. If this argument were valid, it would have dramatic implications for medical care. Since medical care is constantly changing as new forms of care are introduced, it would mean that all clinical trials would need to be repeated at regular intervals to update our medical knowledge to take account of the changing clinical situation. Fortunately, at least in most cases, this is not the case.
The first prerequisite for scientific generalisation is that the results to be generalised are valid and appropriately precise. Results from large-scale clinical trials, such as the CRASH-2 trial, that provide precise estimates of modest treatment effects meet this first criterion. Next, one has to make a scientific judgement about how the treatment worked and whether or not the mechanism of action is likely to be modified by other factors. In the case of the CRASH-2 trial, TXA is a drug that reduces fibrinolysis by inhibition of the blood enzyme plasmin. At least at present, there is no scientific reason to expect that TXA would work differently in different populations of the same species. The basic pathophysiology of fibrinolysis is going to be more or less the same in all members of the human species and it is likely that TXA would inhibit plasmin in a similar way. Of course, we cannot be completely sure that TXA worked through the inhibition of fibrinolysis, but then again we have to bear in mind that any proposed mechanism of action is a causal theory. At least at the moment, inhibition of fibrinolysis seems to be the most reasonable causal theory. In summary, we observe that TXA reduces bleeding in surgical patients most of whom are from HICs, we conduct a large trial and find that TXA reduces mortality in a heterogeneous group of bleeding trauma patients, we suppose that the mechanism of action of TXA is something to do with the inhibition of the enzyme plasmin, that all humans share, so we can reasonably conclude that TXA is likely to work in trauma patients in, for example, the USA, even though there were no patients from the USA included in the CRASH-2 trial.
Of course, at any given level of injury severity, the risk of death following severe trauma in the UK or USA might be different to the risk of death following trauma in, say, Tanzania. But this is not the point. The point is whether or not TXA can reasonably be expected to reduce the risk of death in all these settings. Experience from other large-scale international trials has shown that RRs appear to be remarkably homogeneous even when baseline risk varies. In other words, a treatment that reduces the risk of death by about one-third would reduce the risk of death from 30% to about 20% and from 3% to about 2%. Following the publication of the results of the CRASH-2 trial, several clinicians asked us to examine the extent to which the effect of TXA on the risk of death due to bleeding varied by geographical region. In response, the hospitals participating in the CRASH-2 trial were grouped into four geographical regions: (1) Africa, (2) Asia, (3) Europe, North America and Australia, and (4) South America. We examined the extent to which the effect of TXA on death due to bleeding varied across continents. We found no evidence for heterogeneity in the effect of TXA by region (χ2 = 1.445; p = 0.70).
Cost-effectiveness
Early administration (within 3 hours) of TXA would cost $48, $66 and $64 per LY saved in Tanzania, India and the UK, respectively. According to the WHO Commission on Macroeconomics, health-care interventions costing less than GDP per capita per DALY averted should be considered ‘very cost effective’. 51 DALYs are a measure of the years of life lost from disease and years lived with a disability. According to the World Bank classification, GDP per capita in LICs ranges from $380 to $975, in lower MICs between $976 and $3855, in upper MICs between $3856 and $11,905, and in HICs > $11,906. 52 Thus, if the LYs saved by TXA are spent in perfect health (one LY saved is equal to one DALY averted) then TXA is a highly cost-effective intervention in all the countries considered. The sensitivity analyses presented reinforce this conclusion.
However, a number of limitations should be considered when interpreting these results. It was necessary to model survival over 12 months using data for the first 28 days following the trauma, and different statistical models will produce different estimates of benefit. However, different models were explored and the one selected, as well as providing the best fit to the data, also produced more realistic estimates of the impact on long-term survival. In this evaluation, those predicted to survive the first year following the trauma are assumed then to have the same life expectancy as members of the general population of similar age and sex.
A related limitation is that the measure of incremental cost-effectiveness is cost per LY gained rather than cost per quality-adjusted life-year (QALY; a year in perfect health is considered equal to one QALY) gained or DALY averted. Depending on the extent to which these patients do not enjoy perfect health, the cost per QALY gained or the cost per DALY averted will be higher than the cost per LY saved.
A further potential limitation is that the analysis does not allow for future health service savings. The CRASH-2 trial showed that after 28 days the proportion of patients reporting no symptoms at discharge was significantly higher in the TXA group than in the placebo group. 29 If TXA patients are more likely to survive without disability, then this study undervalues the potential cost saving arising from the administration of TXA as healthier people will have lower future utilisation of health-care services.
Identifying cost-effective interventions to decrease the number of injury-related deaths is a major public health challenge. According to the Disease Control Priority Project, in 2001 unintentional injuries alone accounted for 6% of all deaths worldwide. 23 The majority of unintentional injury-related deaths, > 90%, occur every year in LICs and MICs, posing a further financial burden on these countries' economies and underfinanced health-care systems. 23
This analysis suggests that TXA is a highly cost-effective intervention in three very different settings. Given that TXA is clinically effective and it is of relatively low cost, it is not surprising that it is cost-effective in high-income settings. However, it was important to demonstrate that it was likely to be cost-effective in countries with much more limited health-care budgets, such as in Tanzania and India, where, owing to the high numbers of trauma victims, this simple intervention can avert thousands of deaths every year. Further research is needed to evaluate the effects of TXA on the quality of life of trauma patients.
Future clinical research
The knowledge that TXA is a highly cost-effective treatment to reduce the risk of death from traumatic bleeding raises the possibility that it might also be effective in other situations in which bleeding can be life-threatening or disabling. Traumatic brain injury is commonly accompanied by intracranial bleeding, which can develop or worsen after hospital admission. Traumatic intracranial haemorrhage is associated with an increased risk of death and disability, and, irrespective of location, haemorrhage size is strongly correlated with outcome. If TXA reduced intracranial bleeding after isolated traumatic brain injury, then patient outcomes might be improved. The Clinical Randomisation of an Antifibrinolytic in Significant Head injury-3 (CRASH-3) trial will assess the effect of TXA on patient outcomes after traumatic brain injury (http://crash3.lshtm.ac.uk/).
Tranexamic acid might also have a role in bleeding conditions apart from traumatic injury. Postpartum haemorrhage is a leading cause of maternal mortality, accounting for about 100,000 maternal deaths every year. 53 Although evidence suggests that this drug reduces postpartum bleeding, the quality of the existing trials is poor and none have been large enough to assess the effect of TXA on end points that are important to women. 54 A large trial is currently under way to assess the effect of TXA on the risk of death and hysterectomy in women with postpartum haemorrhage. 53
Chapter 7 Conclusions
The CRASH-2 trial was a publicly funded clinical trial of the effect of the generic drug TXA on mortality in bleeding trauma patients. It recruited 20,211 patients from 274 hospitals in 40 countries. The trial cost the UK taxpayer about £2,500,000. Compared with clinical trials conducted by drug companies, this is remarkably inexpensive. Even compared with publicly funded trials, the cost per randomised participant was less than one-tenth of the average cost. Indeed, it might have cost even less had it not been for the delays imposed by clinical trial regulations, an impediment that the UK government has recently undertaken to tackle. There was also, however, a cost to the environment. The trial emitted about 510 tonnes of carbon dioxide equivalents. 55 Although rapid recruitment helped to reduce its carbon footprint, the environmental impact of health care and health research can no longer be ignored. 56
The results showed that the administration of TXA reduces mortality with no apparent increase in side effects. If given promptly, the treatment reduces the risk of bleeding to death by about one-third. On the basis of these results, we estimate that giving TXA to bleeding trauma patients could save > 100,000 lives per year worldwide. Cost-effectiveness analysis shows that TXA administration is highly cost-effective in HICs, MICs and LICs. 57 The public investment in the CRASH-2 trial has generated knowledge that could, and should, improve human well-being worldwide.
Trauma is a disease of poverty. Although anyone can be the victim of a violent attack or road traffic crash, the risk is higher for those in the most disadvantaged social groups. In the UK, children in the lowest social class are 20 times more likely to be injured as pedestrians than are children in the highest social class. 58 The link between trauma and poverty is seen in HICs, MICs and LICs. 59 Trauma predominantly affects young men. The average age of participants in the CRASH-2 trial was 35 years and 85% of the patients were male. Trauma can be a cause of poverty as well as a consequence. A study in Bangladesh found that many urban households are made destitute by the death or injury of a family member in a road traffic crash. 60 Medical costs, funeral costs and the loss of family income can lead to decreased food consumption, a fall in living standards and increased indebtedness.
The identification of a highly cost-effective treatment for traumatic bleeding could and should benefit some of the most disadvantaged people in the world. Indeed, on the basis of the results of the CRASH-2 trial, TXA has already been included on the WHO list of essential medicines. Nevertheless, the benefits of the new knowledge generated by the CRASH-2 trial will not be fully realised without concerted efforts to ensure that the results are disseminated widely, that the drug is freely available and that all barriers to implementation are addressed.
The traditional way of disseminating new medical knowledge is by publishing a research paper in a medical journal. The main CRASH-2 trial results were published in The Lancet in June 2010 and March 2011. 29,61 Worldwide, traumatic bleeding kills around 2 million people each year, with > 90% of the deaths in LICs and MICs. It is critical therefore that the results are communicated to doctors and patients in these settings. The Lancet helped with this by making the results open access (free of charge), by agreeing to post Chinese, Hindi and Spanish translations of the abstract on its website, and by allowing the investigators in each country to republish the results in national medical journals. On the day of publication a press conference was held and newspapers around the world picked up the results. However, new results are only new for a day. Media coverage is a firestorm that consumes novelty as it rampages. Even if results get extensive news coverage it is unlikely that more than a small fraction of the doctors who treat trauma around the world will have heard the news or will have read the research paper. The real work of dissemination and implementation must now begin.
We were privileged to have been granted > £2,000,000 of public money to search for a safe and effective treatment for bleeding trauma patients, a search that was made possible because of the dedication and trust of a network of clinicians from around the world. We hope that we will continue to enjoy their trust as we move forward with dissemination and implementation.
Acknowledgements
All authors made substantial contributions to conception and design, or acquisition of data, analysis and interpretation of data, all were involved in the drafting of the manuscript or revising it critically for important intellectual content, and all authors approved the final version to be published.
Contribution of authors
Ian Roberts (Professor of Epidemiology) was involved in the design, conduct, analysis and reporting phases.
Haleema Shakur (Senior Lecturer in Clinical Trials) was involved in the design, conduct, analysis and reporting phases.
Tim Coats (Professor of Emergency Medicine) was involved in the design, conduct, analysis and reporting phases.
Beverley Hunt (Professor of Haematology) was involved in the design, conduct, analysis and reporting phases.
Eni Balogun (Assistant Trial Manager) was involved in the acquisition of data, analysis and reporting phases.
Lin Barnetson (Senior Data Manager) was involved in the acquisition of data, analysis and reporting phases.
Lisa Cook (Assistant Trial Manager) was involved in the acquisition of data, analysis and reporting phases.
Taemi Kawahara (Trial Manager) was involved in the acquisition of data, analysis and reporting phases.
Pablo Perel (Clinical Lecturer) was involved in the conduct, analysis and reporting phases.
David Prieto-Merino (Lecturer) was involved in the analysis and reporting phases.
Maria Ramos (Senior Trials Administrator) was involved in the design, conduct, analysis and reporting phases.
John Cairns (Professor of Health Economics) conducted the economic analysis.
Carla Guerriero (Research Fellow) conducted the economic analysis.
Department of Health disclaimer: the views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Department of Health.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
Publications
CRASH-2 trial collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
CRASH-2 trial collaborators. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011;377:1096–101.
References
- Murray CJL, Lopez AD. Global health statistics a compendium of incidence prevalence and mortality estimates for over 200 conditions. Boston, MA: Harvard University Press; 1996.
- Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, et al. Epidemiology of trauma deaths: a reassessment. J Trauma 1995;38:185-93. http://dx.doi.org/10.1097/00005373-199502000-00006.
- The Brain Trauma Foundation. The American Association of Neurological Surgeons. The joint section on neurotrauma and critical care . Hypotension. J Neurotrauma 2000;17:591-5. http://dx.doi. org/10.1089/neu.2000.17.591.
- Lawson JH, Murphy MP. Challenges for providing effective hemostasis in surgery and trauma. Semin Hematol 2004;41:55-64. http://dx.doi.org/10.1053/j.seminhematol.2003.11.012.
- Porte RJ, Leebeek FW. Pharmacological strategies to decrease transfusion requirements in patients undergoing surgery. Drugs 2002;62:2193-211. http://dx.doi.org/10.2165/00003495-200262150-00003.
- Henry DA, Carless PA, Moxey AJ, O’Connell D, Stokes BJ, Fergusson DA, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev 2011;3.
- Kenet G, Walden R, Eldad A, Martinowitz U. Treatment of traumatic bleeding with recombinant factor VIIa. Lancet 2009;354. http://dx.doi.org/10.1016/S0140-6736(99)05155-7.
- Roberts I, Shakur H, Ker K, Coats T. CRASH-2 collaborators . Antifibrinolytic drugs for acute traumatic injury. Cochrane Database Syst Rev 2011;1.
- Aylward GW, Dunlop IS, Little BC. Meta-analysis of systemic anti-fibrinolytics in traumatic hyphaema. Eye 1994;8:440-2. http://dx.doi.org/10.1038/eye.1994.104.
- Kiwanuka N, Gray RH, Serwadda D, Li X, Sewankambo NK, Kigozi G, et al. The incidence of HIV-1 associated with injections and transfusions in a prospective cohort, Rakai, Uganda. AIDS 2004;18:342-4. http://dx.doi.org/10.1097/00002030-200401230-00032.
- Heymann SJ, Brewer TF. The problem of transfusion-associated acquired immunodeficiency syndrome in Africa: a quantitative approach. Am J Infect Control 1992;20:256-62. http://dx.doi.org/10.1016/S0196-6553(05)80199-3.
- Goodnough LT, Shander A, Brecher ME. Transfusion medicine: looking to the future. Lancet 2003;361:161-9. http://dx.doi.org/10.1016/S0140-6736(03)12195-2.
- Fiechtner BK, Nuttall GA, Johnson ME, Dong Y, Sujirattanawimol N, Oliver WC, et al. Plasma tranexamic acid concentrations during cardiopulmonary bypass. Anesth Analg 2001;92:1131-6. http://dx.doi.org/10.1097/00000539-200105000-00010.
- Horrow JC, Van Riper DF, Strong MD, Grunewald KE, Parmet MJ. The dose–response relationship of tranexamic acid. Anaesthesiology 1995;82:383-92. http://dx.doi.org/10.1097/00000542-199502000-00009.
- The CRASH Trials Co-ordinating Centre . Protocol 05PRT 1: The CRASH-2 (Clinical Randomization of an Anti-Fibrinolytic in Significant Haemorrhage) Trial 2005. www.thelancet.com/ protocol-reviews/05PRT-1.
- Peto R, Baigent C. Trials: the next 50 years. Large scale randomised evidence of moderate benefits. BMJ 1998;317:1170-1. http://dx.doi.org/10.1136/bmj.317.7167.1170.
- Perel P, Al-Shahi Salman R, Kawahara T, Morris Z, Prieto-Merino D, . CRASH-2 (Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage) intracranial bleeding study: the effect of tranexamic acid in traumatic brain injury – a nested randomised, placebo-controlled trial. Health Technol Assess 2012;16.
- Perel P, Edwards P, Shakur H, Roberts I. Use of the Oxford Handicap Scale at hospital discharge to predict Glasgow Outcome Scale at 6 months in patients with traumatic brain injury. BMC Med Res Methodol 2008;8. http://dx.doi.org/10.1186/1471-2288-8-72.
- Duley L, Antman K, Arena J, Avezum A, Blumenthal M, Bosch J, et al. Specific barriers to the conduct of randomized trials. Clin Trials 2008;5:40-8. http://dx.doi. org/10.1177/1740774507087704.
- Henry DA, Carless PA, Moxey AJ, O’Connell D, Stokes BJ, McClelland B, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev 2007;4.
- Levy JH. Antifibrinolytic therapy: new data and new concepts. Lancet 2010;376:3-4. http://dx.doi. org/10.1016/S0140-6736(10)60939-7.
- World Bank . GDP (current US$) 2010. http://data.worldbank.org (accessed 2010).
- Norton R, Hyder AA, Bishai D, Peden M. Unintentional injuries: disease control priorities in developing countries. New York, NY: Oxford University Press; 2006.
- Cleves M, Gould WW, Gutierrez RG, Marchenko Y. An introduction to survival analysis using stata. College Station, TX: Stata Press; 2008.
- Tien H, Chu PTY, Brenneman F. Causes of death following multiple trauma. Current Orthopaedics 2004;18:304-10. http://dx.doi.org/10.1016/j.cuor.2004.04.006.
- Updated guide to the methods of technology appraisal. London: NICE; 2008.
- Pennsylvania University . Penn World Table 2010. https://pwt.sas.upenn.edu (accessed 2010).
- Bureau of Labor Statistics . United States Department of Labor. Historical Consumer Price Index U.S. City Average 2009. www.bls.gov/cpi/#tables (accessed 30 January 2009).
- CRASH-2 trial collaborators . Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with Significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23-32. http://dx.doi.org/10.1016/ S0140-6736(10)60835-5.
- Casati V, Guzzon D, Oppizzi M, Crossolini M, Torri G, Calori G, et al. Hemostatic effects of aprotinin, tranexamic acid and epsilon-aminocaproic acid in primary cardiac surgery. Ann Thorac Surg 1999;68:2252-6. http://dx.doi.org/10.1016/S0003-4975(99)00866-8.
- Eaton M P. Antifibrinolytic therapy in surgery for congenital heart disease. Anesth Analg 2008;106:1087-100. http://dx.doi.org/10.1213/ane.0b013e3181679555.
- World Health Organization . CHOsing Interventions That Are Cost Effective. India. Estimates of Unit Costs for Patient Services for India 2005. www.dcp2.org/fle/24/ (accessed 4 December 2012).
- World Health Organization . CHOsing Interventions That Are Cost Effective. United Republic of Tanzania. Estimates of Unit Costs for Patient Services for United Republic of Tanzania 2005. www.who.int/choice/country/tza/cost/en/index.html (accessed 4 December 2012).
- Department of Health . NHS Reference Costs 2008–2009 2010. www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_111591 (accessed 4 December 2012).
- British National Formulary 2009. www.bnf.org/bnf/index.htm (accessed 4 December 2012).
- Private Communication With Muhimbili National Hospital, Tanzania 2010.
- Government Medical College . Chandigarh Hospital India 2010. http://gmch.gov.in/ (accessed 4 December 2012).
- Private Communication With Care Hospital, India 2010.
- Organisation for Economic Co-operation and Development (OECD) . Purchasing Power Parities (PPP) 2008. www.oecd.org/std/pricesandpurchasingpowerparitiesppp/purchasingpowerparities-frequentlyaskedquestionsfaqs.htm (accessed 4 December 2012).
- Curtis L. Unit costs of health and social care. Canterbury: Canterbury Personal Social Services Research Unit, University of Kent; 2009.
- Dzieken G, Chisholm D, Johns B, Rovira J, Hutin YJ. The cost-effectiveness of policies for the safe and appropriate use of injection in healthcare settings. Bull World Health Organ 2003;81:277-85.
- Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ 2009;338. http://dx.doi.org/10.1136/bmj.b2393.
- Rodger A, MacMahon S. Systematic underestimation of treatment effects as a result of diagnostic test inaccuracy: implications for the interpretation and design of thromboprophylaxis trials. Thromb Haemost 1995;73:167-71.
- Yusuf S, Wittes J, Probstfield J, Tyroler HA. Analysis and interpretation of treatment effects in subgroups of patients in randomized clinical trials. JAMA 1991;266:93-8. http://dx.doi.org/10.1001/jama.1991.03470010097038.
- Sun X, Briel M, Walter SD, Guyatt GH. Is a subgroup effect believable? Updating criteria to evaluate the credibility of subgroup analyses. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c117.
- Assmann S F, Pocock SJ, Enos LE, Kasten LE. Subgroup analysis and other (mis)uses of baseline data in clinical trials. Lancet 2000;355:1064-9. http://dx.doi.org/10.1016/S0140-6736(00)02039-0.
- Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfbrinolysis. J Trauma 2008;64:1211-17. http://dx.doi.org/10.1097/TA.0b013e318169cd3c.
- Hess JR, Brohi K, Dutton R P, Hauser CJ, Holcomb JB, Kluger Y, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma 2008;65:748-54. http://dx.doi.org/10.1097/TA.0b013e3181877a9c.
- Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, et al. Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thromb Res 2009;124:608-13. http://dx.doi.org/10.1016/j.thromres.2009.06.034.
- Prentice CR. Basis of antifibrinolytic therapy. J Clin Pathol Suppl 1980;14:35-40. http://dx.doi.org/10.1136/jcp.s3-14.1.35.
- Macroeconomics and health: investing in health for economic development. Geneva: World Health Organization; 2001.
- World Bank . Country Classification 2010. http://data.worldbank.org/about/country-classifcations (accessed 4 December 2012).
- Shakur H, Elbourne D, Gülmezoglu M, Alfrevic Z, Ronsmans C. The WOMAN Trial (World Maternal Antifibrinolytic Trial): tranexamic acid for the treatment of postpartum haemorrhage: an international randomised, double blind placebo controlled trial. Trials 2010;11. http://dx.doi.org/10.1186/1745-6215-11-40.
- Ferrer P, Roberts I, Sydenham E, Blackhall K, Shakur H. Anti-fibrinolytic agents in post partum haemorrhage: a systematic review. BMC Pregnancy Childbirth 2009;9. http://dx.doi.org/10.1186/1471-2393-9-29.
- Subaiya S, Hogg E, Roberts I. Reducing the environmental impact of trials: a comparison of the carbon footprint of the CRASH-1 and CRASH-2 clinical trials. Trials 2011;12.
- Sustainable Trials Study Group . Towards sustainable clinical trials. BMJ 2007;334:671-3. http://dx.doi.org/10.1136/bmj.39140.623137.BE.
- Guerriero C, Cairns J, Perel P, Shakur H, Roberts I. Crash-2 trial collaborators . Cost-effectiveness analysis of administering tranexamic acid to bleeding trauma patients using evidence from the CRASH-2 trial. PLoS One 2011;6. http://dx.doi.org/10.1371/journal.pone.0018987.
- Edwards P, Roberts I, Green J, Lutchmun S. Deaths from injury in children and employment status in family: analysis of trends in class specific death rates. BMJ 2006;333. http://dx.doi.org/10.1136/bmj.38875.757488.4F.
- Nantulya VM, Reich MR. Equity dimensions of road traffic injuries in low- and middle-income countries. Inj Control Saf Promot 2003;10:13-20. http://dx.doi.org/10.1076/icsp.10.1.13.14116.
- Thomas AJ, Jacobs GD, Sexton B, Gururaj G, Rahman, F. The Involvement and Impact of Road Crashes on the Poor: Bangladesh and India Case Studies 2004. www.dfd.gov.uk/r4d/ Output/5429/Default.aspx (accessed 4 December 2012).
- CRASH-2 collaborators . The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011;377:1096-101.
- Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically Significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet 2004;364:1321-8. http://dx.doi.org/10.1016/S0140-6736(04)17188-2.
- Anonymous. Medicines for Human Use (clinical Trials) Amendment (no. 2) Regulations 2006. www.legislation.gov.uk/uksi/2006/2984/contents/made (accessed 7 February 2011).
- Anonymous. Medicines for Human Use (clinical Trials) Regulations 2004. www.legislation.gov.uk/uksi/2004/1031/contents/made (accessed 7 February 2011).
- Anonymous. Protection of Human Subjects: Informed Consent and Waiver of Informed Consent Requirements in Certain Emergency Research. Final Rules 21 n.d. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=50.24 (accessed 4 December 2012).
- CRASH Trial Management Group . Research in emergency situations: with or without relatives consent. Emerg Med J 2004;21. http://dx.doi.org/10.1136/emj.2002.004788.
- World Medical Association . Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects 2008. www.wma.net/en/30publications/10policies/b3/index.html (accessed 7 February 2011).
- Chalmers I. Regulation of therapeutic research is compromising the interests of patients. Int J Pharmaceut Med 2007;21:395-404. http://dx.doi.org/10.2165/00124363-200721060-00004.
Appendix 1 CRASH-2 trial organization
Steering Committee
Professor Ian Franklin (chairperson), University of Glasgow and Scottish Blood Transfusion Service.
Ms Brigitte Chaudhry, RoadPeace.
Professor Tim Coats, University of Leicester.
Dr Charles Deakin, Southampton General Hospital.
Dr Steve Goodacre, University of Sheffield.
Dr Beverley J Hunt, Guy's and St Thomas' Hospital NHS Trust.
Dr David Meddings, World Health Organization.
Professor Sir Richard Peto, University of Oxford.
Professor Ian Roberts, LSHTM.
Professor Peter Sandercock, University of Edinburgh.
Data Monitoring Committee
Professor Rory Collins (chairperson), Professor Adrian Grant and Professor John A Myburgh.
Trial Coordinating Centre Team
Ian Roberts (clinical co-ordinator, chief investigator), Haleema Shakur (trial manager), Pablo Perel (regional co-ordinator), Lin Barnetson (data manager), Maria Ramos (trial administrator), Lisa Cook (assistant trial manager, regional co-ordinator from 2007), Taemi Kawahara (assistant trial manager, regional co-ordinator from 2007), Eni Balogun (regional co-ordinator from 2006), Matthew Berle (trial assistant from 2007), Collette Barrow (assistant administrator from 2008), Tony Brady (programmer to 2006), Chris Rubery (data assistant from 2009), Jackie Wayte (UK nurse co-ordinator from 2008) and Cynthia To (data assistant from 2007 to 2009).
Steering Committee – Ian Franklin (chairperson), Brigitte Chaudhry, Tim Coats, Charles Deakin, Steve Goodacre, Beverley Hunt, David Meddings, Richard Peto, Ian Roberts and Peter Sandercock.
CRASH-2 trial collaborators by country
Albania National Trauma Centre Hospital: Fatos Olldashi, Mihal Kerçi, Tefik Zhurda and Klotilda Ruçi; Spitali Civil Durres: Arben Banushi.
Argentina Hospital Ángel Cruz Padilla: Mario Sardón Traverso and Juan Jiménez; Hospital Regional Rio Grande: Jorge Balbi; Hospital ‘4 de Junio’ Dr Ramon Carrillo: Christian Dellera; Hospital Castro Rendón: Silvana Svampa; Hospital San Martín de La Plata: Gustavo Quintana; Hospital Municipal de Agudos ‘Dr Leonídas Lucero’: Gustavo Piñero; Hospital Interzonal General de Agudos ‘Dr Oscar Alende’: Jorge Teves.
Australia Nepean Hospital: Ian Seppelt; Sir Charles Gairdner Hospital: David Mountain; John Hunter Hospital: Zsolt Balogh.
Bangladesh United Hospital Limited: Maniruz Zaman.
Belgium Sint-Vincentius Hospital: Patrick Druwé and Robert Rutsaert; Centre Hospitalier Regional de Namur: Guy Mazairac.
Cameroon Tombel District Hospital: Fogang Pascal, Zognou Yvette and Djeuchon Chancellin; St Theresa's Catholic Medical Centre: Patrick Okwen; Bamenda Provincial Hospital: Jules Djokam-Liapoe; Bali District Hospital: Ernest Jangwa; Bafut District Hospital: Lawrence Mbuagbaw; Fundong District Hospital: Ninying Fointama; St John of God Medical Centre: Nguemo Pascal.
Canada Hamilton General Hospital: Frank Baillie.
China Renji Hospital: Ji-yao Jiang, Guo-yi Gao and Yin-hui Bao.
Colombia Hospital Universitário San Vicente de Paúl, Universidad de Antioquia: Carlos Morales, Juan Sierra, Santiago Naranjo, Camilo Correa and Carolina Gómez; Hospital Universitário San Jose de Popayan: Jorge Herrera, Liliana Caicedo, Alexei Rojas, Henry Pastas and Hugo Miranda; Hospital Pablo Tobon Uribe: Alfredo Constaín, Mayla Perdomo, Diego Muñoz, Álvaro Duarte and Edwin Vásquez; Hospital San Andrés de Tumaco: Camilo Ortiz, Bernardo Ayala, Hernán Delgado, Gloria Benavides and Lorena Rosero; Fundación Clínica Valle del Lili: Jorge Mejía-Mantilla, Ana Varela, Maríaisabel Calle, José Castillo and Alberto García; Clínica las Americas: Juan Ciro, Clara Villa and Roberto Panesso; Hospital General de Medellin: Luz Flórez and Argemiro Gallego; Hospital San Felix ESE: Fabián Puentes-Manosalva, Leonor Medina and Kelly Márquez; Hospital Universitário del Caribe: Adalgiza Reyes Romero, Ricardo Hernández and Julio Martínez; Hospital Universitário San Jorge: Wilson Gualteros; Hospital San Rafael Tunja: Zulma Urbina and Julio Velandia; Clínica La Estancia SA: Federico Benítez and Adolfo Trochez; Fundación Hospital San José de Buga: Andrés Villarreal and Pamela Pabón; Hospital Civil de Ipiales: Hernán Delgado; Hospital Universitário Departamental Nariño: Héctor López; Hospital Universitário del Valle: Laureano Quintero; Hospital Universitário de Neiva: Andrés Rubiano; Hospital Manuel Uribe Ángel: Juan Tamayo.
Cuba Hospital Clínico-Quirúrgico Docente ‘Saturnino Lora’: Marjoris Piñera, Zadis Navarro, Deborah Rondón and Bárbara Bujan; Hospital General Universitário ‘Carlos Manuel de Céspedes’: Leonel Palacios, Daymis Martínez, Yalisa Hernández and Yaimara Fernández; Hospital Provincial Docente ‘Manuel Ascunce Domenech’: Eugenio Casola; Hospital Universitário ‘Arnaldo Milián Castro’: Rodolfo Delgado, Carlos Herrera, Migdacelys Arbolaéz and Mario Domínguez; Hospital Universitário ‘Dr Gustavo Aldereguía Lima’: Marcos Iraola, Omar Rojas and Alba Enseñat; Hospital Abel Santamaría Cuadrado: Irene Pastrana, Daniel Rodríguez and Sergio Álvarez de la Campa; Hospital Miguel Enríquez: Thorvald Fortún; Hospital General Calixto García: Martha Larrea; Hospital Antonio Luaces Iraola: Lensky Aragón; Hospital Provincial Docente VI Lenin: Aida Madrazo.
Czech Republic Research Institute for Special Surgery and Trauma: Petr Svoboda.
Ecuador Hospital Luis Vernaza: Mario Izurieta, Alberto Daccach, Mónica Altamirano, Antonio Ortega, Bolívar Cárdenas and Luis González; Hospital José Carrasco Arteaga: Marcelo Ochoa, Fernando Ortega, Fausto Quichimbo and Jenny Guiñanzaca; Hospital de Niños Dr Roberto Gilbert Elizalde: Ines Zavala and Sayra Segura; Hospital Naval Guayaquil: Johnny Jerez; Hospital Alcivar: Daniel Acosta; Hospital ‘Dr Rafael Rodríguez Zambrano’: Fabián Yánez; Clínica De Especialidades Medicas ‘San Gregorio’: Rubén Camacho.
Egypt Mataria Teaching Hospital: Hussein Khamis, Hossam Shafei, Ali Kheidr, Hani Nasr, Moetaz Mosaad and Safwat Rizk; Suez Canal University: Hesham El-Sayed, Taha Moati and Emad Hokkam; Aswan Teaching Hospital: Mamdouh Amin, Hany Lowis, Medhat Fawzy, Nabil Bedir and Mohamed Aldars.
El Salvador Hospital Nacional Rosales: Virginia Rodríguez, Juan Tobar and Jorge Alvarenga.
Georgia Tbilisi State University Clinical Hospital ‘I Javakhishvili’: Budu Shalamberidze, Elza Demuria, Nikoloz Rtveliashvili, Gocha Chutkerashvili and David Dotiashvili; Tbilisi First Hospital, University Clinic, Neurosurgery Center: Tamar Gogichaishvili, George Ingorokva, David Kazaishvili, Besik Melikidze and Natia Iashvili; Tbilisi City Hospital #1: Gia Tomadze, Manana Chkhikvadze, Leri Khurtsidze, Zviad Lomidze and Diana Dzagania; Tbilisi State Medical University ER Department: Nikoloz Kvachadze, Giorgi Gotsadze and Vakhtang Kaloiani; Institute of Critical Care Medicine: Nino Kajaia.
Ghana Korle Bu Teaching Hospital: Jonathan Dakubo, Simon Naaeder and Priscilla Sowah; Nyinahin Government Hospital: Adamu Yusuf and Alhaji Ishak; Sogakope District Hospital: Paul Selasi-Sefenu; Methodist Hospital Wenchi: Ballu Sibiri; Effia Nkwanta Regional Hospital: Sampson Sarpong-Peprah; Saint Theresa's Hospital: Theodore Boro.
India Medical Trust Hospital Kochi: Kanjithanda Bopaiah, Kishore Shetty, Raja Subbiah, Lukman Mulla and Anand Doshi; Christian Medical College Ludhiana: Yashbir Dewan, Sarvpreet Grewal, Pradipta Tripathy, Jacob Mathew and Bharat Gupta; Aditya Neuroscience Centre: Anil Lal and Majulie Choudhury; Sri Sai Hospital: Sanjay Gupta, Smita Gupta and Arun Chug; Care Hospital: Venkataramana Pamidimukkala, Palaniappan Jagannath, Mohan Maharaj, Ramaraju Vommi and Naresh Gudipati; North Bengal Neuro Research Centre: W H Chhang; Sheth VS General Hospital and NHL Municipal College: Pankaj Patel, Nilay Suthar, Deepa Banker and Jyotish Patel; LTM Medical College and General Hospital: Satish Dharap, Ranjeet Kamble, Shraddha Patkar and Sushil Lohiya; Government Medical College and Associated Hospitals Jammu: Rakesh Saraf, Dinesh Kumar, Satish Parihar and Rahul Gupta; MKCG Medical College: Rasananda Mangual, Alagumuthu, Don Kooper and Chinmaya Mohapatra; Christian Medical College Hospital Vellore: Suresh David and Wesley Rajaleelan; KLE Hospital and Medical Research Centre: Ashok Pangi, Vivek Saraf and Santhosh Chikareddy; NKP Salve Institute of Medical Sciences and Lata Mangeshkar Hospital: Sushil Mankar, Anil Golhar, Rahul Sakhare and Nilesh Wagh; Sanjivani Diagnostics and Hospital: Anil Lal, Dhiman Hazarika; Parkar Hospital: Pratyush Chaudhuri; Jeevan Jyoti Hospital and Research Centre: Prakash Khetan; Mansarovar Hospital: Govindbhai Purohit, Yogesh Purohit and Mandakini Pandya; Postgraduate Institute of Medical Science Rohtak: Rakesh Gupta, Shashi Kiran and Saurab Walia; Goyal Hospital Jalna: Sonam Goyal, Sidhant Goyal and Satish Goyal; Government Medical College Chandigarh: Sanjay Gupta, Ashok Attri and Rajeev Sharma; Oberai Hospital: Ashok Oberai, Mahesh Oberai and Supriya Oberoi; Rajeev Gandhi Memorial Hospital and Research Centre: Gajendra Kant Tripathi; Calicut Medical College Hospital: Vijayan Peettakkandy, Premkumar Karuthillath and Pavithran Vadakammuriyil; Krishnamai Medical and Research Foundation's NIKOP Hospital: Jalindar Pol, Sunita Pol and Manisha Saste; St Stephen's Hospital: Subrat Raul, Shashi Tiwari and Neileino Nelly; Government Rajaji Hospital: M Chidambaram; Medical College Trivandrum: Viswanathan Kollengode and Sam Thampan; Sanjeevani Hospital: Sunder Rajan and Sushrut Rajan; Kamineni Hospital: Subodh Raju and Renuka Sharma; Sri Sakthi Hospital: Subbiah Venkatesh Babu and Chellappa Sumathi; Bhattacharya Orthopaedic and Related Research Centre: Protyush Chatterjee and Alok Agarwal; Sushrut Hospital: Hemant Magar and Meera Magar; All India Institute of Medical Sciences: Manmohan Singh and Deepak Gupta; GM Hospital (P), Ltd: Anil Lal and Kamal Haloi; Government Medical College and Superspeciality Hospital Nagpur: Varsha Sagdeo and Pramod Giri; Government Medical College New Civil Hospital: Nimesh Verma, Ravi Jariwala and Ashish Goti; Chikitsa Hospital: Aman Prabhu-Gaonkar and Sagar Utagi; Apollo Health City: Mahesh Joshi and Ruchit Agrawal; Apex Neurotrauma and Superspeciality Hospital: Gopal Sharma and Gurvinder Saini; Neuro Centre Gola Ghat: Vinod Tewari; NSCB Medical College: Yad Yadav and Vijay Parihar; BGS Global Hospital: Neelam Venkataramana and Shailesh Rao; Chettinad Hospital and Research Institute: Narayana Reddy and SG Chander; Sir Sayajirao General Hospital and Medical College Baroda: Virsing Hathila; Goyal Hospital and Research Centre Jodhpur: Vithal Das; Krishna Surgical Hospital and Trauma Care Centre: Kantibhai Agaja; Nizam's Institute of Medical Sciences: Aniruddh Purohit; Niramay Hospital: Akilesh Lahari; Apex Hospital Bhopal: Rajesh Bhagchandani; Dr Jeyasekharan Medical Trust: Bala Vidyasagar; Himalayan Institute of Medical Sciences: P K Sachan; Apollo Gleneagles Hospitals: Tanmoy Das; Civil Hospital Gandhinagar: Sharad Vyas; Sukhdev Raj Soin Hospital: Sujoy Bhattacharjee; Sancheti Institute for Orthopaedics and Rehabilitation: Parag Sancheti; St James Hospital: T Manoj; Al Shifa Hospital: Mubarak Moideen; Anant Institute of Medical Sciences: Kailash Pansey; Vinayaka Mission Hospital: V P Chandrasekaran; Gauhati Medical College and Hospital: Kabul Saikia; Krishna Hospital and Medical Research Centre: Hoshedar Tata; Ruby Hall Clinic: Sanjay Vhora; Shreejee Hospital: Aniket Shah; Nazareth Hospital: Gordon Rangad; Ganga Hospital: S Rajasekaran; Makurdi: Andrea Jogo and Terna Bitto; Nnamdi Azikiwe University Teaching Hospital: Stanley Anyanwu and Okechukwu Mbonu; Lagos State University Teaching Hospital: Mobolaji Oludara and Michael Somoye; Usmanu Danfodiyo University Teaching Hospital: Bello Shehu and Nasir Ismail; National Orthopaedic Hospital Enugu: Amechi Katchy; University of Calabar Teaching Hospital: Rowland Ndoma-Egba and Ngim Grace-Inah; University of Abuja Teaching Hospital: Zumnan Songden and Abdulrahman Abdulraheem; University of Uyo Teaching Hospital: Akpan Out and Timothy Nottidge; Federal Medical Centre, Yenagoa: Domingo Inyang and David Idiapho; Seventh Day Adventist Hospital: Herb Giebel; Federal Medical Centre Birnin-Kebbi: Ramatu Hassan; Abia State University Teaching Hospital: Adeyinka Adisa; Wesley Guild Hospital: Akinbolaji Akinkuolie; Federal Medical Centre, Umuahia: Kalu Okam; University of Maiduguri Teaching Hospital: Abubakar Musa; National Orthopaedic Hospital, Igbobi: Ignatius Falope; University of Nigeria Teaching Hospital Enugu: John Eze.
Peru Hospital Regional Docente de Trujillo: José Caballero, Wenceslao Azabache and Oscar Salirrosas; Hospital Nacional Hipolito Unanue: Alonso Soto, Elfi Torres, Gloria Ramírez and Mónica Pérez; Clinica Santa Ana: Cesar Malca; Hospital La Caleta: Juan Velez; Hospital Nacional Sergio E Bernales: Raul Yepez; Hospital de Apoyo de Sullana: Hernan Yupanqui; Hospital IV Essalud Huancayo: Pedro Lagos; Hospital Nacional Arzobispo Loayza: Diana Rodriguez; Hospital Municipal Los Olivos: Jorge Flores; Hospital Jose Cayetano Heredia: Anselmo Moya; Hospital Nacional Carlos Alberto Seguin Escobedo: Alejandro Barrionuevo; Hospital Nacional Dos De Mayo: Marco Gonzales-Portillo; Hospital Nacional Cayetano Heredia: Edgar Nunez.
Saudi Arabia King Khalid University Hospital: Abdelazeem Eldawlatly, Mohammed Al Naami and Bilal Delvi; King Khalid National Guard Hospital: Walid Alyafi.
Serbia Klinicki Centar Srbije: Branko Djurovic.
Singapore National Neuroscience Institute: Ivan Ng.
Slovakia FNsP Ružinov: Aktham Yaghi; NsP Poprad: Anton Laincz; NsP JA Reiman Hospital: Stefan Trenkler; Faculty Hospital FD Roosevelta: Jozef Valky.
South Africa Dr George Mukhari Hospital: Mphako Modiba, Peter Legodi and Thomas Rangaka; George Provincial Hospital: Lee Wallis.
Spain Hospital Universitário Virgen del Roció: Ángeles Muñoz; Hospital Ramón y Cajal de Madrid: Ana Serrano; Hospital Universitário Germans Trias i Pujol: Maite Misis; Hospital Torrecardenas: Martin Rubi; Hospital Universitário Virgen de la Victoria: Victoria de la Torre.
Sri Lanka National Hospital of Sri Lanka: Ranjith Ellawala, Samitha Wijeratna, Lukshrini Gunaratna and Crishantha Wijayanayaka.
Tanzania Muhimbili Orthopaedic Institute: Kitugi Nungu, Billy Haonga and Grenda Mtapa.
Thailand Khon Kaen Regional Hospital: Surakrant Yutthakasemsunt, Warawut Kittiwattanagul, Parnumas Piyavechvirat, Tawatcahi Impool and Santipong Thummaraj; Pattani Hospital: Rusta Salaeh; Suratthani Hospital: Sakchai Tangchitvittaya; Bhumibol Adulyadej Hospital: Kamol Wattanakrai, Chatchai Soonthornthum and Teerasak Jiravongbunrod; Lampang Hospital: Surasak Meephant; Rayong Hospital: Pusit Subsompon; Roi-Et Hospital: Phaiboon Pensuwan; Phrae Hospital: Wicheanrat Chamnongwit.
Tunisia Hospital Habib Thameur: Zouheir Jerbi and Abderraouef Cherif.
UK University Hospital of North Staffordshire: Mark Nash; Royal London Hospital: Tim Harris; Leicester Royal Infirmary: Jay Banerjee; Nottingham University Hospitals NHS Trust: Ramzi Freij; Frenchay Hospital: Jason Kendall; Countess of Chester Hospital: Stephen Moore; Hull Royal Infirmary: William Townend; Royal Sussex County Hospital: Rowland Cottingham; Derby Hospitals NHS Trust: Dan Becker; Bedford Hospital NHS Trust: Stuart Lloyd; Royal Liverpool University Hospital: Peter Burdett-Smith; Colchester General Hospital: Kazim Mirza; Royal Lancaster Infirmary: Andrew Webster; Worthing Hospital: Suzanne Brady and Amanda Grocutt; Darent Valley Hospital: John Thurston; Hope Hospital: Fiona Lecky; Northern General Hospital: Steve Goodacre.
Zambia University Teaching Hospital, Lusaka: Yakub Mulla and Dennis Sakala; Nchanga North General Hospital: Charles Chengo.
Writing committee main analyses Haleema Shakur (chairperson), Ian Roberts (chief investigator), Raúl Bautista (Mexico), José Caballero (Peru), Tim Coats (UK), Yashbir Dewan (India), Hesham El-Sayed (Egypt), Tamar Gogichaishvili (Georgia), Sanjay Gupta (India), Jorge Herrera (Colombia), Beverley Hunt (UK), Pius Iribhogbe (Nigeria), Mario Izurieta (Ecuador), Hussein Khamis (Egypt), Edward Komolafe (Nigeria), María-Acelia Marrero (Cuba), Jorge Mejía-Mantilla (Colombia), Jaime Miranda (Peru), Carlos Morales (Colombia), Oluwole Olaomi (Nigeria), Fatos Olldashi (Albania), Pablo Perel (UK), Richard Peto (UK), PV Ramana (India), RR Ravi (India) and Surakrant Yutthakasemsunt (Thailand). National coordinators – Jonathan Dakubo (Ghana), Tamar Gogichaishvili (Georgia), Nyoman Golden (Indonesia), Mario Izurieta (Ecuador), Hussein Khamis (Egypt), Edward Komolafe (Nigeria), Jorge Loría-Castellanos (Mexico), Jorge Mejía-Mantilla (Colombia), Jaime Miranda (Peru), Ángeles Muñoz (Spain), Vincent Mutiso (Kenya), Patrick Okwen (Cameroon), Zulma Ortiz (Argentina), María Pascual, CENCEC (Cuba), RR Ravi (India), April Roslani (Malaysia), Stefan Trenkler (Slovakia), Annalisa Volpi (Italy) and Surakrant Yutthakasemsunt (Thailand).
Writing committee exploratory analyses Ian Roberts (UK) (chairperson), Haleema Shakur (UK), Adefemi Afolabi (Nigeria), Karim Brohi (UK), Tim Coats (UK), Yashbir Dewan (India), Satoshi Gando (Japan), Gordon Guyatt (Canada), B J Hunt (UK), Carlos Morales (Colombia), Pablo Perel (UK), David Prieto-Merino (UK) and Tom Woolley (UK).
Appendix 2 Trial protocol
A LARGE RANDOMISED PLACEBO CONTROLLED TRIAL AMONG TRAUMA PATIENTS WITH OR AT RISK OF SIGNIFICANT HAEMORRHAGE, OF THE EFFECTS OF ANTIFIBRINOLYTIC TREATMENT ON DEATH AND TRANSFUSION REQUIREMENT.
1. Background
Introduction: For people at ages 5 to 45 years, trauma is second only to HIV/AIDS as a cause of death. Each year, worldwide, about three million people die as a result of trauma, many after reaching hospital. 1 Among trauma patients who do survive to reach hospital, exsanguination is a common cause of death, accounting for nearly half of in-hospital trauma deaths. 2 Central nervous system injury and multi-organ failure account for most of the remainder, both of which can be exacerbated by severe bleeding. 3
Mechanisms: The haemostatic system helps to maintain the integrity of the circulatory system after severe vascular injury, whether traumatic or surgical in origin. 4 Major surgery and trauma trigger similar haemostatic responses and the consequent massive blood loss presents an extreme challenge to the coagulation system. Part of the response to surgery and trauma, in any patient, is stimulation of clot breakdown (fibrinolysis) which may become pathological (hyper-fibrinolysis) in some. 4 Antifibrinolytic agents have been shown to reduce blood loss in patients with both normal and exaggerated fibrinolytic responses to surgery, and do so without apparently increasing the risk of post-operative complications, most notably there is no increased risk of venous thromboembolism. 5
Existing knowledge: Systemic antifibrinolytic agents are widely used in major surgery to prevent fibrinolysis and thus reduce surgical blood loss. A recent systematic review6 of randomised controlled trials of antifibrinolytic agents (mainly aprotinin or tranexamic acid) in elective surgical patients identified 89 trials including 8,580 randomised patients (74 trials in cardiac, eight in orthopaedic, four in liver, and three in vascular surgery). The results showed that these treatments reduced the numbers needing transfusion by one-third, reduced the volume needed per transfusion by one unit, and halved the need for further surgery to control bleeding. These differences were all highly statistically significant. There was also a statistically non-significant reduction in the risk of death (RR = 0.85: 95% CI 0.63 to 1.14) in the antifibrinolytic treated group.
Hypothesis: Because the coagulation abnormalities that occur after injury are similar to those after surgery, it is possible that antifibrinolytic agents might also reduce blood loss, the need for transfusion and mortality following trauma. However, to date there has been only one small randomised controlled trial (70 randomised patients, drug versus placebo: 0 versus 3 deaths) of the effect of antifibrinolytic agents in major trauma. 8 As a result, there is insufficient evidence to either support or refute a clinically important treatment effect. Systemic antifibrinolytic agents have been used in the management of eye injuries where there is some evidence that they reduce the rate of secondary haemorrhage. 9
Need for a trial: A simple and widely practicable treatment that reduces blood loss following trauma might prevent thousands of premature trauma deaths each year and secondly could reduce exposure to the risks of blood transfusion. Blood is a scarce and expensive resource and major concerns remain about the risk of transfusion-transmitted infection. Trauma is common in parts of the world where the safety of blood transfusion is not assured. A recent study in Uganda estimated the population-attributable fraction of HIV acquisition as a result of blood transfusion to be around 2%, although some estimates are much higher. 10,11 Only 43% of the 191 WHO member states test blood for HIV and hepatitis C and B viruses. Every year unsafe transfusion and injection practices are estimated to account for 8–16 million Hepatitis B infections, 2.3–4.7 million Hepatitis C infections and 80,000–160,000 HIV infections. 12 A large randomised trial is therefore needed of the use of a simple, inexpensive, widely practicable antifibrinolytic treatment such as tranexamic acid (aprotinin is considerably more expensive and is a bovine product with consequent risk of allergic reaction and hypothetically transmission of disease), in a wide range of trauma patients who, when they reach hospital are thought to be at risk of major haemorrhage that could significantly affect their chances of survival.
Dose Selection
The systematic review of randomised controlled trials of antifibrinolytic agents in surgery showed that dose regimens of tranexamic acid vary widely. 6 Loading doses range from 2.5 mg/kg to 100 mg/kg and maintenance doses from 0.25 mg/kg/hr to 4 mg/kg/hr delivered over time periods of one to twelve hours. Studies examining the impact of different doses of tranexamic acid on bleeding and transfusion requirements showed no significant difference between a high dose and a low dose.
Studies in cardiac surgery have shown that a 10 mg/kg initial dose of tranexamic acid followed by an infusion of 1 mg/kg/hour produces plasma concentrations sufficient to inhibit fibrinolysis in vitro. 13 The dose-response relationship of tranexamic acid was examined by Horrow et al (1995) who concluded that 10 mg/kg followed by 1 mg/kg/hour decreases bleeding after extracorporeal circulation and that larger doses did not provide any additional haemostatic benefit. 14
In this emergency situation, administration of a fixed dose would be more practicable as determining the weight of a patient would be impossible. Therefore, a fixed dose within the dose range which has been shown to inhibit fibrinolysis and provide haemostatic benefit is being used for this trial. The fixed dose chosen would be efficacious for larger patients (> 100 kgs) but also safe in smaller patients (< 50 kgs), as the estimated dose/kg the latter group would receive has been applied in other trials without adverse effects. The planned duration of administration allows for the full effect of tranexamic acid on the immediate risk of haemorrhage without extending too far into the acute phase response seen after surgery and trauma.
2. Study design
SUMMARY
CRASH 2 is a large pragmatic randomised placebo controlled trial of the effects of the early administration of the antifibrinolytic agent tranexamic acid on death, vascular events and transfusion requirements. Adults with trauma who are within 8 hours of injury and have either significant haemorrhage, or who are considered to be at risk of significant haemorrhage, are eligible if the responsible doctor is for any reason substantially uncertain whether or not to use an antifibrinolytic agent. Numbered drug or placebo packs will be available in each participating emergency department. Randomisation will involve calling a 24-hour freecall randomisation service. The call should last only a minute or two and at the end of it the randomisation service will specify which numbered treatment pack to use. For hospitals where telephone randomisation is not feasible, randomisation will be by taking the next consecutively numbered treatment pack. No extra tests are required but a short form must be completed one month later or on discharge or on death (whichever occurs first).
NUMBER OF PATIENTS NEEDED
Two main factors determine the number of patients needed in a trial. These are the estimated event rate and size of the treatment effect.
Estimated event rate: In the Medical Research Council (MRC) CRASH trial of corticosteroids in head injury, the overall risk of death was 20%. 62 The MRC CRASH trial was the largest international randomised controlled trial in trauma patients and it would be reasonable to expect a similar risk of death in CRASH 2.
Size of treatment effect that should be detectable: Because even a 2% survival advantage for an intervention as simple and widely practicable as tranexamic acid would represent a worthwhile benefit, the current trial has been planned to be able to detect a benefit of this size.
Sample size: If the real mortality difference is 20% versus 18% then there is about an 85% chance that a trial involving 20,000 patients will achieve 2P < 0.01 (and a 95% chance that it will achieve 2P < 0.05). If however, the trial were only half as big then there would be a 50% chance of failing to achieve 2P < 0.01 (and a 28% chance of failing to achieve 2p < 0.05), which is not good enough.
Effects on other outcomes: With such large numbers randomised, even moderate effects on the numbers needing transfusion or on the mean volume per transfusion will be determined very accurately, as will any substantial effects on non-fatal vascular events (haemorrhagic or occlusive).
ELIGIBILITY
Adult trauma patients with ongoing significant haemorrhage or at risk of significant haemorrhage, within 8 hours of injury, except those for whom antifibrinolytic agents are thought to be clearly indicated or clearly contra-indicated.
Inclusion criteria: All trauma patients with ongoing significant haemorrhage (systolic blood pressure less than 90 mmHg and/or heart rate more than 110 beats per minute), or who are considered to be at risk of significant haemorrhage, and are within 8 hours of the injury, are eligible for trial entry if they appear to be at least 16 years old. Although entry is allowed up to 8 hours from injury, the earlier that patients can be treated the better.
Exclusion criteria: The fundamental eligibility criterion is the responsible doctor's ‘uncertainty’ as to whether or not to use an antifibrinolytic agent in a particular adult with traumatic haemorrhage. Patients for whom the responsible doctor considers there is a clear indication for antifibrinolytic therapy should not be randomised. Likewise, patients for whom there is considered to be a clear contraindication to antifibrinolytic therapy (such as, perhaps, those who have clinical evidence of a thrombotic disseminated intravascular coagulation) should not be randomised. Where the responsible doctor is substantially uncertain as to whether or not to use an antifibrinolytic, all these patients are eligible for randomisation and should be considered for the trial. There are no other pre-specified exclusion criteria.
Heterogeneity of the patients entering such a trial is a particular strength in terms of the analysis. If a wide range of patients are randomised then it may be possible for a really big trial such as this one to help determine which (if any) particular types of patient are most likely to benefit from treatment.
Special eligibility considerations: None. In the setting of acute severe haemorrhage the routine exclusion of patients with a history of thrombo-embolic disease is unnecessary, unless the responsible doctor considers these to be a definite contraindication. Brief details of patients assessed but not randomised in the trial will be recorded on a Patient Screening Log at each collaborating unit.
CONSENT
Patients with significant trauma may have impaired consciousness and may be unable to give properly informed consent. In this emergency situation it may not be medically appropriate to delay the start of treatment. Consent will be obtained from either a personal legal representative or a professional legal representative where relevant. The requirements of the relevant ethics committee will be adhered to at all times. An information leaflet on the study for patients (Protocol Appendix 1a) will be available in all drug packs in addition to a form for Legal Representative Consent (Protocol Appendix 1b).
RANDOMISATION
Patients eligible for inclusion should be randomised, and the study treatment started, as soon as possible. Randomisation is done by telephoning a 24-hour freecall service and takes only about two minutes. The patient entry form (Protocol Appendix 2) shows the questions that will be asked by the telephone operator prior to allocation of the treatment pack. The study computer will then randomly assign a treatment pack number that will identify one of the CRASH 2 treatment packs stored in the emergency department. If telephone randomisation is not feasible a local pack system will be used. At such hospitals, baseline information will be collected on the trial entry form and the next consecutively numbered treatment pack taken from a box of eight packs. Once a patient has been randomised, we will definitely wish to learn the outcome in hospital, even if the trial treatment is interrupted or is not actually given.
TREATMENT
Each CRASH 2 treatment pack contains:
-
4 x 500 mg ampoules of Tranexamic Acid or placebo
-
1 x 100 mL bag of 0.9% NaCl (for use with loading dose)
-
Stickers (for attaching to infusion bags and patient notes)
-
Patient information leaflet and Consent forms
-
Patient entry form and Outcome form
TREATMENT
| treatment | ampoules | dose (tranexamic acid or placebo) | infusion rate and duration |
|---|---|---|---|
| Loading | 2 | 1 gram | 100 mL over 10 minutes |
| Maintenance | 2 | 1 gram | 120 mg/hr [60 mL/hr] for about 8 hour |
SERIOUS UNEXPECTED SUSPECTED ADVERSE EVENTS
If a “SUSAR” (Serious Unexpected Suspected Adverse Reaction) occurs and is believed to be related to the study medicine, this should be logged by calling the 24-hour randomisation service, who will inform the Co-ordinating Centre in London. The Co-ordinating Centre will then contact you within 24 hours so that a written SUSAR report can be completed.
EXPECTED SIDE EFFECTS
In general, vascular events such as pulmonary embolism, deep vain thrombosis, stroke, myocardial infarction, gastrointestinal bleeding and multiorgan failure, do not need to be reported in this way because some increase in their incidence might be expected with antifibrinolytic agents. Likewise, the various medical events that are to be expected in severely injured patients do not need to be reported by telephone. However, all such events are routinely monitored among all patients on the outcome form (Protocol Appendix 3).
UNBLINDING
In general there should be no need to unblind the allocated treatment. If some contra-indication to antifibrinolytic therapy develops after randomisation (e.g. clinical evidence of thrombosis), the trial treatment should simply be stopped. Unblinding should be done only in those rare cases when the doctor believes that clinical management depends importantly upon knowledge of whether the patient received antifibrinolytic or placebo. In those few cases when urgent unblinding is considered necessary, the randomisation service should be telephoned, giving the name of the doctor authorising unblinding and the treatment pack number. The caller will then be told whether the patient received antifibrinolytic or placebo.
MEASURES OF OUTCOME
The primary outcome measure is death in hospital within four weeks of injury (causes of death will be described to assess whether deaths were due to haemorrhage or vascular occlusion). Secondary outcome measures are receipt of a blood products transfusion, the number of units of blood products transfused, surgical intervention, and the occurrence of thrombo-embolic episodes (stroke, myocardial infarction, pulmonary embolism, clinical evidence of deep vein thrombosis).
Data collection: In-hospital deaths, transfusion requirement, complications and short-term recovery are to be recorded on the outcome form (Protocol Appendix 3) which can be completed entirely from the hospital notes – no extra tests are needed. The outcome form should be completed at death, discharge or four weeks post randomisation whichever occurs first.
End of trial for patients: Death, discharge or four weeks post randomisation whichever occurs first.
ANALYSIS
Comparisons will be made of the primary outcome measure, comparing all those allocated antifibrinolytic treatment versus those allocated placebo, on an ‘intention to treat’ basis. Analyses will be stratified on time from injury to the initiation of treatment (less than one hour, one to three hours, more than three hours), on severity of haemorrhage as assessed by capillary refill time (0–2, 3–4, ≥ 5 seconds) and systolic blood pressure (< 75, 76–89, > 89 mmHg). Comparisons will also be made of the risks of blood product transfusion, need for operation and thrombo-embolic complications.
3. Organisation
DATA MONITORING COMMITTEE
Professor Rory Collins, chair; Professor Adrian Grant; Professor John A Myburgh
Standard Operating Procedures: The Data Monitoring and Ethics Committee (DMEC) has the responsibility for deciding whether, while randomisation is in progress, the unblinded results (or the unblinded results for a particular subgroup), should be revealed to the Trial Steering Committee (TSC). The DMEC terms of reference state that they will do this if, and only if, two conditions are satisfied: (1) the results provide proof beyond reasonable doubt that treatment is on balance either definitely harmful or definitely favourable for all, or for a particular category of patients, in terms of the major outcome; (2) the results would, if revealed, be expected to substantially change the prescribing patterns of doctors who are already familiar with any other trial results that exist. Exact criteria for “proof beyond reasonable doubt” are not, and cannot be, specified by a purely mathematical stopping rule, but they are strongly influenced by such rules. DMEC members have expressed sympathy with the stopping rule proposed in Part I of the 1976 report to the MRC Leukaemia Committee, whereby an interim analysis of major endpoint would generally need to involve a difference between treatment and control of at least three standard errors to justify premature disclosure. An interim subgroup analysis would, of course, have to be even more extreme to justify disclosure. This rule has the advantage that the exact number and timing of interim analyses need not be pre-specified. In summary, the stopping rules (as successfully applied in other trials including the MRC International Stroke Trial, which randomised 19,436 acute stroke patients) require extreme differences to justify premature disclosure and involve an appropriate combination of mathematical stopping rules and scientific judgement.
STEERING COMMITTEE
Professor Ian Franklin (chair), University of Glasgow and Scottish Blood Transfusion Service
Ms Brigitte Chaudhry, RoadPeace
Professor Tim Coats, University of Leicester
Dr Charles Deakin, Southampton General Hospital
Dr Steve Goodacre, University of Sheffield
Dr Beverley J Hunt, Guy's & St Thomas' Hospital NHS Trust
Dr David Meddings, World Health Organization
Professor Sir Richard Peto, University of Oxford
Professor Ian Roberts, London School of Hygiene & Tropical Medicine
Professor Peter Sandercock, University of Edinburgh
The steering committee consists of respected and experienced trauma and haematology experts, clinical triallists as well as a lay representative. Face to face meetings will be held at regular intervals determined by need but not less than once a year. Routine business is conducted by e-mail and post.
Standard Operating Procedures: The Steering Committee, in the development of this protocol and throughout the trial, will take responsibility for:
-
major decisions such as a need to change the protocol for any reason
-
monitoring and supervising the progress of the trial
-
reviewing relevant information from other sources
-
considering recommendations from the DMEC
-
informing and advising the management group on all aspects of the trial
COLLABORATORS' RESPONSIBILITIES
Co-ordination within each participating hospital will be through a local collaborator who will:
-
Discuss the trial with medical and nursing staff who see trauma patients and ensure that they remain aware of the state of the current knowledge, the trial and its procedures (there are wall charts, pocket summaries and a set of slides to assist with this)
-
Ensure that adults with trauma are considered promptly for the trial
-
Ensure that the patient entry forms (in non-telephone randomising centres) and single sided outcome forms are completed
-
Ensure the trial is conducted in accordance with ICH GCP and fulfils all national and local regulatory requirements
-
Allow access to source data for audit and verification
CO-ORDINATING CENTRE RESPONSIBILITIES
-
Provide study materials and a 24-hour randomisation (and unblinding) service
-
Give collaborators regular information about the progress of the study
-
Help ensure complete data collection at discharge
-
Respond to any questions (e.g. from collaborators) about the trial
-
Assure data security and quality and observe data protection laws
-
Ensure trial is conducted in accordance with ICH GCP
PUBLICATION
The success of CRASH 2 will be dependent entirely upon the collaboration of nurses and doctors in the participating hospitals. Hence, the chief credit for the study will be assigned to the collaborators from each participating centre and they will be named personally in the main publications. The results of the trial will be reported first to trial collaborators. Dissemination of results to patients will take place via the media, trial website (www.crash2@Lshtm.ac.uk) and relevant patient organisations.
INDEMNITY
CRASH 2 is funded by the London School of Hygiene & Tropical Medicine (LSHTM) and the World Health Organization (WHO) and not the manufacturers of tranexamic acid. LSHTM as the Co-ordinating Centre for the trial accepts responsibility attached to its sponsorship of the trial and, as such, would be responsible for claims for any non-negligent harm suffered by anyone as a result of participating in this trial.
FINANCIAL SUPPORT
LSHTM and WHO funding covers meetings and central organisational costs only. The design, management and finance of the study are entirely independent of the manufacturers of tranexamic acid, which is not a new product. Large trials of such drugs, involving many hospitals, are important for future patients but are practicable only if those collaborating in them do so without payment (except for recompense of any minor local costs that may arise).
Protocol Appendix 1a – Patient information sheet and consent form
INFORMATION FOR PATIENTS, INTERNATIONAL STUDY OF BLEEDING AFTER INJURY
This hospital is taking part in a research study to find ways to reduce severe bleeding after serious injury. You have been included in this study.
WHAT YOU SHOULD KNOW ABOUT RESEARCH STUDIES:
This form gives information about the study including the aims, risks and benefits of taking part.
In this hospital, patients with severe bleeding are given the usual emergency treatment for bleeding. The aim of this research study is to find a better treatment. We hope that the study treatment (tranexamic acid) will help clotting and so lessen the amount of blood lost and reduce the need for a blood transfusion. But the study treatment may cause clots where they are not needed. We hope to find that the treatment will do a little more good than harm but we don't yet know this. Please read the information below carefully and ask the doctor looking after you any questions you have.
1) Why is this research being done?
Severe bleeding is a common cause of death after injury and it is important to find better ways of reducing the amount of blood lost.
2) What is the purpose of this study?
Tranexamic acid is often used to reduce bleeding after major surgery such as heart operations. This study is being done to see if it can also reduce bleeding after major injury. Tranexamic acid is not a new drug and is an approved treatment for many common conditions that involve bleeding.
3) Who is doing the study?
{name of doctor} is in charge of this study at this hospital. The study is co-ordinated by doctors at The University of London.
4) A patient cannot be in this study if:
-
he/she is known to be under 16
-
he/she was injured more than 8 hours before arriving in hospital
-
the doctor thinks there is a particular reason why tranexamic acid definitely should not be given
-
the doctor thinks there is a particular reason why tranexamic acid definitely should be given
5) What has happened to you after you were included in this study?
You were given all the usual emergency treatments for bleeding, including fluids to replace the blood that you lost. You were also given a dose of either the active tranexamic acid or an inactive dummy medicine called saline. The dose was given over a period of eight hours. The choice of what to give (active treatment or dummy treatment) was made randomly by a computer at the University of Oxford, UK. The doctors looking after you do not know whether you got the active or the dummy medicine. This information is kept on a confidential list in another hospital. The study involves no extra tests but your doctor will send brief details about how you have been to the Co-ordinating Centre in London. This information will be used in strict confidence by the people working on the study and will not be released under any circumstance.
6) What are the possible risks of being in the study?
Tranexamic acid is widely used and at the moment there is no conclusive evidence of serious side effects with short term use. Tranexamic acid is NOT a new drug.
7) What are the possible benefits of being in the study?
We hope that tranexamic acid may help reduce blood loss. The knowledge that we gain from this study will help people with similar injuries in the future.
8) If you have any questions or problems, who can you call?
If you have any questions you can contact Dr {name of doctor}, {job title e.g. Consultant in Accident & Emergency} by telephoning {telephone number}
9) What information do we keep private?
All information about you and your injury will be kept private. The only people allowed to look at the information will be the doctors who are running the study, the staff at the Co-ordinating Centre and the regulatory authorities who check that the study is being carried out correctly. We will publish the results of the study in a medical journal so that other doctors can benefit from the knowledge, but your personal information will not be included and there will be no way that you can be identified.
10) Can the study end early for the participant?
The study treatment was given in the emergency situation. We hope that you will let us use information about how you got on, but if you do not want us to use it then please tell your doctor.
11) What else do you need to know?
-
The study is funded by the University of London and the World Health Organisation, not the makers of tranexamic acid.
-
The London School of Hygiene & Tropical Medicine (University of London) as the Co-ordinating Centre for the study accepts responsibility attached to its sponsorship of the study and, as such, would be responsible for claims for any non-negligent harm suffered by anyone as a result of participating in this study.
-
We will ask you to sign a separate consent form and give you a copy to keep.
PATIENT CONSENT FORM, INTERNATIONAL STUDY OF BLEEDING AFTER INJURY
-
I confirm that I have read and understood the information sheet Version 2, dated 9 December 2004, for the above study and have had the opportunity to ask questions.
-
I understand that my participation is voluntary and that I am free to withdraw at any time, without giving any reason, without my medical care or legal rights being affected.
-
I understand that sections of any of my medical notes may be looked at by responsible individuals from The London School of Hygiene & Tropical Medicine or from regulatory authorities where it is relevant to my taking part in research. I give permission for these individuals to have access to my records.
-
I agree to take part in the above study/for my information to be used in this trial.
-
I understand that I can withdraw my consent at any time and my medical care will not be affected in anyway by my withdrawal.
| Name of Patient | Date | Signature |
| Name of Person taking consent (if different from researcher | Date | Signature |
| Researcher | Date | Signature |
Protocol Appendix 1b – Representative information sheet and consent form
INFORMATION FOR LEGAL REPRESENTATIVE (PERSONAL/PROFESSIONAL)
INTERNATIONAL STUDY OF BLEEDING AFTER INJURY
This hospital is taking part in a research study to find ways to reduce severe bleeding after serious injury. We are asking for your permission to enrol into a research study or continue study treatment for (the participant). You are being asked because the patient is unable to give consent.
WHAT YOU SHOULD KNOW ABOUT RESEARCH STUDIES:
This form gives information about the study including the aims, risks and benefits of taking part.
In this hospital, patients with severe bleeding are given the usual emergency treatment for bleeding. The aim of this research study is to find a better treatment. We hope that the study treatment (tranexamic acid) will help clotting and so lessen the amount of blood lost and reduce the need for a blood transfusion. But the study treatment may cause clots where they are not needed. We hope to find that the treatment will do a little more good than harm but we don't yet know this. Please read the information below carefully and ask the doctor looking after the participant any questions you have.
1) Why is this research being done?
Severe bleeding is a common cause of death after injury and it is important to find better ways of reducing the amount of blood lost.
2) What is the purpose of this study?
Tranexamic acid is often used to reduce bleeding after major surgery such as heart operations. This study is being done to see if it can also reduce bleeding after major injury. Tranexamic acid is not a new drug and is an approved treatment for many common conditions that involve bleeding.
3) Who is doing the study?
Dr {name of doctor} is in charge of this study at this hospital. The study is co-ordinated by doctors at the University of London.
4) A patient cannot be in this study if:
-
he/she is known to be under 16
-
he/she was injured more than 8 hours before arriving in hospital
-
the doctor thinks there is a particular reason why tranexamic acid definitely should not be given
-
the doctor thinks there is a particular reason why tranexamic acid definitely should be given
5) What will happen to the participant if you decide to allow him or her to be included in this study?
The participant was given all the usual emergency treatments for bleeding, including fluids to replace the blood that he/she lost. He/she was also given a dose of either the active tranexamic acid or an inactive dummy medicine called saline. The dose was given over a period of eight hours. The choice of what to give (active treatment or dummy treatment) was made randomly by a computer in the University of Oxford, UK. The doctors looking after the participant do not know whether he/she got the active or the dummy medicine. This information is kept on a confidential list in another hospital. The study involves no extra tests but the treating doctor will send brief details to the Co-ordinating Centre in London about how the participant has been. This information will be used in strict confidence by the people working on the study and will not be released under any circumstance.
6) What are the possible risks of being in the study?
Tranexamic acid is widely used and at the moment there is no conclusive evidence of serious side effects with short term use. Tranexamic acid is NOT a new drug.
7) What are the possible benefits of being in the study?
We hope that tranexamic acid may help reduce blood loss. The knowledge that we gain from this study will help people with similar injuries in the future.
8) If you have any questions or problems, who can you call?
If you have any questions you can contact Dr {name of doctor}, {job title e.g. Consultant in Accident & Emergency} by telephoning {telephone number}.
9) What information do we keep private?
All information about the participant and his/her injury will be kept private. The only people allowed to look at the information will be the doctors who are running the study, the staff at the Co-ordinating Centre and the regulatory authorities who check that the study is being carried out correctly. We will publish the results of the study in a medical journal so that other doctors can benefit from the knowledge, but personal information will not be included and there will be no way that the participants can be identified.
10) Can the study end early for the participant?
The study treatment can be stopped at any time in the 8 hours if necessary. We hope that you will let us use information about how the participant got on, but if you do not want us to use it then please tell the treating doctor.
11) What else do you need to know?
-
The study is funded by the University of London and the World Health Organisation, not the makers of tranexamic acid.
-
The London School of Hygiene & Tropical Medicine (University of London) as the Co-ordinating Centre for the study accepts responsibility attached to its sponsorship of the study and, as such, would be responsible for claims for any non-negligent harm suffered by anyone as a result of participating in this study.
-
We will give you a copy of this consent form to keep.
LEGAL REPRESENTATIVE CONSENT FORM
INTERNATIONAL STUDY OF BLEEDING AFTER INJURY
1. I confirm that I have read and understood the information sheet Version 2, dated 9 December 2004, for the above study and have had the opportunity to ask questions.
2. The research study described in this consent form, including the risks and benefits, has been explained to me and all of my questions have been answered to my satisfaction.
I consent to the participation of (patient) in this research study. To my knowledge, participation would not conflict with his/her religious or personal beliefs.
3. I may withdraw this consent at any time.
| Name of Patient | Date | Signature |
| Name of Person taking consent (if different from researcher | Date | Signature |
| Researcher | Date | Signature |
Protocol Appendix 2 – Entry form


Protocol Appendix 3 – Outcome Form

Protocol Appendix 4 – Protocol Summary

Appendix 3 Effect of consent rituals on mortality in emergency care research
Authors
Ian Roberts, David Prieto-Merino and Haleema Shakur, Clinical Trials Unit, LSHTM; Iain Chalmers, James Lind Initiative; Jon Nicholl, School of Health and Related Research, University of Sheffield.
Clinical trials are important in improving the safety and effectiveness of emergency care. Many such trials seek to assess the effects of time-critical treatments for life-threatening disorders such as traumatic brain injury, severe haemorrhage or respiratory distress. In general, before patients can be enrolled in such trials, current regulations require that they or their legal representatives provide written informed consent. 63,64 Although the requirement for written informed consent can sometimes be waived65 (e.g. if the patient is unconscious, treatment is urgent, and no relative is available), written consent is usually required in emergency-care research, despite the delays to treatment that this will usually entail.
Analysis of data from the MRC CRASH-1 trial,62 a multicentre randomised controlled trial of corticosteroid administration in acute severe head injury, provides an estimate of the delay associated with the requirement for written consent. On average, compared with hospitals that waived the need for consent, initiation of treatment was delayed by 1.2 (95% CI 0.7 to 1.8) hours in hospitals where written consent from relatives was required. 66 This delay would not occur if the treatment was given (or withheld) outside the context of a clinical trial, in normal clinical practice.
The delay can be life-threatening. The CRASH-2 trial29 showed that giving TXA to trauma patients with bleeds results in a significant and clinically important reduction in overall mortality (RR 0.91; 95% CI 0.85 to 0.97), as well as in mortality specifically ascribed to bleeding (RR 0.85; 95% CI 0.76 to 0.96). Further analyses have shown that these beneficial effects depend importantly on the promptness with which treatment with TXA is started. Taking account of the average delay (1.2 hours) associated with the need to obtain written consent in the MRC CRASH-1 trial, we used CRASH-2 data to provide an estimate of the consequences for survival of a (more conservative) 1-hour delay resulting from the requirement to obtain written consent.
We used a logistic regression model, with mortality due to bleeding as the outcome variable and treatment group, time to treatment, and an interaction term, as explanatory variables. The interaction term from this model estimates how the effect of treatment (OR) changes with time to treatment. We then calculated the risk of death for patients by treatment group, according to time to treatment.
The results are shown in Figure 7. The lines in part (b) of the figure give the risk of death in treated and untreated patients, respectively. The effect of a treatment delay in treated patients was estimated by applying the OR corresponding to a 1-hour treatment delay to the risk of death in the untreated group. The dashed line gives the estimated risk of death in patients in whom treatment is delayed. The point at which the ‘no delay’ and ‘if delay’ lines intersect the ‘risk of death if no intervention’ line gives the time when the trial treatment no longer provides patient benefit.
Using these data and data from the CRASH-2 trial on the proportion of patients who arrive at hospital within a given time since injury, we estimate that a 1-hour treatment delay reduces the proportion of patients who benefit from the trial treatment from 63% to 49%. Whereas the RR of death from bleeding with TXA was estimated in the CRASH-2 trial as 0.85 (95% CI 0.76 to 0.96), the corresponding RR in the presence of a 1-hour delay is 0.96 (95% CI 0.86 to 1.08).
FIGURE 7.
Proportion of patients benefitting from treatment.
The delay from consent rituals in emergency situations has important consequences. First, it results in avoidable mortality and probably morbidity in participants in the trial. Indeed, far from protecting the interests of patients participating in research, requirements for written informed consent and the resultant delay in starting treatment could be lethal. Second, the delay in starting treatment can obscure a real treatment benefit from the administration of a time-critical treatment. In the CRASH-2 trial, the requirement for written informed consent probably means that the trial has underestimated the beneficial effect of TXA in trauma patients with bleeds, which would be given without delay in normal clinical practice.
In the context of research involving people who are incapable of giving informed consent, the Declaration of Helsinki67 states that, if no patient representative is available and the research cannot be delayed, the study may proceed without informed consent provided that the specific reasons for involving patients with a disorder that renders them unable to give informed consent have been stated in the research protocol and that the study has been approved by a research ethics committee.
We argue that the need for an urgent trial treatment, even in patients who are conscious and whose relatives are available, by itself excludes the possibility of fully informed consent. If consent rituals delay the start of a trial treatment such that the treatment effect could be reduced or obscured, we maintain that seeking consent is actually unethical. There might be other treatments whose benefits have been missed or underestimated as a result of insistence on the rituals of informed consent, against the precepts of the Declaration of Helsinki, with resultant avoidable harm to patients. There is little evidence that widely promoted forms of research regulation do more good than harm. 68 Informed consent procedures, like other well-intentioned public health interventions, should be assessed rigorously. The lethal effects we have shown might have been found decades ago had the research ethics community accepted a responsibility to provide robust evidence that its prescriptions are likely to do more good than harm.
Appendix 4 Free Bank of Injury and emergency Research Data – freeBIRD
Why share data?
Data generated through participation of patients and the public should be put to maximum use by the research community and, whenever possible, translated to deliver patient benefit. Data sharing benefits numerous research-related activities: reproducing analyses; testing secondary hypotheses; developing and evaluating novel statistical methods; teaching; aiding design of future trials; meta-analyses; and hopefully helping to prevent error, fraud and selective reporting.
Data sharing achieves many important goals for the scientific community, such as:
-
reinforcing open scientific inquiry
-
encouraging diversity of analysis and opinion
-
promoting new research, testing of new or alternative hypotheses and methods of analysis
-
supporting studies on data collection methods and measurement
-
facilitating education of new researchers.
Free Bank of Injury and emergency Research Data
At the end of the CRASH-2 trial, additional funding was obtained from the NIHR HTA programme for the purpose of launching a data sharing facility for injury and emergency-related research data.
The Free Bank of Injury and emergency Research Data (freeBIRD) website (http://freebird.Lshtm.ac.uk) allows investigators to upload and share data from such trials. Making clinical trial data sets available to investigators beyond the original research team can improve patient care, advance medical knowledge and provide better value for money from health research.
The freeBIRD website aims to facilitate data sharing in the area of injury and emergency research in a timely and responsible manner. It has been launched by providing open access to anonymised data on > 30,000 injured patients (the CRASH-1 and CRASH-2 trials).
We hope that other trial investigators will also upload their datasets and that freeBIRD becomes a valued resource for all those committed to improving injury and emergency care.
Appendix 5 Total randomisations by geographical region
| Geographical region | Number randomised |
|---|---|
| Africa | 4816 |
| Asia | 7366 |
| Europe, Australia and North America | 2218 |
| Caribbean, Central and South America | 5807 |
| Total | 20,207 |
Appendix 6 Economic evaluation
| Variable | Gompertza | Log-normala | Log-logistica | Weibulla |
|---|---|---|---|---|
| Constant | −4.26 (−4.36 to –3.74) | 8.89 (8.09 to 9.69) | 8.36 (7.71 to 9.0) | −3.82 (−4.13 to−3.51) |
| Age | 0.020 (0.016 to 0.024) | 0.83 (0.21 to 1.45) | −0.05 (−0.06 to –0.04) | 0.02 (0.01 to 0.02) |
| Sex | −0.06 (−0.22 to 0.11) | 0.11 (−0.27 to 0.49) | 0.14 (−0.23 to 0.53) | −0.06 (−0.23 to 0.09) |
| GDPlow | −0.31 (−0.57 to –0.08) | 0.83 (0.21 to 1.45) | 0.79 (0.21 to 1.37) | −0.33 (−0.57 to –0.08) |
| GDPmid | −0.61 (−0.81 to –0.41) | 1.46 (0.94 to 1.97) | 1.45 (0.97 to 1.92) | −0.62 (−0.81 to −0.42) |
| Distribution | Observations | AIC | BIC | |
| Gompertz | 9513 | 9425 | 9460 | |
| Log-logistic | 9513 | 10,065 | 10,101 | |
| Log-normal | 9513 | 9954 | 9997 | |
| Weibull | 9513 | 10,088 | 10,130 |
FIGURE 8.
Cox–Snell residuals plot for Gompertz model. H, integrated hazard.
FIGURE 9.
Cox–Snell residuals plot log-logistic model. H, integrated hazard.
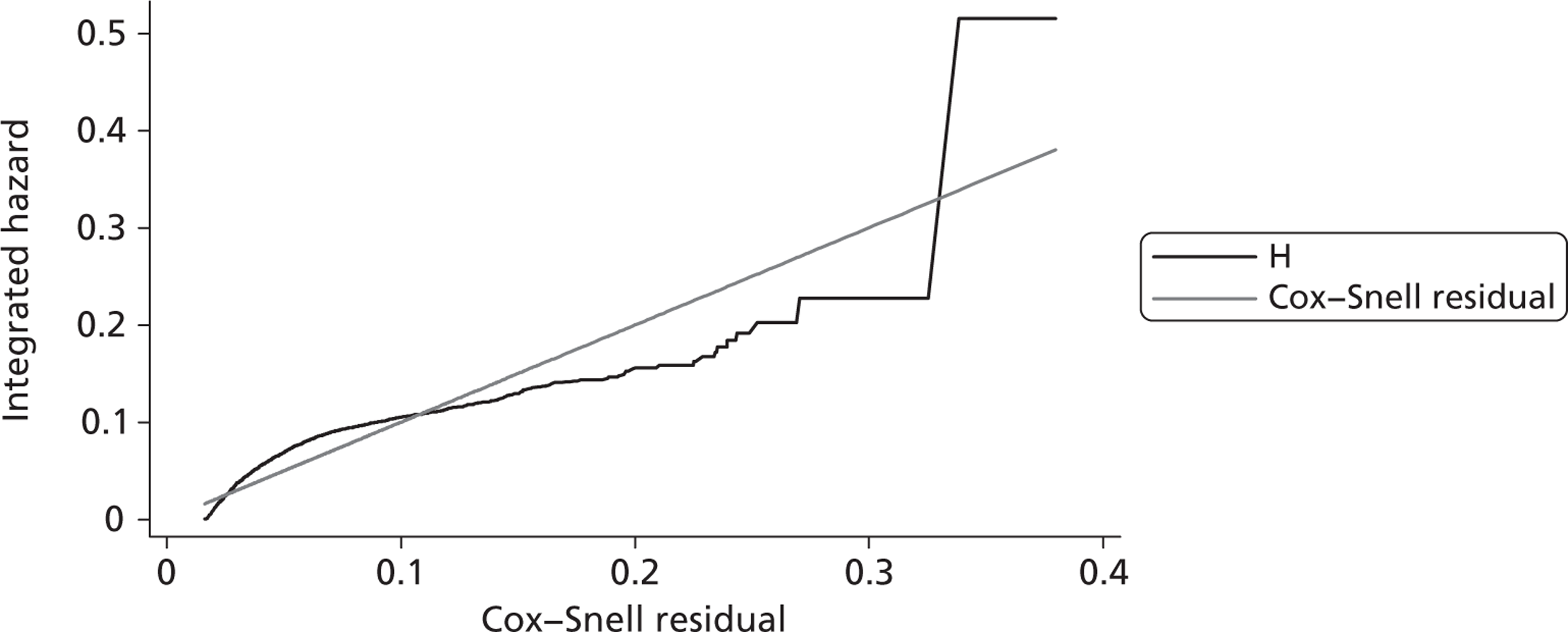
FIGURE 10.
Cox–Snell residuals plot log-normal model. H, integrated hazard.
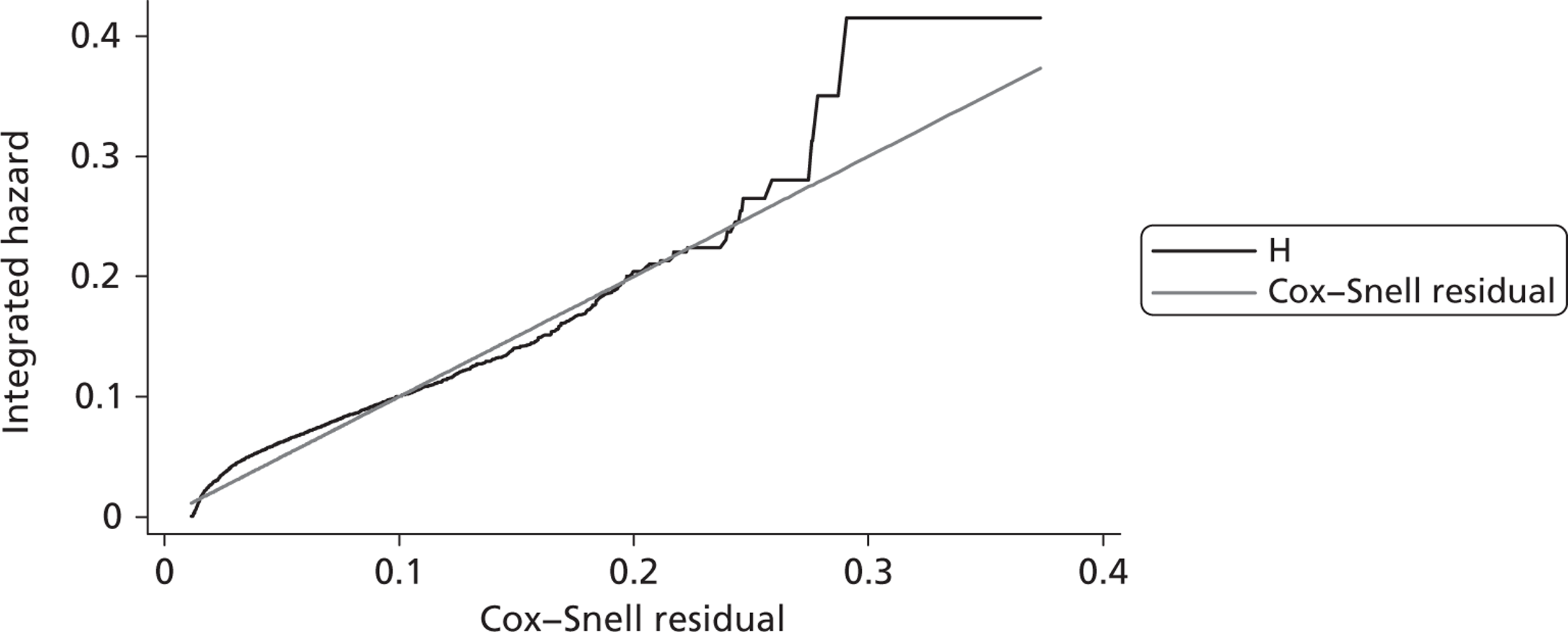
FIGURE 11.
Cox–Snell residuals plot for Weibull model. H, integrated hazard.
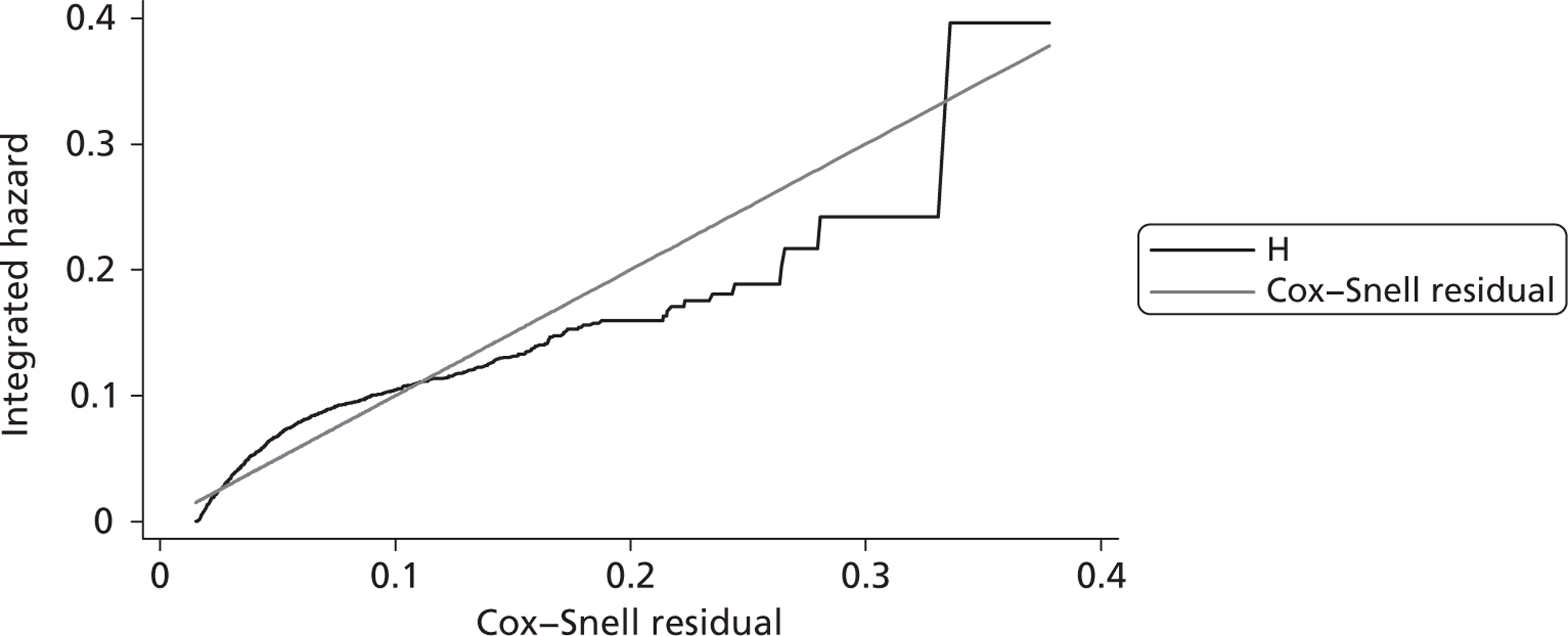
| Parameter name | Values | Distribution | Parameters |
|---|---|---|---|
| RR of death with TXA vs no TXA | 0.87 | Log-normal | σ = 0.156 |
| Additional non-ICU hospital stay for TXA patients | 0.04 | Normal | σ = 2.38 |
| TXA administration cost | |||
| Tanzania | $17 | Gamma | α = 20.54; β = 0.85 |
| India | $19 | Gamma | α = 21.75; β = 0.89 |
| UK | $31 | Gamma | α = 54.11; β = 0.57 |
| Cost of non-ICU hospital stay (per day) | |||
| Tanzania | $13 | Gamma | α = 106.13; β = 0.12 |
| India | $28 | Gamma | α = 10.95; β = 2.55 |
| UK | $429 | Gamma | α = 145.70; β = 0.24 |
FIGURE 12.
Cumulative hazard for Gompertz function by age group in LICs.
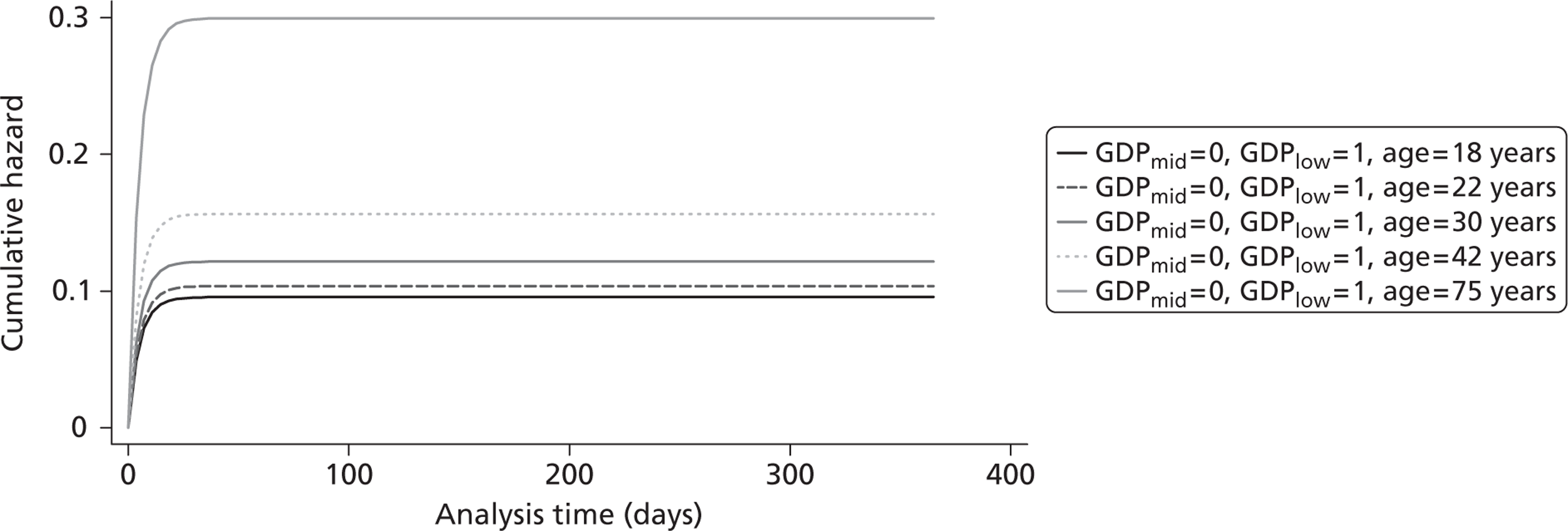
FIGURE 13.
Cumulative hazard for log-logistic function by age group in LICs.
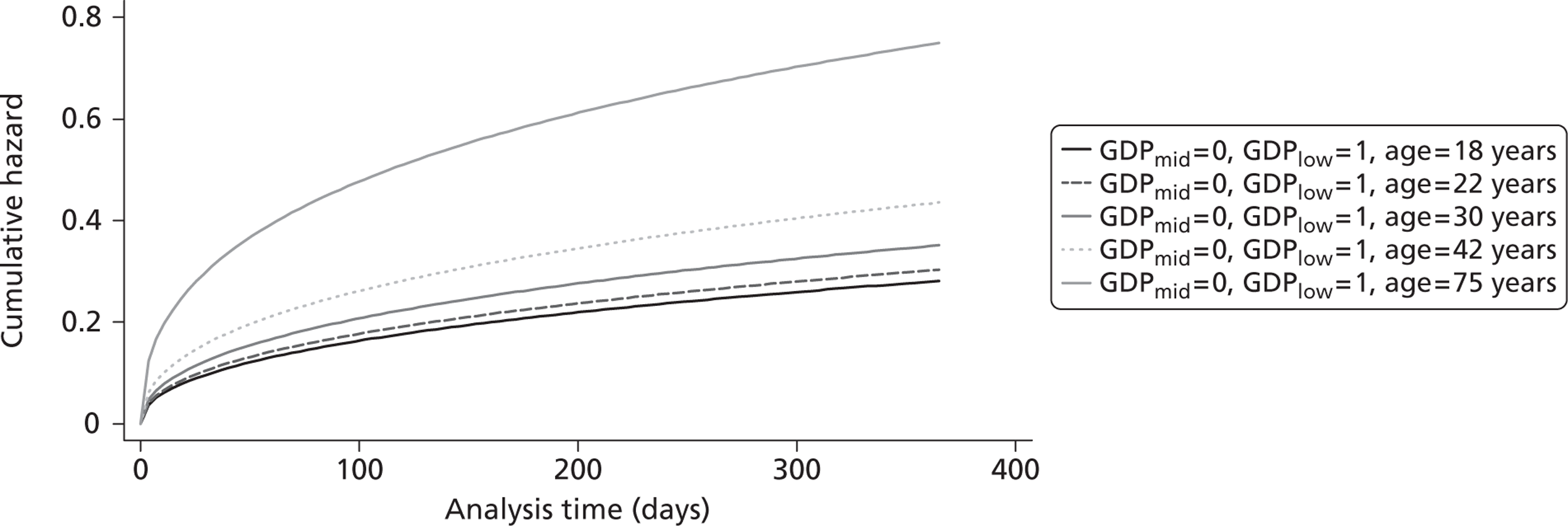
FIGURE 14.
Cumulative hazard for log-normal function by age group in LICs.
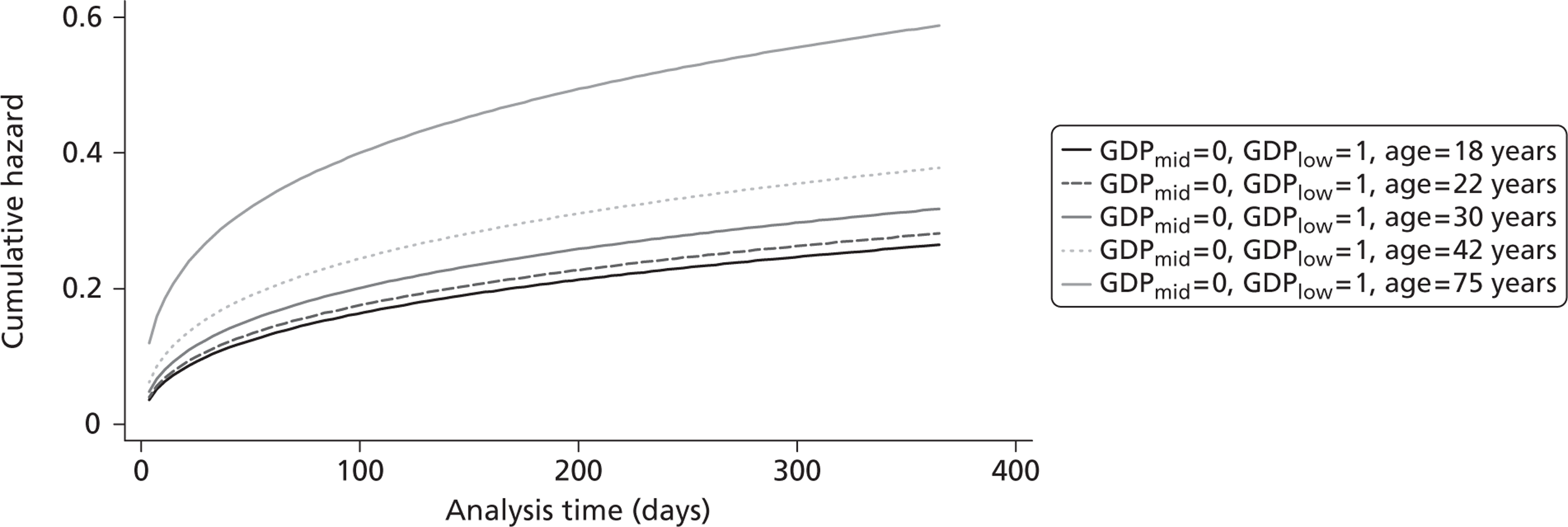
FIGURE 15.
Cumulative hazard for Weibull function by age group in LICs.
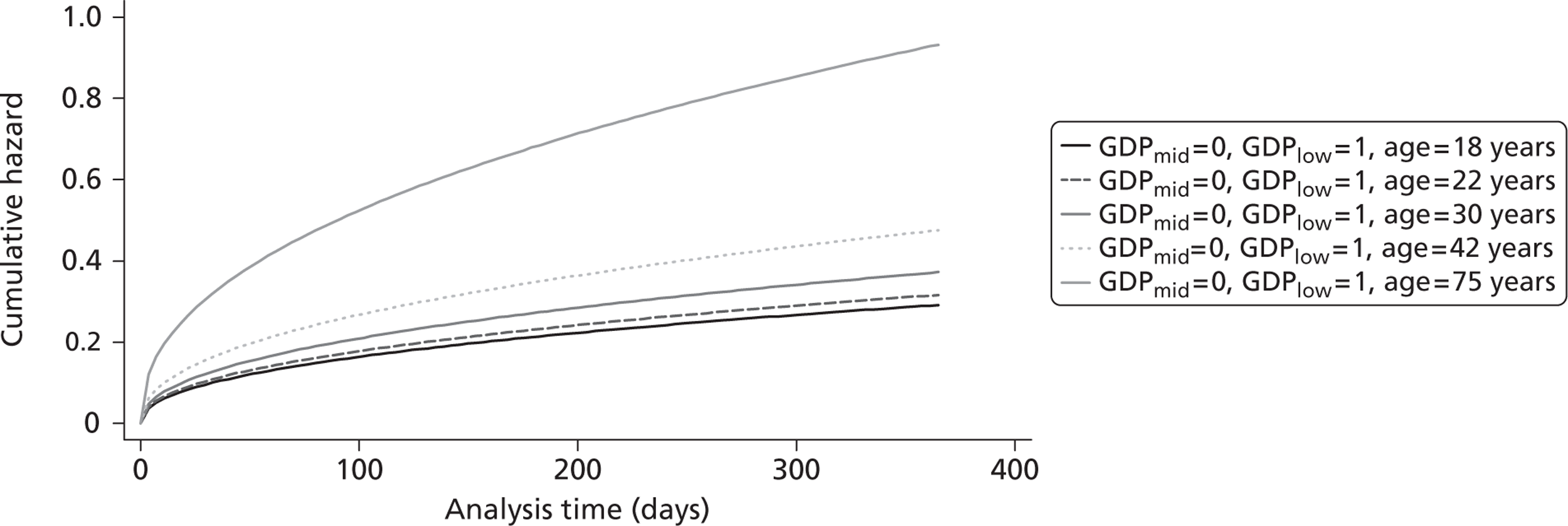
FIGURE 16.
Cumulative hazard for Gompertz function by age group in MICs.
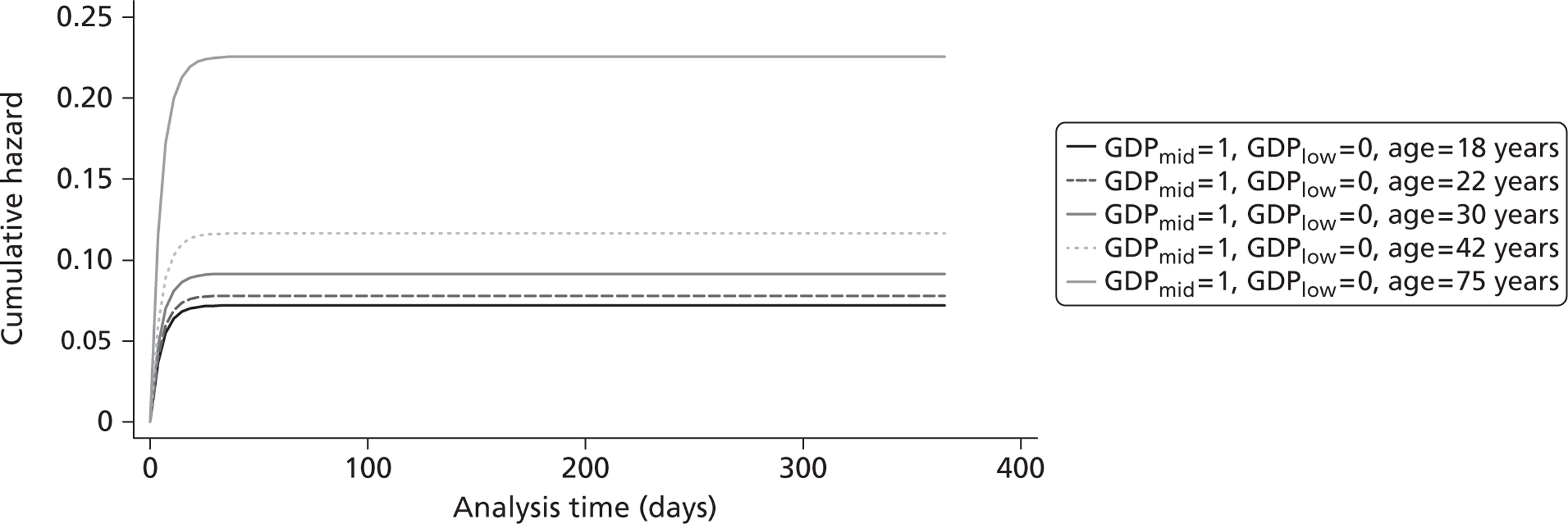
FIGURE 17.
Cumulative hazard for log-logistic function by age group in MICs.
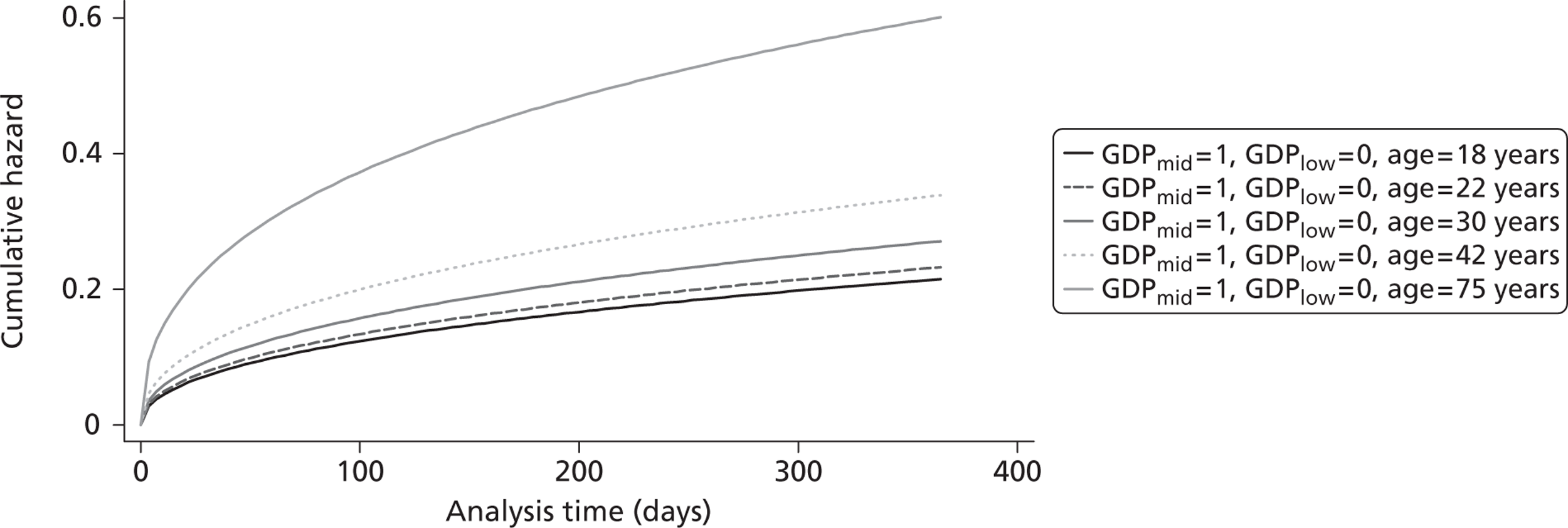
FIGURE 18.
Cumulative hazard for log-normal function by age group in MICs.
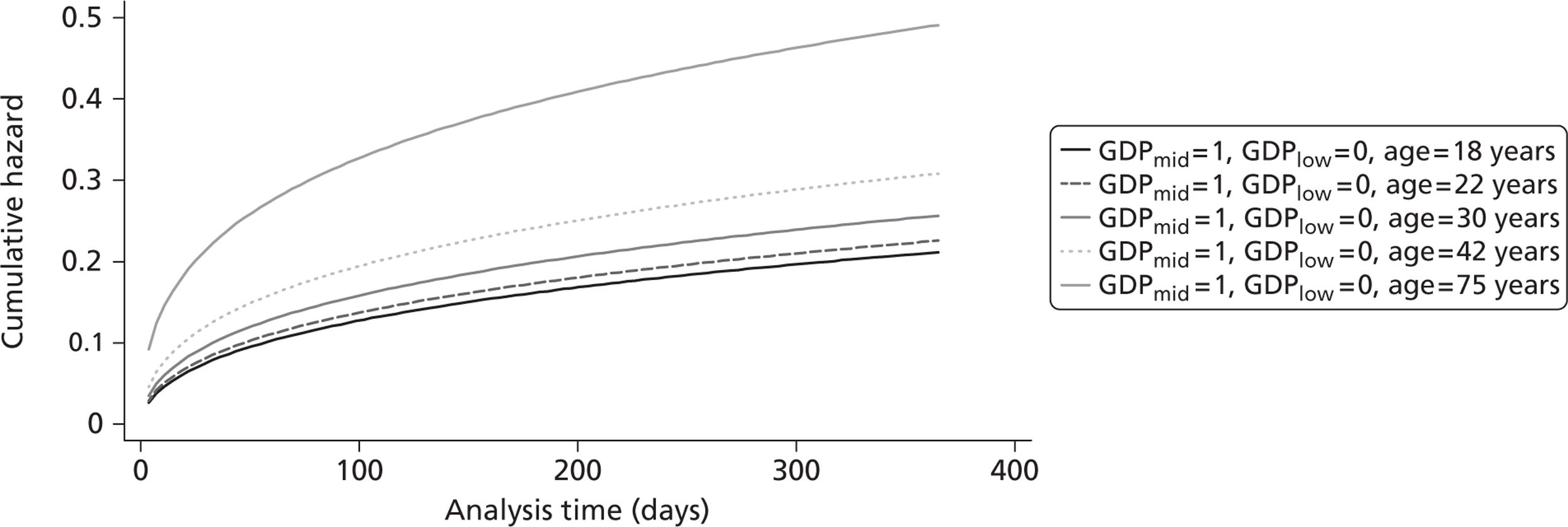
FIGURE 19.
Cumulative hazard for Weibull function by age group in MICs.
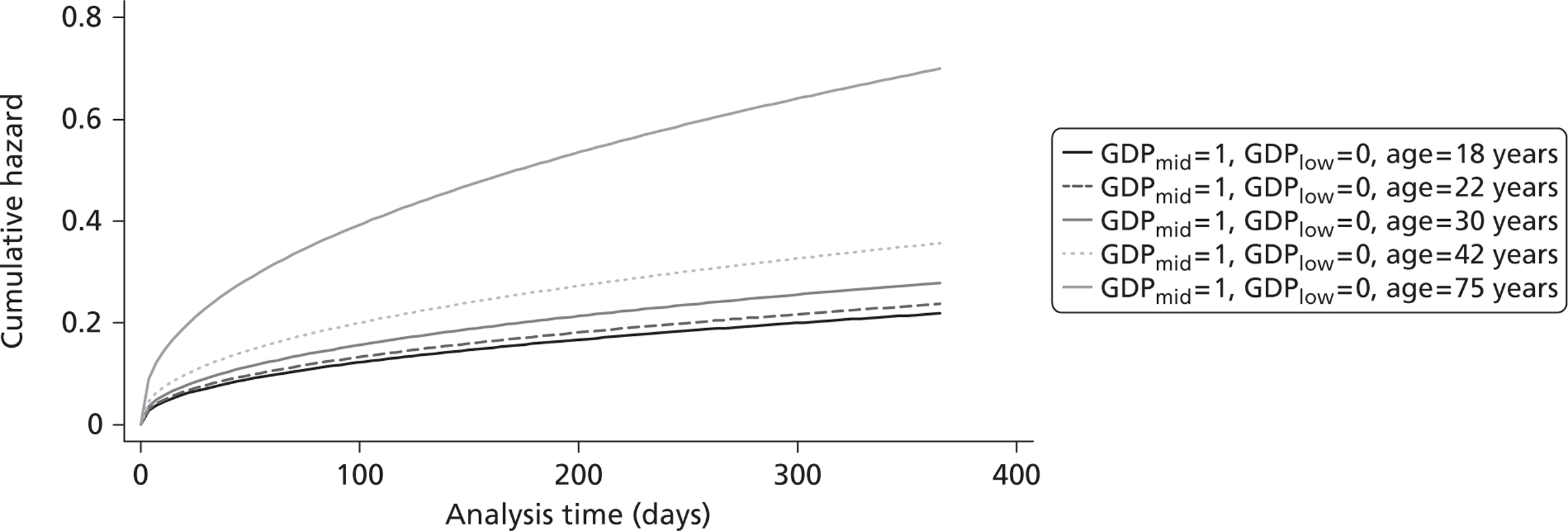
FIGURE 20.
Cumulative hazard for Gompertz function by age group in HICs.
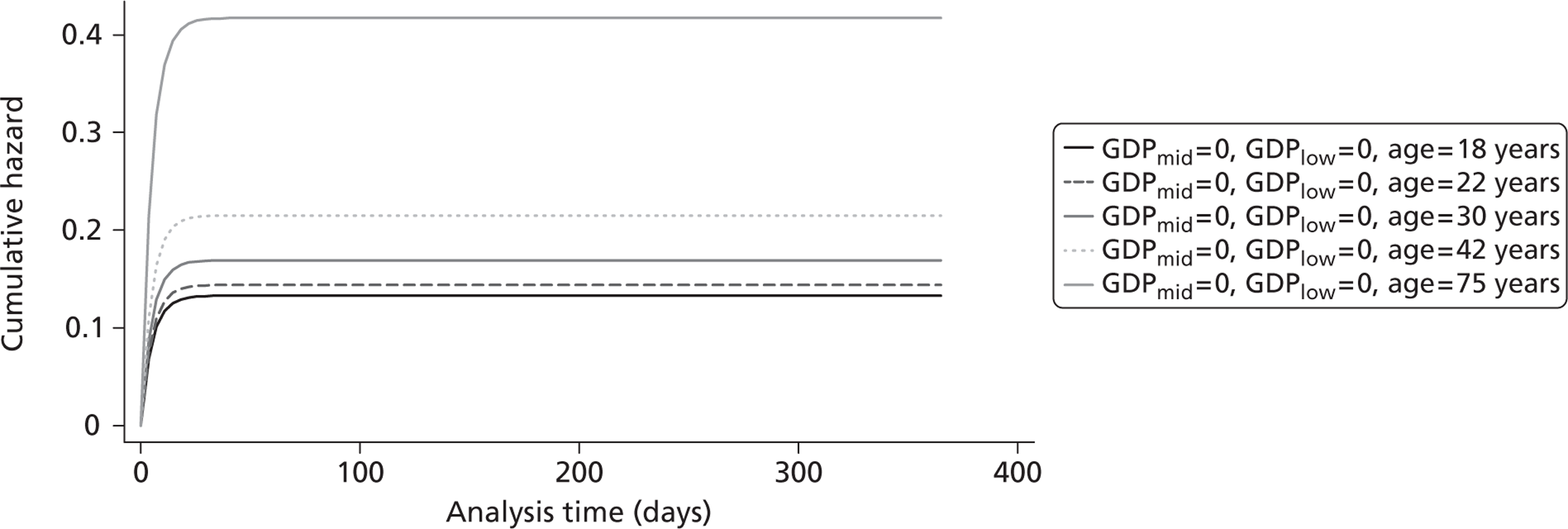
FIGURE 21.
Cumulative hazard for log-logistic function by age group in HICs.
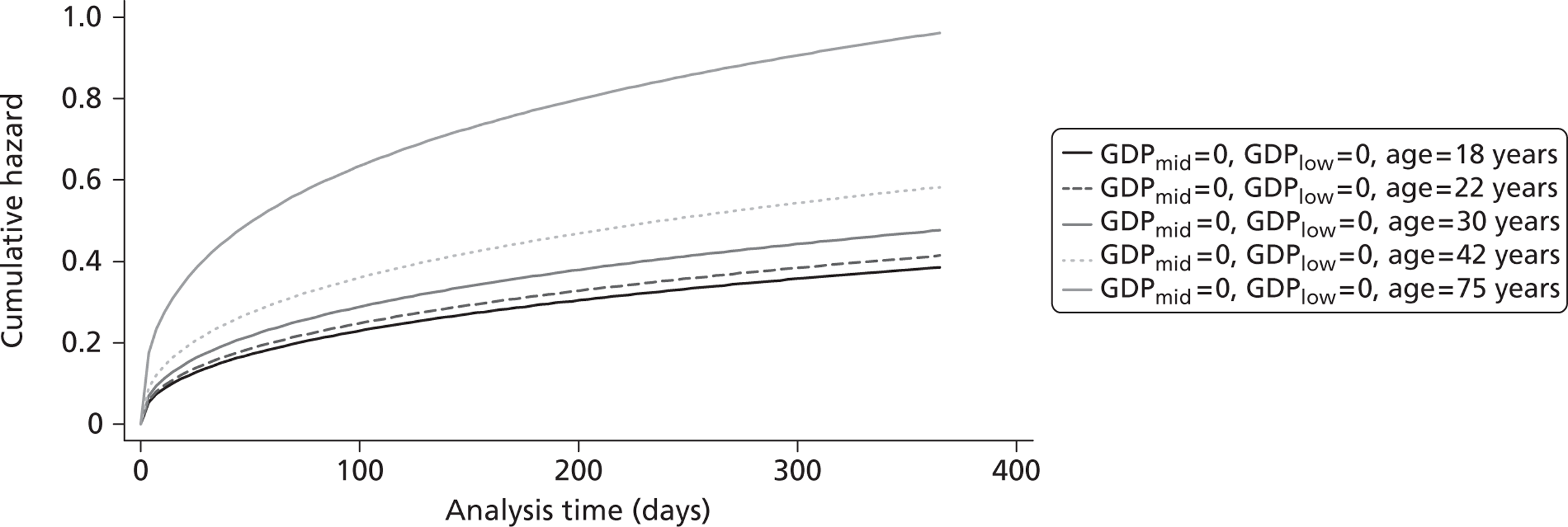
FIGURE 22.
Cumulative hazard for log-normal function by age group in HICs.
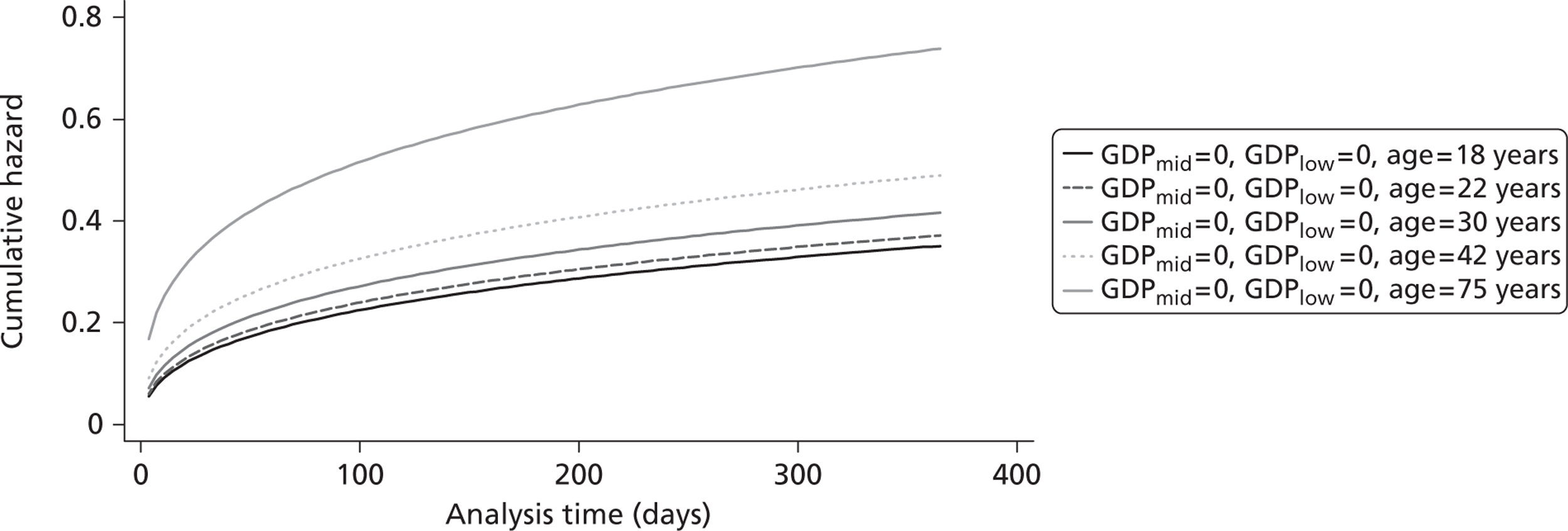
FIGURE 23.
Cumulative hazard for Weibull function by age group in HICs.
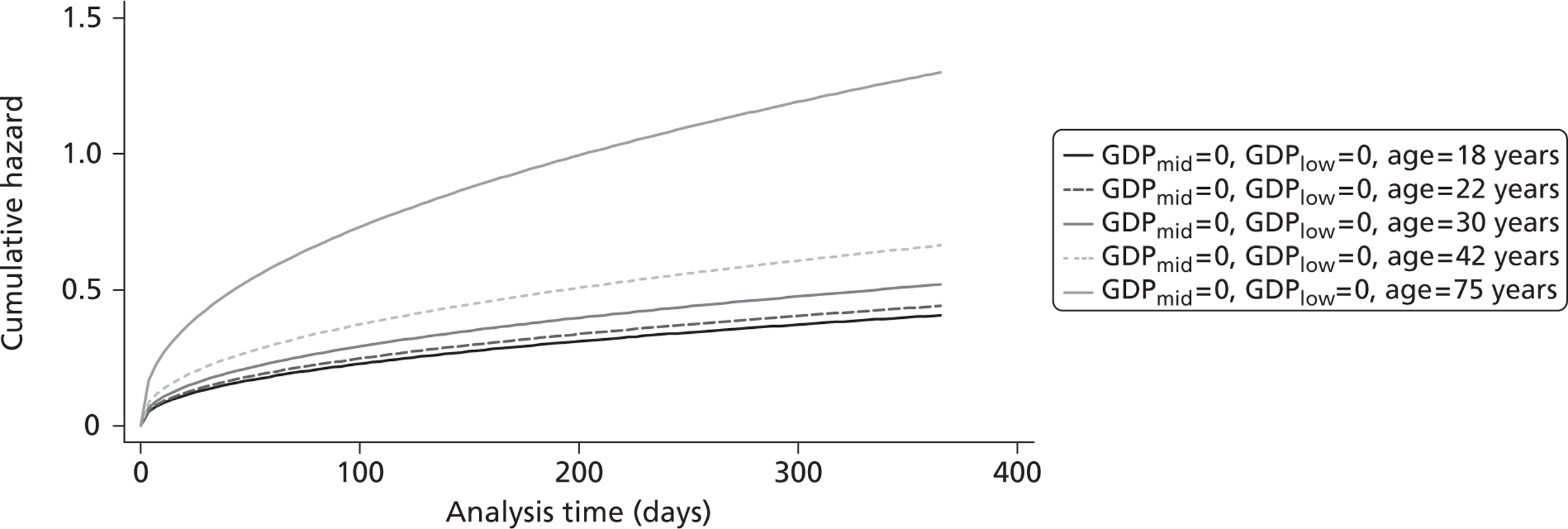
List of abbreviations
- AIC
- Akaike information criterion
- BP
- blood pressure
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CRASH-1
- Corticosteroid Randomisation After Significant Head Injury
- CRASH-2
- Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage-2
- CRASH-3
- Clinical Randomisation of an Antifibrinolytic in Significant Head injury-3
- CT
- computerised tomography
- CTSU
- clinical trial service unit
- DALY
- disability-adjusted life-year factor VIIa activated factor VII
- GCS
- Glasgow Coma Scale
- GDP
- gross domestic product
- HIC
- high-income country
- HIV
- human immunodeficiency virus
- ICER
- incremental cost-effectiveness ratio
- ICU
- intensive care unit
- i.v.
- intravenous
- LIC
- low-income country
- LSHTM
- London School of Hygiene and Tropical Medicine
- LY
- life-year
- MIC
- middle-income country
- MRC
- Medical Research Council
- OR
- odds ratio
- QALY
- quality-adjusted life-year
- rFVIIa
- activated recombinant factor VII
- RR
- relative risk
- SD
- standard deviation
- TXA
- tranexamic acid
- WHO
- World Health Organization
- WTP
- willingness to pay
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.