
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 04/11/02. The contractual start date was in September 2006. The draft report began editorial review in January 2012 and was accepted for publication in May 2012. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors' report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
SB, MD, CB, RB, PB, CF, CH, CK, MK, CL, JL, GL, EM-C, JM, MO, JO'B, AT, KW and AB have received consultancy fees, speakers' fees, research funding or educational support to attend conferences from pharmaceutical companies involved in the manufacture of antidepressants and antidementia drugs. SB and AB have been employed by the Department of Health for England and CB has been employed by the AS.
Permissions
Copyright statement
© Queen's Printer and Controller of HMSO 2013. This work was produced by Banerjee et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Scientific background
Dementia is one of the most common and serious disorders in later life, with a prevalence of 5% and an incidence of 2% per year in the over 65s. 1,2 Worldwide it affects 35 million people, and this will treble by 2050. 3 In the UK there are 750,000 people with dementia currently4 and 200,000 new cases every year. It causes irreversible decline in global intellectual, social and physical functioning. Abnormalities in behaviour, insight and judgement are part of the disorder, as are neuropsychiatric symptoms such as psychosis, anxiety and depression. The economic cost of caring for people with dementia is immense. In the UK, the cost of dementia is around £17B per year,4 greater than stroke (£3B), heart disease (£4B) and cancer (£2B). 5 Worldwide, its annual cost is US$600B, 1% of world gross domestic product,6 and these costs are set to at least triple in the next 20 years. 3,6 The need to improve care for people with dementia is a policy priority. 7–10 More importantly, the negative impacts of dementia on those with the disorder, in terms of deteriorating function, and on carers,11,12 are profound. Dementia has a devastating impact across culture, gender, ethnicity and class.
Depression is common in dementia, with prevalence of > 20%,13,14 causing distress, reducing quality of life, exacerbating cognitive and functional impairment, increasing mortality, and increasing carer stress and depression. 14–16 Treating depression is therefore a key clinical priority to improve the well-being, quality of life and level of function of people with Alzheimer's disease (AD).
We searched the PubMed and The Cochrane Library databases to 1 March 2011, without language restrictions, for full articles reporting randomised controlled trials (RCTs), systematic reviews, and meta-analyses with the search terms ‘depression’, ‘dementia’, ‘Alzheimer's disease’, ‘antidepressant’, ‘meta-analysis’, and ‘CSDD’ (Cornell Scale for Depression in Dementia). We excluded trials without recognised depression outcome measures, placebo controls or specified thresholds for depressive disorder. We identified one Cochrane review17 and three systematic reviews. 18–20
The Cochrane review Antidepressants for treating depression in dementia,17 completed in July 2002, identified six studies with 739 subjects meeting inclusion criteria (‘all relatively unconfounded, double-blind, randomized trials comparing any antidepressant drug . . . with placebo, for patients diagnosed as having dementia and diagnosed as having a depression according to established criteria’). Only three studies, comprising 107 subjects, had data that could be subject to a meta-analysis of efficacy. Petracca et al. 21 studied 24 subjects in a neurological outpatient clinic in Argentina in a double-blind, placebo-controlled crossover trial of clomipramine [a tricyclic antidepressant (TCA)] with two 6-week treatment periods with a 2-week washout period. There was a mean change of −10.7 on the Hamilton Depression Scale in the intervention group and −4.5 in the control group, an equivocal outcome. Reifler et al. 22 selected 61 subjects from two university outpatient clinics in an 8-week, double-blind trial of imipramine (a TCA). The study showed no treatment effect. The third trial23 was an interim analysis of data on 22 subjects, which, subsequently, was reported fully in Lyketsos et al. 24 These final data from the Depression in Alzheimer's Disease Study (DIADS) were not available to the Cochrane review. In the final study, 44 subjects were recruited from a single university outpatient clinic into a 12-week, double-blind, placebo-controlled trial of sertraline [a specific serotonin reuptake inhibitor (SSRI)]. An effect size of 0.51 was reported, with a mean change of −10.5 on the Hamilton Depression Scale in the intervention group and −4.5 in the control group, and −9.9 and −3.2 on the CSDD. 25 Other than the further data on the additional 22 cases reported in Lyketsos et al. ,24 and the group's subsequent DIADS-II study,26 which was negative and which is discussed below, we are not aware of any other studies published since then that would have met the criteria for inclusion in the Cochrane review.
The Cochrane review concluded that, despite its clinical seriousness, there was only weak evidence of the effectiveness of antidepressants in dementia. Two studies used TCAs – ‘drugs not commonly used in this population’ – because anticholinergic side effects may negatively affect cognition, and cardiac side effects; only one used the most commonly used class (SSRIs). None covered newer classes of antidepressants and all were of short duration. Lyketsos et al. 24 acknowledged the need for research into the efficacy of antidepressants in a wider range of depression type and severity, longer-term treatment, and the comparative efficacy of different classes of antidepressants. Therefore, they completed a follow-up study, the DIADS-II study. This compared 67 people who were prescribed sertraline with 64 people who were given placebo. In contrast with DIADS, this study found no benefit whatsoever of sertraline at 12 or 24 weeks, and concluded that this was not a function of depression severity, depression type or severity of dementia. 26,27
One systematic review18 used a different quality assessment and included data from five studies of 165 participants, and concluded that antidepressants were better than was placebo for treatment response [odds ratio (OR) 2.32; 95% confidence interval (CI) 1.04 to 5.16] and remission of depression (OR 2.75; 95% CI 1.13 to 6.65) with rates of discontinuation equivalent to placebo. This did not include the DIADS-II data. The positive message of the meta-analysis of the 2010 systematic review19 is questionable because, although it includes the DIADS-II data,26,27 it seems to count the data from the first DIADS trial twice (i.e. by treating the interim23 and the final trial data24 as separate data sets when the first is a subset of the second). Finally, the 2011 study18 concluded that the efficacy of antidepressants in people with depression and dementia is not established. The reviews and meta-analyses taken together are not conclusive but all reported that the limitations of previous trials were their small sizes, low numbers of participants taking drugs that were used in clinical practice, and short follow-up.
It is clear that the subjects recruited into all of the trials discussed above were highly selected and so there may be limitations in the generalisability of the data derived from them. One element of this is the severity of depression recruited, with Lyketsos et al. 24 and Reifler et al. 22 requiring depression to meet Diagnostic and Statistical Manual (DSM) criteria for major depressive episode. Such disorders form only a small proportion of clinically significant depression requiring intervention in older adults in the community.
All of these studies, except DIADS-II, were of short duration, and so could not tackle the crucial issue of whether or not there is longer-term benefit associated with antidepressant treatment. It is unclear whether the differential efficacy between the published studies relates to the choice of antidepressant, differences in study design and power or chance variation. Importantly, the literature does indicate that the successful resolution of depression may be associated with cognitive and functional improvements. 17 There are, however, several cautions. For example, one study of the TCA imipramine indicated that active treatment increased cognitive impairment and disability, whilst several studies of falls indicate that most antidepressants increase risk of falling. In addition, there have been safety concerns relating to the SSRI sertraline and gastrointestinal bleeding,28 and the SSRI paroxetine and withdrawal.
Despite the equivocal evidence, current practice is to use antidepressants, often sertraline, as a first-line treatment for depression in dementia. The Quality Standards Subcommittee of the American Academy of Neurology29 cited only ‘moderate clinical certainty’ for antidepressants in treating depression in dementia but concluded that ‘SSRIs may offer some benefit with greater tolerability’. A UK primary care guideline suggests antidepressants as the only form of management for depression in dementia30 and the UK National Institute for Health and Clinical Excellence/Social Care Institute for Excellence (NICE/SCIE) clinical guideline on dementia31 also advocates antidepressants for depression in dementia.
Given the limited evidence in this clinically important area, the Health Technology Assessment (HTA) programme of the UK National Institute for Health Research (NIHR) prioritised antidepressant treatment of depression in dementia as an area for primary research. They commissioned the study reported here to fill gaps in the evidence base definitively and enable the formulation of good-quality guidance on care for people with dementia and their carers.
Explanation of rationale
Experimental: inclusion of an arm of the study using tricyclic antidepressants
As discussed above, there are unanswered questions concerning what class of antidepressant to choose and how long to treat. This trial was designed to attempt provide best-quality data on all of these clinically important areas.
One possible area of contention is the appropriateness of including a TCA arm in the trial. This was referred to in the original research brief. Prior to our initial submission we carried out a local consultation with people with dementia, family carers and clinicians in London, Manchester and within the Alzheimer's Society (AS). The findings of this exercise were clear. Patients, carers and clinicians all believed that it would be unacceptable to randomise people with dementia to medication with a predictable set of negative (anticholinergic, e.g. constipation, increased confusion, blurred vision, low blood pressure, drowsiness) side effects, even given the fact that the competing classes of medication had their own profile of side effects.
In addition, clinicians reported to us that their clinical practice was not to use TCAs as a first-line treatment for depression in dementia and that they believed people with dementia to be at a higher risk of harm from TCA side effects than people without dementia. Therefore, they raised questions of the clinical acceptability of a trial that included the possibility of randomisation to a TCA. To be successful we needed a large number of clinical teams to take part in case finding, and if the trial were to generate real effectiveness data then these participants needed to be an unbiased sample of all potential prescribers. On these grounds we therefore decided not to include a TCA arm but instead to compare the clinical effectiveness and cost-effectiveness (including discontinuation and adverse events) of examples of the two classes of antidepressants most in use.
In the subsequent feedback from the HTA Commissioning Board we were invited to reconsider our decision not to include a TCA arm. Therefore, we consulted the AS Quality Research in Dementia (QRD) Network. This was a panel made up of people with dementia and their carers, who advised the UK AS on research issues. The consultation was carried out by the AS Director of Research (Professor Clive Ballard). He consulted regional co-ordinators of the AS QRD and individual members of the network, representing the views of 45 QRD members, most with experience of caring for someone with dementia who has been treated with antidepressants. The purpose was to inform them about key aspects of the study, in particular whether or not it was appropriate to include TCAs as one of the treatments. All but one of the people responding strongly expressed the view that TCAs were an inappropriate treatment for people with dementia, describing a number of personal experiences where serious falls, increased confusion, urinary retention and other adverse events had resulted in a serious detrimental impact to the quality of life of the person with dementia.
We also consulted clinicians through the potential collaborating centres more widely and, again, there was a near unanimous view that it was not clinically supportable to initiate people with depression in dementia on a TCA. They also reported that the existence of such a possibility in randomisation would discourage them from entering patients into the trial. At the very least it was therefore likely that there would be substantial selection bias (both in patient acceptability and clinician referral) introduced by the inclusion of a TCA arm. We therefore decided not to include a TCA arm.
Experimental: choice of antidepressants
The selection of the best candidate antidepressants for this trial is not straightforward. Cost and power considerations dictate that an optimal design should include two active antidepressant treatments and a placebo. However, there are several cautions. One previous small RCT has indicated equivocal benefit with the TCA clomipramine21 but other data indicate marked side effects and exacerbation of disability associated with TCA treatment. For example, one study of the TCA imipramine indicated that active treatment increased cognitive impairment and disability,22 and several studies of falls indicate that most antidepressants increase the risk of falls. 32 In addition, there have been safety concerns with SSRIs, with respect to withdrawal effects and the potential risk of self-harm.
Within this framework, the choice of specific antidepressant agents required careful consideration. For example, the best evidence of efficacy in people with dementia at the time of the trial design was for the SSRI sertraline, as that was the compound used in the original DIADS study. 23 But this was a very small trial and other SSRIs, such as citalopram, have also been reported to be effective in treating depression in later life, including those with dementia, but in less-well-designed studies. 33 Citalopram may have less interactions with other drugs than other SSRIs and people with dementia are usually recipients of polypharmacy. The most effective antidepressant in people without dementia available at that time was probably venlafaxine34 but there are no RCTs in people with dementia and there are potential concerns regarding side effects in these individuals. 35 A newer antidepressant, mirtazapine, appeared to have a good safety profile and a different mode of effect and was becoming widely used in clinical practice to treat depression in people with dementia but had not been evaluated in a RCT for this indication.
In order to design and cost a trial of this sort there is a need to identify the compounds to be tested. We therefore made the decision that our trial design should include sertraline (the SSRI with the best evidence and which would be off licence by the end of the trial) and mirtazapine (the novel antidepressant with the least safety concerns). The doses chosen reflect common clinical practice for the treatment of depression in dementia and (in the case of sertraline) direct trial evidence,24 with higher doses than those suggested here (i.e. > 150 mg of sertraline or 45 mg of mirtazapine) being seen as less appropriate in people with dementia as well as depression.
Controls: use of placebo
The research brief referred to comparison with standard care. Despite the evidence base, standard care for depression in dementia is the provision of antidepressants, with SSRIs being the most commonly used drugs. 29 Standard secondary care (and it was stipulated in the brief that the study should be people referred to secondary care) is, however, much more than just medication. It involves a detailed multidisciplinary assessment of the person with dementia and their family carers with the generation of an individualised care package for each, often with continuing monitoring and follow-up. 36 We therefore developed a study design whereby all participants receive full standard care with only the antidepressant element subject to investigation against placebo and between classes of compound.
We concluded that, at the time of the trial design, there was little convincing evidence that antidepressant treatments were more effective than placebo in treating depression in dementia in real-world clinical practice. As discussed above, the data available were generally from small-scale studies of highly selected groups of patients with depression in dementia. The research brief required a trial that could take the evidence base and clinical practice forward significantly. In these circumstances we came to the belief that a placebo group was not just ethical, but also essential. If antidepressants were indeed not effective then the placebo group might do better, as they should have had fewer side effects. We carried out a further consultation exercise on the acceptability of the inclusion of a placebo group with local people with dementia, family carers and clinicians. They were supportive of the strategy of using placebo in these circumstances, as long as its use was minimised and that the information derived from the trial would yield a definitive answer.
Run-in period
One possible element of a trial such as this is the inclusion of a run-in period. The potential value of this is to identify a group of people more likely to comply with subsequent data collection (i.e. to minimise loss to follow-up) and to identify a group of people with depression who are less likely to spontaneously recover. 37–39 It is also possible that depression scores may be reduced by psychosocial interventions,40 some of which may be provided as part of routine care. The result of these factors is a potentially high placebo response rate in clinical trials. The research brief was clear in its call for an evaluation of antidepressants in routine clinical practice, and it is not routine clinical practice to precede the prescription of antidepressants for depression in dementia with a trial of a non-pharmacological treatment such as exercise. Instead, we proposed the clinically relevant inclusion criterion for the trial that the depression should have been present for at least 4 weeks.
Specific objectives
The primary objective was to determine the clinical effectiveness and cost-effectiveness of a SSRI (sertraline) and a noradrenergic and specific serotonergic antidepressant (NASSA; mirtazapine) in reducing depression (measured by CSDD) 13 weeks post randomisation compared with placebo.
Secondary objectives included clinical effectiveness at 39 weeks; differences in adverse events; other outcomes (e.g. quality of life, cognition, carer burden, carer quality of life); and the influence of clinical characteristics (e.g. dementia severity, dementia type, depression type, depression severity, and neuropsychiatric symptoms).
Chapter 2 Methods
Trial design
A multicentre, parallel group, double-blind placebo-controlled RCT of the clinical effectiveness of two antidepressants, mirtazapine and sertraline, with 13- and 39-week follow-up (1 : 1 : 1 allocation).
Participants
Eligibility
This was a pragmatic trial, with inclusion criteria designed to mirror clinical practice closely. Those eligible met NINCDS/ADRDA criteria for probable or possible AD41 and co-existing depression of at least 4 weeks' duration. A local research worker then assessed their depression severity using the CSDD. 25 Those scoring 8+ were eligible for the trial. The only exclusions were those who were clinically too critical for randomisation (e.g. suicide risk); absolute contraindication to trial medications; those currently taking antidepressants; those in another trial; and those having no family or professional carer to give collateral information.
Settings and location
Participants were recruited from community old-age psychiatry services in nine English centres (Birmingham, Cambridge, Leicester, Liverpool, Manchester, Newcastle, north London, Southampton, and south London and Kent).
Interventions
There were three groups: (1) sertraline, (2) mirtazapine and (3) placebo, all with normal clinical care. The target dose was for all participants was 150 mg of sertraline or 45 mg of mirtazapine per day. Drugs and their placebo were identically presented with participants aiming to take six tablets orally once a day (up to three sertraline 50 mg or sertraline placebo; and up to three mirtazapine 15 mg or mirtazapine placebo).
Outcomes
Co-primary outcomes
Depression in dementia, measured by CSDD,25 and costs measured by the Client Service Receipt Inventory (CSRI)42 at 13 weeks.
Secondary outcomes and moderators
These included: disease-specific health-related quality of life [Dementia Quality of Life (DEMQOL) and DEMQOL-Proxy];43 generic quality of life [European Quality of Life-5 Dimensions (EQ-5D) interview administered to carer]44 withdrawal from treatment; cognitive impairment [Mini Mental State Examination (MMSE)];45 medication adherence; adverse events; carer mental health [General Health Questionnaire (GHQ-12)];46 carer quality of life (SF-12v2)47 and carer burden (Zarit Scale);48 behavioural disorder [Neuropsychiatric Inventory (NPI)];49 and (at baseline) a dementia vascularity index (modified Hachinski scale). 50
Data entry
The data arising from each baseline or follow-up interview were entered at each centre via the internet using the InferMed Macro electronic data capture system by the researchers as the study proceeded. The data entry system used was Macro version 3.0 (InferMed, London, UK). Prior to data base lock, all of the primary outcome measures and 10% of all other outcome measures were source data verified. Table 1 summarises the measures that were used at each assessment time point.
| Assessment | Informant | Screening | Registration | Baseline | Randomisation | Ongoing | Week 4 | Week 8 | Week 13 | Week 39 | Withdrawal |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Verbal consent to referral to recruiting PI | Patient/carer | ✗ | |||||||||
| Eligibility assessment | Referring and recruiting, PI/RW | ✗ | |||||||||
| Exclusions | Patient/carer | ✗ | |||||||||
| Informed consent and registration | Patient/carer | ✗ | |||||||||
| Demographics | Patient/carer | ✗ | |||||||||
| Randomisation | Patient | ✗ | |||||||||
| Carer medication preference | Carer | ✗ | |||||||||
| Pill count | Carer | ✗ | |||||||||
| Medical history | Carer | ✗ | |||||||||
| CSDD | Patient/carer, RW | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| NPI | Carer | ✗ | ✗ | ✗ | |||||||
| DEMQOL-Proxy | Carer | ✗ | ✗ | ✗ | |||||||
| DEMQOL | Patient | ✗ | ✗ | ✗ | |||||||
| BADL | Carer | ✗ | ✗ | ✗ | |||||||
| CSRI | Carer | ✗ | ✗ | ✗ | |||||||
| Zarit Carer Burden Scale | Carer | ✗ | ✗ | ✗ | |||||||
| SF-12 v2 (carer) | Carer | ✗ | ✗ | ✗ | |||||||
| GHQ-12 (carer) | Carer | ✗ | ✗ | ✗ | |||||||
| CGI | Carer | ✗ | ✗ | ||||||||
| MMSE | Patient | ✗ | ✗ | ✗ | |||||||
| EuroQol (participant) | Patient | ✗ | ✗ | ✗ | |||||||
| EuroQol (carer) | Carer | ✗ | ✗ | ✗ | |||||||
| Olin (Depression in Dementia) | Carer | ✗ | ✗ | ✗ | |||||||
| Medication guess | Patient/carer | ✗ | ✗ | ||||||||
| Concomitant therapies | Patient/carer | ✗ | |||||||||
| Concomitant medications | Patient/carer | ✗ | |||||||||
| Trial medication log | Patient/carer | ✗ | |||||||||
| Non-serious adverse event | Patient/carer | ✗ | |||||||||
| Serious adverse event | Patient/carer | ✗ | |||||||||
| Withdrawal form | Patient/carer | ✗ |
Sample size
Initially a sample size of 507 was calculated to provide 90% power to detect a two-point CSDD difference [standard deviation (SD) 5; standardised effect size (SES) 0.4] for the sertraline–placebo and the mirtazapine–placebo comparisons at 13 weeks, and 86% power at 39 weeks. This allowed 20% loss to follow-up, correlation between baseline and outcome CSDD ≥ 0.6, and up to 12.5% of participants to either drop out or drop in using an analysis of covariance with two-sided 5% significance levels. This allowed for two-sided 95% CIs for the difference in the proportion of adverse events between the groups of (a clinically significant) 10%.
Change to protocol
Owing to a call for extra funding following slower recruitment than predicted, the sample size needed for the trial was reassessed by statistical review by the Data Monitoring and Ethics Committee (DMEC) when there were 75 subjects available with 13-week follow-up data. The parameters of the sample size calculation were not changed (SD 5, SES 0.4), but the new target was calculated on the basis of reported values that had greater precision than did the pre-study assumptions. An extended recruitment period was agreed, with a revised target of 339 participants for the sample (113 in every group). This change involved unmasking of a statistician (Clare Rutterford, Clinical Trials Unit, King's College London, UK), who was not involved in the final analyses, to the identity of patients in the placebo group.
Randomisation
Participants were allocated to placebo, sertraline or mirtazapine (1 : 1 : 1) through the Mental Health & Neurosciences Clinical Trials Unit (MH&N CTU) after baseline assessment and obtaining consent. The MH&N CTU database programmer independently undertook treatment allocation. Random allocation was stratified by centre and undertaken with a computer-generated randomisation sequence with randomly varying block sizes (block sizes of three or six). Allocation was physically carried out during working hours from Monday to Friday.
Blinding
The trial was double blind, with medication and placebo identical in appearance for each antidepressant. Referring clinicians and research workers completing baseline and follow-up assessments were kept blind to group allocation, as were patients and pharmacies. Statisticians were blind to group identity until after the analyses were completed.
Statistical methods
The statistical analysis plan was finalised and approved by the Trial Steering Committee and the DMEC. Significance was tested at 5% level for all analyses. Analyses were completed in Stata version 11 (StataCorp LP, College Station, TX, USA). Analyses were pragmatic, based an intention to treat sample.
Descriptive statistics
All baseline data were summarised by treatment groups. Only descriptive statistics were utilised; no formal statistical comparisons were undertaken. Continuous variables were reported as means and SDs, categorical variables are presented as frequencies (n) and percentages (%).
Primary analyses
The CSDD differences between treatment groups (sertraline–placebo and mirtazapine–placebo) were estimated with mixed linear regression models. Covariates were treatment group, baseline CSDD score, time and the stratification factor, and centre. A time-by-treatment interaction term was included to allow estimates at the individual time points to be summarised. The model for the CSDD incorporated random intercepts by participant. Model assumptions were checked by use of diagnostic plots.
We did modelling with the assumption that data were missing at random, and included predictors of missing data (treatment group and centre) in the modelling. We used a logistic model to assess predictors of missing data (examination of all baseline clinical and demographic variables).
Secondary analyses
We compared categorical variables by use of Fisher's exact test. We analysed secondary outcomes with mixed linear regression models with random participant intercepts and a time-by-treatment interaction term; covariates in the model were treatment group, baseline value of outcome, time, and treatment centre. The more detailed NPI analyses utilised the generalised linear model framework, specifying a negative binomial distribution and logit link. The modelling was cross sectional at each time point (13 and 39 weeks); covariates in the model were treatment group, baseline value of outcome and treatment centre. All analyses results are summarised at 13 weeks and 39 weeks with two-sided 95% CIs.
Health economics: methods
Economic evaluation
The primary economic evaluation was a cost-effectiveness analysis comparing differences in treatment costs for patients receiving sertraline, mirtazapine or placebo with differences in effectiveness as measured by the primary outcome, total CSDD score, over two time periods: 0–13 weeks and 0–39 weeks. The secondary analysis was a cost–utility analysis using quality-adjusted life-years (QALYs) computed from the EQ-5D and societal weights over those same periods. Both the primary and secondary economic evaluations were undertaken from the perspective of (a) health and social care agencies and (b) health, social care agencies and informal carers. A measure of quality of life was appropriate for the secondary analysis as it was recognised that trial medication not only has a potential impact on depressive symptoms, but also may affect areas of functioning including self-care and usual activities.
Resource use
Resource-use data for each person were collected over a retrospective period of 6 months before randomisation. At 13 weeks, follow-up data were collected retrospectively for a 3-month period and at 39 weeks for a retrospective period of 6 months. Services and support received by the study participants were recorded on a resource-use questionnaire adapted from the Client Service Receipt Inventory (CSRI),51 including inpatient stays, outpatient attendances, day hospital treatment, visits to social clubs, meals at lunch clubs, day care visits, hours spent in contact with community-based professionals, such as community teams for older people, community psychologists, community psychiatrists, general practitioners (GPs), nurses (either practice, district or community psychiatric), social workers, occupational therapists, paid home help or care workers, and physiotherapists. The study also collected data on the use of voluntary organisation services, such as volunteer support, befriending and telephone care line support, and also on unpaid support provided by friends and relatives. Contacts made with voluntary support and support provided by friends and relatives were also measured in physical units, such as hours of care support time. The prescribed daily doses for the medications were calculated from the trial medication log, and prescribing periods were weighted to the changing dose regime.
Unit costs
All unit costs were estimated at 2009–10 prices and were collected from sources in the public domain. Unit costs are summarised in Table 2. Costs per unit of measurement for each type of service (such as per inpatient day, per appointment, per attendance, per visit or per contact with health and community-based professionals including voluntary services) were taken from Curtis. 52 The National Health Service Schedule of Reference Costs53 was used to estimate the cost of outpatient attendances. The unit cost of medication was obtained from the British National Formulary. 54
| Service | Unit cost (£) | Source |
|---|---|---|
| Inpatient (bed-days) | 299 | Curtis 201052 |
| Day hospital (attendance) | 50–205 | NHS reference costs 2009–10,53 Curtis 2007,55 Curtis 201052 |
| Outpatient (appointment) | 21–165 | NHS reference costs 2009–1053 |
| Accident and Emergency (attendance) | 37, 97 | Curtis 201052 |
| GP (per surgery consultation) | 28 | Curtis 201052 |
| Geriatrician (minutes) | 1.83 | Curtis 201052 |
| Nurse (minutes)a | 0.43–0.52 | Curtis 201052 |
| Occupational therapist (minutes) | 0.65 | Curtis 201052 |
| Community psychiatrist (minutes) | 1.83 | Curtis 201052 |
| Counsellor (minutes) | 0.57 | Curtis 201052 |
| Psychologist (minutes) | 1.20 | Curtis 201052 |
| Chiropodist (contact) | 0.37 | Curtis 201052 |
| Social worker (minutes) | 0.67 | Curtis 201052 |
| Care manager (minutes) | 0.82 | Curtis 201052 |
| Home care worker/care attendant (minutes) | 0.35 | Curtis 201052 |
| Sitting scheme (minutes) | 0.45 | Curtis 201052 |
| Self-help group (minutes) | 0.57 | Curtis 201052 |
| Meals on wheels (meal) | 4.8 | www.ic.nhs.uk/webfiles/publications/009_Social_Care/pss0910expfinal/pss0910updateOct2011/Personal_Social_Services_Expenditure_Report_2009_10.pdf |
| Dentist (minutes) | 2.90 | NHS reference costs 2009–1053 |
| Optician (minutes) | 0.48 | Individual calculationb |
| Day care (day) | 42–66 | Curtis 201052 |
| Lunch club (meal) | 7 | http://cash-online.org.uk/content/1/6/3/; uprated using the Consumer Price Index (CPI) |
| Social club (session) | 5 | Cost of adult social club at 2004–5, uprated using the pay and prices infator (Curtis 201052) |
We collected information on the volume and nature of informal care inputs, mindful of the difficulties of measuring such dimensions and of their interpretation as inputs to the care process. Costs were attached to informal care inputs using a replacement cost – the unit cost of a paid local authority home care worker. 52 This approach allowed us to quantify how much it would cost to replace the informal carer with the services from the market. In sensitivity analyses we examined whether or not the cost-effectiveness results would change under other assumptions.
Cost estimation
Three main categories of costs were analysed: medication costs, aggregated health and social care costs (primary care and hospital outpatient visits, inpatient admissions and community-based health and social care), and cost of time spent care-giving by relatives and friends. Costs were categorised in this way to facilitate the comparison of costs alongside measures of effectiveness from the perspectives of the economic analysis previously defined. The costs of services and support used by patients were derived by combining medication, health and social care resource utilisation data with estimated unit costs. Costs were calculated for the periods 0–13 weeks and 0–39 weeks.
Statistical analysis
Cost data were analysed in a similar way to the effectiveness data. Health and social care costs for 0–13 months and 0–39 months (and health, social care and costs of informal care costs for the parallel analysis from the broader perspective for the same time periods) were regressed in turn on treatment allocation, baseline cost, baseline CSDD and centre. To mitigate the effects of skewness, non-parametric bootstrapping methods – which avoid the distributional assumptions of parametric testing by use of resampling – were used to estimate 95% CIs for mean costs. Where the bias-corrected 95% CIs of between-group change scores excluded zero, they could be judged to be significant at p = 0.05.
Estimates of bootstrapped mean cost and effectiveness were used to estimate an incremental cost-effectiveness ratio (ICER) for each analysis. The incremental cost-effectiveness ratio for each replication was calculated as [(costb − costa)/(effectb − effecta)], which summarises the cost difference between two treatments per incremental difference in the outcome (CSDD and EQ-5D in turn). The EQ-5D was measured directly from patients – as recommended by NICE guidelines (2008) – and weighted by a valuation of changes in quality of life reported from UK population data. The value of health effects was then expressed in terms of QALYs. The ratio statistic compared the treatments in terms of observed differences in costs and effects, regardless of whether or not the difference in costs and effects was statistically significant.
Uncertainty around the costs and effectiveness estimates was addressed by plotting cost-effectiveness acceptability curves (CEACs). A CEAC assesses trade-offs between costs and outcomes, showing the likelihood of each of the two medications in turn being seen as cost-effective relative to the other or relative to placebo, given different (implicit monetary) values placed on incremental outcome improvements. In this net benefit (NB) approach, monetary values of incremental effects and incremental costs for each case are combined, and the net benefit derived as:
where a = control, b = drug treatment, NB = net benefit and λ = willingness to pay for unit improvement in CSDD-depression severity score or an additional QALY.
The impact on costs given uncertainty around the value attached to informal care inputs was assessed in one-dimensional sensitivity analysis.
All analyses were carried out in Stata version 11 and SPSS 17 (SPSS Inc., Chicago, IL, USA).
Chapter 3 Results
Participant flows
The CONSORT (Consolidated Standards of Reporting Trials) diagram, below, shows the flow of participants through the trial (Figure 1).
FIGURE 1.
Participant flows in the Health Technology Assessment-Study of Antidepressants for Depression in Dementia (HTA-SADD) trial. RW, research worker.
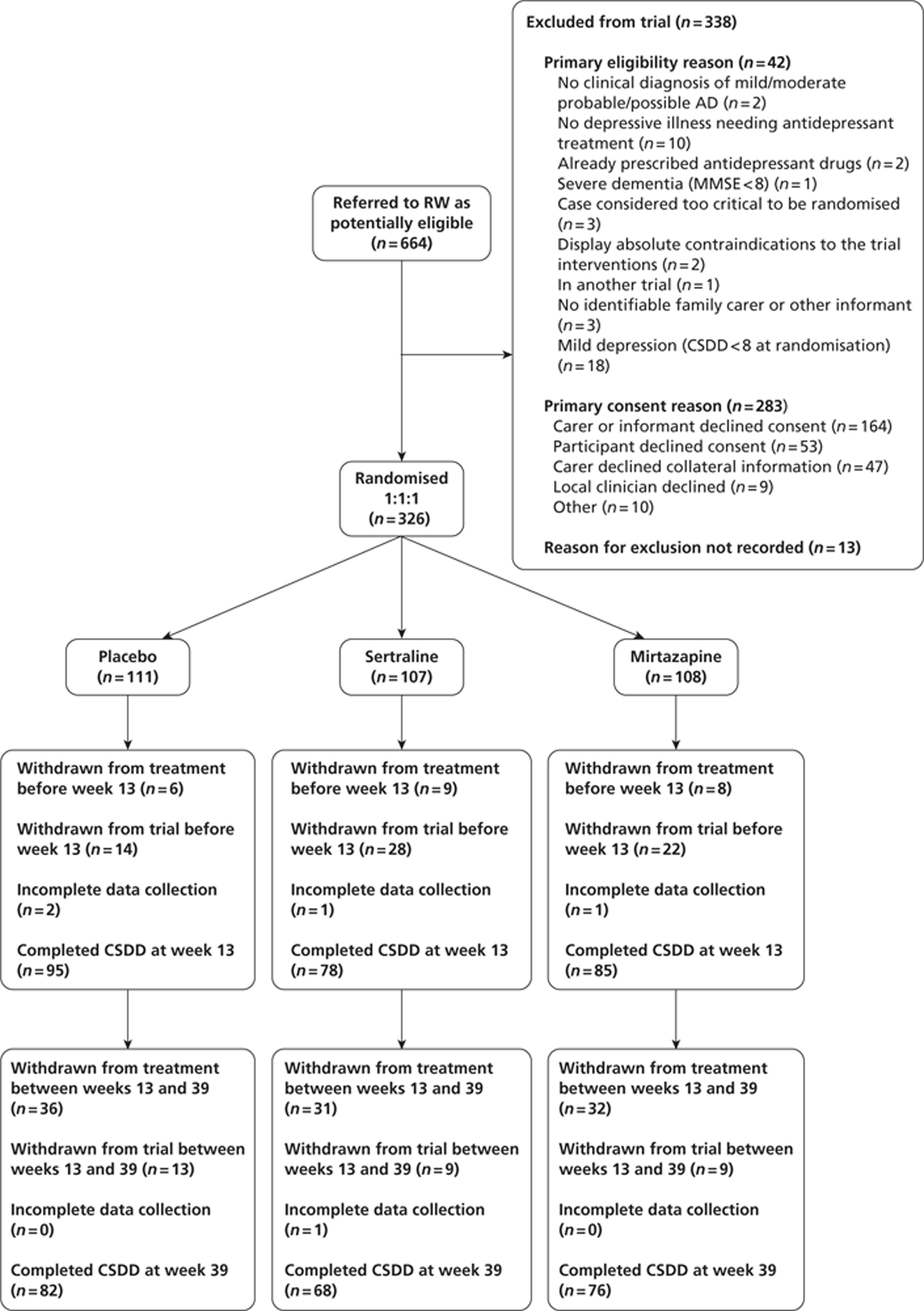
Withdrawal from treatment implies that the participant remains in the trial. Withdrawal from the trial implies the participant withdrawal from the trial and treatment. These two categories are mutually exclusive.
Trial recruitment
Over the course of the trial there were nine recruiting sites, all in the UK. These were Birmingham, Cambridge, Leicester, Liverpool, Manchester, Newcastle, north London, Southampton and south London and Kent. Randomisation was stratified by site; all sites successfully recruited. Recruitment began in December 2006 and ended in January 2010. Follow-up interviews were completed by October 2010. In total, 664 individuals were screened as potential participants; of these 326 (49%) were randomised. The overall recruitment rate is shown in Figure 2. The number of participants recruited per site ranged from 7 to 60.
FIGURE 2.
Cumulative recruitment over the trial period (revised target).
Baseline data
In total 326 participants were randomised in the trial; 111 to the placebo arm, 107 to the sertraline arm and 108 to the mirtazapine group. All participants completed the CSDD baseline questionnaire. Baseline demographics are summarised for participant and carers. The total number of collected questionnaires completed is featured. Table 3 shows the participant and carer demographics, whereas data collected on clinical characteristics is summarised in Table 4. Groups were evenly matched.
| Characteristic | Participant | Carer | ||||
|---|---|---|---|---|---|---|
| Placebo (n = 111) | Sertraline (n = 107) | Mirtazapine (n = 108) | Placebo (n = 111) | Sertraline (n = 107) | Mirtazapine (n = 108) | |
| Age (years) | 79 (8.8) | 80 (8.4) | 79 (8.4) | 59 (14.8) | 61 (13.9) | 61 (17.1) |
| Sex (male) | 40 (36) | 34 (32) | 31 (29) | 46 (30) | 37 (30) | 48 (35) |
| Ethnicity (white) | 104 (94) | 98 (92) | 101 (94) | 119 (79) | 109 (89) | 119 (86) |
| Marital status (married) | 48 (43) | 51 (48) | 60 (56) | 93 (62) | 82 (67) | 85 (61) |
| Residence (lives in care home) | 20 (18) | 13 (12) | 17 (16) | – | – | – |
| Relation to participant (paid carer) | – | – | – | 40 (26) | 19 (15) | 34 (24) |
| Clinical characteristics | Placebo | Sertraline | Mirtazapine |
|---|---|---|---|
| Duration of depression | |||
| Data available | 111 (100%) | 102 (95%) | 106 (98%) |
| 1 month | 7 (6%) | 3 (3%) | – |
| 1–2 months | 4 (4%) | 6 (6%) | 10 (9%) |
| 2–6 months | 24 (22%) | 18 (17%) | 26 (25%) |
| > 6 months | 76 (68%) | 75 (71%) | 70 (66%) |
| Severity of depression | |||
| CSDD 8–11 | 43 (39%) | 45 (42%) | 54 (50%) |
| CSDD ≥ 12 | 68 (61%) | 64 (58%) | 54 (50%) |
| Dementia vascularity | 2.1 (1.3) | 2.2 (1.3) | 2.2 (1.3) |
| Carer-rated scores | |||
| Participant SF-12 | |||
| Data available | 103 (93%) | 101 (94%) | 96 (89%) |
| Physical component (0–100) | 43.2 (10.6) | 45.2 (11.2) | 44.9 (12.4) |
| Mental component (0–100) | 50.1 (11.8) | 47.9 (11.1) | 46.1 (12.5) |
| Participant generic quality of life | |||
| Data available | 109 (99%) | 106 (99%) | 105 (97%) |
| EuroQoL VAS (0–100) | 52.3 (21.1) | 53.8 (19.6) | 51.9 (22.4) |
| Participant depression | |||
| Data available | 111 (100%) | 107 (100%) | 108 (100%) |
| CSDD (0–38) | 13.6 (5.2) | 12.8 (3.6) | 12.5 (3.7) |
| Participant activity limitation | |||
| Data available | 111 (100%) | 106 (99%) | 107 (99%) |
| BADL (0–60) | 18.2 (11.1) | 16.6 (11.2) | 18.4 (10.9) |
| Participant quality of life | |||
| Data available | 91 (82%) | 97 (91%) | 91 (84%) |
| DEMQOL-Proxy (31–124) | 88.4 (15.3) | 86.5 (15.6) | 86.9 (13.1) |
| Carer mental health | |||
| Data available | 105 (95%) | 103 (96%) | 98 (91%) |
| GHQ-12 (0–36) | 12.6 (5.1) | 12.5 (4.9) | 13.0 (5.9) |
| Carer burden | |||
| Data available | 87 (78%) | 93 (87%) | 91 (84%) |
| Zarit Score (0–88) | 27.2 (16.6) | 27.8 (14.7) | 26.1 (16.0) |
| Participant neuropsychiatric symptoms | |||
| Data available | 106 (95%) | 104 (97%) | 108 (100%) |
| NPI (0–144) | 30.2 (17.6) | 26.9 (16.8) | 29.9 (20.9) |
| Participant-rated scores | |||
| Participant cognition | |||
| Data available | 82 (74%) | 79. (74%) | 90 (83%) |
| Standardised MMSE (0–33) | 18.2 (7.4) | 18.5 (6.7) | 17.6 (6.0) |
| Participant generic quality of life | |||
| Data available | 92 (83%) | 86 (80%) | 91 (84%) |
| EuroQoL VAS (0–100) | 60.3 (24.1) | 66.6 (17.8) | 66.9 (18.5) |
| Participant generic quality of life | |||
| Data available | 87 (78%) | 82 (77%) | 91 (84%) |
| DEMQOL (28–122) | 83.7 (17.2) | 82.5 (14.3) | 85.1 (12.8) |
The majority of participants were female, and had a mean age of 79 years, ranging from 47 to 98 years. In total, 146 (45%) of participants were married and the majority ethnicity was white.
The carers ranged in age from 22 to 95 years, with a mean age of 61 years. Again, the majority were female, but a higher proportion of the carers were married (63%) compared with the participants. On average 23% of carers were paid workers.
Clinical characteristics of participants and carers are shown in Table 4. Completeness of data varies from 74% MMSE to 100% CSDD, primary outcome measure. Clinical characteristics are balanced over treatment arms. Participant qualities of life have worst outcomes when rated by carers in comparison with the participant rating of equivalent scales.
The mean overall dosages (including participants who withdrew from medication) were 70 mg (SD 52) per day for sertraline and 24 mg (16) per day for mirtazapine. For participants who remained on prescribed medication the mean dose was 95 mg (36) per day for sertraline and 30 mg (23) per day for mirtazapine.
Data collection
The mean (SD) time interval between randomisation and completion of questionnaires was 18.2 (14.19) weeks at the 13-week assessment time point and 42.15 (10.7) weeks at the 39-week assessment. The distribution of initiation of treatment following randomisation is shown in Figure 3. Out of the 326 participants randomised, 321 received allocated treatment.
FIGURE 3.
Distribution of initiation of treatment from randomisation.
Numbers analysed
In total, 111 participants were randomised to placebo, 107 to sertraline, and 108 to mirtazapine. The number of participants included in each analysis is indicated in the tables.
Outcomes and estimation
Primary outcome: Cornell Scale for Depression in Dementia
In total, 258 participants completed the research worker rated CSDD at 13 weeks post randomisation, with 226 participants going on to complete the measure at 39 weeks. As measured by the CSDD severity of depression was shown to decrease in all three intervention groups, compared with baseline, the results of which can be seen in Figure 4. The absolute change from baseline at 13 weeks was greatest for placebo −5.6 (SD 4.7), compared with −3.9 (5.1) for sertraline and −5.0 (4.9) for mirtazapine. This difference was maintained through to 39 weeks, with change scores of −4.8 (5.5) for placebo, −4.0 (5.2) for sertraline and −5.0 (6.1) for mirtazapine.
FIGURE 4.
The CSDD scores by treatment group, unadjusted means with 95% CI (a lower CSDD score means fewer depressive symptoms).
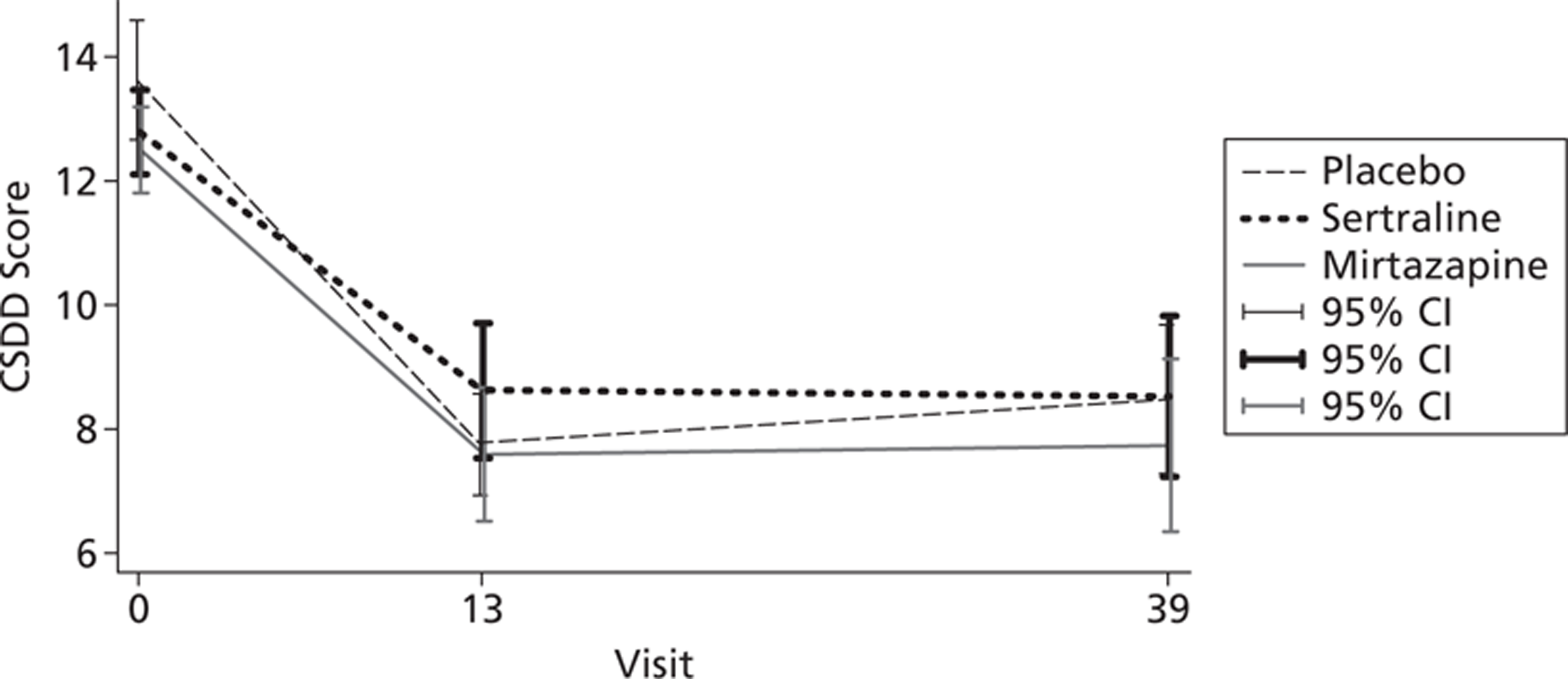
The results from the linear-mixed modelling (Table 5), after adjusting for baseline depression and the stratification factor centre, highlighted that there was no evidence of a difference between sertraline and placebo or mirtazapine and placebo, on the CSDD score at 13 or 39 weeks. This analysis provides robust evidence of an absence of clinical effectiveness of the antidepressants tested here compared with placebo.
| Time point | CSDD score | ||
|---|---|---|---|
| Placebo | Sertraline | Mirtazapine | |
| Baseline: mean (SD), n | 13.6 (5.2), 111 | 12.8 (3.6), 107 | 12.5 (3.7), 108 |
| Week 13: mean (SD), n | 7.8 (4.1), 95 | 8.6 (4.9), 78 | 7.9 (5.0), 85 |
| Week 39: mean (SD), n | 8.5 (5.5), 82 | 8.6 (5.5), 68 | 7.7 (6.2), 76 |
| Mean difference from placebo | |||
| 13 weeks: mean difference (SE); (95% CI); p-value; n | – | 1.17 (0.72); (−0.23 to 2.78); 0.10; 173 | 0.01 (0.70); (−1.37 to 1.38); 0.99; 180 |
| 39 weeks: mean difference (SE); (95% CI); p-value; n | – | 0.38 (0.76); (−1.12 to 1.87); 0.63; 150 | −0.67 (0.74); (−2.12 to 0.79); 0.37; 158 |
| Mean difference from mirtazapine | |||
| 13 weeks: mean difference (SE); (95% CI); p-value; n | – | 1.16 (0.72); (−0.25 to 2.57); 0.11; 163 | – |
| 39 weeks: mean difference (SE); (95% CI); p-value; n | – | 1.04 (0.76); (−0.48 to 2.56); 0.18; 144 | – |
On exploratory analysis of the primary outcome measure, we classified the participants as suffering from depression based on the CSDD; a score of ≥ 8 is the threshold for depression (Table 6). As part of our eligibility criteria all participants at baseline had depression. This criteria was examined at 13 and 39 weeks. By 13 weeks the same proportion of participants in all treatment arms had moved to the no depression classification (49%); the main deviation from this trend was seen at 39 weeks when mirtazapine showed the largest percentage of non-depressive arm (55%). On evaluation with a generalised linear-mixed model (using the a logit link for the dichotomous data), adjusting for baseline depression and the stratification factor centre, there was no evidence for a difference in the proportion of participants classified with depression by the CSDD between treatment arms at 13 or 39 weeks (Table 7).
| Treatment | Visit | |||||
|---|---|---|---|---|---|---|
| Baseline | Week 13 | Week 39 | ||||
| No depression | Depression | No depression | Depression | No depression | Depression | |
| Placebo | 111 | 47 (49%) | 48 | 40 (49%) | 42 | |
| Sertraline | 107 | 38 (49%) | 40 | 33 (47%) | 37 | |
| Mirtazapine | 108 | 42 (49%) | 43 | 42 (55%) | 34 | |
| Total | 326 | 127 | 131 | 115 | 111 | |
| Comparison | OR | SE | p-value | (95% CI) | |
|---|---|---|---|---|---|
| Week 13 | Sertraline vs placebo | 1.050 | 0.349 | 0.883 | 0.547 to 2.016 |
| Mirtazapine vs placebo | 1.008 | 0.327 | 0.979 | 0.534 to 1.906 | |
| Sertraline vs mirtazapine | 1.042 | 0.351 | 0.904 | 0.538 to 2.018 | |
| Week 39 | Sertraline vs placebo | 1.077 | 0.408 | 0.845 | 0.512 to 2.263 |
| Mirtazapine vs placebo | 0.692 | 0.256 | 0.319 | 0.335 to 1.428 | |
| Sertraline vs mirtazapine | 1.557 | 0.596 | 0.247 | 0.736 to 3.296 |
Secondary outcomes
Table 8 shows the effectiveness of the medications compared with placebo and between each other on secondary outcomes in participants and carers. Again, few differences can be attributed to the antidepressants. However, there were fewer neuropsychiatric symptoms and higher carer-rated participant scores for health-related quality of life (HRQL) (DEMQOL-Proxy) in participants given mirtazapine compared with sertraline, but these differences did not persist to 39 weeks (see Table 4).
| Outcome | Week 13 | Week 39 | |||||
|---|---|---|---|---|---|---|---|
| Sertraline vs placebo: coefficient (SE); 95% CI; (p-value) | Mirtazapine vs placebo: coefficient (SE); 95% CI; (p-value) | Sertraline vs mirtazapine: coefficient (SE); 95% CI; (p-value) | Sertraline vs placebo: coefficient (SE); 95% CI; (p-value) | Mirtazapine vs placebo: coefficient (SE); 95% CI; (p-value) | Sertraline vs mirtazapine: coefficient (SE); 95% CI; (p-value) | ||
| Cognition; MMSE | −0.22 (0.65); −1.50 to 1.05; (0.73) | −0.27 (0.61); −1.48 to 0.94; (0.66) | 0.05 (0.64); −1.21 to 1.31; (0.94) | −0.55 (0.68); −1.89 to 0.79; (0.42) | −1.71 (0.67); −2.48 to 0.14; (0.08) | 0.62 (0.69); −0.73 to 1.97; (0.37) | |
| Activity limitation; BADL | 1.40 (1.26); −1.07 to 3.88; (0.27) | −0.04 (1.23); −2.44 to 2.36); (0.97) | 1.44 (1.30); −1.10 to 3.99; (0.27) | 1.63 (1.35); −1.01 to 4.27; (0.26) | 1.19 (1.30); −1.37 to 3.75; (0.36) | 0.44 (1.38); −2.26 to 3.14; (0.75) | |
| Behaviour, problems; NPI | 2.72 (2.41); −2.01 to 7.45; (0.26) | −3.56 (2.30); −8.07 to 0.96; (0.12) | 6.28 (2.42); 1.53 to 11.03; (0.010) | 2.02 (2.53); −2.94 to 6.97; (0.43) | −1.51 (2.42); −6.25 to 3.24; (0.53) | 3.53 (2.53); −1.44 to 8.49; (0.164) | |
| Depression severity | Low CSDD score: 8–11 | 1.12 (1.01); −0.85 to 3.10; (0.26) | −0.30 (0.98); −2.21 to 1.61; (0.76) | 1.43 (0.99); −0.51 to 3.36; (0.15) | 0.33 (1.04); −1.72 to 2.37; (0.76) | −0.99 (1.02); −2.98 to 1.00; (0.33) | 1.31 (1.04); −0.72 to 3.34; (0.20) |
| High CSDD score: 12+ | 1.18 (0.91); −0.60 to 2.96; (0.34) | 0.27 (0.89); −1.47 to 2.01; (0.76) | 0.91 (0.91); −0.95 to 2.77; (0.34) | 0.38 (0.94); −1.47 to 2.23; (0.69) | −0.41 (0.91); −2..20 to 1.37; (0.65) | 0.0.80 (0.97); −1.10 to 2.69; (0.41) | |
| Life quality; DEMQOL | 0.30 (1.89); −3.40 to 4.01; (0.87) | −0.06 (1.76); −3.52 to 3.39; (0.97) | 0.37 (1.89); −3.52 to 3.39; (0.85) | −1.76 (2.04); −5.75 to 2.23; (0.39) | −0.03 (1.92); −3.80 to 3.75; (0.99) | −1.74 (2.07); −5.79 to 2.32; (0.40) | |
| Life quality; DEMQOL-Proxy | −1.98 (2.14); −6.16 to 2.21; (0.36) | 3.13 (2.15); −1.09 to 7.35; (0.15) | −5.11 (2.22); −9.45 to −0.76; (0.021) | 2.69 (2.28); −1.77 to 7.15; (0.24) | 3.69 (2.28); −0.77 to 8.16; (0.11) | −1.00 (2.35); −5.61 to 3.60; (0.67) | |
| Life quality; self rated; EQ-5D | −3.44 (3.78); −10.86 to 3.98; (0.36) | 2.00 (3.67); −5.18 to 9.19; (0.59) | −5.44 (3.72); −5.18 to 9.19; (0.14) | −4.34 (4.19); −12.56 to 3.88; (0.30) | −1.18 (4.12); −9.25 to 6.89; (0.78) | −3.16 (4.21); −9.25 to 6.89; (0.45) | |
| Life quality; carer rated; EQ-5D | 0.61 (3.05); −5.38 to 6.59; (0.84) | 3.62 (3.03); −2.31 to 9.55; (0.23) | −3.02 (3.17); −9.23 to 3.20; (0.34) | −0.27 (3.32); −6.77 to 6.24; (0.94) | −1.11 (3.23); −7.44 to 5.21; (0.73) | 0.85 (3.42); −5.86 to 7.56; (0.80) | |
| Carer burden; Zarit | −0.50 (1.93); −4.28 to 3.27; (0.80) | −1.14 (1.83); −4.93 to 0.65; (0.56) | 0.64 (1.98); −3.23 to 4.51; (0.75) | −0.09 (2.07); −4.15 to 3.98; (0.97) | −2.80 (2.14); −6.99 to 1.38; (0.19) | 2.71 (2.13); −1.45 to 6.88; (0.20) | |
| Carer mental health; GHQ | 1.47 (0.72); 0.06 to 2.89; (0.042) | −0.57 (1.23); −0.84 to 1.98; (0.43) | 0.90 (0.75); −0.56 to 2.37; (0.23) | 0.43 (0.77); −1.09 to 1.95; (0.58) | −0.61 (0.77); −2.12 to 0.90; (0.43) | 1.04 (0.80); −0.53 to 2.61; (0.20) | |
| Life quality; SF-12 physical component score | 1.28 (1.40); −1.48 to 4.03; (0.36) | −0.53 (1.39); −2.20 to 3.26; (0.70) | 0.75 (1.45); −2.10 to 3.59; (0.61) | −1.68 (1.48); −4.58 to 1.22; (0.26) | 0.02 (1.46); −2.84 to 2.88; (0.99) | −1.70 (1.53); −2.84 to 2.88; (0.27) | |
| Life quality; SF-12 mental component score | −2.99 (1.47); −5.87 to −0.11; (0.042) | 0.52 (1.45); −2.31 to 3.36; (0.72) | −3.52 (1.52); −6.50 to −0.54; (0.021) | 0.09 (1.54); −2.94 to 3.11; (0.96) | −0.31 (1.51); −3.28 to 2.66; (0.84) | 0.40 (1.60); −2.74 to 3.54; (0.80) | |
Our findings did not differ in subgroup analyses examining outcomes by baseline depression severity (CSDD score 8–11 vs ≥ 12). All but eight participants (one in the placebo group, three in the sertraline group and four in the mirtazapine group) met criteria for categorical diagnosis of depression in AD as per Olin criteria. 56 Sensitivity analyses with the Olin criteria56 as a moderator were not appropriate because of the low frequency of participants who did not meet Olin criteria. 56 However, this gives reassurance of the clinical significance of the depression in dementia investigated here.
Carers whose relatives were receiving placebo had higher quality-of-life scores at 13 weeks (SF-12 mental component score) and higher mental-health scores (GHQ-12) than those whose relatives were on sertraline (see Table 8). Finally, carers of participants in the mirtazapine group had higher quality-of-life scores (SF-12 mental component score) at 13 weeks than the carers of participants in the sertraline group. However, these differences did not persist at 39 weeks.
Neuropsychiatric Inventory
The distributional assumptions of the regression model were improved by specifying a negative binomial distribution for the NPI data. The data were examined cross-sectionally, thus missing data were accounted for by inverse probability weighting. The subscales of the NPI can be combined to yield four factors: factor 1, agitation, disinhibition and irritability; factor 2, delusions, depression and anxiety; factor 3, hallucinations, aberrant motor behaviour and sleep; and factor 4, elation, apathy and appetite. The summaries for the factors came be seen in Table 9. Under the new regression model, there is evidence for a beneficial effect of mirtazapine in comparison with sertraline. The difference in odds between sertraline and placebo, although non-significant, trends towards a better outcome in placebo. These differences are seen at 13 weeks (Table 10).
| Time | Placebo: data available; mean (SD) | Sertraline: data available; mean (SD) | Mirtazapine: data available; mean (SD) |
|---|---|---|---|
| Baseline | |||
| Factor 1 | 110; 6.89 (6.44) | 107; 5.87 (5.53) | 108; 6.5 (6.67) |
| Factor 2 | 111; 9.18 (6.65) | 105; 8.76 (6.33) | 108; 9.42 (7.55) |
| Factor 3 | 111; 6.41 (6.73) | 106; 5.71 (5.57) | 108; 6.36 (7.21) |
| Factor 4 | 107; 7.43 (6.25) | 105; 6.62 (5.18) | 108; 7.58 (6.44) |
| Week 13 | |||
| Factor 1 | 97; 4.30 (4.65) | 79; 4.58 (5.32) | 88; 4.28 (5.68) |
| Factor 2 | 96; 5.21 (5.59) | 79; 5.85 (6.62) | 88; 4.17 (5.54) |
| Factor 3 | 97; 4.41 (5.14) | 79; 5.43 (6.02) | 88; 4.02 (5.74) |
| Factor 4 | 93; 5.53 (5.56) | 75; 5.25 (5.65) | 88; 4.42 (4.73) |
| Week 39 | |||
| Factor 1 | 84; 5.08 (5.58) | 70; 4.91 (6.03) | 78; 4.36 (5.88) |
| Factor 2 | 84; 5.37 (5.48) | 69; 5.61 (6.44) | 78; 5.32 (7.08) |
| Factor 3 | 83; 3.39 (4.83) | 70; 4.46 (5.35) | 78; 4.34 (6.22) |
| Factor 4 | 81; 5.59 (5.57) | 68; 5.68 (5.38) | 78; 5.19 (6.45) |
| NPI factor: OR (SE); 95% CI; (p-value) | Week 13 | Week 39 | ||||
|---|---|---|---|---|---|---|
| Sertraline vs placebo | Mirtazapine vs placebo | Sertraline vs mirtazapine | Sertraline vs placebo | Mirtazapine vs placebo | Sertraline vs mirtazapine | |
| Total score | 1.098 (0.12); 0.884 to 1.363; (0.397) | 0.790 (0.10); 0.618 to 1.009; (0.059) | 1.390 (0.18); 1.084 to 1.782; (0.009) | 1.150 (0.15); 0.884 to 1.495; (0.298) | 0.933 (0.14); 0.693 to 1.256; (0.647) | 1.232 (0.18); 0.926 to 1.640; (0.151) |
| Factor 1 | 1.104 (0.19); 0.782 to 1.558; (0.573) | 1.013 (0.21); 0.678 to 1.516; (0.948) | 1.090 (0.24); 0.706 to 1.681; (0.698) | 1.101 (0.233); 0.728 to 1.666; (0.649) | 0.913 (0.19); 0.608 to 1.372; (0.663) | 1.206 (0.28); 0.760 to 1.911; (0.427) |
| Factor 2 | 1.203 (0.22); 0.840 to 1.723; (0.313) | 0.738 (0.13); 0.516 to 1.054; (0.095) | 1.631 (0.30); 1.141 to 2.331; (0.007) | 0.996 (0.18); 0.697 to 1.425; (0.984) | 0.886 (0.17); 0.609 to 1.287; (0.524) | 1.125 (0.21); 0.775 to 1.634; (0.536) |
| Factor 3 | 1.395 (0.26); 0.964 to 2.018; (0.078) | 0.716 (0.17); 0.451 to 1.138; (0.158) | 1.946 (0.45); 1.239 to 3.056; (0.004) | 1.663 (0.42); 1.020 to 2.713; (0.042) | 1.398 (0.35); 0.856 to 2.283; (0.181) | 1.190 (0.26); 0.779 to 1.819; (0.422) |
| Factor 4 | 1.064 (0.18); 0.762 to 1.486; (0.715) | 0.853 (0.13); 0.633 to 1.148; (0.294) | 1.248 (0.22); 0.887 to 1.755; (0.204) | 1.141 (0.21); 0.786 to 1.656; (0.488) | 0.842 (0.17); 0.570 to 1.244; (0.387) | 1.355 (0.27); 0.912 to 2.015: (0.133) |
The significant odds seen for the total score (OR 1.39; 95% CI 1.08 to 1.78; p = 0.009), is supported by factors 2 and 3. These effects are not continued into week 39.
Safety data
In total, 119 participants reported 240 adverse reactions. Table 11 shows adverse reactions by week 39. 29 of 111 participants (26%) in the placebo group had adverse reactions, compared with 46 of 107 (43%) in the sertraline group (p = 0.010) and 44 of 108 (41%) in the mirtazapine group (p = 0.031; overall p-value for placebo vs either drug 0.017). Gastrointestinal reactions were most common with sertraline (usually nausea) and psychological reactions were most common with mirtazapine (usually drowsiness and sedation). At 13 weeks, there were 15 serious adverse events in the placebo group of which three (20%) were rated severe, compared with 12 in the sertraline group [eight severe (67%)] and 14 [10 severe (71%)] in the mirtazapine group. Overall, the number of serious adverse events reported did not differ between groups but more of these events were severe in those on antidepressants compared with placebo (p = 0.003). Mortality did not differ between groups (five deaths in each group by 39 weeks).
| Classifcation | Treatment group | Total events | ||
|---|---|---|---|---|
| Placebo | Sertraline | Mirtazapine | ||
| Psychological | 10 (22) | 9 (18) | 24 (44) | 53 (84) |
| Neurological | 8 (9) | 16 (25) | 18 (21) | 42 (55) |
| Gastrointestinal | 7 (7) | 20 (24) | 11 (13) | 38 (44) |
| Other | 2 (2) | 5 (5) | 3 (3) | 10 (10) |
| Genitourinary | 4 (4) | 3 (3) | 2 (3) | 9 (10) |
| Musculoskeletal | 2 (3) | 3 (3) | 3 (3) | 8 (9) |
| Dermatological | 3 (4) | 3 (3) | 2 (2) | 8 (9) |
| Respiratory | 2 (2) | 1 (1) | 2 (2) | 5 (5) |
| Cardiovascular | 1 (1) | 0 (0) | 2 (4) | 3 (5) |
| Infection | 1 (1) | 1 (1) | 1 (1) | 3 (3) |
| ENT | 2 (2) | 1 (1) | 0 (0) | 3 (3) |
| Haematological | 1 (1) | 1 (1) | 0 (0) | 2 (2) |
| Endocrine | 0 (0) | 1 (1) | 0 (0) | 1 (1) |
| Totala | 29 (58) | 46 (86) | 44 (96) | 119 (240) |
Health economic results
Baseline comparisons
At baseline, full service use data were available for 326 participants (111 placebo, 107 sertraline, 108 mirtazapine). At 13 weeks, economic data were available for 97 participants in the placebo group, 78 in the sertraline group and 88 in the mirtazapine group. By 39 weeks, there were 84 participants in the placebo group, 69 in the sertraline group and 77 in the mirtazapine group.
Service use and support
Contacts made by patients with services and support over weeks 0–13 and 0–39 are shown in Table 12. There were few differences between the three patient groups in either time period, except when mirtazapine and sertraline were compared with placebo and mirtazapine was compared with sertraline. There were no statistically significant differences between the groups in the number of contacts with any services.
| Time period | Placebo (n = 97) | Sertraline (n = 78) | Mirtazapine (n = 88) | |||
|---|---|---|---|---|---|---|
| No. using | Meana (SD) | No. using | Meana (SD) | No. using | Meana (SD) | |
| Weeks 0–13 | ||||||
| Hospital-based care | ||||||
| Inpatient (bed-day)b | 8 | 1.65 (7.98) | 5 | 1.58 (6.82) | 5 | 0.49 (2.19) |
| Outpatient (attendance) | 33 | 0.53 (1.08) | 25 | 0.60 (1.10) | 26 | 0.53 (1.10) |
| Accident and Emergency (attendance) | 8 | 0.12 (0.48) | 5 | 0.08 (0.27) | 4 | 0.57 (0.28) |
| Day hospital (contact) | 3 | 0.23 (1.44) | 6 | 0.83 (4.11) | 4 | 0.32 (1.66) |
| Community-based care | ||||||
| GP (contact) | 57 | 1.36 (2.36) | 44 | 1.09 (1.44) | 49 | 1.22 (2.84) |
| Geriatrician (contact) | 3 | 0.03 (0.17) | 0 | 0 | 88 | 0.03 (0.18) |
| Nursec (contact) | 41 | 0.87 (1.49) | 29 | 2.50 (10.83) | 43 | 1.56 (3.71) |
| Occupational therapist (contact) | 11 | 0.21 (0.66) | 7 | 0.35 (1.70) | 5 | 0.08 (0.38) |
| Community psychiatrist (contact) | 21 | 0.26 (0.54) | 14 | 0.24 (0.63) | 19 | 0.27 (0.58) |
| Psychologist (contact) | 2 | 0.82 (0.64) | 3 | 0.06 (0.37) | 2 | 0.09 (0.62) |
| Counsellor (contact) | 1 | 0.01 (0.10) | 3 | 0.36 (2.94) | 2 | 0.17 (1.32) |
| Care manager (contact) | 7 | 0.10 (0.42) | 1 | 0.01 (0.11) | 4 | 0.05 (0.21) |
| Social worker (contact) | 15 | 0.21 (0.69) | 10 | 0.19 (0.58) | 12 | 0.28 (0.87) |
| Home care worker/care attendant (contact) | 19 | 18.57 (60.57) | 17 | 21.92 (72.77) | 22 | 28.33 (72.19) |
| Chiropodist (contact) | 33 | 0.43 (0.710 | 16 | 0.26 (0.57) | 23 | 0.40 (0.88) |
| Sitting scheme (contact) | 5 | 1.21 (6.71) | 5 | 0.68 (4.29) | 3 | 0.59 (3.75) |
| Self-help group (contact) | 0 | 0 | 0 | 0 | 1 | 0.03 (0.32) |
| Meals on wheels (contact) | 3 | 0.30 (1.77) | 3 | 5.82 (33.95) | 4 | 2.32 (12.74) |
| Dentist (contact) | 10 | 0.13 (0.49) | 10 | 0.15 (0.43) | 15 | 0.23 (0.58) |
| Optician (contact) | 10 | 0.12 (0.39) | 13 | 0.19 (0.46) | 12 | 0.15 (0.39) |
| Day services | ||||||
| Day services (day) | 15 | 4.15 (11.95) | 17 | 6.50 (15.64) | 16 | 5.47 (13.33) |
| Lunch club (visit) | 3 | 1.88 (15.92) | 0 | 0 | 3 | 1.18 (8.51) |
| Social club (visit) | 2 | 0.27 (1.86) | 4 | 0.67 (2.89) | 2 | 0.44 (3.08) |
| Informal care | ||||||
| Care giving (hours/week) | 45 | 10.05 (17.65) | 37 | 11.63 (21.59) | 42 | 9.84 (23.85) |
| Weeks 0–39 | ||||||
| Hospital-based care | ||||||
| Inpatient (bed-day)b | 9 | 3.05 (10.48) | 11 | 2.55 (9.26) | 14 | 4.54 (15.08) |
| Outpatient (attendance) | 44 | 0.83 (1.15) | 33 | 0.90 (1.41) | 29 | 0.69 (1.15) |
| Accident and Emergency (attendance) | 13 | 0.17 (0.41) | 8 | 0.25 (0.86) | 7 | 0.10 (0.35) |
| Day hospital (contact) | 1 | 0.01 (0.11) | 8 | 2.61 (9.42) | 3 | 0.56 (3.30) |
| Community-based care | ||||||
| GP (contact) | 57 | 1.51 (1.83) | 40 | 1.52 (2.15) | 55 | 1.88 (2.40) |
| Geriatrician (contact) | 4 | 0.05 (0.21) | 0 | 0 | 2 | 0.03 (0.16) |
| Nursec (contact) | 37 | 1.24 (2.34) | 33 | 5.84 (29.57) | 34 | 1.46 (3.53) |
| Occupational therapist (contact) | 9 | 0.17 (0.53) | 8 | 0.45 (2.23) | 5 | 0.10 (0.44) |
| Community psychiatrist (contact) | 22 | 0.33 (0.67) | 15 | 0.26 (0.53) | 29 | 0.60 (1.48) |
| Psychologist (contact) | 5 | 0.21 (1.34) | 2 | 0.03 (0.17) | 1 | 0.01 (0.11) |
| Counsellor (contact) | ||||||
| Care manager (contact) | 6 | 0.52 (2.87) | 3 | 0.04 (0.21) | 5 | 0.10 (0.44) |
| Social worker (contact) | 12 | 0.58 (2.98) | 15 | 0.42 (0.98) | 17 | 0.44 (1.47) |
| Home care worker/care attendant (contact) | 16 | 33.56 (107.73) | 19 | 38.07 (95.60) | 17 | 38.95 (110.10) |
| Chiropodist (contact) | 35 | 0.88 (1.37) | 20 | 0.53 (1.52) | 32 | 1.11 (1.89) |
| Sitting scheme (contact) | 0 | 0 | 5 | 1.23 (5.49) | 4 | 0.76 (3.69) |
| Meals on wheels (contact) | 2 | 0.63 (5.67) | 2 | 3.77 (21.70) | 2 | 3.14 (19.49) |
| Dentist (contact) | 18 | 0.33 (0.96) | 18 | 0.47 (1.25) | 19 | 0.34 (0.81) |
| Dietician (contact) | 0 | 0 | 0 | 0 | 1 | 0.01 (0.11) |
| Day services | ||||||
| Day services (day) | 16 | 5.57 (14.31) | 18 | 7.26 (15.13) | 16 | 5.17 (12.63) |
| Lunch club (visit) | 1 | 0.31 (2.84) | 1 | 0.38 (3.15) | 3 | 0.83 (4.84) |
| Social club (visit) | 2 | 0.62 (4.47) | 3 | 0.57 (2.69) | 1 | 0.33 (2.94) |
| Informal care | ||||||
| Care giving (hours per week) | 40 | 12.27 (21.24) | 34 | 12.32 (24.07) | 33 | 6.74 (11.82) |
Costs
Daily medication costs of £0.05 for sertraline 50 mg and £0.23 for mirtazapine 15 mg were applied. Mean cost of medication per person was estimated to be £7 (CI £6 to £9) and £37 (CI £32 to £41).
Mean total costs over 0–13 weeks and 0–39 weeks are detailed in Table 13. Pair-wise comparisons were made between the two antidepressants and placebo using regression analysis and bootstrapping. There were no statistically significant differences between the groups in either of the time periods, either when health and social care service costs only were included, or when health and social care services and informal care costs are summed.
| Weeks | Placebo | Sertraline | Mirtazapine | Bootstrapped mean difference (95% CI) | |||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD), £ | n | Mean (SD), £ | n | Mean (SD), £ | Sertraline – placebo | Mirtazapine – placebo | Mirtazapine – sertraline | |
| (a) Medication costs | |||||||||
| 0–13 | 97 | 0 | 78 | 7 (5) | 88 | 37 (22) | 7 (6 to 8) | 37 (32 to 41) | 30 (25 to 34) |
| 0–39 | 84 | 0 | 69 | 7 (5) | 78 | 37 (22) | 7 (6 to 8) | 37 (32 to 41) | 30 (25 to 34) |
| (b) Health and social care costs | |||||||||
| 0–13 | 97 | 1438 (3339) | 78 | 1434 (2326) | 88 | 1094 (1871) | −4 (−900 to 798) | −344 (−1207 to 322) | −340 (−1049 to 283) |
| 0–39 | 84 | 2146 (4402) | 69 | 2832 (4111) | 78 | 2513 (4290) | 686 (−630 to 1973) | 367 (−977 to 1596) | −319 (−1643 to 1023) |
| (c) Informal care cost | |||||||||
| 0–13 | 97 | 2744 (4819) | 78 | 3175 (5897) | 88 | 2687 (6511) | 431 (−1000 to 2242) | −57 (−1686 to 1537) | −488 (−2380 to 1470) |
| 0–39 | 84 | 3351 (5799) | 69 | 3363 (6573) | 78 | 1841 (3228) | 12 (−1940 to 2256) | −1510 (−3088 to −136) | −1522 (−3398 to −72) |
| Total costs excluding informal care inputs (a + b) | |||||||||
| 0–13 | 97 | 1438 (3339) | 78 | 1441 (2327) | 88 | 1131 (1869) | 3 (−893 to 806) | −307 (−1172 to 358) | −310 (−910 to 299) |
| 0–39 | 84 | 2146 (4402) | 69 | 2839 (4112) | 78 | 2550 (4289) | 693 (−622 to 1980) | 404 (−972 to 1626) | −289 (−1545 to 1151) |
| Total costs including informal care inputs (a + b + c) | |||||||||
| 0–13 | 97 | 4182 (5821) | 78 | 4616 (6488) | 88 | 3818 (7060) | 434 (−1340 to 2356) | −365 (−2212 to 1560) | −798 (−2754 to 1498) |
| 0–39 | 84 | 5497 (7922) | 69 | 6202 (8241) | 78 | 4391 (5285) | 705 (−1855 to 3234) | −1106 (−3137 to 970) | −1811 (−4048 to 543) |
| Depression score (CSDD) | |||||||||
| 13 | 95 | 7.8 (4.1) | 78 | 8.6 (4.9) | 85 | 7.9 (5.0) | 0.84 (−0.60 to 2.14) | 0.16 (−1.53 to 1.11) | −0.7 (−0.57 to 2.52) |
| 39 | 82 | 8.5 (5.5) | 68 | 8.6 (5.5) | 76 | 7.7 (6.2) | 0.05 (−1.83 to 1.67) | −0.80 (−2.55 to 1.21) | −0.9 (−1.10 to 2.73) |
| QALY 39 weeks (EQ-5D) | 57 | 0.55 (0.17) | 53 | 0.57 (0.14) | 52 | 0.60 (0.13) | 0.03 (−0.09 to 0.03) | 0.05 (−0.10 to 0.10) | 0.02 (−0.03 to 0.07) |
After adjustment for baseline costs, CSDD score at baseline and site, there were no statistically significant differences in health and social care costs – or in health, social care and informal care costs – in any pair-wise comparison in either time period.
In terms of observed mean differences, aggregated health and social care service costs per patient over 0–13 weeks were £3 between sertraline and placebo, £307 between placebo and mirtazapine and £310 between sertraline and mirtazapine. In each case, the first named treatment was the more costly. In the 6 months leading up to 39 weeks, mean difference in health and social care costs was £693 between sertraline and placebo, £404 between mirtazapine and placebo, and £289 between sertraline and mirtazapine. Again, in each case the first-named treatment was more costly.
Informal care costs exceeded health and social care costs by a factor of 1.2–1.7. Including informal care costs results in a change in the ranking of total costs, with mirtazapine being the least expensive of all treatments in both periods.
Cost-effectiveness
As noted earlier, the primary economic evaluation was a cost-effectiveness analysis with CSDD as the outcome over, first, the period 0–13 weeks after randomisation and, second, the period 0–39 weeks after randomisation. A secondary analysis was a cost–utility analysis using QALYs computed from the EQ-5D and societal weights over the same periods. Data used in the estimation of the ICERs are shown in Table 14. An ICER was calculated for each analysis comparing sertraline and mirtazapine against placebo and comparing mirtazapine against sertraline.
| Comparison | Sertraline–placebo | Mirtazapine–placebo | Mirtazapine–sertraline | |||
|---|---|---|---|---|---|---|
| 0–13 weeks | 0–39 weeks | 0–13 weeks | 0–39 weeks | 0–13 weeks | 0–39 weeks | |
| Incremental cost (£, 2009–10): mean (95% CI) | ||||||
| Health and social care | 3 (−893 to 806) | 693 (−622 to 1980) | −307 (−1172 to 358) | 404 (−972 to 1626) | −310 (−910 to 299) | −289 (−1545 to 1151) |
| Health and social care and informal care | 434 (−1340 to 2356) | 705 (−1855 to 3234) | −365 (−2212 to 970) | −1106 (−3137 to 970) | −798 (−2754 to 1498) | −1811 (−4048 to 543) |
| Incremental effect: mean (95% CI) | ||||||
| (a) CSDD scorea | 0.84 (−0.60 to 2.14) | 0.05 (−1.83 to 1.67) | 0.16 (−1.53 to 1.11) | −0.80 (−2.55 to 1.21) | −0.7 (−0.57 to 2.52) | −0.9 (−1.10 to 2.73) |
| (b) QALY (EQ-5D); meanb | – | 0.03 (−0.09 to 0.03) | – | 0.05 (−0.10 to 0.10) | – | 0.02 (−0.03 to 0.07) |
| Incremental cost-effectiveness (£) – health and social care and: | ||||||
| (a) CSDD score | 4 | 13,860 | 1919 | −505 | 443 | 321 |
| (Dominated) | (Dominated) | (Lower costs; worse outcome) | (Higher costs; better outcome) | (Mirtazapine dominant) | (Mirtazapine dominant) | |
| (b) QALY (EQ-5D) | – | 23,100 | – | 8080 | – | −14,450 |
| – | (Higher costs; better outcome) | – | (Higher costs; better outcome) | – | (Mirtazapine dominant) | |
| Incremental cost-effectiveness (£) – health and social care and informal care costs and: | ||||||
| (a) CSDD score | 517 | 14,100 | 2281 | 1382 | 1140 | 2012 |
| (Dominated) | (Dominated) | (Lower costs; worse outcome) | (Mirtazapine dominant) | (Mirtazapine dominant) | (Mirtazapine dominant) | |
| (b) QALY (EQ-5D)b | – | 23,500 | – | −22,120 | – | −90,550 |
| – | (Higher costs; better outcome) | – | (Mirtazapine dominant) | – | (Mirtazapine dominant) | |
As reported previously, there were no significant differences in CSDD scores or QALYs in any of the pair-wise comparisons between sertraline, mirtazapine and placebo. There were also no significant pair-wise differences in costs from either perspective between the treatment groups.
Given uncertainty surrounding the choice of treatment when incremental costs are higher and incremental outcome better (or when incremental costs are lower and incremental outcome also lower), CEACs were used to aid decision-making. Probability estimates were plotted for a range of implicit monetary values attached to improvements in depression score and QALY gain. We are not aware of any studies that have attached monetary values to incremental changes in CSDD.
In Figure 5, we see that mirtazapine has a low probability (around 30%) of being more cost-effective than placebo if society was not willing to pay anything for a unit improvement in the CSDD depression score. The probability rose to 80% if society was willing to pay £5000 for a unit improvement in CSDD score, and stays at 80% over values of willingness to pay for an improvement in CSDD score up to £30,000. Sertraline had a < 20% chance of being cost-effective compared with placebo, with the probability increasing moderately to about 42% if society was willing to pay £5000 for each point improvement in CSDD score, and stayed below 50% for willingness-to-pay values greater than £5000 and up to £30,000 for a point improvement in CSDD score.
FIGURE 5.
Probability that treatment is cost-effective at 0–39 weeks: health, social care costs and depression score (CSDD).
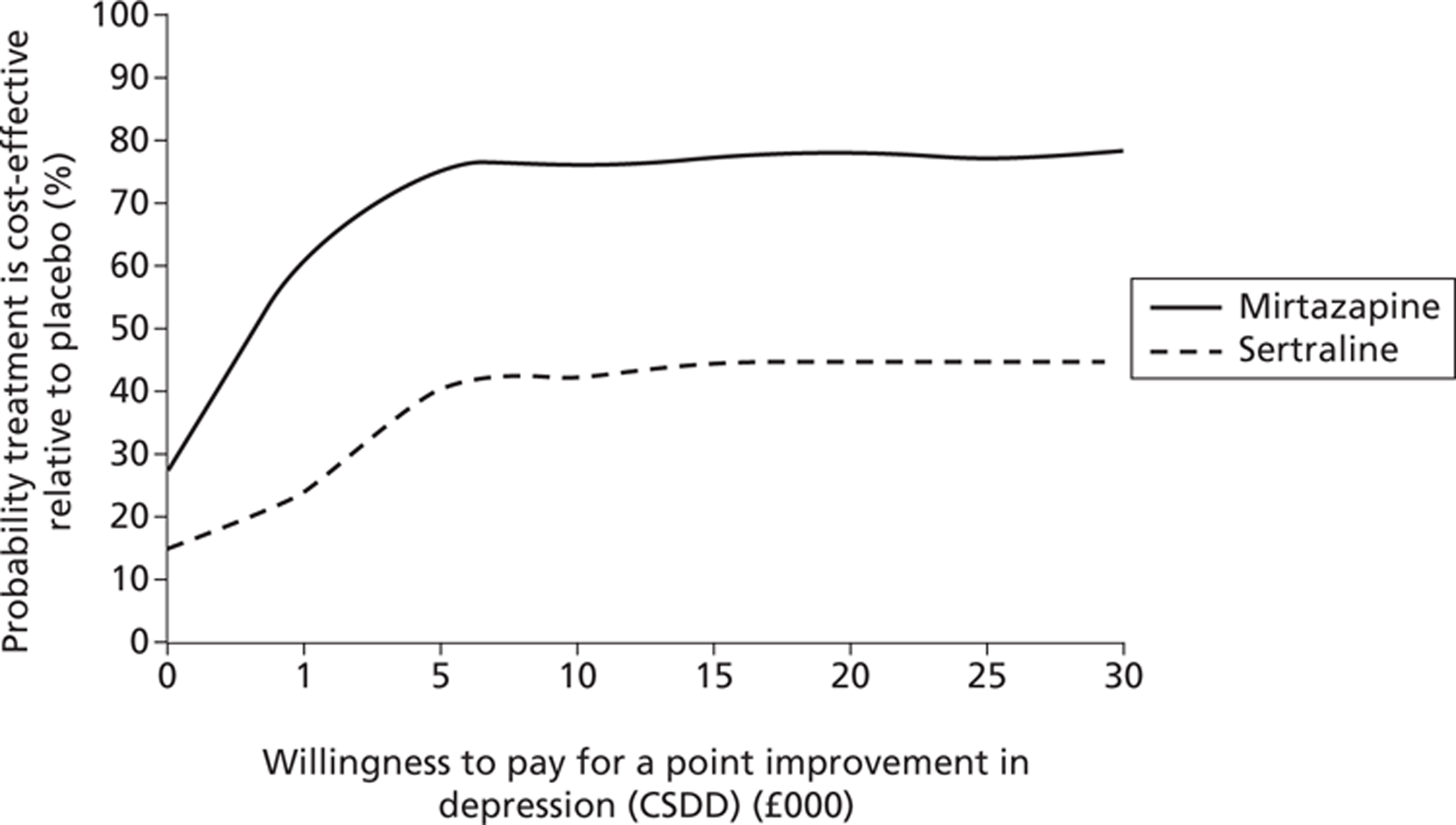
When both active treatments – sertraline and mirtazapine – were compared against each other the likelihood that treatment with mirtazapine would be seen as more cost-effective than sertraline would be over 60% from a health and social care perspective (and over 90% from a health, social care and informal care costs perspective).
Figures 5 and 6 show the CEACs from the secondary economic evaluation, where costs were considered alongside QALYs. Although we found no significant differences in QALY gain in any of the pair-wise comparisons between sertraline, mirtazapine and placebo, we see a trend towards marginally higher QALY gains (using the EQ-5D measured directly from patients) for the active treatments.
FIGURE 6.
Probability that mirtazapine is cost-effective relative to sertraline at 0–39 weeks: costs and depression score (CSDD).
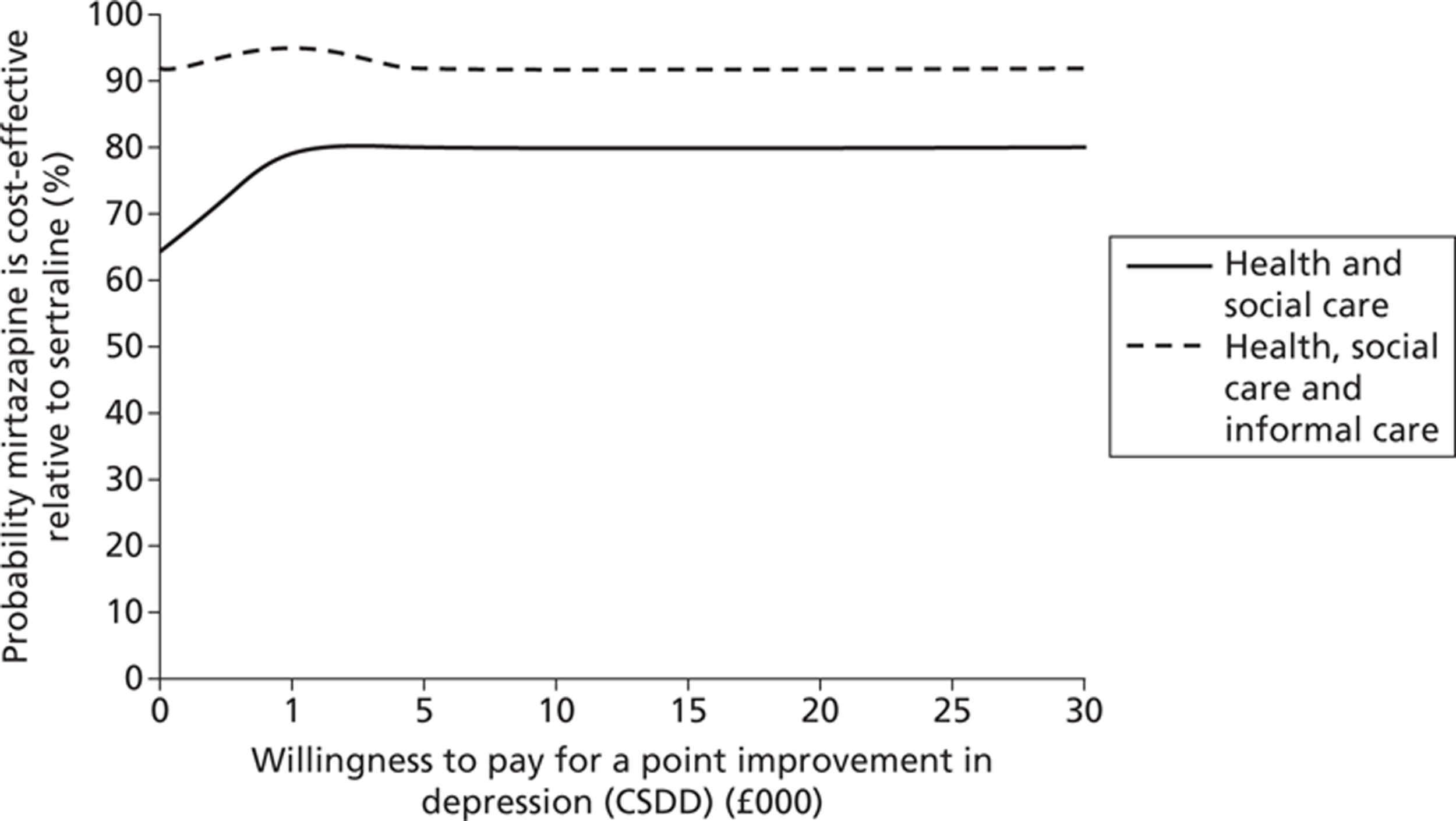
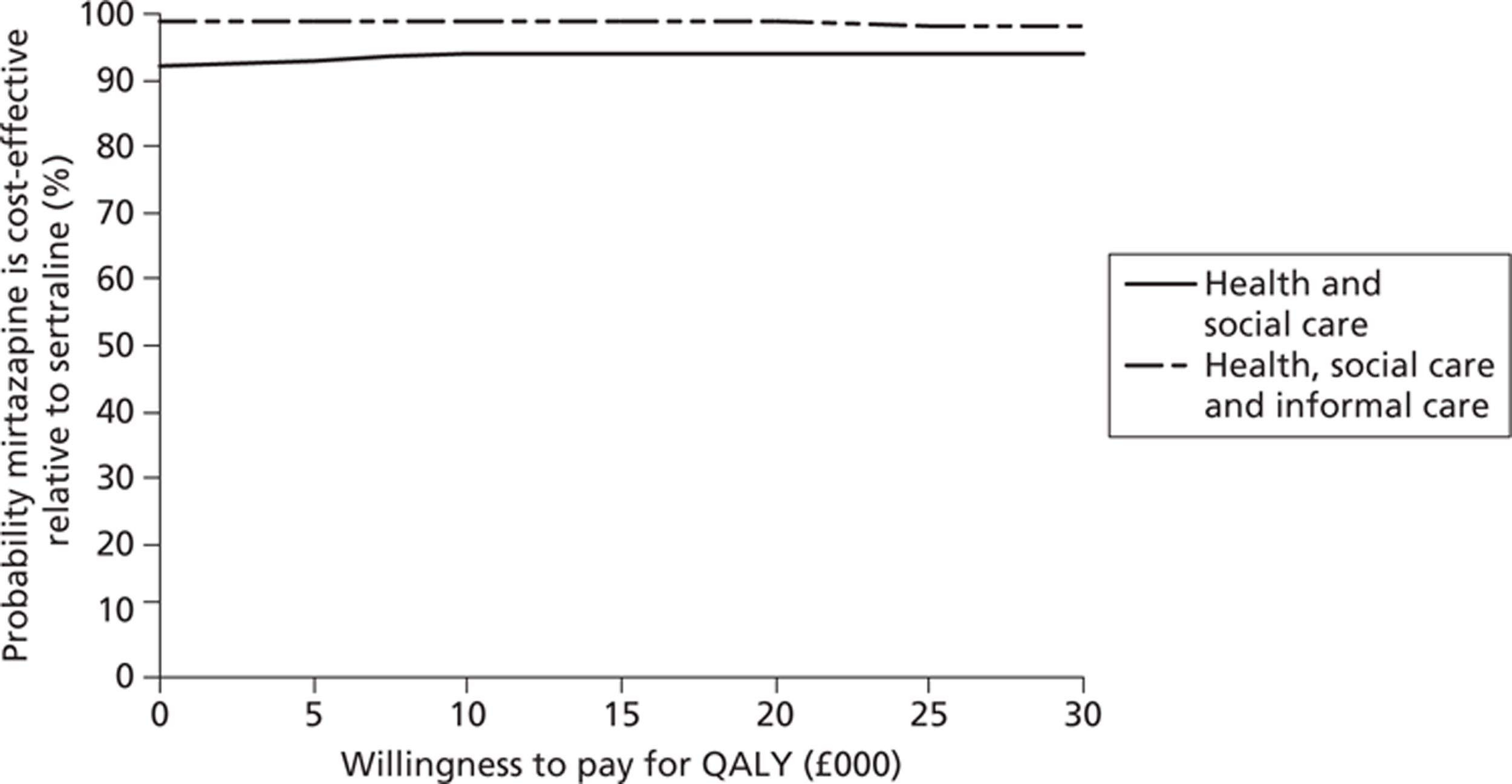
Figure 7 suggests that the probability that mirtazapine is more cost-effective than placebo was 89%, and increased to over 90% for a willingness to pay of £30,000 for a QALY. The likelihood of sertraline being more cost-effective than placebo was just over 50% and rose to just over 70% over higher values of willingness to pay for a QALY. Figure 8 shows that mirtazapine had a higher probability of being more cost-effective than sertraline (over a range of willingness-to-pay values from £0 to £30,000) when health and social care costs are considered on their own, and also when considering health, social care and informal care costs.
FIGURE 7.
Probability that treatment is cost-effective relative to placebo: health, social care and informal care costs and QALYs.
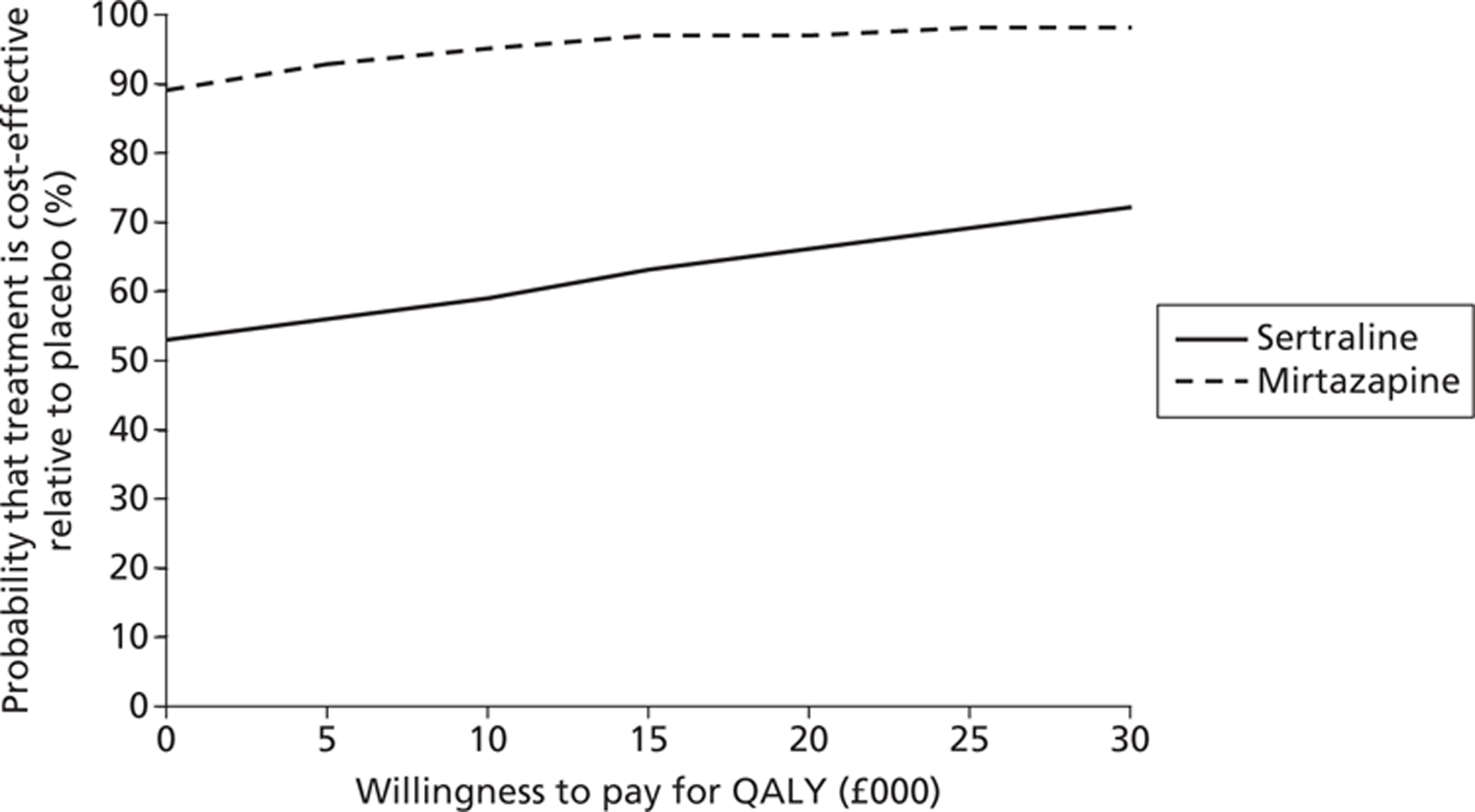
FIGURE 8.
Probability that mirtazapine is cost-effective relative to sertraline: costs and QALYs.
In addition to assessing the uncertainty surrounding the cost-effectiveness of the antidepressants, we also assessed uncertainty around parameter estimates included in the cost analysis. For the main analyses, informal care costs were based on hourly cost of a home care worker. This hourly value for the care-giving inputs by friends and family was replaced in sensitivity analysis by an opportunity cost estimate, calculated as the gross hourly wage of a carer in paid employment and zero for a carer not in paid employment. Using alternative values of caregiver time inputs did not alter the findings (Table 15).
| Analysis | Placebo: mean (SD) | Sertraline: mean (SD) | Mirtazapine: mean (SD) | Sertraline–placebo: mean difference (95% CI) | Mirtazapine–placebo: mean difference (95% CI) | Mirtazapine–sertraline: mean difference (95% CI) |
|---|---|---|---|---|---|---|
| Main analysis 0–13 weeks (total cost including informal care) | 4182 (5821) | 4616 (6488) | 3818 (7060) | 434 (−1340 to 2356) | −365 (−2212 to 1560) | −798 (−2754 to 1498) |
| Applying gross wage for informal care inputs | 3368 (4769) | 3663 (5008) | 3592 (5461) | 322 (−1081 to 1797) | −353 (−1778 to 1087) | −71 (−1588 to 1588) |
| Main analysis 0–39 weeks (total cost including informal care) | 5497 (7922) | 6202 (8241) | 4391 (5285) | 705 (−1855 to 3234) | −1106 (−3137 to 970) | −1811 (−4048 to 543) |
| Applying gross wage for informal care inputs | 4476 (6512) | 5177 (6574) | 3830 (4777) | 702 (−1313 to 2751) | −645 (−2415 to 986) | −1347 (−3368 to 280) |
Chapter 4 Discussion
This is a trial with negative findings but important clinical implications. The data suggest clearly that antidepressants, given with normal care, are not clinically effective when compared with placebo for the treatment of clinically significant depression in dementia. This implies a need to change the current clinical practice of prescribing antidepressants as the first-line treatment of depression in dementia due to AD.
Limitations
First, the dropout will have introduced bias if those dropping out had a different response to the trial interventions or placebo compared with those completing the trial. However, this was designed as a pragmatic trial, with few exclusions to mirror closely real clinical populations, and the levels of disengagement are similar to those experienced in clinical settings. Strenuous efforts were made to follow up and obtain outcome data on all those randomised but who defaulted from either the trial compound or clinical services.
A second putative limitation is the revision during the trial of the target sample size. Because of slower than forecast recruitment, we sought an extension and therefore further funding. The funder ordered interim analyses to determine the numbers needed using the data available on 75 cases followed up at 13 weeks. The new target set was 339. We recruited 326, falling short of the new target by 13. Nevertheless, this is the largest ever RCT of depression in dementia with unequivocal findings showing no effect of either antidepressant compared with placebo. Had the pattern of change seen in those recruited been continued, the extra precision in estimates that would have come from either another 13 cases, or even achieving the original trial target of 507, would not have generated a statistically significant positive result for either antidepressant.
Third, measurement error caused by the effect of cognitive impairment on domains such as memory, language and reasoning is a potential limitation. However, the study included only those measures best validated for use in dementia. Our primary outcome, the CSDD, is the most robust available measure of depression in dementia57 incorporating data from the carer, the person with dementia, and the rater. Finally, we did not capture elements of intervention by the clinical teams other than the group to which they were randomised. Had we been able to characterise these non-drug elements of treatment then we might have been able to investigate their role in patient recovery. However, there is no suggestion that these would have varied across the three groups, so again the results would not have changed.
Finally, it might be considered a limitation that we did not adjust the results for the multiple comparisons made in the secondary analyses. The data are presented as they are so that the reader can interpret the actual findings as they find best. The work should be reviewed considering a 1% significance level for all secondaries.
Generalisability
This study was designed to reflect real clinical populations and interventions as closely as possible. To this end we minimised exclusions and had permissive inclusion criteria. However, the findings may not generalise to those too critically ill to risk randomisation (chiefly those with high suicide risk). Only three potential participants were excluded on this criterion but there will have been more not referred into the trial. Equally, outcomes of those with depression but a CSDD score of < 8 would not be covered by this study. In practice, however, very few people with a CSDD score at this level would be considered to have clinically significant depression, so the effect on generalisability will be limited.
One of the strengths of this study is its size and the broad nature of the study group, both by the range of depressive symptoms and the severity of dementia, neither of which appeared to influence outcomes. We included not only those with narrowly defined AD but also those with probable and possible AD. This is closer to the population encountered in clinical practice where there is often mixed dementia (i.e. those with a vascular component to their dementia). However, prudence would limit generalisability to AD and mixed dementia only and not to other subtypes, such as vascular dementia, dementia with Lewy bodies or frontotemporal dementia.
The one major limit to generalisability comes from all cases being drawn from referrals to old-age psychiatry services. Such services are designed to deal with complex clinical situations but there will be instances in which people with depression in dementia are not referred to specialist services but remain either treated or untreated in primary care. Possibly, such cases would respond differently to antidepressants. However, finding unrecognised and untreated cases in primary care is difficult and referral of such cases to specialist services is good practice. Given the participants were not drawn from specialist research clinics or tertiary care but from nine geographically diverse areas and a large number of clinicians representative of services in general (see Acknowledgements), the external validity of the results reported here will be maximised.
The drugs used in this study represent the two most used classes of antidepressants but the extent to whether or not other classes [e.g. dual-acting antidepressants, such as venlafaxine (Effexor®, Pfizer)] might have an effect is unclear; however, it would be reasonable to expect broadly similar responses in drugs of the same general class.
Interpretation
The main message from this study is that the drugs from the two classes of antidepressants most likely to be prescribed for depression in AD appear to be no more effective than placebo. This negative finding does not seem attributable to the type or the severity of depression in dementia included, or to the severity and vascularity of the dementia included. In this, our results are in line with those of the DIADS-II study. 26,27 It is, however, encouraging for people with depression in dementia that there was a strong consistent pattern of improvement in the depression at 3- and 9-month follow-up for this group of people referred to old age psychiatric services. This study gives strong evidence that this improvement is not attributable to antidepressants. What this study cannot tell us is if this improvement is a function of the non-drug ‘treatment as usual’ by these old age psychiatric services, or due to artefact such as regression of the mean, the Hawthorne effect, or part of the natural history of depression in dementia. The last is perhaps made less likely by the finding that 221/326 (68%) had been depressed for > 6 months prior to randomisation.
In terms of harms from medication, there were more adverse reactions in those treated with antidepressants compared with placebo as in other studies. 26,27 It is important to be cautious about drawing conclusions from the analyses of secondary outcomes; the key message remains that there is no positive effect of the antidepressants on any of the pre-specified comparisons compared with placebo. There is, however, a signal in the data that is consistent with the pattern of adverse reactions observed. There were fewer neuropsychiatric symptoms, and there was higher carer-rated participant quality of life, and higher carer quality of life in those treated with mirtazapine compared with sertraline. Also, carers of those receiving placebo had higher quality of life themselves and better mental health compared with those caring for people on sertraline. Taken together, even though these differences did not persist at the 39-week follow-up, they may suggest that sertraline has more negative impacts than mirtazapine. This is of clinical importance since it is common clinical practice to use sertraline following the positive results of the first DIADS study. 26
One of the unique elements of this study is the simultaneous evaluation of cost-effectiveness as well as clinical effectiveness. As far as we are aware, this is the first RCT with an economic evaluation of the use of pharmacotherapy for older people with dementia and depression. Because of the lack of significant pair-wise differences in costs or outcomes (CSDD score) between sertraline, mirtazapine and placebo, the active treatments mirtazapine and sertraline have a low probability of being cost-effective compared with placebo. However, it is interesting that when both active treatments are compared against each other, treatment with mirtazapine has a high probability of being cost-effective compared with sertraline.
Care professionals, policy-makers and people with dementia and their families are primarily interested in quality of life, and so a secondary cost-effectiveness analysis examined pair-wise cost differences between the three treatments relative to the incremental difference in QALY gain. There were non-significant pair-wise differences in costs or outcomes (QALY gains) between sertraline, mirtazapine and placebo. Sertraline had a low probability of being cost-effective compared with placebo. However, treatment with mirtazapine had a high probability of being cost-effective compared with placebo or when compared against sertraline.
This seems counterintuitive given the lack of clinical effectiveness demonstrated in the primary analyses. We considered possible reasons for this finding: first, there was a trend towards lower incremental costs and higher incremental QALY gains for mirtazapine when compared with sertraline and placebo, in turn. The trends observed towards lower costs were due to the significantly lower informal care inputs when patients treated with mirtazapine were compared with those treated with placebo or sertraline. The differences in improvements in quality of life could perhaps be explained in part by the effects of treatment with mirtazapine, such as amelioration of sleep disturbances or anxiety state not explored in this study. 58,59 Improvements in sleep could potentially enhance mood not captured by the CSDD and mood has been shown to be correlated with patient-reported EQ-5D scores. 60 In this way mirtazapine might have a more general effect that was beneficial for both the patient and the carer.
When looking at our secondary outcomes (such as quality of life and NPI) it may well be that the amendments to protocol in terms of sample size resulted in a loss of power for secondary analyses. As discussed above, during the study, the protocol needed to be amended after slower than expected recruitment. An interim analysis was completed of the primary outcome and the sample size was recalculated based on the estimates from the interim analysis. The variance in these CSDD scores was smaller than previously expected. So under the new calculation (of a smaller sample size), there was enough power to show the potential differences in the CSDD but there were no such analyses for the secondary outcomes. The study may therefore not have been sufficiently powered to test the patterns of response observed in the secondary outcomes.
In any case it is striking that in the long run those randomised to mirtazapine appear to use half as much carer time as those randomised to sertraline or placebo. Likewise the pattern of dominance of mirtazapine over sertraline is maintained in these analyses. This all provides further evidence that sertraline may not be a good choice for the treatment of depression in dementia. The extent to which this is generalisable to other SSRIs is not clear from our study. The potential positive effects of mirtazapine seem more general than specific and may act more in the realm of general behavioural and psychological symptoms in dementia (BPSD) than depression per se. It is possible, for example, that a positive effect on sleep or agitation in the person with dementia may result in relief, not only for the person with dementia but also the carer in terms of hours of care needed.
The development of BPSD (e.g. agitation, aggression, wandering, shouting, repeated questioning, depression and sleep disturbance) is common in dementia occurring at some stage in up to 90% of cases. These symptoms cause problems in themselves, which complicate care, and they can occur at any stage of the illness. They are a legitimate object for intervention to decrease distress and harm and increase quality of life for the person with dementia and their carers. One area for concern is the reflexive use of antipsychotic drugs to treat these symptoms. A ministerial enquiry into the use of antipsychotic drugs in dementia concluded that ‘. . . current systems appear to deliver a largely antipsychotic-based response’. 61 It is clear that these medications are being prescribed to deal with BPSD rather than just for psychosis.
The evidence includes gaps, contradictions and complexity but there is emerging consensus with respect to the level of use and risk of antipsychotic drugs for people with dementia. Reviewing the evidence, these drugs appear to have only a limited positive effect in treating these symptoms but can cause significant harm to people with dementia. On balance, it appears that around 180,000 people with dementia are treated with antipsychotic medication across the country per year. Of these, up to 36,000 may derive some benefit from the treatment. In terms of negative effects that are directly attributable to the use of antipsychotic medication, use at this level equates to an additional 1800 deaths, and an additional 1620 cerebrovascular adverse events, around half of which may be severe, per year. 61
Despite the limited evidence base, the use of non-pharmacological interventions as the first-line treatment for BPSD reflects ‘best practice’ when taking into account safety considerations and the high rates of resolution of symptoms with placebo in pharmacological trials. 31 The main reason for the widespread use of antipsychotic drugs is the limited evidence for alternative treatments. Other pharmacological treatments used include anticonvulsants (carbamazepine and sodium valproate), and antidepressants (trazadone and citalopram). The best evidence is for carbamazepine, which has been shown to be better than placebo for agitation in several small placebo-controlled trials,62 but there is limited information about long-term safety in people with dementia. A recent meta-analysis concluded that sodium valproate was effective only at high doses that were associated with unacceptable side effects. 63 The results of double-blind placebo-controlled trials of trazadone have been disappointing. 64
In a double-blind placebo-controlled trial of people with AD that predominantly focused on depression, citalopram was also associated with improvement in a number of other behavioural and psychiatric symptoms, including irritability and restlessness. 65 However, as neuropsychiatric symptoms were not the main focus of the study, only a modest proportion of participants had clinically significant behavioural and psychiatric symptoms at baseline and so the results are difficult to interpret. In a definitive trial of a cholinesterase inhibitor for the treatment of clinically significant agitation in people with AD, donepezil showed no advantage over placebo. 66 One recent reanalysis of a placebo-controlled trial of memantine in people with moderate to severe AD suggested that patients with neuropsychiatric symptoms benefited from treatment. 67
The data presented here suggest that there may well be value in conducting a RCT of mirtazapine for the treatment of BPSD; no such trial has ever been completed. One small-scale open-label pilot study gives supportive evidence for the potential of a trial in this area (those on mirtazapine did better). 68 Given the paucity of alternatives and the priority of finding safe and effective treatments for BPSD, these data suggest that a placebo-controlled trial of mirtazapine would be of value.
Chapter 5 Conclusions
Implications for health care
So what can be concluded? This study finds no evidence to support the use of antidepressants as as a first-line treatment for people with depression in AD who are referred to old-age psychiatry services as many cases will resolve with usual care and without sertraline or mirtazapine. An important exclusion to this are the most critical of cases (by reason for example of self-harm or other risk) which were not included in this study.
Stepped care, with ‘watchful waiting’, is advocated currently for the general treatment of depression (without dementia) in the community. The first step is provision of ‘low-intensity psychosocial interventions’ with more complex psychosocial interventions an alternative to antidepressants at the next stage of severity. 69 Those recruited into the trial will have received non-drug ‘treatment as usual’ provided by the community mental-health teams to whom they were referred. This will have included a broad range of supportive and problem-solving interventions, commonly delivered by a community psychiatric nurse, often in their own household. This will have focused on problems encountered by the person with dementia and the carer, covering aspects of dementia as well as depression, and ranging in intensity from low to high as needed. Identifying which components of ‘usual care’ may be effective is an important area for future research. Compared with this personalised care the Hawthorne effect of the study assessments is likely to have had only a minor impact. These data suggest that having depression in dementia may be an appropriate trigger for referral to specialist services where non-drug treatments can be deployed, perhaps avoiding the use of medication with potential for adverse reactions.
In summary, the practical implications of this study are that we should reframe the way we think about the treatment of people with dementia who are depressed, as the evidence does not support the routine prescription of antidepressants for depression in dementia. As we find no evidence to support use of antidepressants, it suggests that potential cases might be more appropriately managed by specialist services that are able to offer non-drug interventions for depression and case management, which may not be available in primary care. Based on the data (a decrease at 13 weeks and this then maintained), except for those in whom medication is indicated by risk or extreme severity, and in the absence of evidence to the contrary, it might be appropriate to reconsider antidepressant prescribing for those who have not responded within a 3-month period (Figure 9).
FIGURE 9.
Management of depression in dementia.
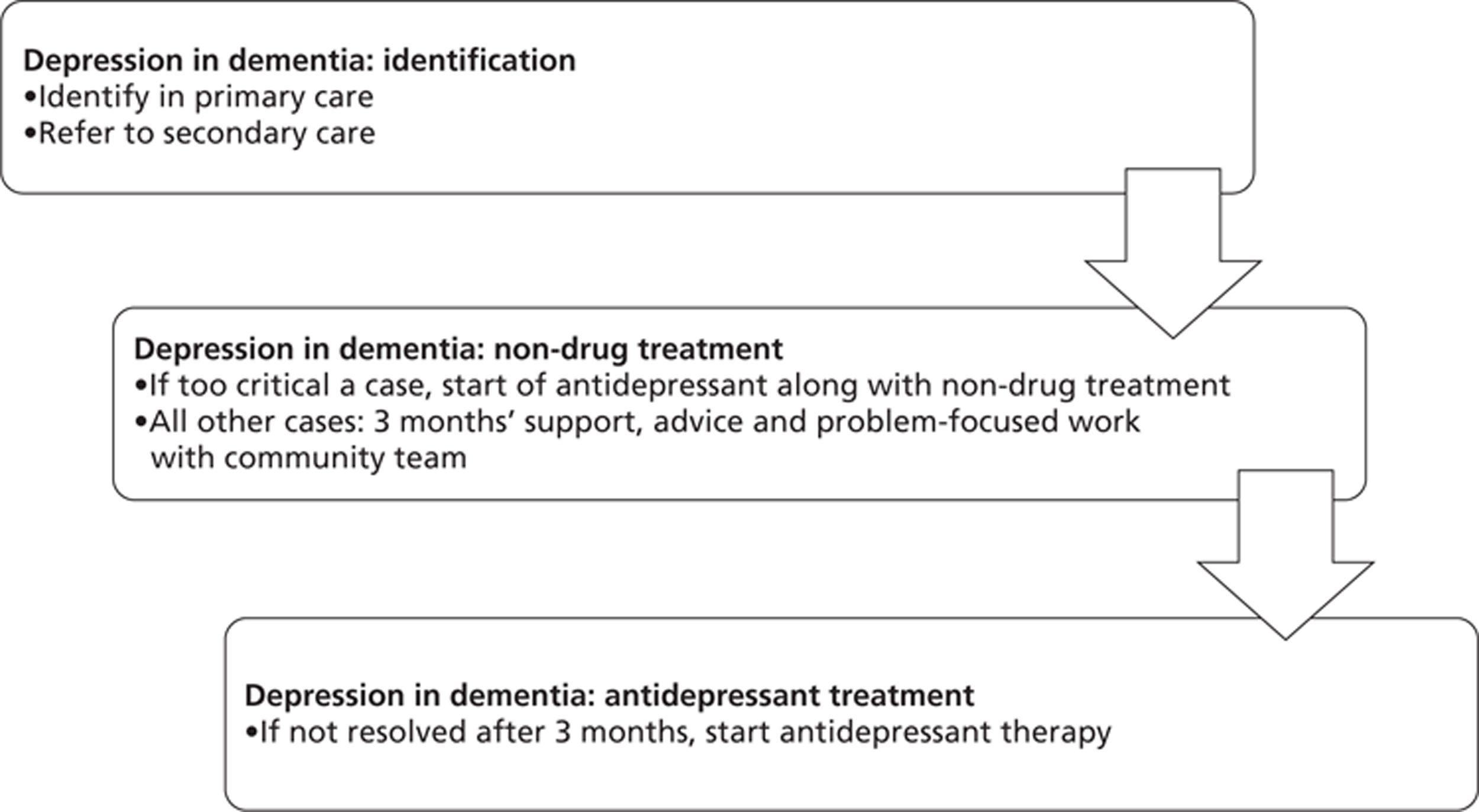
Recommendations for research
-
The secondary analyses presented here suggest that there would be value in carrying out a placebo-controlled trial of the clinical effectiveness and cost-effectiveness of mirtazapine in the management of BPSD.
-
A conclusion from this study is that it remains both ethical and essential for trials of new medication for depression in dementia to have a placebo arm.
-
Further research is required to evaluate the impact that treatments for depression in people with dementia can have on their carers, not only in terms of any impacts on their quality of life, but also the time they spend care-giving.
-
There is a need for research into alternative biological and psychological therapies for depression in dementia. These could include evaluations of new classes of antidepressants (such as venlafaxine) or antidementia medication (e.g. cholinesterase inhibitors).
-
Research is needed to investigate the natural history of depression in dementia in the community when cases are not referred to secondary care services.
-
Further work is needed to investigate the cost modelling results in this rich data set, investigating carer burden and possible moderators to the treatment effects.
-
There is scope for reanalysis of the primary outcome in terms of carer and participant CSDD results.
Acknowledgements
We thank all the participants and carers who gave their time to be part of this study; Georgina Charlesworth, Tom Dening and David Wilkinson for invaluable scientific advice, support and input into the project at key stages from planning onwards; Pfizer for their donation of the sertraline and sertraline placebo for this trial; members of the HTA-SADD DMEC and the HTA-SADD Trial Steering Committee (TSC) [Peter Connelly (chairperson DMEC), Robin Jacoby (chairperson TSC), Rowan Harwood, Cornelius Kelly, Angela Clayton-Turner, Craig Ritchie and Ed Juszczak]; the AS for providing patient and public involvement support into the study; the NIHR Mental Health Research Network (MHRN) and Dementia and Neurodegenerative Disease Research Network (DeNDRoN) for practical help; and referring clinicians in every area.
Contribution of authors
SB was the chief investigator for the study, and designed and managed the study with input from the group. JH and MD undertook the statistical analyses. All authors participated in data interpretation. SB drafted the first and subsequent versions of this report, with input and key revisions by all authors, who reviewed and approved the final submitted report.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
HTA-SADD recruitment group (in addition to the authors)
Birmingham Abdul Patel, Chris Vasillas, George Tadros, Martin Curtice, Alison Taylor, Avtar Singh Dhariwal, Seng E Goh, Deepak Kumar Shukla, William John Creaney, Rafi Arif, Karim Saad, Lucy Caswell, Bart Sheehan and Pravir Sharma.
Cambridge Carol Gregory, Rob Butler, Ehab Hegazi and Shamim Osmani Ruhi.
Leicester Ann Boyle, Ban Al-Kaissy and Saminathan Anand.
Liverpool Lisa Beddoes, Tafika Chowdhury, Mavis Evans, Sumanth Kumar, Javier de Arcaute, Peter Metcalfe, Jane Devaney, Andrew Chatfield, Ashley Baldwin, Sudip Sikdar, Jukanti Raju, Frances Lindon, Mark Theophanous, John Glyn Thomas, Maryyum Hussain, Miranda Conway and Emad Salib.
Manchester Sean Lennon, Harry Allen.
Newcastle Andrew Teodorczuk, Akshya Vasudev, Jonathan Richardson, John-Paul Taylor, Jane Newby, Mani Santhanakrishnan, Rod Gallagher, Julian Hughes, Adedayo Sobowale, Darren Craddock, Frances Dobie, Peter Howorth, Rory O'Shea, Apsara Panikkar, Anitha Howard and Richard Harrison.
North London Robert Tobiansky, Vincent Kirchner, Elizabeth Sampson, Anthony Katz, Lucy Watkin, Theofanis Vorvolakos, Jegathesvary Thirunathan, Hilary Kinsler, Shakil Khawaja, Andrew Winnet, Mohan Bhat, Amod Dalvi, Rajeeva Abeysuriya, Zuzana Walker, Beverly Louis, Gareth O'Leary, Simon Adelman, Pushpa Naveenan and Domi Gnanenthiran.
Southampton Vicky Banks, Karen Cotton, Janet Daoud, Valerie Hall and June Salkeld.
South London and Kent Patricia Irogeme, Tim Helme, John Besson, Naheed S Khan, Jenifer Chan, Kompancariel K Kuruvilla, Ananth Puranik, Carl Beckley, Justin Sauer and Suki Greaves.
Research workers and Mental Health Research Network and Dementias and Neurodegenerative Diseases Research Network clinical study officers
Birmingham Analisa Smythe, Jan Wright, Divya Chadha, Mohammed Shabbir and Siobhan Keogh.
Cambridge Angela Lynch, Kathryn Betts, Jane Addison, Fiona McDougal, Angela Browne, Regina Mello- Barreto and Freya Mellor.
Leicester Sarah Baillon, Penny Wakefield, Alex Satchwell, Anne Chafer, Tracy McCranor, Rumun Sandhu and Shaukat Desai.
Liverpool Lisa Douglas, Helen Newell, Samantha Fitzpatrick, Rachel Whalley, Leann Westmoreland, Maggie Lo, Caroline Mogan and Helen Beaumont-Kellner.
Manchester Jacqueline Crowther, Stephen Chew-Graham, Octavia Smart, Emma Oughton, Jonathan Bowker, Katrina Wade, Ann Morrow, Gemma Woods, Helen Williams, Maria Kaltsi, Magdalen Fiddler, Nichola Verstraelen, Rebecca Rowles and Lindsey Copeland.
Newcastle June Pearson, Jill Davison, Suzanne Humphrey, Joshua Wood, Saffra Knox, Jessica McClosky, Katherine Richardson, Karen Anne Morgan and Vanessa Waggott.
North London Ryan Li, Sharmila Logathas, Stephanie Habermann, Kofi Kramo, Shilpa Bavishi, Patricia Ndhlovu, Sarah Dickens, Khodayar Shahriyarmolki, Emily Dixon, Maria Sampson, Gemma Hardy and Bertha Mangunda.
Southampton Christine Dean, Annette Stevens and Laura Wolfe.
South London and Kent Michaela Poppe, Thandiwe Mtendera, Gaby Illingworth, Susan Thompson, Mohamed Pujeh and Alex Quigley.
Mental Health & Neurosciences Clinical Trials Unit Joanna Kelly, Caroline Murphy, Clare Rutterford and Rajesh Shah.
Publication
Banerjee S, Hellier J, Dewey M, Romeo R, Ballard C, Baldwin R, et al. Study of the use of anti-depressants for depression in dementia: the HTA-SADD Trial – a multicentre randomised double-blind, placebo-controlled trial of the clinical effectiveness of sertraline and mirtazapine. Lancet 2011;378:403–11.
References
- Hofman A, Rocca WA, Brayne C, . The prevalence of dementia in Europe. Int J Epid 1991;20:736-48.
- Launer LJ, Brayne C, Dartigues J-F, . Epilogue. Neuroepidemiology 1992;11:119-21.
- Prince M, Jackson J. World Alzheimer's Report 2009. London: Alzheimer's Disease International; 2009.
- Knapp M, Prince M, Albanese E, Banerjee S, Dhanasiri S, . Dementia UK: the full report. London: Alzheimer's Society; 2007.
- Lowin A, Knapp M, McCrone P. Alzheimer's disease in the UK: comparative evidence on cost of illness and volume of research funding. Int J Geriatr Psychiatry 2001;16:1143-8.
- Wimo A, Prince M. World Alzheimer's Report 2010: the global economic impact of dementia. London, UK: Alzheimer's Disease International; 2010.
- National service framework for older people. London: DoH; 2001.
- Everybody's business. London: CSIP; 2005.
- Everybody's business. London: CSIP; 2005.
- National Audit Office (NAO) Report . Improving Services and Support for People With Dementia 2007.
- Murray J, Schneider J, Banerjee S, Mann A. EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: II – A qualitative analysis of the experience of caregiving. Int J Geriatr Psych 1999;14:662-7.
- Schneider J, Murray J, Banerjee S, Mann A. EUROCARE: a cross national study of co-resident spouse carers for people with Alzheimer's disease: I – Factors associated with carer burden. Int J Geriatr Psychiatry 1999;14:651-61.
- Burns A, Jacoby R, Levy R. Psychiatric phenomena in Alzheimer's disease III: Disorders of mood. Br J Psychiatry 1990;157:81-6.
- Ballard CG, Bannister C, Oyebode F. Depression in Dementia Sufferers – A Review. Int J Geriatr Psychiatry 1996;11:507-15.
- Burns A. Affective symptoms in Alzheimer's Disease. Int J Geriatr Psychiatry 1991;6:371-6.
- Greenwald BS, Kramer-Ginsberg E, Marin DB, Laitman LB, Hermann CK, Mohs RC, et al. Dementia with coexistent major depression. Am J Psychiatry 1989;146:1472-8.
- Bains J, Birks JS, Dening TR. The efficacy of antidepressants in the treatment of depression in dementia. Cochrane Database Syst Rev 2002;4.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in people with depression and dementia. J Am Geriatr Soc 2011;49:577-85.
- Modrego PJ. Depression in Alzheimer's Disease. Pathophysiology, diagnosis, and treatment. J Alzheimers Dis 2010;21:1077-87.
- Thompson S, Herrmann N, Rapoport MJ, Lanctôt KL. Efficacy and safety of antidepressants for treatment of depression in Alzheimer's disease: a meta-analysis. Can J Psychiatry 2007;52:248-55.
- Petracca G, Teson A, Chemerinski E, Leiguarda R, Starkstein SE. A double-blind placebo-controlled study of clomipramine in depressed patients with Alzheimer's disease. J Neuropsychiatr Clin Neurosci 1996;8:270-5.
- Reifler BV, Teri L, Raskind M, Veith R, Barnes R, White E, et al. Double-blind trial of imipramine in Alzheimer's disease patients with and without depression. Am J Psychiatry 1989;146:45-9.
- Lyketsos C, Sheppard J, Steele C, Steele C, Brandt J, Steinberg M, et al. A randomized placebo-controlled, double-blind, clinical trial of sertraline in the treatment of depression complicating Alzheimer's Disease. Am J Psychiatry 2000;157:1686-9.
- Lyketsos CG, DelCampo L, Steinberg M, Miles Q, Steele CD, Munro C, et al. Treating depression in Alzheimer disease: efficacy and safety of sertraline therapy, and the benefits of depression reduction: the DIADS. Arch Gen Psychiatry 2003;60:737-46.
- Alexopoulos GS, Abrams RC, Young RC, Shamoian C. Cornell scale for depression in dementia. Biol Psychiatry 1988;23:271-84.
- Rosenberg PB, Drye LT, Martin BK, Frangakis C, Mintzer JE, Weintraub D, et al. Sertraline for the treatment of depression in Alzheimer disease. Am J Geriatr Psychiatry 2010;18:136-45.
- Weintraub D, Rosenberg PB, Drye LT, Martin BK, Frangakis C, Mintzer JE, et al. Sertraline for the treatment of depression in Alzheimer disease: week-24 outcomes. Am J Geriatr Psychiatry 2010;8:332-40.
- Anonymous. Do SSRIs cause gastrointestinal bleeding?. Drugs Ther Bull 2004;42:17-8.
- Doody R, Stevens J, Beck C, Dubinsky RM, Kaye JA, Gwyther L, et al. Practice parameter: management of dementia (an evidence based review). Neurology 2000;56:1156-66.
- Eccles M, Clarke J, Livingston M, Freemantle M, Mason J. North of England evidence based guidelines development project: guideline for the primary care management of dementia. BMJ 1998;317:802-8.
- Dementia: supporting people with dementia and their carers in health and social care. London: Department of Health; 2006.
- Ensrud KE, Blackwell TL, Mangione CM, Bowman PJ, Whooley MA, Bauer DC, et al. Study of Osteoporotic Fractures Research Group. Central nervous system-active medications and risk for falls in older women. J Am Geriatr Soc 2002;50:1629-37.
- Nyth A, Gottfries C, Luby K, Smedegaard-Andersen L, Gylding-Sabroe J, Kristensen M, et al. A controlled multicenter clinical study of citalopram and placebo in elderly depressed patients with and without dementia. Acta Psychiatry Scand 1991;86:138-45.
- Stahl SM, Entsuah R, Rudolph RL. Comparative efficacy between venlafaxine and SSRIs: a pooled analysis of patients with depression. Biol Psychiatry 2002;52:1166-74.
- Oslin D, Ten Have T, Streim J, Datto CJ, Weintraub D, DiFilippo S, et al. Probing the safety of medications in the frail elderly: evidence from a randomized controlled clinical trial of sertraline and venlafaxine in depressed nursing home residents. J Clin Psychiatry 2003;64:875-82.
- Banerjee S, Thornicroft G, Smuzkler G. Textbook of community psychiatry. London: Oxford University Press; 2001.
- Ballard CG, Patel A, Solis M, Lowe K, Wilcock G. A one year follow up study of depression in dementia sufferers. Br J Psychiatry 1996;68:287-91.
- Ballard CG, O’Brien JT, Swann AG, Thompson P, Neill D, McKeith IG. The natural history of psychosis and depression in dementia with Lewy bodies and Alzheimer's disease: persistence and new cases over 1 year of follow-up. J Clin Psychiatry 2001;62:46-9.
- Ballard C, O’Brien J, Swann A, Fossey J, Lana M. A one year follow-up study of behavioural and psychological symptoms in dementia (BPSD) amongst people in care environments. J Clin Psychiatry 2001;62:631-6.
- Teri L, Gibbons LE, McCurry SM, Logsdon RG, Buchner DM, Barlow WE, et al. Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003;290:2015-22.
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM, et al. Clinical diagnosis of Alzheimer's Disease: report of the NINCDS-ADRDA work group. Neurology 1984;34:939-44.
- Netten A, Netten A, Beecham J. Costing community care: theory and practice. Aldershot: Ashgate; 1993.
- Smith SC, Lamping DL, Banerjee S, Harwood R, Foley B, Smith, et al. Measurement of health-related quality of life for people with dementia: development of a new instrument (DEMQOL) and an evaluation of current methodology. Psychol Med 2006;37:737-46.
- The EuroQoL Group . EuroQoL – a new facility for the measurement of health-related quality of life. Health Policy 1990;16:199-208.
- Folstein MF, Folstein SE, McHugh PR. Mini Mental State. J Psychiatric Res 1975;12:189-98.
- Goldberg D, Williams P. A user's guide to the General Health Questionnaire. Windsor: NFER-NELSON; 1988.
- Ware JE, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care 1996;34:220-33.
- Zarit SH, Reever KE, Bach-Peterson J. Relatives of the impaired elderly: correlates of feelings of burden. Gerontologist 1980;20:649-55.
- Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308-14.
- Rosen WG, Terry RD, Fuld PA, . Pathologic verification of ischemic score in differentiation of Dementias. Ann Neurol 1980;7:486-8.
- Beecham J, Knapp M, Thornicroft G. Measuring Health Needs. London: Gaskell; 2001.
- Curtis L. Unit costs of health and social care. Canterbury: PSSRU, University of Kent; 2010.
- Department of Health (DoH) . National Health Service Schedule of Reference Costs 2010 n.d. www.doh.gov.uk/nhsexec/refcosts.htm (accessed May 2010).
- British national formulary. London: BMA and RPS; 2007.
- Curtis L. Unit costs of health and social care. Canterbury: PSSRU, University of Kent; 2007.
- Olin JT, Katz IR, Meyers BS, Schneider LS, Lebowitz BD. Provisional diagnostic criteria for depression of Alzheimer Disease. Am J Geriatr Psychiatry 2002;10:129-41.
- Moniz-Cook E, Vernooij-Dassen M, Woods R, Verhey F, Chattat R, De Vugt M, et al. A European consensus on outcome measures for psychosocial intervention research in dementia care. Aging Ment Health 2008;12:14-29.
- Schittecatte M, Dumont F, Machowski R, Cornil C, Lavergne F, Wilmotte J. Effects of mirtazapine on sleep polygraphic variables in major depression. Neuropsychobiology 2002;46:197-201.
- Muehlbacher M, Nickel MK, Nickel C, Kettler C, Lahmann C, Pedrosa Gil F, et al. Mirtazapine treatment of social phobia in women: a randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol 2005;25:580-3.
- Naglie G, Tomlinson G, Tansey C, Irvine J, Ritvo P, Black SE, et al. Utility-based Quality of Life measures in Alzheimer's disease. Qual Life Res 2006;15:631-43.
- Banerjee S. Use of antipsychotics for people with dementia – time for action. London: Department of Health; 2009.
- Tariot PN, Erb R, Podgorski CA, Cox C, Patel S, Jakimovich L, et al. Efficacy and tolerability of carbamazepine for agitation and aggression in dementia. Am J Psychiatry 1998;155:54-61.
- Lonergan E, Luxenberg J. Valproate preparations for agitation in dementia. Cochrane Database Syst Rev 2009;3.
- Teri L, Logsdon RG, Peskind E, Raskind M, Weiner MF, Tractenberg RE, et al. Alzheimer's Disease Cooperative Study. Treatment of agitation in AD: a randomized, placebo-controlled clinical trial. Neurology 2000;55:1271-8.
- Nyth AL, Gottfries CG. The clinical efficacy of citalopram in treatment of emotional disturbances in dementia disorders. A Nordic multicentre study. Br J Psychiatry 1990;157:894-901.
- Howard RJ, Juszczak E, Ballard CG, Bentham P, Brown RG, Bullock R, et al. CALM-AD Trial Group. Donepezil for the treatment of agitation in Alzheimer's disease. N Engl J Med 2007;357:1382-92.
- Gauthier S, Wirth Y, Möbius HJ. Effects of memantine on behavioural symptoms in Alzheimer's disease patients: an analysis of the Neuropsychiatric Inventory (NPI) data of two randomised, controlled studies. Int J Geriatr Psychiatry 2005;20:459-64.
- Cakir S, Kulaksizoglu IB. The efficacy of mirtazapine in agitated patients with Alzheimer's disease: a 12-week open-label pilot study. Neuropsychiatr Dis Treat 2008;4:963-6.
- Depression – the treatment and management of depression in adults. London: NICE; 2009.
Appendix 1 Project protocol
Project protocol (PDF download)
1 GENERAL INFORMATION
1.1 Protocol Information
1.1.1 Compliance
The trial will be conducted in compliance with the protocol, the European Union Clinical Trials Directive (2001/20/EC), the associated UK Medicines for Human Use (Clinical Trials) Regulations (2004) and Medicines for Human Use (Clinical Trials) Amendment Regulations 2006, the Data Protection Act (1998), Ethics Committee and MHRA approvals, the principles of ICH Good Clinical Practice (GCP) guidelines (CPMP/ICH/135/95), the principles of the Declaration of Helsinki (1996) and other requirements as appropriate.
1.1.2 Name of person/s authorised to sign the final protocol and protocol amendments for the sponsor
The sponsor of the trial is the Kings College London and the nominated individual authorised to sign the protocol on behalf of the sponsor is Dr Gill Dale.
1.1.3 Peer-Review
This study has been subject to intensive independent anonymous peer review by the Health Technology Assessment Programme prior to their making their decision to fund this study.
Main Contacts
1.2.1 Sponsor
Dr Gill Dale
Director of Research Quality
Research & Development Department
Box P005
Institute of Psychiatry Kings College London De Crespigny Park London SE5 8AF
Email gill.dale@iop.kcl.ac.uk
Tel +44 (0)20 7848 0675
1.2.2 Central Medical Advisors
Professor Sube Banerjee
Professor of Mental Health and Ageing P026, Section of Mental Health and Ageing Health Services
Research Department
The David Goldberg Centre
The Institute of Psychiatry
De Crespigny Park
London SE5 8AF
Email S.Banerjee@iop.kcl.ac.uk
Tel +44 (0) 20 7848 0012
Fax +44 (0) 20 7848 5056
Professor Alistair Burns
Department of Old Age Psychiatry
2nd Floor, Education and Research Centre
Wythenshawe Hospital
Manchester M23 9LT
Email: alistair.burns@manchester.ac.uk
Tel: 0161 291 5887
Fax: 0161 291 5882
Professor Clive Ballard
The Wolfson CARD, The Wolfson Wing, Hodgkin Building, Guy's Campus, London, SE1 1UL
Email: clive.ballard@kcl.ac.uk
Tel: 020 7848 8054
Fax: 020 7848 6145
1.2.3 Chief Investigator
Professor Sube Banerjee
Professor of Mental Health and Ageing P026, Section of Mental Health and Ageing Health Services
Research Department
The David Goldberg Centre, The Institute of Psychiatry, De Crespigny Park
London SE5 8AF
Email S.Banerjee@iop.kcl.ac.uk
Tel +44 (0) 20 7848 0012
Fax +44 (0) 20 7848 5056
1.2.3.1 Other Lead Investigators
Professor Alistair Burns, Community Based Medicine, Psychiatry Research Group, Floor 3 East, Room 306, University Place,
Oxford Road, Manchester, M13 9PL
Email: alistair.burns@manchester.ac.uk
Tel: 0161 306 7913
Fax: 0161 306 7945
Mobile: 07917 277628
Professor Clive Ballard
The Wolfson CARD The Wolfson Wing Hodgkin Building Guy's Campus London SE1 1UL
Email: clive.ballard@kcl.ac.uk
Tel: 020 7848 8054
Fax: 020 7848 6145
1.2.4 Principal investigators
There will be 9 recruiting PI sites, where Research Workers will be employed. These 9 Recruiting PI sites are listed here. Each of these Recruiting PI's will have an Investigator Site File, managed by the Research Worker for that site. All participants will be registered as patients at the recruiting NHS Trust and that NHS Trust pharmacy will dispense study medication for that participant. The Recruiting PI will list all doctors, nurses, psychologists and other staff within that site on a ‘delegation of authority’ form, which will clearly identify responsibilities within the study. Only authorised medical doctors within that site (i.e. those holding substantive or honorary contracts within that NHS Trust) may prescribe study medication.
There are also Referring Investigators, who will identify suitable potential participants and refer them to the Recruiting PI. Because the Referring Investigators will undertake assessments that will not be repeated by the Recruiting PI, all the Referring Investigators must be ‘part’ of the study.
Therefore, each NHS Trust from which participants are referred to a Recruiting PI site will have an identified ‘Referring PI’ who will be on the ethics application for that site and will hold a ‘Referring Investigator Site File’. Any other clinician within that NHS Trust who is also willing to refer participants to the study must be listed on a ‘delegation of authority form’ for that referring site. When the Research Worker receives a referral for the study from any authorised Referring Investigator within that site, he or she will copy the referral back into the ‘Referring Investigator Site File’ for completeness of the NHS Trust records.
Copies of all delegation of authority forms for all sites must be sent to the Trial Manager, along with CVs for all those listed.
01 Birmingham
Dr Peter Bentham
Consultant/Senior Lecturer in Old Age Psychiatry
Mental Health Services for the Older Adult
Queen Elizabeth Psychiatric Hospital
Birmingham B15 2QZ
Email Pwblmb@aol.com
Tel +44 (0) 121 301 2070
Fax +44 (0) 121 301 2071
02 Cambridge
Dr Claire Lawton
Consultant Psychiatrist & Clinical Director
Older People's Mental Health Services
Cambs & P'boro Mental Health Partnership NHS Trust
Beechcroft, Box 311
Fulbourn Hospital
Cambridge CB1 5EF
Email Claire.Lawton@cambsmh.nhs.uk
Tel +44(0)1223 218 890
Fax +44(0)1223 218 992
03 Leicester
Professor James Lindesay
Professor of Psychiatry for the Elderly
Psychiatry for the Elderly
Leicester General Hospital
Gwendolen Road
Leicester LE5 4PW
Email jeb1@le.ac.uk
Tel +44 (0)116 258 8161
Fax +44 (0)116 273 1115
04 Liverpool
Professor Kenneth Wilson
Professor of Old Age Psychiatry
University Department of Psychiatry
University of Liverpool
Royal Liverpool University Hospital
Liverpool L69 3GA
Email K.C.M.Wilson@liverpool.ac.uk
Tel +44 (0)151 706 4149
Fax +44 (0)151 706 3765
05 Manchester
Professor Alistair Burns
Community Based Medicine
Psychiatry Research Group
Floor 3 East, Room 306
University Place
Oxford Road
Manchester, M13 9PL
Tel: 0161 3067913
Fax 0161 3067945
Mobile 07917 277628
06 Newcastle
Professor John O'Brien
Professor of Old Age Psychiatry
Wolfson Research Centre Institute for Ageing and Health
University of Newcastle
Newcastle General Hospital
Newcastle-Upon-Tyne NE4 6BE
Tel +44 (0)191 256 3323
Fax +44 (0)191 219 5051
07 North London
Professor Gillian Livingston
Reader in Psychiatry of Older People
Department of Mental Health Sciences
University College London
Archway Campus
Holborn Union Building
Highgate Hill
London N19 5NL
Email g.livingston@ucl.ac.uk
Tel +44 (0)20 7561 4218
Fax +44 (0) 20 75614236
08 Southampton
Professor Clive Holmes
Professor in Old Age Psychiatry
Memory Assessment and Research Centre
Moorgreen Hospital Botley Road
West End
Southampton SO30 3JB
Email Clive.Holmes@wht.nhs.uk
Tel +44 (0)23 80475216
Fax +44 (0)23 80463022
09 South London & Kent
Professor Sube Banerjee
Professor of Mental Health and Ageing
P026, Section of Mental Health and Ageing
Health Services Research Department
The David Goldberg Centre
The Institute of Psychiatry
De Crespigny Park
London SE5 8AF
Email S.Banerjee@iop.kcl.ac.uk
Tel +44 (0) 20 7848 0012
Fax +44 (0) 20 7848 5056
1.2.5 User/Consumer Lead
Mrs Shirley Nurock
London Regional Co-ordinator
Consumer Involvement in Dementia
Quality Research in Dementia
Alzheimer's Society
Gordon House
10 Greencoat Place
London SW1P 1PH
Email s_nurock@hotmail.com
Tel +44 (0)20 7306 0606
Fax +44 (0)20 7306 0808
1.2.6 Collaborative Investigators
Robert Baldwin
Jayne Byrne
David Wilkinson
Georgina Charlesworth
Gordon Wilcock
Martin Orrell
George Fox
Cornelius Katona
Dolores Moniz-Cook
Joanna Murray
1.2.7 Trial Management
Niall McCrae
SADD Trial Manager
P026, Section of Mental Health and Ageing
Health Services Research Department
The David Goldberg Centre The Institute of Psychiatry De Crespigny Park
London SE5 8AF
Email HTASADD@iop.kcl.ac.uk
Tel +44 (0) 20 7848 0012
Fax +44 (0) 20 7848 5056
1.2.8 Data Management
SADD Data Management c/o Niall McCrae
P026, Section of Mental Health and Ageing
Health Services Research Department
The David Goldberg Centre The Institute of Psychiatry De Crespigny Park
London SE5 8AF
Email HTASADD@iop.kcl.ac.uk
Tel +44 (0) 20 7848 0012
Fax +44 (0) 20 7848 5056
1.2.9 Trial Statisticians
Rebecca Walwyn
Statistician
Mental Health & Neuroscience Clinical Trials Unit
Box P064
Institute of Psychiatry
London SE5 8AZ
Email R.Walwyn@iop.kcl.ac.uk
Tel +44 (0) 20 7848 5424
Fax +44 (0) 20 7848 5229
Dr Michael Dewey
Senior Lecturer in Statistics
PO60, Section of Epidemiology
Institute of Psychiatry, King's College London
De Crespigny Park
London SE5 8AF
Email m.dewey@iop.kcl.ac.uk
Tel +44 (0)20 7848 0136
Fax +44 (0)20 7277 0283
Clare Rutterford
Statistician
Mental Health & Neuroscience Clinical Trials Unit
Box P064
Institute of Psychiatry
London SE5 8AZ
Email clare.rutterford@iop.kcl.ac.uk
Tel +44 (0) 20 7848 0679
Fax +44 (0) 20 7848 5229
1.2.10 Health Economists
Professor Martin Knapp
Professor of Social Policy and Health Economics
Cowdray House
London School of Economics and Political Science
Houghton Street
London WC2A 2AE
Email m.knapp@lse.ac.uk
Tel +44 (0)20 7955 6840
Fax +44 (0)20 7955 6803
Dr Linda Davies
Director of Health Economics Education & Research Centre, Wythenshawe Hospital, Manchester M23 9LT
Email Linda.Davies@man.ac.uk
Tel +44 (0)161 291 5886
Fax +44 (0)161 291 5882
Renee Romeo
Honorary Lecturer
Centre for the Economics of Mental Health (CEMH) PO24, Institute of Psychiatry, King's College London
De Crespigny Park
London SE5 8AF
Email r.romeo@iop.kcl.ac.uk
Tel +44 (0)20 7848 0588
Fax +44 (0)20 7701 7600
1.2.11 Randomisation Centre
Mental Health & Neurology Clinical Trials Unit
103 Denmark Hill, Institute of Psychiatry London SE5 8AF
Email randomization_request@iop.kcl.ac.uk
Tel +44 (0) 20 7848 5282
Fax +44 (0) 20 7848 5229
1.2.12 Study Medication Manufacture & Distribution
Manufacture of Mirtazapine
Genus Pharmaceuticals
As of 1st November 2009: Arrow Pharmaceuticals
Manufacture of Sertraline & Matching Placebo
Pfizer UK Limited
Manufacture of Mirtazapine Placebo, Central Packaging & Labelling & Distribution to Local Pharmacies
Catalent
Clinical Supply Services Wingates Industrial Park Westhoughton
Bolton
Lancs, BL5 3XX
Email Karl.Jones@catalent.com
Tel +44 (0)1942 790000
Fax +44 (0)1942 799799
1.3 Trial Committees
1.3.1 Trial Steering Committee (TSC)
The Trial Steering Committee (TSC) is responsible for the independent oversight of the progress of the trial, investigation of serious adverse events, and determining the future progress of the trial in light of regular reports from the DMC. The TSC has the power to prematurely close the trial. The TSC will meet annually or more often if the chair determines a reason for doing so and is composed of:
Professor Robin Jacoby, Professor of Old Age Psychiatry, University of Oxford (Chair)
Dr Cornelius Kelly, Consultant Old Age Psychiatrist, Central & North West Mental Health Trust
Dr Craig Ritchie, Clinical Research Fellow in Old Age Psychiatry, Imperial College London
Angela Clayton-Turner, Alzheimer's Society/Carer Representative
Professor Sube Banerjee (Chief Investigator)
Ms Rebecca Walwyn (Trial Statistician)
Niall McCrae (Trial Manager; Secretary to the TSC).
Invited observers include: NHS HTA, Sponsor, applicants.
Membership has been approved by the sponsor.
1.3.2 Data Monitoring Committee (DMC)
The Data Monitoring Committee (DMC) is independent and is responsible for monitoring progress of the trial and serious adverse events and reactions. The DMC will meet annually or more often if the chair determines a reason for doing so. They will provide a confidential trial progress report at the end of each meeting which will be sent to the TSC. The DMC will agree their structure and organisation in an IDMC Charter (DAMOCLES Study Group, 2005) before randomisation commences. The DMC can recommend premature closure of the trial to the TSC in accordance with the IDMC charter. The DMC is composed of:
Dr Peter Connolly, Consultant Old Age Psychiatrist, Murray Royal Hospital, Perth (Chair)
Dr Rowan Harwood, Medicine and Rehabilitation, Nottingham City Hospital
Dr Pat Shariatmadari, Alzheimer's Society/Carer Representative
Ed Juszczak, Senior Medical Statistician, Centre for Statistics in Medicine, Oxford.
1.3.3 Trial Management Group (TMG)
The Trial Management Group (TMG) is responsible for the day-to-day running and management of the trial. The full TMG will meet quarterly in the first year and biannually thereafter. It is composed of:
Professor Sube Banerjee (Chair)
All Investigators
Trial statisticians
Health economists User/Consumer representative Trial manager
Data manager (Secretary to the TMG)
Other HTA-SADD team members may attend as observers with the permission of the Chief Investigator.
Sub-committees may be formed from the full TMG for specific purposes (e.g. protocol development, writing papers, etc.). These committees will be appointed by the full TMG and will meet as necessary.
1.4 Staff Training Programme
All staff employed on the grant and all Investigators will be trained in:
-
GCP
-
Use of the assessment tools
-
Trial standard operating procedures.
Up-to-date CVs of all staff working on the trial will be kept in the Trial Office along with a log of all trial training received by staff.
1.5 Declarations of Competing Interests
All Investigators have received support from pharmaceutical companies for example to attend conferences, for giving lectures, for the provision of consultancy, or for the conduct of research. No Investigator or member of staff employed on the grant has any shareholding in any company that might gain from the subject of this study.
2 ABBREVIATIONS
| AE | Adverse Event | NASSA | Noradrenergic and Specific Serotonergic Antidepressant |
| AR | Adverse Reaction | NHS | National Health Service |
| ANCOVA | Analysis of Covariance | NINCDS- ADRDA | National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association |
| BADL | Bristol Activities of Daily Living (scale) | NPI | Neuro Psychiatric Inventory |
| CALM-AD | Cholinesterase Inhibitor in the Management of Agitation in Dementia | OCD | Obsessive Compulsive Disorder |
| CI | Chief Investigator | PI | Principal investigator |
| CIN | Carer Identification Number | PTSD | Post-Traumatic Stress Disorder |
| CIOMS | Council for International Organisation of Medical Sciences | QALY | Quality Adjusted Life Years |
| CSDD | Cornell Scale for Depression in Dementia | QRD | Quality Research in Dementia |
| CSRI | Client Service Receipt Inventory | R&D | Research & Development |
| CSO | Clinical Studies Officer | RCT | Randomised Controlled Trial |
| CTA | Clinical Trial Authorisation | REC | Research Ethics Committee |
| CTIMP | Clinical Trial of an Investigational Medicinal Product | RW | Research Worker |
| DEMQOL | Dementia Quality of Life | SADD | Study of Antidepressants in Dementia |
| DMC | Data Monitoring Committee | SAE | Serious Adverse Event |
| DSM-IV | Diagnostic & Statistical Manual, version 4 | SAR | Serious Adverse Reaction |
| EC | European Community | SD | Standard Deviation |
| eCRF | Electronic Case Report Form | SES | Standardised Effect Size |
| EQ5D | EuroQol version 5D | SF-12 | Short Form 12 version 2 (health survey) |
| GCP | Good Clinical Practice | SGOT | Serum Glutamic Oxaloacetic Transaminase |
| GHQ-12 | General Health Questionnaire version | 12 | SGPT |
| GP | General Practitioner | SDW | Source Data Worksheet |
| HTA | Health & Technology Assessment | SmPC | Summary of Product Characteristics |
| IDMC | International Data Monitoring Committee (Charter) | SOP | Standard Operating Procedure |
| IMP | Investigational Medicinal Product | SNRI | Selective Noradrenergic Reuptake |
| LREC | Local Research Ethics Committee | SSRI | Selective Serotonin Reuptake Inhibitors |
| LSE | London School of Economics | SUSAR | Suspected Unexpected Serious Adverse Reaction |
| MH&N CTU | Mental Health & Neurology Clinical Trials Unit | TCA | TriCyclic Antidepressant |
| MHRA | Medicines & Health Care Products Regulatory Agency | TMF | Trial Master File |
| MHRN | Mental Health Research Network | TMG | Trial Management Group |
| MMSE | Mini-Mental State Examination | TSC | Trial Steering Committee |
| MRC | Medical Research Council | UK | United Kingdom |
3 SUMMARY
3.1 Structured Synopsis
Primary Objective
-
To determine the clinical and cost effectiveness of two classes of antidepressants for depression in dementia (compared with placebo).
-
To determine whether an SSRI (sertraline) is i) more clinically effective and ii) more cost effective than placebo in reducing Cornell depression score 13 weeks post randomisation.
-
To determine whether a NASSA (mirtazapine) is i) more clinically effective and ii) more cost effective than placebo in reducing Cornell Depression score 13 weeks post-randomisation.
-
Secondary Objectives
-
2. To investigate differences in the clinical and cost effectiveness, and, in terms of adverse events, withdrawals from treatment and adherence to treatment between mirtazapine and sertraline for depression in dementia at 13 and 39 weeks post-randomisation.
-
3. To investigate differences in the clinical and cost effectiveness of mirtazapine or sertraline compared with placebo on patient (e.g. quality of life, cognition) and family carer (e.g. carer burden, carer quality of life) outcomes at 13 and 39 weeks post-randomisation.
-
4. To investigate the influence on clinical and cost effectiveness of clinical characteristics including: dementia severity, dementia type, depression type, depression severity, care arrangements, neuropsychiatric symptoms, and physical illness.
Design
A multicentre double-blind placebo-controlled RCT of the clinical and cost effectiveness of two classes of antidepressants, and more specifically, mirtazapine and sertraline, from baseline to 3 months (13 weeks) and 9 months (39 weeks) enabling estimation of short and long-term impacts of these antidepressants on depression in dementia. Participants will remain on blinded study medication for a total of 10 months to allow time for data entry prior to routine unblinding.
Setting
Secondary care, referrals to old age psychiatric services and memory clinics in 9 regional sites each covering a catchment area of 100,000 older people (Birmingham, Cambridge, Leicester, Liverpool, Manchester, Newcastle, North London, Southampton and South London) aided by the Department of Health Mental Health Research Network (MHRN).
Target Population
People with probable and possible dementia of the Alzheimer type and co-existing depression.
Eligibility
This is a pragmatic trial. The criteria for inclusion are as close to clinical practice as possible. We will recruit those where a secondary care doctor makes a clinical diagnosis of mild to moderate probable or possible Alzheimer's Disease and a co-existing depressive illness of at least four weeks duration, likely to need treatment with antidepressants. The local research worker (RW) will then assess the patient's depression severity and those with a Cornell Scale for Depression in Dementia (CSDD) of 8+ will be eligible for entry into the trial. The other trial exclusions will be: the case being too critical to be randomised; absolute contra-indications to trial medications, being on another trial, and no family or professional carer to give collateral information.
Health Technologies Being Assessed
There will be three groups: 1. a Selective Serotonin Reuptake Inhibitor (SSRI), sertraline, with normal clinical care; 2. a Noradrenaline and Selective Serotonin Antidepressant (NASSA) mirtazapine, with normal clinical care; and 3. a control group, placebo, with normal clinical care. Interventions will be presented in an identical double dummy form with all participants taking up to six capsules: up to three sertraline 50mgs or sertraline placebo; and up to three mirtazapine 15mgs or mirtazapine placebo.
Randomisation
Patients will be allocated to placebo, sertraline or mirtazapine (ratio 1:1:1) by the Mental Health & Neurology Clinical Trials Unit based at the Institute of Psychiatry. Allocation will be stratified by centre by stratified block randomisation with randomly varying block sizes. Allocation will be physically carried out during weekdays by phone, email or fax within 24 hours of a request.
Measurement Of Cost And Outcome
Cases identified will be assessed by a local research worker (G grade CPN or equivalent) who will collect baseline and follow-up data (0m, 3m, and 9m). The primary outcomes will be depression score - Cornell Scale for Depression in Dementia (CSDD) and cost - Client Service Receipt Inventory (CSRI). Secondary outcomes will include: adverse events, compliance, patient quality of life (disease-specific DEMQOL, generic EQ5D), cognition (MMSE), behavioural and psychological symptoms (NPI), carer burden (Zarit), carer stress (GHQ12), and carer quality of life (SF12 v2). The analysis of the economic impact of the interventions is a central, fully integrated element of the proposed study. The comprehensive costs of care for all participants will be calculated (including the costs of formal care such as that provided by health and social services and also the costs of informal care) using data gathered using the CSRI completed by key workers or family carers at baseline, 13w and 39w. Unit costs will be best national estimates of the long-run marginal opportunity costs. Informal care will be costed.
Sample Size
An overall sample size of 507 patients will provide 90% power to detect a 2 point difference in CSDD (SD 5; SES 0.4) for the primary comparisons of mirtazapine vs placebo and sertraline vs placebo at 13 weeks and 86% power for the secondary analysis of these comparisons at 39 weeks. This allows for 10% loss to follow-up at 13 weeks and 20% loss to follow-up at 39 weeks, correlation between baseline and outcome CSDD> 0.6, and up to 12.5% of those randomized (per comparison) to be either drop-outs or drop-ins using an analysis of covariance with 2-sided 5% significance levels. Allowing for the same levels of loss to follow-up, an overall sample of 507 patients would also enable us to calculate 2-sided 95% confidence intervals for the difference in the proportion of pre-specified adverse events between the antidepressant arms of (a clinically significant) 10% (i.e. 5% vs 15%) ± 6% at 13 weeks and ± 7% at 39 weeks.
Statistical Analyses
Primary Analyses
CSDD score at 13 weeks will be analysed by ANCOVA adjusted for baseline CSDD and centre with contrasts for (a) sertraline vs placebo and (b) mirtazapine vs placebo.
Secondary Analyses
The ANCOVA of CSDD score at 13 weeks will further include a contrast for mirtazapine vs sertraline. CSDD score at 39 weeks will be analysed by ANCOVA adjusted for baseline CSDD and centre with contrasts for (a) sertraline vs placebo; (b) mirtazapine vs placebo, and (c) mirtazapine vs sertraline. Secondary outcomes will be compared using the same contrasts as above within a [longitudinal] generalised linear model framework adjusting for the respective baseline scores and centre. The significance level will be 5% (2-sided) for all specified analyses of the primary outcome variable and 1% (2-sided) for all specified analyses of secondary outcome variables.
Economics
From the cost and the outcome data, we will compare total and component (by service or agency) costs, incremental cost-effectiveness ratios and net benefits (using the primary outcome measure CSDD), cost–utility ratios (using utility scores computed from the EQ-5D and societal weights) and cost-consequences results (using all non-cost outcomes measures). The primary evaluation will be the cost effectiveness analyses with CSDD change as the outcome. The evaluation will include the plotting of cost-effectiveness acceptability curves generated from bootstrap analyses. Sensitivity analyses will explore the impact of differences in key costs and outcome assumptions. Modelling will be conducted to predict costs and outcomes beyond the duration of the trial. The evaluation will be conducted from (a) societal, (b) public sector and (c) NHS perspectives.
Project Timetable
| Month −6 to 0 | development and finalisation of full protocol and CRFs, trial approvals sought; |
| Month 1 to 3 | trial systems set up; |
| Month 1 to 3 | manufacture and packaging of medications and placebo; |
| Month 3 | training RWs, centres set up and priming; |
| Month 4 to 33 | recruitment of patients, randomisation (30m); |
| Month 7 to 42 | follow-up interviews (3m and 9m); |
| Month 43 to 45 | final analyses and study closeout. |
3.2 Flowchart of Trial Design
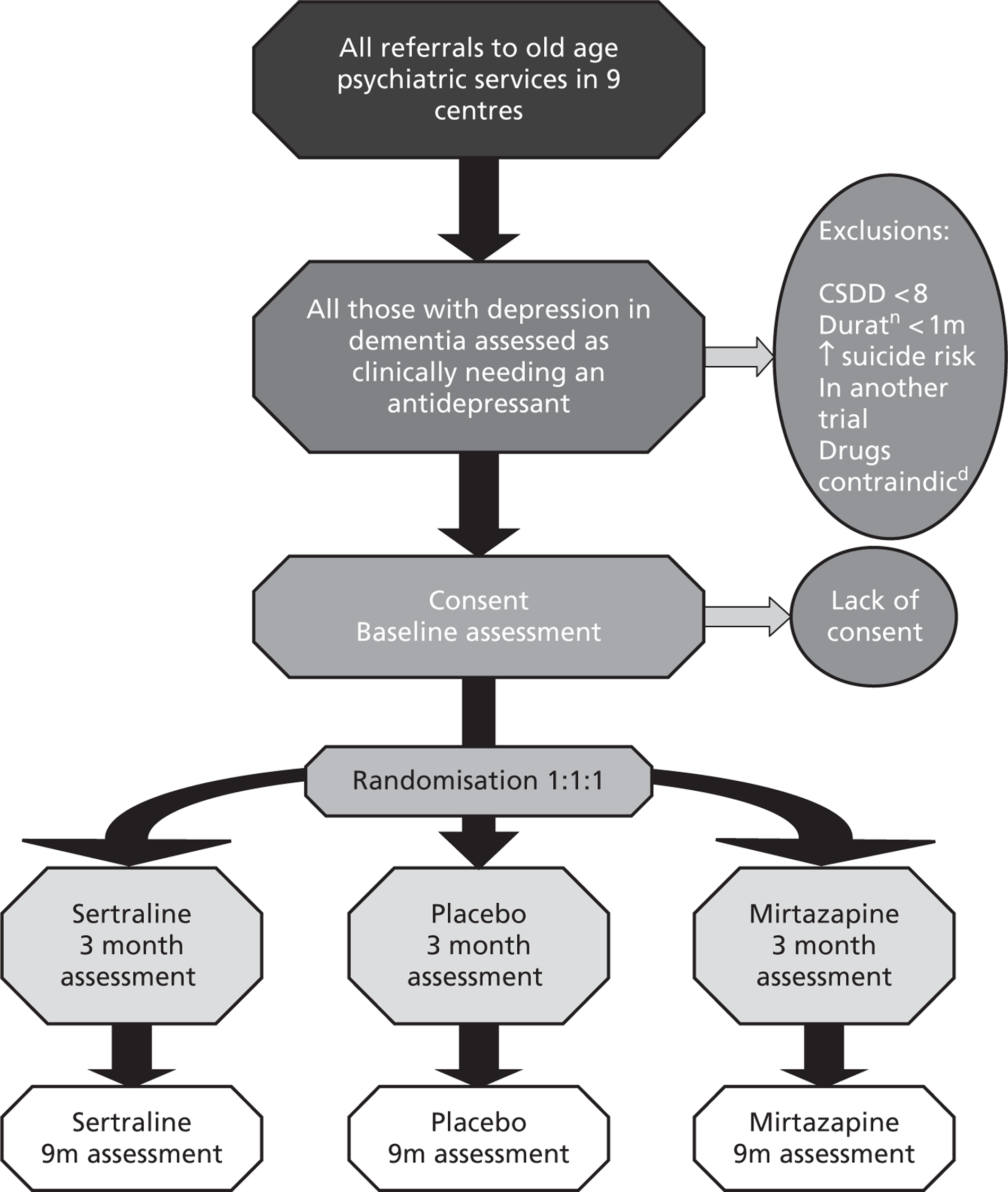
4 BACKGROUND INFORMATION
4.1 Introduction including Relevant Studies
Depression occurs in at least 20% (Burns et al 1990; Ballard et al 1996a) of people with Alzheimer's disease (AD) in whom it causes considerable distress (Burns et al 1991), reduces quality of life (Burns 1991), exacerbates cognitive and functional impairment (Greenwald et al 1989), increases mortality (Burns et al 1991), and is associated with added carer stress and depression (Ballard et al 1996b). Treating depression is therefore a key clinical priority to improve the well-being, quality of life and level of function of people with Alzheimer's disease.
A Cochrane review completed in July 2002 Antidepressants for treating depression in dementia addresses directly the questions raised in the research brief (Bains et al. 2003); one of the applicants (TD) is an author of this review. The review identified six studies with 739 participants meeting inclusion criteria (“all relatively unconfounded, double-blind, randomized trials comparing any antidepressant drug… with placebo, for patients diagnosed as having dementia and diagnosed as having a depression according to established criteria”). Only three studies, comprising 107 participants, had data that could be subject to a meta-analysis of efficacy. Petracca et al (1996) studied 24 participants in a neurological out-patent clinic in Argentina in a double-blind placebo controlled crossover trial of clomipramine (a tricyclic antidepressant [TCA]) with two 6 week treatment periods with a 2 week washout period. There was a mean change of -10.7 on the Hamilton depression scale in the intervention group and –4.5 in the control group. Reifler et al (1989) selected 61 participants from two university outpatient clinics in an 8 week double-blind trial of imipramine (a TCA). The study showed no treatment effect. The third trial included was Lyketsos et al (2000), which is an interim analysis of data on 22 participants that subsequently were reported fully in Lyketsos et al (2003). These final data were not available to the Cochrane review. In the final study 44 participants were recruited from a single university out-patient clinic into a 12 week double-blind placebo controlled trial of sertraline (a specific serotonin reuptake inhibitor [SSRI]). An effect size of 0.51 was reported with a mean change of -10.5 on the Hamilton depression scale in the intervention group and –4.5 in the control group and –9.9 and –3.2 in on the Cornell Scale for Depression in Dementia (CSDD; Alexopoulos et al 1988). Other than the further data on the additional 22 cases reported in Lyketsos et al (2003), we are not aware of any other studies published since that would have met the criteria for inclusion in the Cochrane review.
The main finding of the Cochrane review was that despite the clinical seriousness of the condition, there was only weak evidence available of the effectiveness of antidepressants in dementia. They noted that two of the studies used TCAs “drugs not commonly used in this population”, that only one used the most commonly used class of drugs, the SSRIs, and that there were no studies of the newer classes of antidepressants such as selective noradrenergic reuptake inhibitors. The review concluded that there was a need for further definitive research of “modern frequently used drugs”. In addition they identified the need for trials to use instruments to measure outcome which have been validated for use in depression in dementia such as the CSDD.
It is clear that the participants recruited into all the trials discussed above were highly selected and so there may be limitations in the generalisability of the data derived from them. One element of this is the severity of depression recruited, with Lyketsos et al (2003) and Reifler et al (1989) requiring depression to meet DSM criteria for major depressive episode. Such disorders form only a small proportion of clinically significant depression requiring intervention in older adults in the community (Copeland et al 1990; Schaub et al 2003). Lyketsos et al (2003) acknowledged the need for research into the efficacy of antidepressants in a wider range of depression type and severity, longer-term treatment, and the comparative efficacy of different classes of antidepressants.
The Quality Standards Subcommittee of the American Academy of Neurology (Doody et al 2001) found that there was evidence of only “moderate clinical certainty” for antidepressants in the treatment of depression in dementia, concluding that “SSRIs may offer some benefit with greater tolerability than other antidepressants”. They too reported the need for further research into the treatment of depression in dementia.
All of the studies to date are of short duration, and none tackle the crucial issue of whether there is longer term benefit associated with antidepressant treatment. It is unclear whether the differential efficacy between the published studies relates to the choice of antidepressant, differences in study design and power or chance variation. Importantly, the literature does indicate that the successful resolution of depression is associated with cognitive and functional improvements (Greenwald et al 1989). There are however several cautions. For example, one study of the tricyclic antidepressant imipramine indicated that active treatment increased cognitive impairment and disability, whilst several studies of falls indicate that most antidepressants increase falls risk. In addition, there have been recent safety concerns relating to the SSRI sertraline and gastrointestinal bleeding (Anonymous 2004) and the SSRI paroxetine and withdrawal.
Depression is a major issue for the function and quality of life of people with dementia. A well- powered large randomised controlled trial (RCT) is crucial to determine the long-term clinical effectiveness, benefit to harm ratio and cost-effectiveness of antidepressant therapy in the treatment of depression in dementia, and to inform the optimal choice of antidepressant agent to enable best clinical practice and maximum benefit for people with dementia and their carers.
The HTA therefore prioritised this as an area for primary research and this protocol was successful in the competitive tendering process for a study that would fill these major gaps in the evidence base definitively.
4.2 Consumer Involvement
This study has been developed in collaboration with the Alzheimer's Society. The consultations that have been conducted prior to the generation of this protocol are detailed below. The Alzheimer's Society is the leading care and research charity for people with Alzheimer's disease and other forms of dementia, their families and carers in the UK. It is a national membership organisation and works through nearly 250 branches and support groups. The Society is also a member of Alzheimer's Disease International and it works closely with dementia charities and organisations in other countries.
The Alzheimer's Society has an active research programme (Quality Research in Dementia - QRD), which is an active partnership between carers, people with dementia and the research community. The heart of Quality Research in Dementia is the QRD Advisory network of 150 carers, former carers and people with dementia who play a full role in all areas of setting priorities for research. They are involved in selecting and then commenting on grant applications and project monitoring.
The Alzheimer's Society, utilizing the QRD framework is therefore in an ideal position to act as an effective partner in the current project, having made an important contribution to our pre-trial consultation. One of the three co-PIs (Professor Clive Ballard) on this application is the Director of Research at the Alzheimer's Society and another (Professor Alistair Burns) is the chair of the Alzheimer's Society's Scientific Advisory panel. One of the applicants is a nominee of the Alzheimer's Society (Mrs Shirley Nurock). All the centres have close and active links with their local Alzheimer's Society branches and consultation and collaboration on this project will take place on a local as well as a national level.
The QRD network has expressed enthusiasm and emphasised the importance of a strong consumer involvement in all key aspects of the study. QRD will be an integral part of the whole research process, from pre-trial design through project monitoring as a whole including the Trial Steering Committee, the Data Monitoring and Ethics Committee and the Trial Management Group with a remit for study monitoring and governance, concluding in the analyses, interpretation and dissemination of data generated. However, we will also look beyond QRD to also involve local user and carer groups in the study process and monitoring. This integration will enable broad and innovative dissemination of the results to ensure that the important elements are communicated to people with dementia, carers and the general public as well as health care professionals, to enable effective implementation.
4.3 Choice of Trial Population
We have designed this study as a pragmatic trial of effectiveness in routine clinical practice. We wish to minimise exclusions from the study in order to maximise the generalisability of the data generated.
We are not intending to exclude participants on the basis of their taking concomitant psychotropic medication e.g. hypnotics, antipsychotics or cholinesterase inhibitors. These medications will be commonly prescribed in our study group and any such exclusions would limit the generalisability of the data generated, so compromising the pragmatic nature of the trial. Management of the participants in this study will therefore mimic true clinical practice with the sole exception of the trial medication.
4.4 Choice of Investigational Interventions
Inclusion of a TCA Arm
As discussed above and in the research brief, there are unanswered questions concerning what class of antidepressant to choose and how long to treat. We have designed this trial to attempt provide best-quality data on all these clinically important areas.
One possible area of contention is the appropriateness of including a tricyclic antidepressant (TCA) arm in the trial. This was referred to in the research brief. Prior to our initial submission we carried out a local consultation with people with dementia, family carers and clinicians in London, Manchester and within the Alzheimer's Society. The findings of this exercise were clear. Patients, carers and clinicians all believed that it would be unacceptable to randomise people with dementia to medication with a predictable set of negative (anticholinergic e.g. constipation, increased confusion, blurred vision, low blood pressure, drowsiness) side effects even given the fact that the competing classes of medication have their own profile of side effects. In addition clinicians reported to us that their clinical practice was not to use TCAs as a first line treatment for depression in dementia and that they believed people with dementia to be at a higher risk of harm from TCA side effects than people without dementia. They therefore raised questions of the clinical acceptability of a trial that included the possibility of randomisation to a TCA. To be successful we will need a large number of clinical teams to take part in case finding and if the trial is to generate real effectiveness data then these participants need to be an unbiased sample of all potential prescribers. On these grounds we therefore decided not to include a tricyclic antidepressant arm but instead to compare the clinical effectiveness and cost-effectiveness (including discontinuation and adverse events) of examples of the two classes of antidepressants most in use.
In the subsequent feedback from the HTA Commissioning Board we were invited to reconsider our decision not to include a TCA arm. We therefore consulted the Alzheimer's Society Quality Research in Dementia (QRD) Network. This is a panel made up of people with dementia and their carers that advises the UK Alzheimer's Society (AS) on research issues. The consultation was carried out by the AS Director of Research (Prof. Clive Ballard). He consulted regional co-ordinators of the Alzheimer Society's (QRD) and individual members of the network, representing the views of 45 QRD members; most with experience of caring for someone with dementia who has been treated with antidepressants. The purpose was to inform key aspects of the study, in particular whether it was appropriate to include TCAs as one of the treatments. All but one of the people responding strongly expressed the view that TCAs were an inappropriate treatment for people with dementia, describing a number of personal experiences where serious falls, increased confusion, urinary retention and other adverse events had resulted in a serious detrimental impact to the quality of life of the person with dementia.
We also consulted clinicians through the potential collaborating centres more widely and again there was a near unanimous view that it was not clinically supportable to initiate people with depression in dementia on a TCA. They also reported that the existence of such a possibility in randomisation would discourage them from entering patients into the trial. At the very least it is therefore likely that there would be substantial selection bias (both in patient acceptability and clinician referral) introduced by the inclusion of a TCA arm. We therefore decided not to include a TCA arm.
Choice of Antidepressants
The selection of the best candidate antidepressants for this trial is not straightforward. Cost and power considerations dictate that an optimal design should include two active antidepressant treatments and a placebo. There are however several cautions. One previous small RCT has indicated benefit with the tricyclic antidepressant clomipramine (Petracca et al 1996), but other data indicate marked side effects and exacerbation of disability associated with TCA treatment. For example, one study of the tricyclic antidepressant imipramine, indicated that active treatment increased cognitive impairment and disability (Reifler et al 1989), whilst several studies of falls indicate that most antidepressants increase falls risk (e.g. Ensrud et al 2002). In addition, there have been recent safety concerns with SSRIs, particularly with respect to withdrawal effects and the potential risk of self harm (currently under review by the Committee for the Safety of Medicines).
Within this framework, the choice of specific antidepressant agents requires careful consideration. For example, the best evidence of efficacy in people with dementia is for the SSRI sertraline since that was the compound used in the Lyketsos et al (2003) RCT. But this was a very small trial and other SSRIs such as citalopram have also been reported to be effective in treating depression in later life including those with dementia but in less well designed studies (Nyth et al, 1992). Citalopram may have less interactions with other drugs than other SSRIs and people with dementia are usually recipients of polypharmacy. The most effective antidepressant in people without dementia is probably venlafaxine (Stahl et al 2002), but there are no RCTs in people with dementia and there are potential concerns regarding side effects in these individuals (Oslin et al 2003). A newer antidepressant, mirtazapine, has a good safety profile and is widely used in clinical practice to treat depression in people with dementia, but has not been evaluated in a RCT for this indication.
In order to design and cost a trial of this sort there is a need to identify the compounds to be tested. We have therefore made the decision that our working trial design should include sertraline (the SSRI with the best evidence and which will be off licence by the end of the trial) and mirtazapine (the novel antidepressant with the least safety concerns). The doses chosen reflect common clinical practice for the treatment of depression in dementia and (in the case of sertraline) direct trial evidence (Lyketsos et al 2003), with higher doses than those suggested here (i.e. over 150mg of sertraline or 45mg of mirtazapine) being seen as less appropriate in people with dementia as well as depression.
Controls – Use of Placebo
The research brief referred to comparison with standard care. Standard care for depression in dementia is generally the provision of antidepressants with SSRIs the most commonly used drugs (Doody et al, 2001). Standard secondary care is however much more than just medication. It involves a detailed multidisciplinary assessment of the person with dementia and their family carers with the generation of an individualised care package for each, often with continuing monitoring and follow-up (Banerjee 2001). We have therefore developed a study design whereby all participants receive full standard care with only the antidepressant element subject to investigation against placebo and between classes of compound.
Currently there is little convincing evidence that anti-depressant treatments are more effective than placebo in treating depression in dementia in real-world clinical practice. As discussed above, the data available are generally from small-scale studies of highly selected groups of patients with depression in dementia. The research brief requires a trial which can take the evidence base and clinical practice forward significantly. In these circumstances a placebo group is not just ethical, but probably essential. If antidepressants are indeed not effective, then the placebo group may do better as they should have fewer adverse events. The 1:1:1 randomisation results in a third of the participants receiving placebo. We carried out a further consultation exercise on the acceptability of the inclusion of a placebo group with local people with dementia, family carers and clinicians. They were supportive of the strategy of using placebo in these circumstances as long as its use was minimised and that the information derived from the trial would yield a definitive answer.
Run-in Period
One possible element of a trial such as this is the inclusion of a run in period. The potential value of this is to identify a group of people more likely to comply with subsequent data collection (i.e. to minimise loss to follow-up) and to identify a group of people with depression who are less likely to spontaneously recover (Ballard et al 1996c, Ballard et al 2001a,b). It is also possible that depression scores may be reduced by psychosocial interventions (Teri et al 2003), some of which may be provided as part of routine care. The result of these factors is a potentially high placebo response rate in clinical trials. The research brief was clear in its call for an evaluation of antidepressants in routine clinical practice and it is not routine clinical practice to precede the prescription of antidepressants for depression in dementia with a trial of a non-pharmacological treatment such as exercise. Instead we propose to include the clinically relevant inclusion criterion for the trial that the depression should have been present for at least 4 weeks.
The large sample size in this trial allows for the possibility of a high response in the placebo group. The placebo group also enables us to estimate the 13 and 39 week recovery rate with normal clinical care. We will be recruiting from a wide range of teams with heterogeneity in what constitutes “normal clinical care”. We will catch this variation by applying a typology of team intervention to identify those elements the team intervention offered and delivered as part of normal clinical care. We will then be able to complete secondary exploratory analyses to investigate the determinants of positive and negative outcome, controlling for the effect of antidepressants. Also we will have data from the Client Service Receipt Inventory (CSRI) on the services received by each patient so we can also include such “input” data into secondary analyses to test their influences on the outcomes.
4.5 Choice of Primary Outcomes
The outcome measures have been chosen on the basis of their being the best-validated instruments available for the domains of function and activity of prime importance. We have balanced comprehensiveness with minimising respondent burden. The interview schedule is based on other successful trials in dementia (e.g. MRC CALM-AD) and designed to be completed in one session with the person with dementia and their carer lasting no more than 60 minutes.
4.6 Risks and Benefits
4.6.1 Potential Risks
There are potential side effects of the medications but as these are being used within their licensing terms, the risks are well known.
Currently there is little convincing evidence that anti-depressant treatments are more effective than placebo in treating depression in dementia in real-world clinical practice. The data available are generally from small-scale studies of highly selected groups of patients with depression in dementia. The research brief required a trial which can take the evidence base and clinical practice forward significantly. In these circumstances a placebo group is not just ethical, but probably essential. If antidepressants are indeed not effective, then the placebo group may do better as they should have fewer adverse events. The 1:1:1 randomisation results in a third of the participants receiving placebo. We carried out a further consultation exercise on the acceptability of the inclusion of a placebo group with local people with dementia, family carers and clinicians. They were supportive of the strategy of using placebo in these circumstances as long as its use was minimised and that the information derived from the trial would yield a definitive answer.
The research assessments can take a considerable amount of time, but will take place in the participants' homes to minimise inconvenience.
The placebo group will have untreated depression for the duration of the trial but this is justified in section 4.4 and all participants will be closely monitored and can withdraw at any time.
4.6.2 Potential Benefits
Participants will potentially benefit from an improvement in their symptoms.
5 TRIAL OBJECTIVES AND PURPOSE
5.1 Aims
To conduct a multicentre double-blind placebo-controlled RCT of the clinical and cost- effectiveness of two classes of antidepressants, and more specifically mirtazapine and sertraline, at 3 months (13 weeks) and 9 months (39 weeks) post randomisation. The primary outcome will be the 13 week outcome with assessment of long term outcome at 39 weeks.
5.2 Objectives
5.2.1 Primary Objectives
5.2.1.1
To determine the clinical and cost effectiveness of two classes of antidepressants for depression in dementia (compared with placebo).
-
To determine whether a Selective Serotonin Reuptake Inhibitor (SSRI, sertraline) is (i) more clinically effective and (ii) more cost-effective than placebo in reducing Cornell depression score 13 weeks post randomisation.
-
To determine whether a Noradrenaline and Selective Serotonin Antidepressant (NASSA, mirtazapine) is (i) more clinically effective and (ii) more cost-effective than placebo in reducing Cornell Depression score 13 weeks post-randomisation.
5.2.2 Secondary Objectives
5.2.2.1
To investigate differences in the clinical and cost effectiveness, and in terms of adverse events, withdrawals from treatment and adherence to treatment between mirtazapine and sertraline for depression in dementia at 13 and 39 weeks post-randomisation.
5.2.2.2
To investigate differences in the clinical and cost effectiveness of mirtazapine/sertraline and placebo on patient (e.g. quality of life, cognition) and family carer (e.g. carer burden, carer quality of life) outcomes at 13 and 39 weeks post-randomisation.
5.2.2.3
To determine the influence on clinical effectiveness and cost-effectiveness of clinical characteristics of importance including: dementia severity, dementia type, depression type, depression severity, care arrangements, neuropsychiatric symptoms, and physical illness.
-
To investigate what baseline factors (other than randomised treatment) predict a reduction in Cornell Depression Score at i) 13 weeks and ii) 39 weeks.
-
To investigate whether there are any differential predictors of response to the antidepressants (both vs placebo) (i.e. treatment-covariate interactions).
6 TRIAL DESIGN
6.1 Description of Overall Trial Design and Plan
We propose to conduct a multicentre double-blind placebo-controlled RCT of the clinical effectiveness and cost-effectiveness of two classes of antidepressants, and more specifically mirtazapine and sertraline, at 3 months (13 weeks) and 9 months (39 weeks) post randomisation. The primary outcome will be 13 week outcome with assessment of long term outcome at 39 weeks.
6.2 Schematic Trial Flow Diagram
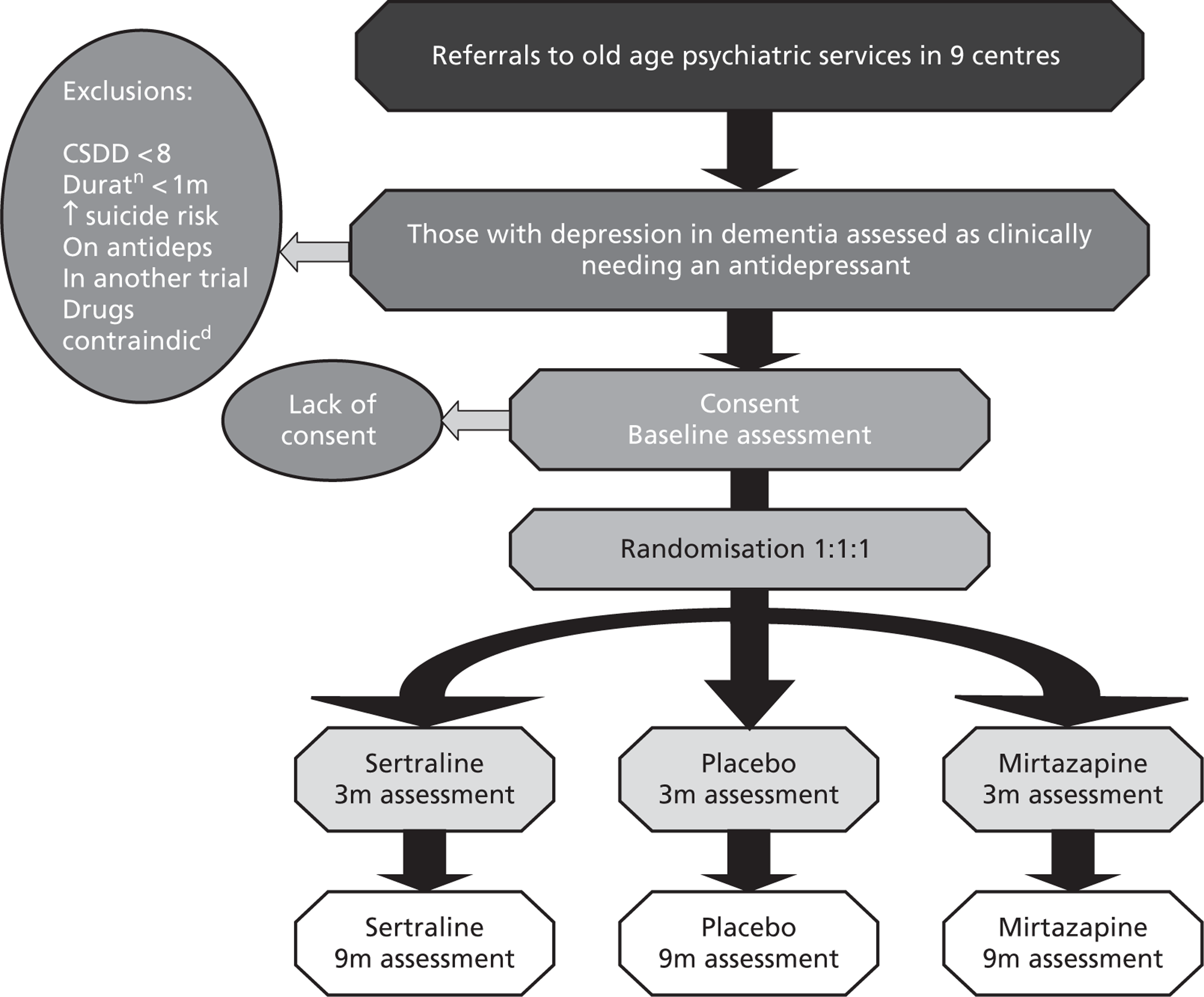
6.3 Trial Duration
6.3.1 Duration of the treatment period
Ten months. Nine months defined as 39 calendar weeks post randomisation to final follow up plus one month further randomised treatment to allow for clinical transfer of care and database closure prior to routine unblinding.
6.3.2 Duration of the follow-up period
Short-term outcomes will be ascertained at 3 months (13 calendar weeks), long term outcomes will be ascertained at 9 months (39 calendar weeks) post randomisation. Safety outcomes will also be collected at 10 months, as participants come off the trial medications. Any ongoing serious adverse events will be tracked until closed.
6.3.3 Definition of completion of the trial for an individual participant
Completion of 10 months on the trial medication or withdrawal from follow-up for any cause before. Participants may withdraw from the trial medication but remain in follow-up. Participants may not formally withdraw from follow-up and remain on the trial medication.
6.3.4 Definition of the end of the trial
In ethics and regulatory terms, the end of the trial is defined as the end of data collection i.e. 10 months after the randomisation of the last patient into the trial (to allow for the collection of adverse events and concomitant medications until all patients have stopped taking the trial medication). In terms of the funder, the end of the trial is defined as the provision of the final report to the HTA.
6.4 Overview of Data Recording and Case Report Forms
An overview of data recording and the content of case report forms is given below in table 6.4.1 (research assessments by timepoint) and table 6.4.2 (other trial forms by timepoint).
| Informant | Screening | Baseline & randomisation | Wk 13 | Wk 39 | Treatment discontinn | Trial drop out | |
|---|---|---|---|---|---|---|---|
| Consent to pre-trial | Patient/Carer | ✗ | |||||
| Eligibility assessment | Doctor/RW | ✗ | |||||
| Informed consent | Patient/Carer | ✗ | |||||
| NINCSD-ADRDA (for dementia) | Doctor | ✗ | |||||
| Modifed Hachinski Ischemic Scale (MHIS; vascularity) | Carer | ✗ | |||||
| DSM-IV (for Depression) | Carer | ✗ | ✗ | ✗ | |||
| Olin (Depression in Dementia) | Carer | ✗ | ✗ | ✗ | |||
| Cornell Scale for Depression in Dementia (CSDD) | Patient/Carer | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Demographics – participant | Carer | ✗ | |||||
| Demographics – carer | Carer | ✗ | |||||
| Client Service Receipt Inventory (CSRI) | Carer | ✗ | ✗ | ✗ | |||
| DEMQOL | Patient | ✗ | ✗ | ✗ | |||
| DEMQOL-Proxy | Carer | ✗ | ✗ | ✗ | |||
| EQ5D (Patient) | Patient | ✗ | ✗ | ✗ | |||
| EQ5D (Proxy) | Carer | ✗ | ✗ | ✗ | |||
| SF-12 v2 (Carer) | Carer | ✗ | ✗ | ✗ | |||
| Mini-Mental State Examination (MMSE) | Patient | ✗ | ✗ | ✗ | |||
| GHQ-12 (Carer) | Carer | ✗ | ✗ | ✗ | |||
| Zarit Carer Burden Scale (ZCBS) | Carer | ✗ | ✗ | ✗ | |||
| Neuropsychiatric Inventory (NPI) | Carer | ✗ | ✗ | ✗ | |||
| Bristol Activities of Daily Living (BADL) | Carer | ✗ | ✗ | ✗ | |||
| Carer global impression of change | Carer | ✗ | ✗ | ||||
| Medical history | Carer | ✗ | |||||
| Adverse Events Checklist | Carer | ✗ | ✗ | ✗ | ✗ | ||
| Adverse Events Log | Carer | ✗ | ✗ | ✗ | ✗ | ||
| Pill Count | RW | ✗ | ✗ | ✗ | ✗ | ||
| Medication Preference | ✗ | ||||||
| Treatment Guess | Carer/RW | ✗ | ✗ | ✗ | ✗ | ||
| Concomitant Medications | Carer | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Concomitant Treatments | Carer | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Trial Treatment Log | Carer | ✗ | ✗ | ✗ | ✗ |
| Informant | Screening | Baseline & randomisation | Wk 13 | Wk 39 | Treatment discontinn | Trial drop out | |
|---|---|---|---|---|---|---|---|
| Registration form | RW | ✗ | |||||
| Exclusion Form | RW | ✗ | |||||
| Randomisation Request Form | RW | ✗ | |||||
| Serious Adverse Event Report Form | RW/Doctor | ✗ | ✗ | ✗ | ✗ | ||
| Withdrawal Form | RW/PI | ✗ | ✗ | ✗ | ✗ | ||
| Unblinding Request Form | RW/PI | ✗ | ✗ | ✗ | ✗ |
6.5 Research Setting
Participants will be drawn from secondary care as stipulated in the research brief. These will be referrals to and other contacts with old age psychiatric services and memory clinics in 9 regional sites each covering a catchment area of at least 100,000 older people each (Birmingham, Cambridge, Leicester, Liverpool, Manchester, Newcastle, North London, Southampton, South London/Kent).
The applicants are at the centre of networks of old age psychiatric and memory services in their regions. The study has been adopted by the DH-funded Mental Health Research Network (MHRN) and will benefit from its resources in facilitating trial approvals and recruitment in the study sites. Support will also be sought from the emergent Dementia and Neurodegenerative Disease Research Network (DeNDRoN).
7 SELECTION AND WITHDRAWAL OF PARTICIPANTS
7.1 Number and Source of Participants
General Issues
In order to succeed this trial needs to recruit a large number of people with depression in dementia in a relatively short period of time (one year) and then follow them up over 9 months. Also, given that this is an effectiveness trial, these participants need to be representative of all people with depression in dementia presenting to secondary care (as stipulated in the research brief). The need to recruit quickly and broadly requires a multicentre approach. However, both criteria also require that our participants are not simply drawn from highly specialist research centres first because of the need to maximise generalisability but also because the number needed could not be generated to time by existing research facilities (e.g. university memory clinics).
A final cardinal design issue consequent to this is that the recruitment of participants will require the active and prolonged collaboration of numerous old age psychiatrists and their teams. For this to work requires as little a burden on these teams as possible. After considerable consultation, drawing on the experience of other successful trials in dementia (e.g. MRC CALM-AD) and the comments of reviewers and the Commissioning Board, we have designed a robust multicentre recruitment and follow-up strategy which will interfere as little possible with routine clinical care.
Establishing the Multi-Site Recruitment Frame
Our participants will be drawn from referrals to and other contacts with old age psychiatric services in England; these will include community mental-health teams and their associated memory clinics. Each centre has well developed successful research links with a network of such local service providers. The local PIs in each university centres will establish and co-ordinate a local network of service providers in their area participating in this trial. Old age psychiatric services are provided on “catchment area” basis with individual consultants and their teams responsible for a geographically defined area. These catchment areas are typically described in terms of the numbers of older people (i.e. over 65) falling within the area and so the responsibility of the consultant and team. The size of these catchment area varies from 7,000 to 20,000 older people per full time consultant.
Each local PI will establish a local network for the trial covering at least 100,000 older people. Depending on local configuration of teams and trusts this will represent the catchment areas of 7-14 community mental health for older adults teams provided by 2 to 6 NHS Trusts. This creates exactly equivalent areas for recruitment in each centre, enabling equal recruitment from each site and so requiring equal resource for recruitment in each area. In addition to this a further “reserve list” of potential local teams will be identified covering a further 50,000 older adults to enable substitution or addition of teams if services withdraw from the study or if recruitment fails to meet target levels.
Planned Recruitment Rate and Feasibility
There are nine centres each expected to recruit 57 patients. One RW is employed at each site, who will assess patients referred to the trial from clinical old age psychiatric services.
Recruitment will be pursued through all psychiatric services, particularly focusing on new referrals to outpatient clinics, community teams and memory clinics, but also screening other secondary contacts including care homes. It was originally anticipated that a catchment area of 100,000 would yield at least 100 referrals of people with dementia per month, and that on a conservative estimate 20% of these would have depression, equating to 240 potential cases per centre per year.
Initially the recruitment period was 12 months, but as the target transpired as rather more difficult to achieve than anticipated, partly because many patients have already been prescribed anti-depressants by GPs prior to referral, an extension to the recruitment period was sought. The HTA agreed to this request on 15th May 2008. The recruitment period is 30 months. The HTA have agreed to a minimum recruitment target of 1.3 per site per month, although sites will continue to aim for 2 recruits per month. Recruitment rates of 1.3 and 1.5 per site per month would generate 351 and 405 participants respectively. Recruitment will be monitored formally on a monthly basis centrally and if any site fails to achieve its minimum recruitment target, extension funding may be transferred to another site. Sites have been allocated extension funding beyond the original period to employ a RW on a half-time basis. Note that the Leicester site will not receive extension funding, the allocation has gone to Birmingham which has recently expanded to trusts across the West Midlands.
7.2 Recruitment Strategies
We will employ a single local RW in each site to carry out all study-related work. This will include publicising the trial and maintaining awareness, but the major role of the local RW will be to carry out recruitment and follow-up interviews.
Referring Investigators will identify cases meeting study criteria and will document in their medical notes that they have obtained verbal consent for the RW to contact cases to discuss the study and obtain written consent to the trial. We will recruit those in whom a secondary care doctor makes a clinical diagnosis of mild to moderate probable or possible Alzheimer's Disease (MMSE>8) and a co-existing depressive illness likely to need treatment with antidepressants with a duration over 4 weeks as detailed below. The RW will actively promote the study with the participating Referring Investigators to help maximise referrals into the study.
When a case is identified the RW will then assess the patient within one week at a place of the patient and carer's choosing. Our experience suggests that this will most commonly be the person with dementia's household rather than a clinic or GP surgery. This is a function of the age and frailty of the population under study. This accords with normal old age psychiatric practice where home assessment and delivery of care is the norm. The RW will extract data from the participants' NHS notes in order to minimise duplication. The assessment interview will ascertain type of dementia and depression according to set diagnostic criteria: NINCDS-ADRDA [McKhann et al 1984] for dementia; DSM-IV for depression (American Psychiatric Association 1994); the Olin criteria specifically designed for depression in dementia (Olin et al 2002); and depression severity (CSDD). The purpose of this diagnostic work is not to exclude further individuals from the study (this would limit the generalisability of the findings) but instead to closely characterise the cases on the basis of diagnoses and severity to enable us to be able to describe the study group in detail and to be able to investigate as secondary analyses the effect of diagnostic group and severity on subsequent outcome.
The local RW will complete a semi-structured interview with the person with dementia and their main carer. This interview will include the primary and secondary outcome measures (please see below) and possible moderating variables including behavioural and psychological disturbance (Neuropsychiatric Inventory, NPI, Cummings et al, 1994]), physical illness, and severity of cognitive impairment (MMSE Folstein et al 1975).
In industry efficacy studies recruitment is often managed by the use of payment by case recruited. In this existing resource (often the highly selected participating consultant or worker in a specialised clinic) is used to carry out the trial assessments. This is not possible within this trial since there are no local research resources that could be used in this way to carry out the detailed and systematic assessments required at baseline and follow-up. It would not be feasible to expect the wide range of local consultants needed in this effectiveness study to complete these assessments and it would be very difficult to control and assure data quality.
We will work closely with the MHRN as a local and national partner. We have discussed their possible role. They will play a vital role in expediting local R&D approvals and ethical approvals. They will promote the study within the mental health trusts they cover and will help with recruitment monitoring and problem-solving if needed. What they are unable to provide is direct help with recruitment or individuals to carry our assessments and recruit to the study. This is not their role. The DeNDRoN will be setting up through the life of the trial but will also have no resource to help directly recruit to the study.
If payment by case is not possible then specific resource needs to be made available in each recruiting site. We estimate that the minimum level of staffing needed to complete these tasks is 1.0WTE (whole time equivalent) RW in each site. The rationale for the equality of provision over the nine sites is that the work demanded is equal over the nine sites.
Monitoring and Ensuring Recruitment to the Trial
Recruitment will be monitored by the TMG, the TSC and the DMC as well as the MHRN. The intention will be to identify problems early and problem solve to bring recruitment back on track. We propose that centres are given 6 months funding for a full time RW in the first instance with continuation of funding depending on satisfactory recruitment. If recruitment is low then only 0.5WTE will be continued in that site and the resource freed (i.e. funding rather than a person) will be used to extend or bolster a centre with effective recruitment although we hope that this will not be necessary.
The local recruitment frames are the same size and there will be careful monitoring and support to maximise recruitment. We believe that all these factors will minimise the likelihood of failure to recruit in individual centres and overall.
7.3 Consent Procedures
The main potential ethical issue in this study is that dementia itself may interfere with an individual's ability to give their informed consent, especially in more severe stages of the illness. Carers and people with dementia have contributed to finalising the information sheets and consent forms. We estimate on the basis of previous experience that less than 20% of potential participants will lack capacity to give informed consent.
The issue of informed consent in people with Alzheimer's disease is complex. Full informed consent will be obtained where possible. If the person with dementia does not have the capacity to consent, then the next of kin or primary carer of the patient will be asked to act as the personal legal representative to the person being enrolled in the trial. This person would also be expected to act as caregiver informant on the study. We will rely on them to use their previous knowledge of the individual in terms of any stated preference for research, to assess whether they would have agreed to take part if they had capacity.
The study RWs will be trained in issues of obtaining consent by the local Recruiting PI and will only be delegated to undertake this task if their skills in this area are satisfactory. The Referring Investigator will obtain verbal consent for the potential participant to be approached by the RW and will document this in their medical notes. The RW will telephone the potential participant and their caregiver to confirm their agreement to be approached and to arrange a screening visit appointment. The RW will send them each a pack containing all of the following documents to read and consider prior to the screening visit:
-
‘Information and Consent Form for Patient (full version)’
-
‘Information and Assent Form for Patient (shortened version)’
-
‘Information Sheet and Consent Forms for Carer’.
At the screening visit, if the patient has capacity to consent, they will be asked to read and sign an ‘Information and Consent Form for Patient (full version)’. If they lack capacity, they will be given an ‘Information and Assent Form for Patient (shortened version)’ and if possible they will sign the form to indicate their assent. If this is not possible and they can only give verbal assent, the caregiver will be asked to sign the form to witness the patient's verbal assent.
The caregiver will be asked to read the ‘Information and Consent Forms for Carer’. Within this document there are two consent forms. As data will be collected directly from carers about their experiences and health status, a separate consent form will be signed by the carer to cover this data. Therefore if the patient has capacity to consent, the caregiver will be asked to sign the ‘Carer Consent for Carer Participation’ form only. However, if the participant lacks capacity and has only given been able to give their assent to participate, the carer must also sign the ‘Carer Consent for Patient Participation’ form.
In practical terms, when the participant is approached to be interviewed or to take the study medication, that individual will be able to indicate whether he or she wishes to be interviewed or take the medication. The interviews and recruitment will be completed only if there is no sign of distress in the person with dementia. This is an approach that has been used successfully in trials and other descriptive and evaluative studies.
The study RW will discuss the study in detail with participants and carers and will obtain consent as described above. Participants will be given as long as they wish to consider participating before the end of the recruitment phase, but a minimum of 24 hours. It is expected that it will normally take at least a week between the initial approach by the Referring Investigator and the taking of consent by the RW.
7.4 Eligibility Criteria
7.4.1 Inclusion Criteria
We have designed this study as a pragmatic trial of effectiveness in routine clinical practice. We wish to minimise exclusions from the study in order to maximise the generalisability of the data generated.
The criteria for inclusion are set to be as close to clinical practice as possible. For this reason we do not specify the use of anything other than clinical diagnoses of dementia and depression since standardised instruments (other than the MMSE as a measure of severity) are not used in routine practice. A detailed characterisation of cases using standardised tools will be completed at the research assessment. We will recruit those in whom a secondary care doctor makes at the point of referral to the RW:
-
a clinical diagnosis of mild to moderate probable or possible Alzheimer's Disease,
-
a co-existing depressive illness likely to need treatment with antidepressants, and
-
that depression should have a duration of more than four weeks.
7.4.2 Exclusion Criteria
Again we wish to minimise exclusions. We will exclude from the trial those in whom a secondary care doctor finds at the point of referral to the RW are:
-
currently taking antidepressants;
-
those with severe dementia (defined as the participant being unable to contribute to theCSDD);
-
the case is considered as being too critical to be randomised (e.g. because of suicide risk);
-
displays absolute contraindications to one or more of the trial treatments;
-
they are on another trial; and
-
those where there is no identifiable family carer or other informant (e.g. a formal/professional carer who spends sufficient time with the person with dementia to be able to give an informed opinion) to give collateral information.
We will further exclude from the trial those in whom the RW finds have:
-
a Cornell score < 8 at the point of randomisation.
The impact of these exclusions is likely to be small with our estimate that around 10% would be excluded by reason of severity and 10% by reason of lack of identified carer. The carer exclusion is needed because our primary outcome measure, the Cornell, is a carer report instrument. However, we will not require carers to be co-resident or to be providing hands-on care (many will see themselves as supporters or simply family members rather than carers per se), also information can be obtained by friends and neighbours or professional carers who take on a caring or support role.
Given our intention to ensure that the trial follows routine clinical practice as closely as possible, we would seek to recruit patients for whom switching of anti-depressants has been deemed necessary by their referring clinicians, after allowing an existing therapy a reasonable chance to work. Timing of trial initiation would be determined by normal clinical practice for the initiation of sertraline or mirtazapine. An existing regimen may continue up to the point of referral, depending on the drug and prescribing guidelines. Patients must not be taking another anti- depressant while participating in the trial.
We are not intending to exclude participants on the basis of their taking concomitant psychotropic medication e.g. hypnotics, antipsychotics or cholinesterase inhibitors. These medications will be commonly prescribed in our study group and any such exclusions would limit the generalisability of the data generated, so compromising the pragmatic nature of the trial. Management of the participants in this study will therefore mimic true clinical practice with the sole exception of the trial medication.
The Referring Investigator will refer potentially eligible patients to the Recruiting PI for eligibility assessment. The RW will review the case notes for eligibility criteria. The RW will contact the patient and carer by phone to ask if they would like an appointment to be considered for the trial and if the carer is, in principle, willing to participate as the participant's informant. An ‘Information and Consent Form for Patient (Full version)’, an ‘Information and Assent Form for Patient (Shortened version)’ and an ‘Information and Consent Forms for Carer’ should then be posted to both the potential participant and the carer. Patients cannot participate in the trial without a carer informant to complete the assessments. After screening, the RW will review the patient according to the eligibility criteria above and will then discuss and review the eligibility criteria and case notes for each participant that appears to meet eligibility criteria with the Recruiting PI. This discussion will be documented and signed by the Recruiting PI prior to randomisation.
7.5 Screening/Baseline Procedures
7.5.1 Time Periods
Referrals to the trial will be randomised within a maximum of 28 days of having been seen by the Referring Investigator, although it is expected that they will be randomised within 14 days of having been seen by the Referring Investigator.
In order to ensure study medication availability and to ensure statistical credibility, follow-up interviews must be completed within strict timelines.
The week 4 phone contact must be completed between 21 and 28 days from treatment start date in order to decide whether to dose increase at Day 28. The week 8 contact, if needed, should be completed between 49 and 56 days from treatment start date.
The 13 week (3 calendar month) and 39 week (9 calendar month) assessments must be completed at those timepoints +/- 7 days from randomization
7.5.2 Informed Consent for Eligibility/Baseline Assessment
A log will be kept by the RWs of everyone referred to them and their path through the trial for the purposes of the CONSORT diagram (see Appendix 5). Research workers will be assisted by MHRN clinical study officers (CSOs) in identifying and recruiting participants to the study. RWs and CSOs will work together to decide how trial related activities are shared within their area. Where RW is specified in this section, both the RW and CSO are included.
Referring Investigators will identify cases meeting study criteria and will obtain verbal consent to refer to the Recruiting PI. The referral letter will be copied to the RW. The Recruiting PI and the RW will work together to recruit the patient to the trial. We will recruit those who meet the eligibility criteria as defined in Section 7.4 above. The RW will actively promote the study with the Referring Investigators to help maximise referrals into the study.
Once a referral is received, the RW will then telephone the potential participant and their carer to arrange an appointment and to inform them that a long patient information sheet, a short patient information sheet and a carer information sheet will be posted to the potential participant and participant's carer in advance of the appointment so that they have time to read them through and consider whether they wish to participate. The RW should explain that there is a long and a short patient information sheet so that on a case by case basis the patient and/or carer can decide which is more appropriate for each patient.
The RW will assess the patient within 28 days of receiving the referral letter at a place of the patient and carer's choosing. Our experience suggests that this will most commonly be the person with dementia's household rather than a clinic or GP surgery. The assessment interview will ascertain type of dementia and depression according to set diagnostic criteria: NINCDS- ADRDA [McKhann et al 1984] for dementia; DSM-IV for depression (American Psychiatric Association 1994); the Olin criteria specifically designed for depression in dementia (Olin et al 2002); and depression severity (CSDD).
The local RW will complete a semi-structured interview with the person with dementia and their main carer. This interview will include the primary and secondary outcome measures (please see sections 10.3.1.1 and 10.3.1.2) and possible moderating variables including behavioural and psychological disturbance (Neuropsychiatric Inventory, NPI, Cummings et al, 1994]), physical illness, and severity of cognitive impairment (MMSE Folstein et al, 1975).
7.6 Randomisation and Enrolment Procedure
7.6.1 Method of Identification of Participants and Carers
The local RWs will assign Participant Identification Numbers (PINs) and Carer Identification Numbers (CINs) to each patient–carer dyad once they receive a referral letter from the Referring Investigator. The PIN will start with a “P” (to indicate that it refers to a patient), will be followed by a two-digit number to indicate the centre (Birmingham = 01; Cambridge = 02; Leicester = 03; Liverpool = 04; Manchester = 05; Newcastle = 06; North London = 07; Southampton = 08; and South London/ Kent = 09) and then a three-digit number indicating the number within the centre. The CIN will be formatted in the same manner except that it will start with a “C” (to indicate that it refers to a carer) and it will end with an ‘A’ to indicate that they are the original carer. These identification numbers will be unique to an individual and will remain with the patients and carers throughout the trial. New carers who may join during the trial (if, for example, the original carer becomes incapacitated) will be assigned their own unique CIN, which will be the same as the original carers CIN but with the final character of the CIN changing (A, B or C). Using this system, data management can see when a new informant carer has become involved. This is very important as it may have a significant impact on the assessments.
7.6.2 Method of Randomisation (inc. Allocation Concealment)
Patients will be allocated to placebo, mirtazapine or sertraline (ratio 1:1:1) by the Mental Health & Neurology Clinical Trials Unit (MH&N CTU) based at the Institute of Psychiatry. Allocation will be stratified by centre (Birmingham, Cambridge, Leicester, Liverpool, Manchester, Newcastle, North London, Southampton, South London/Kent) by stratified block randomisation with randomly varying block sizes.
7.6.3 Implementation Procedures
The medication will be sent to an independent company (Catalent, Bolton) for manufacture of placebo to mirtazapine and this company will handle the pre-packaging and labelling of all the study medications. The MH&N CTU (while keeping the Trial Statistician blind) will communicate the randomisation sequence to this company so that they can package the study medications accordingly. The study medications will then be sent to the relevant pharmacies (ensuring that the pharmacies remain blind). Once an eligible patient–carer dyad has completed the baseline assessment and provided written informed consent, a member of the local trial team will complete a “Randomisation Request Form” and contact the MH&N CTU via email, phone or fax to register the request. Once the MH&N CTU are happy that the patient is eligible and that minimal baseline data is available, they will notify the local pharmacy, the Recruiting PI and the RW within 24 hours of the request (Mondays to Fridays, 9am to 5pm, except bank holidays) which treatment pack number to dispense to the patient and copy this communication to the Trial Managers Office. The local pharmacy will then acknowledge to the MH&N CTU that they have received this information. The RW will ensure that this number is entered correctly on the trial specific prescription and signed by the Recruiting PI. When pharmacy receives the prescription they will cross check the prescription with the fax from the MH&N CTU to ensure there has been no error.
7.7 Withdrawal of Participants from the Trial
It is the aim of the trial to minimise withdrawal of participants from treatment and follow-up. Withdrawal may be initiated by the participant, their carer, the Recruiting PI or their Referring Investigator. Withdrawal from treatment is separated from withdrawal from follow-up assessments.
7.8 Loss to Follow-Up
We estimate a loss to follow up of 10% at 13 weeks and 20% at 39 weeks. One of the features of the natural history of dementia is that this is a substantial mortality associated with the disorder. We estimate there will be a 3% mortality at 13 weeks and a 9% mortality at 39 weeks with the rest of the loss to follow up contributed by refusal, loss of carer, or death of carer.
Loss of data by to follow up other than by death will be minimised by all means including the following: carrying out assessments at the subject's home; prioritising order of collection of follow-up data to safeguard primary endpoints; and using telephone interviewing of carers if necessary.
Loss to follow-up will be monitored locally and centrally. If a participant or carer is identified as potentially lost to follow-up, procedures will be put in place to avoid loss to follow-up and to obtain data. They will only be regarded as lost to follow-up following agreement with the Recruiting PI and the Trial Manager. The Patient and Carer Information Sheets will state explicitly that participants contact details and GP details will be collected centrally by the Trial Manager. In the event of loss to follow-up the Trial Manager will use this information to track participants via the NHS system.
8 TREATMENT OF PARTICIPANTS
8.1 Description of Randomised Treatments
8.1.1 Placebo
Matching placebo tablets will be manufactured for both the 15mg mirtazapine and the 50mg sertraline tablets. These will be identical to the active tablets in all respects.
8.1.2 Mirtazapine and Sertraline
The experimental interventions are:
-
5. mirtazapine (a NASSA) with normal clinical care
-
6. sertraline (an SSRI) with normal clinical care.
Interventions will be available in 15mg tablets for mirtazapine and 50mg tablets for sertraline.
8.1.3 Double-Dummy Design
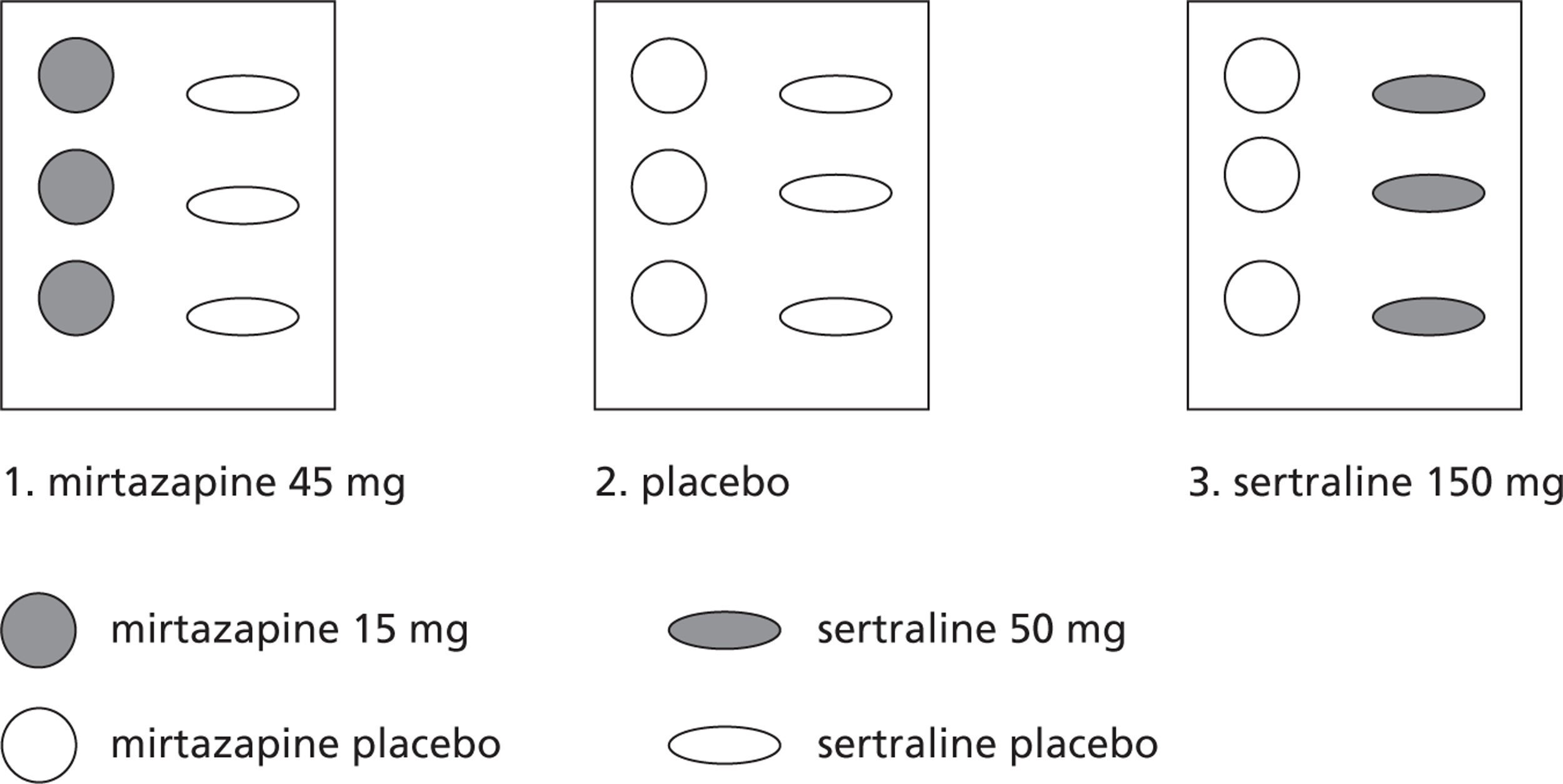
8.2 Selection of Doses for the Trial
These study medications are being used within their recommended doses for their licensed indication.
8.3 Selection & Timing of Dose for Each Participant
Interventions will be available in 15mg tablets for mirtazapine and 50mg tablets for sertraline. The design will be a double dummy with each participant taking:
-
sertraline plus placebo of mirtazapine, or
-
mirtazapine plus placebo of sertraline, or
-
placebo of mirtazapine plus placebo of sertraline.
For the first two weeks of treatment, participants will receive:
-
sertraline 50mg plus a placebo mirtazapine tablet
-
mirtazapine 15mg plus a placebo sertraline tablet
-
a placebo sertraline tablet and a placebo mirtazapine tablet.
For the second two weeks, participants will receive:
-
sertraline 100mg (2 tablets) plus two placebo mirtazapine tablets
-
mirtazapine 30mg (2 tablets) plus two placebo sertraline tablets
-
two placebo sertraline tablets and two placebo mirtazapine tablets.
At week 4 (indexed on treatment start date) carers will be contacted by telephone and the CSDD completed. Those who score less than 4 will remain on the above dose and those scoring 4 or more will move to the higher dose below. The carers of those who remain on the middle dose will be contacted by telephone and the CSDD completed in the same way after 8 weeks (defined as day 49 to 56) and if CSDD is 4 or more at this time they will be placed on the higher dose.
With the above exceptions, from week 4 until the end of the trial (nine months in total), participants will receive:
-
sertraline 150mg (3 tablets) plus three placebo mirtazapine tablets
-
mirtazapine 45mg (3 tablets) plus three placebo sertraline tablets
-
three placebo sertraline tablets and three placebo mirtazapine tablets.
Dose adjustments can be made by reducing back to 2 of each tablet daily or to 1 of each tablet daily in participants experiencing troublesome side effects.
8.4 Blinding of Investigational Medicinal Products
Active study medications and placebos for each will be identical.
Mirtazapine and matching placebo will be different to sertraline and matching placebo.
8.5 Identity & Supply of Investigational Medicinal Products
Mirtazapine – Genus Pharmaceuticals (from 1st November 2009 : Arrow Pharmaceuticals) Sertraline – Pfizer UK Ltd.
8.6 Packaging & Labelling of Investigational Medicinal Products
Active study medications and placebo will be bottled in pots of 100 tablets. One months supply will be one pot of each allocated treatment. Packaging and labelling will be completed in accordance with Good Manufacturing Practice (GMP) and GCP by Catalent in Bolton.
8.7 Prescription of Investigational Medicinal Products
A trial specific prescription will be designed for use by all centres. This will be completed by Recruiting PIs or other medically qualified doctors with a substantive or honorary contract with the recruiting NHS Trust and who have signed the ‘Recruiting Investigator site delegation of authority form’. If prescriptions are faxed to pharmacy in advance of collection by the RW, the original prescription with the Recruiting PIs signature (or another authorised doctor within the site) must be given to the pharmacist before study medication is dispensed.
RWs will fax the site delegation of authority forms to the Trial Manager whenever it is updated. From these delegation forms, the Trial Manager will create a list of authorised prescribers for the trial in that site. This list will be provided to the site pharmacy and the pharmacist will be instructed to only dispense if the medication is prescribed by an authorised person.
8.8 Dispensing & Distribution of Investigational Medicinal Products
Study medication will be distributed to the nine study site pharmacies by Catalent. Study medication receipt will be recorded in the study pharmacy file. A study medication dispensing and return log will be maintained by the site pharmacies. RWs will deliver the study medications to the participants.
Supplies of the study medications will be dispensed to the patient on a three monthly basis up to the nine month assessment, when they will be given a final one month supply of trial medication.
8.9 Administration of Investigational Medicinal Products
These will be taken by participants using their normal methods for medicines management in a single dose at night. They and their carers will be provided with written information from the RW detailing the dose to be taken.
8.10 Unused Study Medication & Study Medication Accountability
Used treatment packs will be obtained from the patient by visit. Pharmacy departments in each site will maintain a study medication dispensing and returns log, including date dispensed, batch number, expiry date, number of tablets dispensed, study medication return date and amount of study medication returned. In addition, the study specific prescriptions will be maintained in the pharmacy file for audit purposes. Study medications supplies will not be destroyed until the end of the trial analysis, they will be sent back to Catalent. The RW will count the medication returns and enter the information on the eCRF. The Trial Manager will cross check this information with the pharmacy records during site visits and re-count pill bottles where there is any discrepancy. The pharmacist or RW will then be asked to amend whichever record was incorrect.
8.11 Prior & Concomitant Interventions
All concomitant drug and non-drug interventions received will be recorded at baseline and followup assessment.
8.12 Departures from Randomised Treatment
8.12.1 Treatment Compliance/Adherence
This trial is a pragmatic trial and non-compliance and attempts to promote compliance are part of routine clinical practice. Our approach will essentially be to observe this and to compare compliance between the three study groups.
The treatment packs dispensed during the previous visit will be collected, inspected, and stored for inspection by the Trial Manager. Tablet counts will be completed with the number of capsules returned per bottle recorded in the case report forms. Reasons for significant instances of non- compliance with the dosing regimen will be recorded in the electronic case report form and source file. Details on how individuals receive their medication (e.g. self managing, prompted by carer, or given by carer) will also be noted at each interview. Carers will be asked to make a note of the dates of any occasions when patients missed their medication and the reason for missing it. The research worker will note this information during each visit.
8.12.2 Treatment Preference/Guess
The primary effectiveness outcome (CSDD) is a subjective outcome completed by the RW after an interview with the participant and the participant's carer. While the trial is double-blind, and therefore the participant, carer and RW are blind to treatment status, the success of this blinding will be evaluated by collecting data on medication preferences and guesses. The participant and the Recruiting PI must be in equipoise regarding the three trial interventions for the participant to be randomised into the trial. Carers will be asked at baseline to rate their preference for antidepressants versus nothing and mirtazapine versus sertraline. Carers and RWs will be asked at 13 and 39 weeks post randomisation to guess whether the participant was randomly prescribed an antidepressant versus placebo and mirtazapine versus sertraline.
8.12.3 Emergency Unblinding
Emergency code break envelopes will be distributed by Catalent to Guy's Toxicology Unit, where an emergency unblinding service will be available 24 hours a day. The pharmacy site files will contain an emergency unblinding SOP. In office hours site pharmacists will direct requests for unblinding to the Recruiting PI, who will contact the unblinding service if needed. Out of hours Guy's Toxicology Unit will be responsible for requests for emergency unblinding. Each participant or their carer will be given an emergency card to carry for the participant for the duration of the trial. Depending on the participants circumstances, the RW will work with the participant and carer to decide who the most appropriate person to carry the card is, or whether it should be kept in a specific location where carers can access it if needed (e.g. with the medication). In addition, the emergency unblinding number will be printed on the boxes of study medication.
The toxicology unit must notify the Trial Manager on the next working day of any requests for unblinding, whether they were unblinded or not. Only requests to unblind from a medical doctor will be accepted (e.g. A&E doctor, GP). The Trial Manager will inform the Recruiting PI of an unblinding where appropriate. If the participant has been unblinded, the Recruiting PI may not be informed of the treatment allocation unless that information is needed for the participant's medical care. The decision on whether to inform the Recruiting PI will be made by the CI in conjunction with the Trial Statistician.
Where possible participants will be advised to omit the study medication rather than unblind. Code break envelopes will be collected and reconciled by the Trial Manager at the end of the trial.
8.13 Modification of Trial Treatment
The design allows for modification of the dose of medication where there are concerns about side effects that have not remitted. Under these circumstances the dose can be decreased (to mirtazapine 30mg, sertraline 100mg or placebo or, in exceptional circumstances, to mirtazapine 15mg, sertraline 50mg or placebo).
8.14 Treatment at the End of the Trial
The arrangements for continued provision of the trial medication at the end of the trial will be made on an individual basis by the Referring Investigator or other clinician responsible for the patients care at the end of the trial. At the 39 week assessment participants will be given a further four weeks supply of their medication. Further prescriptions will be the responsibility of the Referring Investigator or any other clinician who has taken over the participants care during the study. That person will be informed of the treatment group to which the participant was randomised.
The Recruiting PI must inform the Referring Investigator or other clinician taking over the participant's ongoing care of their treatment allocation between month 9 and month 10, as no further blinded study medication will be available to the participant. Since the clinician will need time to review the patient clinically, make a decision on ongoing treatment for depression and have a prescription issued, it is vital that the system of data entry, review and database lock on a participant by participant basis happens promptly after their week 39 assessment, so that their treatment allocation can be revealed (see section 12.5).
Routine unblinding will be requested by the Trial Manager once the data has been monitored, using the ‘Unblinding Request Form’. This will be sent to the MH&N CTU who will then inform the Trial Manager of the treatment allocation.
9 ADVERSE EVENTS
9.1 Adverse Events
9.1.1 Adverse Events and Adverse Reactions
Adverse Event (AE)
An adverse event is any untoward medical occurrence in a participant to whom a medicinal product has been administered, including occurrences which are not necessarily caused by or related to that product.
Note: An adverse event can therefore be any unfavourable and unintended sign (including abnormal lab results), symptom or disease temporally associated with the use of the medicinal product, whether or not considered to be related to the medicinal product.
Adverse Reaction (AR)
An adverse reaction is any untoward and unintended response in a participant to an investigational medicinal product which is related to any dose administered to that participant.
Note: Any adverse event judged by either the reporting Investigator or the sponsor as having a reasonable causal relationship to an IMP qualifies as an AR; there is evidence or argument to suggest a causal relationship.
All adverse reactions are adverse events.
Unexpected Adverse Reaction
An unexpected adverse reaction is an adverse reaction, the nature and severity of which is not consistent with the applicable product information:
-
the summary of product characteristics for that product (for an approved investigational medicinal product) or
-
the Investigator's brochure (for an unapproved investigational product).
Note: Reports which add significant information on specificity or severity of a known, already documented serious adverse reaction constitute unexpected events. For example, when the outcome of an expected adverse reaction is not consistent with the relevant product information, the event may be considered unexpected.
9.1.2 Serious Adverse Events (SAEs)
An adverse event or adverse reaction is defined as serious if it:
-
results in death
-
is life-threatening1
-
requires hospitalisation
-
prolongs a current hospitalisation
-
results in persistent or significant disability or incapacity
-
consists of a congenital anomaly or birth defect
-
deliberate self harm
-
other (please specify). 2
1 Life threatening in the definition of an SAE or SAR refers to an event in which the participant was at risk of death at the time of the event; not an event that hypothetically might have caused death if it were more severe .
2 Medical judgement should be exercised in deciding whether other AEs may be considered serious because they jeopardize the patient or may require intervention to prevent one of the other outcomes. Examples include blood dyscrasias or convulsions that do not result in hospitalisation, or development of drug dependency or cancer.
9.1.3 Serious Adverse Reactions (SARs)
A suspected serious adverse reaction, the nature and severity of which is consistent with information about the IMP in question presented in either:
-
the summary of product characteristics for that product (in the case of a product with a marketing authorisation) or
-
the Investigator's brochure relating to the IMP in question (in the case of any other IMP).
9.1.4 Suspected Unexpected Serious Adverse Reactions (SUSARs)
A Suspected Unexpected Serious Adverse Reaction (SUSAR)
All adverse events that are suspected to be related to an investigational medicinal product and that are both unexpected and serious are considered to be SUSARs.
Not all adverse events are adverse reactions but all adverse reactions (including those that are unexpected) are adverse events.
9.1.5 Assessment of Severity and Causality
Each AE should be evaluated for seriousness, causality, expectedness and intensity. This evaluation may be performed by both the Recruiting PI and the Sponsor (or CI acting on behalf of the Sponsor). In this trial, the Recruiting PI will assess an event for seriousness, causality and intensity and the CI will assess for expectedness.
Intensity (severity)
The assessment of intensity will be based on the Investigator's clinical judgement using the following definitions:
-
Mild: An event that is easily tolerated by the participant, causing minimal discomfort and not interfering with everyday activities.
-
Moderate: An event that is sufficiently discomforting to interfere with normal everyday activities.
-
Severe: An event that prevents normal everyday activities.
Note: Severity is often used to describe the intensity of a specific event. This is not the same as ‘seriousness’, which is based on participant/event outcome or action criteria.
Seriousness
An adverse event, adverse reaction or is defined as serious if it:
-
results in death
-
is life-threatening1
-
requires hospitalisation
-
prolongs a current hospitalisation
-
results in persistent or significant disability or incapacity
-
consists of a congenital anomaly or birth defect
-
deliberate self harm
-
other (please specify). 2
1 Life threatening in the definition of an SAE or SAR refers to an event in which the participant was at risk of death at the time of the event; not an event that hypothetically might have caused death if it were more severe .
2 Medical judgement should be exercised in deciding whether other AEs may be considered serious because they jeopardize the patient or may require intervention to prevent one of the other outcomes. Examples include blood dyscrasias or convulsions that do not result in hospitalisation, or development of drug dependency or cancer.
Causality
The relationship between the study medication and the occurrence of each adverse event will be assessed and categorised (as detailed below). The Investigator will use clinical judgement to determine the relationship. Alternative causes, such as natural history of the underlying diseases, concomitant therapy, other risk factors etc. will also be considered. The Investigator will also consult the SmPC or other product information.
-
Not related Temporal relationship of the onset of the event, relative to administration of the product, is not reasonable or another cause can by itself explain the occurrence of the event.
-
Remote Temporal relationship of the onset of the event, relative to administration of the product, is likely to have another cause which can by itself explain the occurrence of the event.
-
*Possibly related Temporal relationship of the onset of the event, relative to administration of the product, is reasonable but the event could have been due to another, equally likely cause.
-
*Probably related Temporal relationship of the onset of the event, relative to administration of the product, is reasonable and the event is more likely explained by the product than any other cause.
-
*Definitely related Temporal relationship of the onset of the event, relative to administration of the product, is reasonable and there is no other cause to explain the event, or a re-challenge (if feasible) is positive.
*Where an event is assessed as possibly, probably, or definitely related , the event is an adverse reaction .
Expectedness
The expectedness of an adverse reaction shall be determined according to the summary of product characteristics (SmPC).
-
Expected Reaction previously identified and described in protocol and/or reference documents, e.g. SmPC.
-
Unexpected Reaction not previously described in the protocol or reference documents.
NB Adverse reactions must also be considered as unexpected if they add significant information on the specificity or severity of an expected adverse reaction.
It is most appropriate for the Recruiting PI at each centre to evaluate each event before reporting it to the Sponsor (or CI acting on behalf of the Sponsor). The Recruiting PI's causality assessment should not be downgraded by the Sponsor (or CI acting on behalf of the Sponsor).
If a Sponsor (or CI acting on behalf of the Sponsor) disagrees with the Recruiting PI's assessment, further clarification and discussion should take place to reach a consensus. If a consensus cannot be reached, both the opinion of the Recruiting PI and the Sponsor (or CI acting on behalf of the Sponsor) should be provided if the report requires expedited reporting to the MHRA and REC.
9.1.6 Reporting Adverse Events
All adverse events occurring in the trial will be recorded in the participant's source data worksheet and filed in their medical records at the end of the trial. They will also be transcribed onto the electronic Case Record Form (eCRF). Data on adverse events will be collected by the RW from participants and their carers at weeks 4, 13 and 39. Any events reported by participants or their clinical teams will also be reported and followed up between visits. The RW will then review the adverse events immediately to ascertain whether they meet the criteria for ‘serious’ (see section 9.1.2). If the event is assessed as being an SAE, see section 9.1.7. For events that are not defined as serious, the RW will review the events for each participant with the Recruiting PI, prior to completing the eCRF, in order to assess and record intensity and causality of the event. The intensity, causality and seriousness of each event will be recorded on the eCRF and can be amended if new information about the event later emerges. All adverse events will be monitored until resolution or until month 10. At month 10, the Recruiting PI should formally write to the clinician assuming responsibility for the participants' ongoing clinical management, informing him or her of any ongoing unresolved adverse events.
Please note, when the RW meets with the Recruiting PI to review the non-serious adverse events, the Recruiting PI may decide at this point that an event should have been considered an SAE, based on their clinical judgement and the event would at that point be reported as an SAE.
It is expected that the RW would only be expected to assess events as being serious if they result in death, are immediately life threatening, require hospitalisation or a prolonged hospitalisation. Medical judgement should be exercised in deciding whether other AEs may be considered serious because they jeopardize the patient or may require intervention to prevent one of the other outcomes. Examples include blood dyscrasias or convulsions that do not result in hospitalisation, or development of drug dependency or cancer.
9.1.7 Reporting SAEs and SARs
King's College London (Institute of Psychiatry), as sponsor, have delegated the delivery of the sponsor's responsibility for pharmacovigilance, as defined in Regulation 5 of the Medicines for Human Use (Clinical Trials) Regulations 2004 to the Joint Clinical Trials Office (JCTO).
If an event is assessed as ‘serious’ (see 9.1.2), the RW and Recruiting PI will complete an SAE form and enter it onto the eCRF within 24 hours of becoming aware of the event. The SAE report form should be signed electronically by the Recruiting PI or a doctor within the research team delegated to undertake this task. In the absence of the Recruiting PI or another trial doctor being available to assess an adverse event that qualifies as an SAE, the RW should complete the eCRF form with as much information as possible. The Recruiting PI should then review the event at the earliest opportunity, make changes to the assessments as appropriate on the eCRF. It will then enter the pharmacovigilance system as a follow-up report. The RW should assess causality to the best of their ability but should seek assistance from the Trial Manager if necessary, in order that the medical advisor to the trial can assist if the Recruiting PI is unavailable. Any SAE received by the CI will be assumed to be definitely study medication related if no causality assessment is completed and may then enter the SUSAR reporting system.
The Trial Manager will enter a unique number on the eCRF to identify each SAE and will issue queries on the eCRF to collect follow up information until event resolution
Every event (new and follow up) received by the CI on the eCRF must be reviewed within 24 hours. The eCRF will trigger an email to the Recruiting PI, the CI and the Trial Manager informing them that an SAE has been entered or altered on the eCRF.
The CI (or a doctor nominated by the CI) must review every event within 1 working day of it being received. The review will consist of a review of the seriousness, causality and intensity. If there is disagreement about the assessments, there should be a discussion between the Recruiting PI and the CI to resolve the discrepancy and any changes sent added to the eCRF and signed off by the Recruiting PI. The CI, acting on behalf of the sponsor, is at liberty to upgrade the intensity or causality of an event without the Recruiting PIs agreement, but may not downgrade that assessment. Only the Recruiting PI can downgrade the event based on further follow up information. The CI, acting on behalf of the Sponsor, must assess and document the expectedness of the event (see 9.1.5).
Once the event has been signed off by the CI, a report will be generated from the eCRF and forwarded by fax from the Trial Manager to the JCTO and the pharmacovigilance department at Pfizer UK Ltd. They will issue queries about the event to the Trial Manager, who will relay them to sites via the eCRF.
Serious Adverse Reactions will be extracted from the eCRF for the annual MHRA safety report.
In the event that the RW is away and another member of site staff without access to the eCRF needs to report an SAE, the paper form should be faxed to the Trial Manager. It is a legal requirement that the site informs the CI within 24 hours of becoming aware of the event.
9.1.8 Reporting SUSARs
The JCTO will report SUSARs and other SARs to the regulatory authority (MHRA). The Chief Investigator will report to the relevant ethics committees. Reporting timelines are as follows:
-
SUSARs that are fatal or life-threatening must be reported not later than 7 days after the sponsor is first aware of the reaction. Any additional relevant information must be reported within a further 8 days.
-
SUSARs that are not fatal or life-threatening must be reported within 15 days of the sponsor first becoming ware of the reaction.
-
The Chief Investigator will provide an annual report of all SARs (expected and unexpected) and SAEs, which will be distributed to the sponsor (JCTO, MHRA and ethics committee)
Reporting Other Safety Issues
In addition, other safety issues also qualify for expedited reporting (15 day timeframe) where they might alter the current risk-benefit assessment of the IMP or would be sufficient to consider changes in the IMP administration or overall conduct of the trial, for example:
-
single case reports of an expected serious adverse reaction with an unexpected outcome (e.g. death);
-
an increase in the rate of occurrence of an expected serious adverse reaction, which is judged to be clinically important;
-
post-study SUSARs that occur after the participant has completed a trial;
-
a new event, related to the conduct of the trial or the development of the investigational medicinal product (IMP), that is likely to affect the safety of participant;
-
a serious adverse event which could be associated with the trial;
-
procedures and which could modify the conduct of the trial; a significant hazard to the participant population such as lack of efficacy of an IMP used for the treatment of a life-threatening disease;
-
a major safety finding (e.g. carcinogenicity) from a newly completed animal study.
These safety issues must be reported to the MHRA and the main REC in the format of a letter titled “Safety Report”.
The CI, acting on behalf of the sponsor, should retain a copy of the expedited report and associated documentation in the TMF
The sponsor (via the DMC) will perform an integrated safety analysis of all adverse event information reported and ensure discussions are held and actions undertaken to secure the safety of all participants. Discussions may result in the expedited reports being submitted and/or the discontinuation of the trial.
9.2 Expected Adverse Reactions to the Trial Medications
As per Summary of Product Characteristics (SmPC):
MIRTAZAPINE
Depressed patients display a number of signs and symptoms associated with the illness itself. It is therefore sometimes difficult to ascertain which symptoms are a result of the illness itself and which are a result of mirtazapine treatment.
Blood and the Lymphatic System Disorders
Rare >1/10000, <1/1000
Acute bone marrow depression (eosinophilia, granulocytopenia, agranulocytosis, aplastic anaemia, thrombocytopenia).
Metabolism and Nutrition Disorders
Common >1/100, <1/10.
Increased appetite and weight gain
Psychiatric Disorders
Rare >1/10000, <1/1000.
Mania, confusion, hallucinations, anxiety*, insomnia*, nightmares/ vivid dreams. (*Anxiety and insomnia, which may be symptoms of depression, can develop and become aggravated - under treatment with mirtazapine, development or aggravation of anxiety and insomnia has been reported very rarely).
Nervous System Disorders
Common >1/100, <1/10.
Somnolence (which may impair alertness), usually occurring during the first few weeks of therapy (NB. dose reduction does not generally lead to less sedation but can jeopardize antidepressant efficacy); dizziness, headache.
Rare >1/10000, <1/1000.
Convulsions (seizures), tremor, myoclonus, paraesthesia, restless legs.
Cardiac Disorders
Rare >1/10000, <1/1000 (Orthostatic) hypotension, syncope.
Gastrointestinal Disorders
Uncommon >1/1000, <1/100.
Nausea.
Rare >1/10000, <1/1000.
Dry mouth, diarrhoea.
Hepato-biliary Disorders
Rare >1/10000, <1/1000.
Elevations of hepatic transaminase levels.
Skin and Subcutaneous Tissue Disorders
Rare >1/10000, <1/1000.
Exanthema.
Musculoskeletal, Connective Tissue and Bone Disorders
Rare >1/10000, <1/1000.
Arthralgia, myalgia.
General Disorders
Common >1/100, <1/10.
Generalized or local oedema and accompanying weight gain, fatigue.
Although mirtazapine does not cause dependence, post-marketing experience shows that abrupt termination of treatment after long-term administration may sometimes result in withdrawal symptoms. The majority of withdrawal reactions are mild and self-limiting. Among the various reported withdrawal symptoms, nausea, anxiety and agitation are the most frequently reported. Treatment with mirtazapine should be discontinued gradually.
SERTRALINE
Side-effects which occurred significantly more frequently with sertraline than placebo in multiple dose studies were: nausea, diarrhoea/loose stools, anorexia, dyspepsia, tremor, dizziness, insomnia, somnolence, increased sweating, dry mouth and sexual dysfunction (principally ejaculatory delay in males). The side-effect profile commonly observed in double-blind, placebo-controlled studies in patients with obsessive compulsive disorder (OCD) and post-traumatic stress disorder (PTSD) was similar to that observed in patients with depression. In paediatric OCD patients, side-effects which occurred significantly more frequently with sertraline than placebo were: headache, insomnia, agitation, anorexia, tremor. Most were of mild to moderate severity. Post-marketing spontaneous reports include the following:
Cardiovascular
Blood pressure disturbances including postural hypotension, tachycardia.
Eye Disorders
Abnormal vision.
Gastro-intestinal
Vomiting, abdominal pain.
Nervous System
Amnesia, headache, drowsiness, movement disorders, paraesthesia, hypoaesthesia, depressive symptoms, hallucinations, aggressive reaction, agitation, anxiety, psychosis, depersonalisation, nervousness, panic reaction and signs and symptoms associated with serotonin syndrome which include fever, rigidity, confusion, agitation, diaphoresis, tachycardia, hypertension and diarrhoea. There have also been reports of manic reaction, although this phenomenon may be part of the underlying disease.
Convulsions (Seizures)
Sertraline should be discontinued in any patient who develops seizures (See ‘Special warnings and special precautions for use’).
Musculoskeletal
Arthralgia, myalgia.
Hepatic/Pancreatic
Rarely, pancreatitis and serious liver events (including hepatitis, jaundice and liver failure). Asymptomatic elevations in serum transaminases (SGOT and SGPT) have been reported in association with sertraline administration (0.8 – 1.3%), with an increased risk associated with the 200mg daily dose. The abnormalities usually occurred within the first 1 to 9 weeks of study medication treatment and promptly diminished upon study medication discontinuation.
Renal & Urinary Disorders
Urinary retention.
Reproductive
Hyperprolactinemia, galactorrhoea, menstrual irregularities, anorgasmy.
Skin and Allergic Reactions
Rash (including rare reports of erythema multiforme, photosensitivity), angioedema, ecchymoses, pruritus and anaphylactoid reactions.
Metabolic
Rare cases of hyponatremia have been reported and appeared to be reversible when sertraline was discontinued. Some cases were possibly due to the syndrome of inappropriate antidiuretic hormone secretion. The majority of reports were associated with older patients, and patients taking diuretics or other medications.
Haematologic
There have been rare reports of altered platelet function and/or abnormal clinical laboratory results in patients taking sertraline. While there have been reports of thrombocytopenia, abnormal bleeding or purpura in several patients taking sertraline, it is unclear whether sertraline had a causative role. See also ‘Special warnings and special precautions for use’.
General
Malaise.
Other
Withdrawal reactions have been reported with sertraline. Common symptoms include dizziness, paraesthesia, headache, anxiety and nausea. Abrupt discontinuation of treatment with sertraline should be avoided. The majority of symptoms experienced on withdrawal of sertraline are non-serious and self- limiting.
Suicidal thoughts and suicide attempts were mainly observed in clinical trials with Major Depressive Disorder.
9.3 Emergency Unblinding Procedure
Please see 8.12.3.
9.4 Study ID Cards
Due to the participants having dementia, it may be considered impractical to expect participants to carry cards during the study, with emergency unblinding procedures detailed on them. A card will be provided with the contact details of the research team and out of hours emergency procedures, to show medical staff in the event of an emergency, as they may need to contact Guy's Toxicology Unit for unblinding.
It is anticipated that the RW will decide on a case by case basis with the participants and carers who needs to have this information for that particular participant. In addition, the emergency unblinding telephone number will be printed on the study medication boxes to ensure that in an emergency where the carer may not be present, a health care professional would have access to that information.
10 VISIT ASSESSMENTS
10.1 Assessments and Procedures
10.1.1 Assessment Schedule
The main trial assessments will be completed at baseline, 13 weeks and 39 weeks. Data on adverse events will also be collected by the RW from participants and their carers monthly for the first three months (when medication is dispensed), and three monthly thereafter. There will be a final phone call at 10 months to the participant/carer to review any new or outstanding adverse events and to record concomitant medication for safety monitoring.
10.1.2 Flexibility of Visit Assessments
The baseline assessment will take place within 28 days of the patient having been referred by the Referring Investigator. It is intended that the follow up assessments will be completed within +/− 7 days of the 13 and 39 calendar weeks from randomisation. Baseline assessments will take place prior to randomisation. Follow-up assessments will take place after randomisation.
10.1.3 Unscheduled Assessments
These will be sought at treatment discontinuation or if loss to follow-up is anticipated. Please see table 6.5.1. Patients can be seen by the Recruiting PI at any time if they experience troublesome adverse events that require assessment.
10.1.4 Details of Assessments
Please see table 6.5.1.
Participants will be visited in their own homes and they and their carer will be interviewed by a research worker who will complete the assessments due at that time. The assessments are a mixture of direct assessment of the person with dementia (e.g. MMSE, DEMQOL); proxy report by the carer of the person with dementia (e.g. CSRI, NPI, DEMQOL-Proxy, EQ5D, adverse events, adherence); and self-report from the carer (e.g. GHQ, SF-12, Zarit). Instruments completed will be interview administered. The full set of assessments will be completed at baseline and at 13 and 39 week follow-up onto Source Data Worksheets (SDWs). During the trial these will be kept in the participants medical notes or similar files as arranged by the Recruiting PI. At the end of the trial they will be stored in the participants medical records as source data in case of future audit.
10.1.5 Premature Trial Closure
The trial may be stopped by the Trial Steering Committee. The Data Monitoring Committee in accordance with the IDMC charter, may recommend to the Trial Steering Committee that the trial be stopped.
10.2 Visit Procedures
10.2.1 Baseline Visit
The RW will arrange a meeting with the person with dementia and their carer. The RW will describe the trial and attempt to obtain consent as per the trial consent procedure (see Section 7.3). This will include the person with dementia and their carer. Where this is forthcoming the baseline assessment will be completed.
The assessment interview will ascertain type of dementia and depression according to set diagnostic criteria: NINCDS-ADRDA [McKhann et al 1984] for dementia; DSM-IV for depression (American Psychiatric Association 1994); the Olin criteria specifically designed for depression in dementia (Olin et al 2002); depression severity (CSDD); and vascularity (MHIS). The purpose of this diagnostic work is not to exclude further individuals from the trial (this would limit the generalisability of the findings) but instead to closely characterise the cases on the basis of diagnoses and severity to enable us to be able to describe the trial group in detail and to be able to investigate as secondary analyses the effect of diagnostic group and severity on subsequent outcome.
The local RW will complete a semi-structured interview with the person with dementia and their carer. This interview will include the measures used during follow-up as primary and secondary outcome measures and possible moderating variables including behavioural and psychological disturbance (Neuropsychiatric Inventory, NPI, Cummings et al, 1994]), physical illness, and severity of cognitive impairment (MMSE Folstein et al 1975).
10.2.2 Week 4 Follow-Up Visit
At week 4 (indexed on treatment start date) carers will be contacted by telephone and the CSDD completed. Those who score less than 4 will remain on the middle dose (2 mirtazapine/placebo and 2 sertraline/placebo) and those scoring 4 or more will move to the higher dose.
10.2.3 Week 8 Follow-Up Visit
The carers of those who remain on the middle dose will be contacted by telephone and the CSDD completed in the same way after 8 weeks (indexed on treatment start date) and if CSDD is 4 or more at this time they will be placed on the higher dose.
10.2.4 Week 13 Follow-Up Visit
Please see Table 6.5.1. The interview will include the primary and secondary outcome measures and AEs will be assessed using the aide memoire.
10.2.5 Week 39 Follow-Up Visit
Please see Table 6.5.1. The interview will include the longer-term outcome measures and AEs will be assessed using the aide memoire.
10.3 Measures
10.3.1 Baseline measures
The outcome measures have been chosen on the basis of their being the best-validated instruments available for the domains of function and activity of prime importance. We have balanced comprehensiveness with minimising respondent burden. The interview schedule is designed to be completed in one session with the person with dementia and their carer lasting no more than 60 minutes.
10.3.1.1 Participant measures
Primary Outcomes:
Depression in dementia – CSDD (Alexopoulos et al 1988) The CSDD was designed specifically for the measurement of depression in dementia. It is widely used and well validated with acceptable reliability and feasibility. It has been shown to be responsive to change in previous trials.
Costs:
Client Service Receipt Inventory (CSRI; Beecham et al 2001) This schedule measures service use and informal care input. It allows for the comprehensive costs of care for all participants to be calculated (including the costs of formal care such as that provided by health and social services and also the costs of informal care) using data gathered from carers.
Secondary Outcomes:
-
Disease specific quality of life – DEMQOL and DEMQOL-Proxy (Smith et al 2005).
-
Generic measure of quality of life – interview administered to carer (Coucill et al 2001) EQ-5D (EuroQoL Group 1990).
-
Withdrawal from treatment arm.
-
Cognitive impairment – MMSE (Folstein et al 1975) Medication adherence.
-
Adverse events.
10.3.1.2 Carer measures
Secondary Outcomes:
-
Carer mental health – General Health Questionnaire – 12 (GHQ-12; Goldberg et al 1988) Carer quality of life -- SF-12 v2 (Ware et al 1996).
-
Carer burden – Zarit Carer Burden Scale (Zarit 1980).
10.4 Safety Monitoring
All adverse events occurring in the trial will be recorded in the participants' source data worksheets and filed in their medical records at the end of the trial. They will also be transcribed onto the eCRF. Data on adverse events will be collected by the RW from participants and their carers monthly for the first three months (when medication is dispensed), and three monthly thereafter. Any events reported by participants or their clinical teams will also be reported and followed up between visits.
Reports of serious adverse events will be forwarded to the DMEC members as requested. Any recommendations or additional information required by the DMEC will be actioned. Non-serious unexpected adverse events will be collated and sent to DMEC members in advance of scheduled meetings.
11 STATISTICS
11.1 Sample Size
11.1.1 Assumptions
Based on a review of previous studies (e.g. Alexopoulos et al, 1988; Katona et al, 1998; Lyketsos et al, 2003; Teri et al, 2003), the sample sizes used in estimation, and the broad eligibility criteria of the proposed trial we assume that the common standard deviation (SD) of the CSDD scores at baseline and follow-up will be approximately 5 points. We propose that a clinically important difference between the antidepressant and placebo groups would be 2 points on the CSDD observable at 13 weeks, and maintained at 39 weeks. This equates to a moderate standardised effect size (SES) of 0.4. We have further assumed that loss to follow-up (including by death of participant or carer) will be 10% at 13 weeks increasing to 20% at 39 weeks. We believe these estimates are realistic based on the existing literature and given the measures we will take to minimise loss to follow-up (e.g. active follow-up in the home, minimising the burden of data collection) in the trial design.
11.1.2 Power Analyses
Our primary intention to treat analyses will compare i) sertraline against placebo and ii) mirtazapine against placebo at 13 weeks post-randomisation. Allowing for 10% loss to follow-up at 13 weeks, an overall sample of 444 patients (randomised 1:1:1 to placebo: sertraline: mirtazapine) would provide 90% power to detect a 2 point difference in CSDD (SD 5; SES 0.4) for the sertraline vs placebo and the mirtazapine vs placebo comparisons using independent sample t-tests with 2-sided 5% significance levels. This is equivalent to assuming a zero correlation between baseline and outcome CSDD in an analysis of covariance adjusting for baseline CSDD (Machin et al, 1997). Machin et al (1997) suggest that correlations of 0.6 to 0.75 between baseline and outcome measurements are common. Assuming a conservative correlation of at least 0.6, the overall sample size required based on an analysis of covariance with 2-sided 5% significance levels reduces to 285 patients (using a multiplying factor of 0.64; Machin et al, 1997) but making no particular adjustment for drop-outs (patients allocated to sertraline or mirtazapine withdrawing from treatment and effectively shifting to placebo) or drop-ins (patients allocated to placebo withdrawing from placebo and effectively shifting to sertraline or mirtazapine). It is important to adjust the power calculation for such drop-outs and drop-ins. Therefore, additionally allowing up to 12.5% of those randomized (per comparison) to be either drop-outs or drop-ins (e.g. 10% drop-outs and 15% drop-ins) the overall sample size required becomes 507 patients (i.e. 169 patients in each arm) (using a multiplying factor of 1.78; Friedman et al 1998).
An overall sample size of 507 patients will therefore provide 90% power to detect a 2 point difference in CSDD (SD 5; SES 0.4) for the sertraline vs placebo and the mirtazapine vs placebo comparisons at 13 weeks and an 86% power at 39 weeks. This allows for 20% loss to follow-up, correlation between baseline and outcome CSDD 0.6, and up to 12.5% of those randomized (per comparison) to be either drop-outs or drop-ins using an analysis of covariance with 2-sided 5% significance levels.
Allowing for the same levels of loss to follow-up, an overall sample of 507 patients would also enable us to calculate 2-sided 95% confidence intervals for the difference in the proportion of pre-specified adverse events between the antidepressant arms of (a clinically significant) 10% (i.e. 5% vs 15%) 6% at 12 weeks and 7% at 39 weeks.
11.2 Data Monitoring & Interim Analyses
We have no planned interim analyses. One analysis of the data will be conducted once the trial database has closed. We have not planned an interim analysis of the data primarily because the first 39 week outcome data will only become available as recruitment into the trial is coming to an end (i.e. after 39 weeks with only 13 weeks of recruitment to follow). The Data Monitoring Committee will wish to collate effectiveness and safety data during the trial to inform their recommendations to the Trial Steering Committee but we do not anticipate a formal analysis of the data mid-trial at this stage.
11.3 Brief Analysis Plan
11.3.1 General Considerations
The analyses of effectiveness will be pragmatic, based on intention to treat, and will utilise all available follow-up data from all randomised patients. A full Analysis Strategy will be developed, independently of looking at the trial data, and before undertaking any analysis, about 6 months after the start of randomisation. This will be approved by the TMG and the TSC before any analysis is undertaken. The Trial Statistician will remain blind wherever possible until the main analyses are complete.
11.3.2 Analyses of Effectiveness
Primary Effectiveness Analyses
The primary outcome of symptoms of depression on the CSDD (CSDD, continuous score) at 13 weeks post randomisation will be analysed by ANCOVA adjusted for baseline CSDD and centre with contrasts for (a) sertraline vs placebo and (b) mirtazapine vs placebo.
Secondary Effectiveness Analyses
Change in CSDD score from baseline to 13 weeks will further include a contrast for mirtazapine vs sertraline. CSDD score at 39 weeks will be analysed by ANCOVA adjusted for baseline CSDD and centre with contrasts for (a) sertraline vs placebo; (b) mirtazapine vs placebo and (c) mirtazapine vs sertraline. Secondary outcomes will be compared using the same contrasts as above within a longitudinal generalised linear model framework adjusting for the respective baseline scores and centre. Results will be summarised as mean differences together with 95% confidence limits.
The significance level will be 5% (2-sided) for specified analyses of the primary outcome variable and 1% (2-sided) for specified analyses of secondary outcome variables. Sensitivity analyses will be used to assess the robustness of conclusions to missing outcome data and to departures from randomised treatment. Loss to follow-up, departures from randomised treatment and the prevalence of serious adverse events will be reported at 13 and 39 weeks post randomisation.
Missing Data
Missing data will be considered according to type in the analyses. It is anticipated that there will be no missing scale covariate data for the primary analyses as copies of the relevant data (e.g. CSDD) collected at baseline will be required at the point of randomisation. Missing covariate item data will be imputed using mean imputation per patient (pro-rating) if no more than 5% of items are missing across all of the data collected and using multiple imputation (Schafer 1999) if more than 5% of items are missing on at least some of the data collected. Missing primary outcome data is anticipated from three sources: missing items; missing scale due to loss to follow-up; and missing scale due to death. As the primary analysis is of CSDD at 3 months there will only be one assessment of CSDD post-randomisation to include in this analysis. To guard against missing post randomisation CSDD data for patients withdrawing from treatment before 3 months, the CSDD will also be collected at the point of withdrawal if consent for this is available. Missing outcome item and scale data will be imputed using multiple imputation.
Three month outcome data will not be imputed, however, if the patient has died between randomisation and 3 months. In addition, sensitivity analyses will be constructed using methods to assess the robustness of the conclusions to the method used (Little & Rubin 2002). The secondary analysis of CSDD at 39 weeks will utilise pattern mixture models (Little & Wang 1996; Hedeker & Gibbons 1997) to handle any missing 13 or 39 week outcome scale data and multiple imputation to handle any 13 or 39 week outcome item data. Thirty nine week outcome data will not be imputed if the patient has died between the 13 and 39 week assessments. Again several sensitivity analyses will be conducted using a range of methods.
Non-Compliance/Non-Adherence
The primary ‘intention to treat’ analysis is intended to provide inferences regarding the effectiveness of the three interventions overall. It is not primarily intended to provide inferences regarding the causal effect of the interventions themselves, but on the interventions as deployed in as ‘real life’ a setting as possible. As such, compliance information is not necessary to ensure that the ‘intention to treat’ analysis is valid. The implications of treatment non-compliance for the ‘intention to treat’ analysis were handled within the power calculation by adjusting the sample size for drop-outs and drop-ins, effectively reducing the expected effect size to allow for a degree of non-compliance.
Our secondary analyses will include an assessment of the causal effect of the trial interventions and will be detailed in the full analysis strategy. Two aspects of treatment compliance will be considered: the proportion of the intended dose actually taken per patient; and the impact of non-randomised concomitant medications and treatments received. The expected magnitude of the placebo effect in this trial makes the methods described by Nagelkerke et al (2000) inappropriate. We will therefore draw on the methods described by Dunn et al (2003), White et al (2003), Kenna & Sheiner (2004) and Levy et al (2004).
11.3.3 Analyses of cost-effectiveness
The economic evaluation will be led by Professor Martin Knapp and his group at the Institute of Psychiatry with input from the London School of Economics (LSE) (linking to long term care financing projections work and social care studies for older people) and Dr Linda Davis at Manchester. The analysis of the economic impact of the interventions is a central, fully integrated element of the proposed study. The study design as presented in the proposal overall has been constructed to meet the needs of economic and well as clinical evaluations of the interventions to be studied. In an earlier draft of this proposal the economic funding was mistakenly included as ‘consultancy’, this has been corrected.
Costs of formal and informal care
The comprehensive costs of care for all participants will be calculated (including the costs of formal care such as that provided by health and social services and also the costs of informal care) using data gathered using the CSRI completed by key workers or family carers at baseline, 13 weeks and 39 weeks. Unit costs will be best national estimates of the long-run marginal opportunity costs, built up from both national unit costs compendia (Curtis et al 2004), NHS specialty costs and specific care homes (costs or charges, depending on availability).
Informal care will be costed (Netten 1993). We will collect information on the volume and nature of informal care inputs, mindful of the difficulties of measuring such dimensions and of their interpretation as inputs to the care process. We will attach costs to informal care inputs using two or three approaches (opportunity cost of lost work/leisure; replacement cost with a (paid) home carer or similar; zero value; and possibly some blending of these approaches to reflect different informal care tasks and circumstances, along the lines of Brouwer's approach [van den Berg et al 2004]) and examine the cost-effectiveness consequences of these different approaches (one dimension of sensitivity analysis). Aggregate and agency-specific costs will be reported.
Analyses
From these costs and the outcomes data, we will compare total and component (by service or agency) costs, incremental cost-effectiveness ratios and net benefits (using the primary outcome measure CSDD), cost–utility ratios (using utility scores computed from the EQ-5D and societal weights) and cost-consequences results (using all non-cost outcomes measures). The primary evaluation will be the cost-effectiveness analysis using CSDD change as the outcome. Missing data will be addressed using multiple imputation. The evaluation will include the plotting of cost- effectiveness acceptability curves generated from bootstrap analyses. Sensitivity analyses will explore the impact of differences in key costs and outcome assumptions. Given the nature of the study aims and the data to be generated we are unlikely to use probabilistic sensitivity analysis. Evaluation will be conducted from (a) societal, (b) public sector and (c) NHS perspectives, and comparisons made between the results.
Quality Adjusted Life Years (QALYs)
We will calculate QALYs from the EQ5D, though recognizing that there remains uncertainty about the validity of this measure with a population of older people with dementia and depression. Societal weights will be employed. The QALYs will not include ‘carer quality of life (QOL) issues’, as described by one of the reviewers, but will focus exclusively on the patient. Technically it would be complex and speculative to merge patient and carer QOL measures into a composite QALY representation especially in dementia where there are so few relevant comparative data available. The study will however assess a number of aspects of carer QOL and experience (directly using the SF-12; and indirectly via the GHQ 12 [carer mental health] and Zarit [carer burden]). This will enable a full evaluation of the impact of the intervention on carers of people with dementia. Dementia studies have a good track record of assessing carer impacts including economic impacts, and we as individual researchers have similarly given this attention and emphasis over many years and studies (Murray et al 1999; Schneider et al 1999, 2001, 2002; Banerjee et al 2003).
Beyond Trial Modelling
We will seek alternate funding for this further economic exploitation of data from this study.
11.3.4 Analyses of Safety
All cause withdrawal from randomised treatment will be reported at 13 and 39 weeks post randomisation. Withdrawal rates will be compared at 13 and 39 weeks across the three trial arms (as randomised) using Chi square tests. The prevalence of specific adverse events will be reported descriptively at 13 and 39 weeks post randomisation. The prevalence of patients experiencing one or more serious adverse events will be compared at 13 and 39 weeks post randomisation across the three trial arms (as randomised) using Chi Square tests. We will calculate 2-sided 95% confidence intervals for the difference in the proportion of pre-specified adverse events between the antidepressant arms.
11.3.5 Other Exploratory Analyses
Associations between post-treatment outcomes and baseline predictor variables (including dementia severity, dementia type, depression type, depression severity, care arrangements; type of “normal clinical care” received; behavioural and psychological symptoms in dementia, and physical illness) will be examined using multiple linear and logistic regression modelling techniques, including a limited examination of first order interactions. We will also explore the baseline “predictors” of costs measured for the first 13 weeks and the full 39 weeks post randomisation.
11.4 Changes to the Analysis Plan
Any significant changes made to the Statistical Analysis Strategy (see Appendix 6) after approval by the TMG and the TSC will be taken back to the TMG and the TSC for their approval before the changes are put into effect.
12 DATA MANAGEMENT & MONITORING PROCEDURES
12.1 Direct Access to Trial Data & Documents
The principal investigators, carers and participants will permit trial-related monitoring, audits, ethics committee review and regulatory inspections by providing direct access to source data/documents.
During the course of the trial, only the principal investigators, their staff and the coordinating research team will have access to data generated by the trial. Other researchers may submit an application to the Trial Management Group at a later date for access to the anonymised master database.
12.2 Confidentiality
The confidentiality of participant and carer identification details will be protected according to the Data Protection Act. Participants will be identified by their PIN, initials and date-of-birth only. The one exception will be a separate list, held by the Trial Manager at the South London & Kent Centre only, of name, postal address, email address and telephone numbers of participant/carer, date of birth, PIN/ CIN, medication pack number, date of randomisation, GP contact details and NHS number (to enable the trial team to track the participant for follow-up assessments). Participant names, addresses, and other contact details will be written in the CRF for identification and contact purposes. The CRFs will be regarded as confidential, and kept in locked filing cabinets in the local centre and the coordinating centre (South London & Kent).
12.3 Record Keeping
12.3.1 Custodian of the Data
The Chief Investigator will have control of and act as custodian for the data generated by the trial (i.e. the source data and the Trial Master Database) on behalf of the Trial Management Group.
The Trial Statisticians and Health Economists will be responsible for all analyses covered by the Statistical Analysis Strategy (see Appendix 7). Any further analyses will be conducted by researchers with the approval of the Trial Management Group (and the Trial Steering Committee prior to the publication of the main papers).
12.3.2 Format of Records
The majority of the source data will be collected on the paper source data worksheets (SDW). These will be entered at each centre encrypted via the internet using the InferMed Macro electronic data capture and stored on a dedicated secure server within the MH&N Clinical Trials Unit at King's College London. The system ensures confidentiality.
A proportion of the assessment interviews with the participants and carers may be audiotaped in order to assess the interrater reliability of the RWs in their ratings of the primary outcome.
These will be collected using audio devices and will be stored centrally.
12.3.3 Duration & Location
The eCRF will be archived in accordance with the EU Directive by Recruiting PIs on behalf of the Trial Management Group. The MH&N CTU will provide each site with a disk containing their data at the end of the trial.
The electronic Trial Master Database will be stored on a dedicated server within the MH&N CTU, King's College London indefinitely so that it is available should the Trial Management Group wish to access it for further analysis.
Copies of the Trial Master Database will be kept securely on university computers and/or laptop computers.
12.4 Trial Data Management System
12.4.1 eCRF
An eCRf will be created using the InferMed Macro system. This system is regulatory compliant (GCP, 21CRF11, EC Clinical Trial Directive). The eCRF will be created in collaboration with the Trial Manager, the Trial Statisticians and the Health Economists and maintained by the MH&N Clinical Trials Unit. It will be hosted on a dedicated secure server within KCL.
12.4.2 Training and User Support
The Trial Manager will be trained in the data entry and data query modules of the MACRO system by the MH&N CTU. He will then train the RWs and Recruiting PIs on the data entry and data query modules during site initiation.
Sites will seek support with the eCRF from the Trial Manager in the first instance and problems that cannot be resolved at that level will be passed by the Trial Manager to the MH&N CTU. There will be only one point of contact between the Trial Team and the MH&N CTU (Trial Manager to Database Programmer).
12.5 Entry of Data by Local Research Assistants
RWs will complete participant data using source data worksheets provided to the sites at site initiation. Data will be entered onto the eCRF from these source data worksheets. The source data will be filed in the participants clinical records (or within the Recruiting PIs department if this is not possible) at the end of the trial.
Each RW and Recruiting PI will have a unique username and password for the eCRF provided by the MH&N CTU. Passwords must not be shared and if new researchers join the study, a personalised username and password should be requested via the Trial Manager. Data, once entered, can subsequently be changed. The system will maintain a clear audit trail of when and who entered the original data, what that data was, what it was changed to, by whom, when and why. It is a legal requirement that passwords to the eCRF are not shared and that only those authorised to access the system are allowed to do so. It is the responsibility of each individual issued with a password to keep it secret and ensure nobody else uses it.
RWs will transcribe data from the SDWs at their own centre (which will act as the source data for the trial) onto the eCRF. This should be done within one working week of a participant assessment. A proportion of these will be checked by the Trial Manager during on-site monitoring visits. Queries will be resolved on the eCRF by referring back to the SDWs.
After the week 39 assessment, the RW must enter the participant data as soon as possible, in order to allow time for the Trial Manager and Trial Statistician to review the data and issue any queries promptly. These queries must then be answered by the RW before the participant's data is locked within the database. After that time, changes cannot be made to the database by sites. The participant's treatment allocation cannot be revealed until this process has been completed. The Recruiting PI must inform the Referring Investigator or other clinician taking over the participant's ongoing care of their treatment allocation between month 9 and month 10, as no further blinded study medication will be available to the participant. Since the clinician will need time to review the patient clinically, make a decision on ongoing treatment for depression and have a prescription issued, it is vital that the system of data entry, review and database lock on a participant by participant basis happens promptly after their week 39 assessment, so that their treatment allocation can be revealed.
At the end of the trial, the Recruiting PI will review all the data for each participant and provide and electronic signoff to verify that all the data is complete and correct. The electronic signoff is the legal equivalent to a paper signature.
At the end of the trial each centre will be supplied with a CD-ROM containing the eCRF data for their centre which must be filed in the Recruiting PI site file in case of future regulatory or internal audits.
12.6 Trial Monitoring Procedures
12.6.1 Quality Assurance
12.6.1.1 Selection of Centres/Sites
The nine trial centres/Recruiting PIs were selected because of their track record and experience in other large multicentre dementia trials.
12.6.1.2 Training of Trial Personnel
All staff employed on the grant and all Investigators will be trained in:
-
GCP
-
Use of the assessment tools
-
Trial standard operating procedures.
Interrater reliability of the CSDD will be assessed prior to the start of data collection and reassessed during the course of the trial.
12.6.1.3 On-Site Monitoring
The Trial Manager will conduct a minimum of three on-site monitoring visits to each site and will complete a Site Monitoring Report for each site at each visit.
During monitoring visits he will check and update the Investigator Site Files and will visit pharmacies to check the Pharmacy Files.
He will examine the consent forms for 100% of the participants during site monitoring visits.
He will examine the source data worksheets on a pre-defined subset of participants (to be detailed in the monitoring SOP) to check for accuracy and completeness against the eCRF. He will check all reported SAE data and discuss any follow-up data needed with the site and he will also talk through with the RW the progress of each participant, double checking that there have been no unreported SAEs.
He will meet with the RW and Recruiting PI at regular intervals to discuss the progress of the trial on the site to check if there are any significant problems.
He will check the study medication dispensing and returns data for all participants against the eCRF data. The current stock held in pharmacy should be checked in order to ensure the study medication distribution system is working. He will re-count all the medications returned to pharmacy, check his pill count against the pharmacists' pill count and the pill count recorded in the eCRF by the RW and resolve any discrepancies. He will then complete a study medication returns inventory, package up the study medication returns and notify Catalent that they are ready for collection from site. Once they are received back at Catalent, a warehouse number will be issued for that shipment and this must be retained with the details of the study medication returns shipped, in order that they can be identified and retrieved if this is ever required during the trial.
12.6.1.4 Essential Documentation
The Trial Manager will maintain a Trial Master File containing the essential trial documents in accordance with GCP and the EC Clinical Trial Directive. In addition, he will supply each site with an Investigator Site File and a Pharmacy File, which will contain the some of the essential documents.
12.6.2 Quality Control
12.6.2.1 Data Checking & Verification Procedures
Where possible, the eCRF system will generate automatic queries to the RW if data fields are being completed with illogical data. These will appear as pop up boxes as the data is entered.
The Trial Manager will monitor the data electronically for completeness and will generate queries back to the site, via the eCRF, if there is data that looks as if it may be erroneous or is unclear.
The RW will respond to the queries and the Trial Manager will review the response. The query will then be closed by the Trial Manager, though it can be re-issued if the matter is not resolved satisfactorily. All queries raised and their responses will be retained as an audit trail.
The Trial Manager or Statistician may also identify data fields that should be checked against the source data during site monitoring visits. These can then be listed per site while the Trial Manager is there and ‘signed off’ electronically as they are checked. Likewise, if any source data is missing or incorrect, this will be noted in the eCRF.
Detailed standard operating procedures will be established for data checking once the eCRF has been created.
12.7 Data Locking Procedures
Trial data will be locked in stages. Once a participant has completed the 39 week follow-up assessments, or these have formally been lost to follow-up, all outstanding queries on their eCRF will be resolved before their data is locked. A period of one month following participants 39 week assessment has been allowed for this. The Recruiting PI will sign off each participant's eCRF prior to unblinding. Once all data collection to the trial has been completed any further outstanding queries will be resolved and the entire database will be locked prior to its transfer to the Trial Statisticians and Health Economists. The Trial Statisticians and Health Economists will conduct the final checking of the data and any queries generated will be resolved at site by the Trial Manager and RWs before the Master Trial Database is frozen for analysis.
13 ETHICAL CONSIDERATIONS
The main potential ethical issue in this study is that dementia itself may interfere with an individual's ability to give their informed consent, especially in more severe stages of the illness. Given that this is an effectiveness study it is very desirable that all potential participants with depression in dementia are included if the overall impact of the medications are to be ascertained. This is an important issue in this study's design since compromised capacity and lack of insight may be a source of significant variation (e.g. via compliance).
The trial group has considerable experience in the ethical issues raised by obtaining consent for treatment trials in dementia, including severe dementia. One strength of the study is the level and integral nature of consumer involvement at all stages which means that carers and people with dementia will contribute to finalising information sheets and consent forms. The methods of obtaining consent for the study proposed here follow COREC guidance for information sheets and consent forms and specific guidance for incapacitated adults in CTIMPs to ensure that they are compliant with the Medicines for Human Use (Clinical Trial) 2004 Regulations. However, for the purposes of this trial our design minimises the likely impact of lack of capacity by having an informant caregiver involved throughout who will act as the participant's personal legal representative.
Full informed consent will be obtained where possible. If the person with dementia does not have the capacity to consent, then their assent will be sought, the consent of an appropriate caregiver will be sought and the interviews and recruitment will be completed only if there is no sign of distress in the person with dementia. This is an approach that has been used successfully in trials and other descriptive and evaluative studies.
That said, the giving and discussion of information to people with dementia to enable them to make an informed decision with respect to consent is more complex and time consuming than for people without cognitive impairment. The study RWs will be trained in issues in obtaining consent and will only be deployed if their skills in this area are satisfactory. Also for this reason, the Referring Investigator will obtain verbal consent, not for entry into the study, but only for the potential participant to be approached by the RW. The study RW will then discuss the study with participants and carers, providing information, and will obtain consent or assent as described in section 7.3. Participants will be given a 7 day period to consider the information given and their willingness to participate.
14 REGULATORY AND ETHICS APPROVAL
14.1 Research Ethics Approval
We have submitted the protocol for consideration by the Manchester Research Ethics Committee (REC) which is a Type 3 ethics committee and therefore has the authority to approve a multiple domain CTIMP (clinical trial of an investigational medicinal product).
Ethics Reference: 06/Q1407/66.
See also sections 17, 18 and 19.
14.2 Local Research Ethics Approvals (LREC)
Site specific assessments (SSAs) will be undertaken by local ethics committees, the submission of which are being coordinated by the Mental Health Research Network.
Within each of the 9 recruiting sites, the Recruiting PI will be named on the local ethics SSA.
For each NHS Trust with Referring Investigators, one lead ‘Principal Referring Investigator’ will be identified and named on the SSA. Other Referring Investigators within that site will be listed on a delegation of authority form which is filed in the ‘Referring Investigator Site File’ that is held by the Principal Referring Investigator. The RW, in collaboration with the Trial Manager, will be responsible for ensuring the site files for the Recruiting PI and all the Principal Referring PIs contain all essential documents.
See also sections 17, 18 and 19.
14.3 Medicines and Healthcare Products Regulatory Agency Approval (MHRA; CTA)
CTA application has been submitted by the Chief Investigator on behalf of the sponsor and has been approved. The Qualified Person (QP) is Dr Ian Scully at Catalent.
See also sections 17, 18 and 19.
14.4 R&D Approvals and Research Governance
The study will be approved by all local NHS Trust R&D Departments involved prior to recruitment at each site or referral by a Referring Investigator. This will be facilitated by the Mental Health Research Network.
15 FINANCIAL AND INSURANCE MATTERS
15.1 Funding Arrangements
15.1.1 Contact Details of Funding Bodies
Liz Dunn
Commissioning Manager
NHS Health Technology Assessment
University of Southampton
Biomedical Sciences Building (Mailpoint 728) Bassett Crescent East
Southampton SO16 7PX
Email hta@soton.ac.uk
Tel +44 (0)23 80 595770
Fax +44 (0)23 80 595639.
Sertraline and sertraline placebo are being donated by Pfizer UK Ltd. (Total value c. £25,000).
15.1.2 Duration of Grant
Twenty-seven months, from 1st August 2006 to 31st October 2008.
15.1.3 Grant Summary
Total grant: £1.6 million
Funders reference number: 04/11/02
15.2 Indemnity/Compensation/Insurance Arrangements
The Chief Investigator is covered under the Kings College London no-fault liability insurance for clinical trials. Catalent, Pfizer UK, Genus Pharmaceuticals and Arrow Pharmaceuticals are covered by their own appropriate insurances and contracts will be agreed between them and KCL.
The study site staff will be covered by NHS indemnity from the principal investigators' employers for negligent harm.
15.3 Site Agreements
These have been drawn up by sub-contracts between KCL and the participating sites.
16 PUBLICATION POLICY
It is intended that the trial results will be disseminated in peer-reviewed scientific journals, an internal report to the HTA, conference presentations, written feedback to trial participants and carers, and presentations to relevant community groups.
A publication policy will be developed by the TMG and all data from the study will only be disseminated with the prior agreement of the TMG.
The results will be made available by a newsletter to the research participants. If any news comes to light during the trial itself about the treatments involved, these will be conveyed to the research participant, their GP and their family and carer.
17 ETHICS SUBMISSIONS
| Ethics application submitted 24.03.06 | Approved 12.07.06 |
| Substantial amendment 1 submitted 23.08.06 | Approved 14.09.06 |
| Substantial amendment 2 submitted 04.10.06 | Approved 20.10.06 |
| Non-substantial amendment 1 submitted 20.12.06 | Noted 17.01.07 |
| Non-substantial amendment 2 submitted 02.05.07 | Noted 10.05.07 |
| Non-substantial amendment 3 submitted 08.06.07 | Noted 27.06.07 |
| Substantial amendment 3 submitted 14.09.07 | Approved 2.10.07 |
| Substantial amendment 4 submitted 5.12.08 | Approved 9.01.09 |
| Substantial amendment 5 14th September 2009 | Approved 2.11.09. |
18 MHRA SUBMISSIONS
| CTA application submitted 21.06.06 | Approved 04.08.06 |
| Substantial amendment 1 submitted 23.08.06 | Approval not required |
| Substantial amendment 2 submitted 04.10.06 | Approval not required |
| Substantial amendment 3 submitted 14.09.07 | Approval not required |
| Substantial amendment 4 submitted 5.12.08 | Approval not required |
| Substantial amendment 5 14th September 2009 | Approved October 2009. |
19 AMENDMENTS
19.1 Non substantial amendments
Non-substantial amendment 1 was a protocol amendment (protocol 2.1)
Minor changes made to sections 1.1.1, 1.1.2, 1.2.4, 6.2 and 15.2.
Non-substantial amendment 2 was not a protocol amendment
Non-substantial amendment 3 was a protocol amendment (protocol 2.2)
Minor changes made to sections 3.1 and 7.4.2.
19.2 Substantial amendments
Substantial amendment 1 (ethics) was not a protocol amendment
Key changes:
-
Original ethics application stated incorrect CI. Ethics application revised. Lock code AB/88518/1.
Substantial amendment 2 (ethics & MHRA) was a protocol amendment (v2.3)
Key changes:
-
Principal investigator in Cambridge changed from Tom Dening to Claire Lawton (MHRA also informed) (section 1.2.4. (02))
-
Addition of Appendix 3: Source Data Worksheets (data entered on eCRF)
-
Clarification of study medication dispensing regimen (sections 8.7. – 8.10.)
-
Participant & Carer Information & Consents revised to provide additional information to participants and carers and protocol amended to make clearer the situation for consent of incapacitated adults in CTIMPs, as required by the Sponsor (sections 7.3. and 7.5.2.)
-
Clarification on the roles & responsibilities of Referring Investigators and Recruiting Investigators (section 1.2.4.)
-
Confirmation that Referring Investigators will document that the patient has given verbal consent to be referred to the Recruiting Investigator, rather than written consent (sections 7.2., 7.3. and 7.5.2.)
-
List of abbreviations added (section 2.)
-
Update of contact details to the study including DMC members and minor administrative changes to improve readability of protocol (section 1.3.2 and throughout protocol)
-
Pfizer UK Ltd. pharmacovigilance department to have access to anonymised SAE data for information only, as they are now donating sertraline and matching placebo tablets in bulk for the study (sections 9.1.7. and 15.1.1.)
-
Protocol amended to reflect the addition of week 4 and week 8 data collection
-
points as agreed in the original ethics approval (sections 7.5.1, 8.3, 10.2.2 10.2.3)
-
Protocol amended to reflect alterations to the system of adverse event data collection (sections 9.1.1. – 9.1.8.)
-
Addition of information regarding 24 hour emergency unblinding service via
-
Guy's Toxicology Unit (section 8.12.3)
-
More detailed procedure for data management prior to routine unblinding at end of study added to protocol, to ensure both continuity of prescribing and integrity of trial data (sections 8.1.4. and 12.5.)
-
Addition of section in protocol to document all substantial and non-substantial amendments to the study, for administrative purposes (sections 17, 18 and 19.)
Substantial amendment 3 (ethics & MHRA) was a protocol amendment
Key changes:
-
Change of sponsor (sections 1.1.2 and 1.2.1)
-
Change of sponsor representative (section 1.2.1)
-
Update of section on submissions to ethics committee and regulatory authority.
Substantial amendment 4 (ethics & MHRA) was a protocol amendment (v2.4)
Key changes:
-
Extension of recruitment period (sections 7.1 and 7.2)
-
Change of address of Manchester PI, change of telephone and fax numbers for
-
Birmingham PI (1.2.4)
-
Change of role title for health economist Renee Romeo (1.2.10)
-
Change of name of drug supplier (1.2.12)
-
Changes in membership of Trial Steering Committee (1.3.1)
-
Addition of Clare Rutterford as statistician (1.2.9)
-
Week 4 and 8 assessments indexed on treatment start date rather than randomisation (7.5.1, 8.3, 10.2.2, 10.2.3)
-
Adverse events checklist replaced by aide memoire (Appendix 3).
Substantial amendment 5 (ethics & MHRA) was a protocol amendment (v2.5)
Key changes:
-
Supplier of mirtazapine changed from Genus to Arrow (pages 15, 43, 72)
-
Section on reporting procedure for SAEs, SARs and SUSARs (9.1.8, p52) revised to describe new arrangements
-
Line stating that consent forms are sent to the Chief Investigator deleted.
20 ANCILLARY STUDIES
These will be agreed by the TMG and the TSC.
21 REFERENCES
- Alexopoulos GS, Abrams RC, Young RC, . Cornell scale for depression in dementia. Biol Psychiatr 1998;23:271-84.
- Diagnostic and Statistical Manual of Mental Disorders. Washington DC: APA; 1994.
- Anonymous. Do SSRIs cause gastrointestinal bleeding?. Drug Therapeut Bull 2004;42:17-8.
- Bains J, Birks JS, Dening TR. The efficacy of antidepressants in the treatment of depression in dementia. Cochrane Database Syst Rev 2002.
- Ballard CG, Bannister C, Oyebode F. Depression in dementia sufferers: a review. Int J Geriatr Psychiatry 1996;11:507-15.
- Ballard C, Eastwood C, Gahir M, Wilcock G. A follow-up study of depression in the carers of dementia sufferers. BMJ 1996;312:947-8.
- Ballard CG, Patel A, Solis M, Lowe K, Wilcock G. A one year follow up study of depression in dementia sufferers. Br J Psychiatry 1996;68:287-91.
- Ballard CG, O’Brien JT, Swann AG, Thompson P, Neill D, McKeith IG. The natural history of psychosis and depression in dementia with Lewy bodies and Alzheimer's disease: persistence and new cases over 1 year of follow-up. J Clin Psychiatry 2001;62.
- Ballard C, O’Brien J, Swann A, Fossey J, Lana M. A One Year Follow-up study of behavioural and psychological symptoms in dementia (BPSD) amongst people in care environments. J Clin Psychiatry 2001;62:631-6.
- Banerjee S, Thornicroft G, Smuzkler G. Textbook of Community Psychiatry. London: Oxford University Press; 2001.
- Banerjee S, Murray J, Foley B, Atkins L, Schneider J, Mann A. Predictors of institutionalisation in older people with dementia. J Neurol Neurosurg Psychiatr 2003;74:1315-516.
- Beecham J, Knapp M, Thornicroft G. Measuring Mental Health Needs. London: Gaskell; 2001.
- van den Berg B, Brouwer WB, Koopmanschap MA. Economic valuation of informal care. An overview of methods and applications. Eur J Health Econ 2004;5:36-45.
- Burns A, Jacoby R, Levy R. Psychiatric phenomena in Alzheimer's disease III: Disorders of mood. Br J Psychiatry 1990;157:81-6.
- Burns A. Affective symptoms in Alzheimer's Disease. Int J Geriatr Psychiatry 1991;6:371-6.
- Burns A, Lewis G, Jacoby R. Factors affecting survival in Alzheimer's disease. Psychol Med 1991;21:363-70.
- Coucill W, Stirling W, Bentham P, . EQ-5D in patients with dementia an investigation of inter-rater agreement. Med Care 2001;39:760-71.
- Copeland JRM, Dewey ME, Griffiths-Jones HM. Dementia and depression in elderly persons: AGECAT compared with DSM-III and pervasive illness. Int J Geriatr Psychiatry 1990;5:47-51.
- Cummings J, Mega M, Gray K, . The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308-14.
- Curtis L, Netten A. Unit Costs of Health and Social Care 2004. Canterbury: Personal Social Services Research Unit, University of Kent; 2004.
- Doody R, Stevens J, Beck C, . Practice parameter: management of dementia (an evidence based review). Neurology 2000;56:1156-66.
- Dunn G, Maracy M, Dowrick C, Ayuso-Mateos JL, Dalgard OS, Page H, et al. Estimating psychological treatment effects from a randomised controlled trial with both non-compliance and loss to follow-up. Br J Psychiatry 2003;185:323-31.
- Detailed guidance on the European database of Suspected Unexpected Serious Adverse Reactions (Eudravigilance – Clinical Trial Module). Brussels: EC; 2004.
- Detailed guidance on the collection, verification and presentation of adverse reaction reports arising from clinical trials on medicinal products for human use. Brussels: EC; 2004.
- European Union . Directive 2001/20/EC Of The European Parliament And Of The Council of 4 April 2001on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Official Journal of the European Communities 2001;121:34-4.
- The EuroQoL Group . EuroQoL-a new facility for the measurement of health-related quality of life. Health Policy 1990;16:199-208.
- Ensrud KE, Blackwell TL, Mangione CM, Bowman PJ, Whooley MA, Bauer DC, et al. Study of Osteoporotic Fractures Research Group. Central nervous system-active medications and risk for falls in older women. Journal of the American Geriatrics Society 2002;50:1629-37.
- Folstein MF, Folstein SE, McHugh PR. Mini Mental State. Journal of Psychiatric Research 1975;12:189-98.
- Friedman LM, Furberg CD, DeMets DL. Fundamentals of Clinical Trials. New York: Springer; 1998.
- Goldberg D, Williams P. A user's guide to the General Health Questionnaire. Windsor: NFER- NELSON; 1988.
- Greenwald BS, Kramer-Ginsberg E, Marin DB, Laitman LB, Hermann CK, Mohs RC, et al. Dementia with coexistent major depression. Am J Psychiatry 1989;146:1472-8.
- Hedeker D, Gibbons RD. Application of random-effects pattern-mixture models for missing data in longitudinal studies. Psychol Meth 1997;2:64-78.
- Katona, . A double-blind comparison of the efficacy and safety of paroxetine and imipramine in the treatment of depression with dementia. International Journal of Geriatric Psychiatry 1998;13:100-8.
- Kenna LA, Sheiner LB. Estimating treatment effect in the presence of non-compliance measured with error: precision and robustness of data analysis methods. Stat Med 2004;23:3561-80.
- Levy DE, O’Malley AJ, Normand ST. Covariate adjustment in clinical trials with non-ignorable missing data and non-compliance. Stat Med 2004;23:2319-39.
- Little RJA, Rubin DB. Statistical Analysis with Missing Data. New York: John Wiley & Sons; 2002.
- Little RJA, Wang Y. Pattern-mixture models for multivariate incomplete data with covariates. Biometrics 1996;52:98-111.
- Lyketsos CG, DelCampo L, Steinberg M, Miles Q, Steele CD, Munro C, et al. Treating depression in Alzheimer disease: efficacy and safety of sertraline therapy, and the benefits of depression reduction: the DIADS. Arch Gen Psychiatr 2003;60:737-46.
- Lyketsos C, Sheppard J, Steele C, . A randomized placebo-controlled, double-blind, clinical trial of sertraline in the treatment of depression complicating Alzheimer's Disease. Am J Psychiatry 2000;157:1686-89.
- Machin D, Campbell M, Fayers P, Pinol A. Sample Size Tables for Clinical Studies. Oxford: Blackwell Science; 1997.
- McKhann G, Drachman D, Folstein M, . Clinical diagnosis of Alzheimer's Disease: report of the NINCDS-ADRDA work group. Neurology 1984;34:939-44.
- Medical Research Council . MRC Guidelines for Good Clinical Practice in Clinical Trials 1998.
- Murray J, Schneider J, Banerjee S, Mann A. EUROCARE: a cross national study of co-resident spouse carers for people with Alzheimer's Disease II - a qualitative analysis of the experience of caregiving. Int J Geriatr Psychiatry 1999;14:662-67.
- Nagelkerke N, Fidler V, Bernsen R, Borgdorff M. Estimating treatment effects in randomizes clinical trials in the presence of non-compliance. Stat Med 2000;19:1849-64.
- Netten A, Netten A, Beecham J. Costing community care: theory and practice. Aldershot: Ashgate; 1993.
- Nyth A, Gottfries C, Luby K, . A controlled multicenter clinical study of citalopram and placebo in elderly depressed patients with and without dementia. Acta Psychiatr Scand 1991;86:138-45.
- Olin JT, Katz IR, Meyers BS, Schneider LS, Lebowitz BD. Provisional diagnostic criteria for depression of Alzheimer Disease. Am J Geriatr Psychiatry 2002;10:129-41.
- Oslin D, Ten Have T, Streim J, . Probing the safety of medications in the frail elderly: evidence from a randomized controlled clinical trial of sertraline and venlafaxine in depressed nursing home residents. J Clin Psychiatry 2003;64:875-82.
- Petracca G, Teson A, Chemerinski E, Leiguarda R, Starkstein SE. A double-blind placebo-controlled study of clomipramine in depressed patients with Alzheimer's disease. J Neuropsychiatry Clin Neurosci 1996;8:270-5.
- Ratcliffe J. Public preferences for the allocation of donor liver grafts for transplantation. Health Econ 2000;9:137-48.
- Reifler BV, Teri L, Raskind M, Veith R, Barnes R, White E, et al. Double-blind trial of imipramine in Alzheimer's disease patients with and without depression. Am J Psychiatry 1989;146:45-9.
- Ryan M, McIntosh E, Dean T, Old P. Trade-offs between location and waiting times in the provision of health care: the case of elective surgery on the Isle of Wight. J Publ Health Med 2000;22:202-10.
- Schafer JL. Multiple imputation: a primer. Stat Meth Med Res 1999;8:3-15.
- Schaub RT, Linden M, Copeland JRM. A comparison of GMS/AGECAT, DSM-III-R for dementia and depression, including subthreshold depression (SD) – results from the Berlin Aging Study (BASE). Int J Geriatr Psychiatry 2003;18:109-17.
- Schneider J, Hallam A, Murray J, Foley E, Atkin L, Banerjee S, et al. Formal and informal care for people with dementia: factors associated with service receipt. Ageing Ment Health 2002;6:255-65.
- Schneider J, Hallam A, Murray J, Foley E, Atkin L, Banerjee S, et al. Formal and informal care for people with dementia: investigating changes in costs over time. Ageing Soc 2003;23:203-326.
- Schneider J, Murray J, Banerjee S, Mann A. EUROCARE: a cross national study of co-resident spouse carers for people with Alzheimer's Disease I - factors associated with carer burden. Int J Geriatr Psychiatry 1999;14:665-1.
- Stahl SM, Entsuah R, Rudolph RL. Comparative efficacy between venlafaxine and SSRIs: a pooled analysis of patients with depression. Biol Psychiatry 2002;52:1166-74.
- Smith SC, Lamping DL, Banerjee S, Harwood R, Foley B, Smith P, et al. Measurement of health-related quality of life for people with dementia: development of a new instrument (DEMQOL) and an evaluation of current methodology. Health Technol Assess 2005;9.
- Teri L, Gibbons LE, McCurry SM, Logsdon RG, Buchner DM, . Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003;290:2015-22.
- Ware JE, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: Construction of scales and preliminary tests of reliability and validity. Med Care 1996;34:220-33.
- White IR, Carpenter J, Pocock SJ, Henderson RA. Adjusting treatment comparisons to account for non-randomised interventions: an example from an angina trial. Stat Med 2003;22:781-93.
- Zarit SH, Reever KE, Bach-Peterson J. Relatives of the impaired elderly: correlates of feelings of burden. Gerontologist 1980;20:649-55.
22 APPENDICES
22.1 Appendix 1 Participant and Carer Information
| Ethics approved 12 July 2006 | ||
|---|---|---|
| Participant information sheet | Version 1 (shortened version) | 29.03.06 |
| Participant information sheet: Patient | Version 2 | 28.06.06 |
| Participant information sheet: Carer | Version 2 | 28.06.06 |
| Ethics submitted 28 September 2006 | ||
|---|---|---|
| Information and consent form for patient (full version) | Version 3 | 28.09.06 |
| Information and assent form for patient (shortened version) | Version 2 | 28.09.06 |
| Information and consent forms for carer | Version 3 | 28.09.06 |
22.2 Appendix 2 Letters
Standard letters will be contained within the Trial SOPs. These will include:
-
letter of referral from Referring Investigator to Recruiting Investigator
-
letter from Research Worker to GP informing them of Participants inclusion.
22.3 Appendix 3 Source Data Worksheets and Electronic Case Report Forms)
A3.1 Additional Diagnostic Questions
A3.2 Adverse Events Aide Memoire
Adverse Events
Adverse events will be recorded throughout the trial, with information systematically collected at weeks 4, 13 and 39. Adverse events should be recorded if they have prevented the participant from continuing with his / her normal activities, or if they have significantly affected the participant's well being. Any adverse events should be discussed with the principal recruiting investigator, to consider causality and seriousness. Adverse events are recorded in the eCRF on the basis of these discussions.
The following list is an aide memoire only. All adverse events should be recorded, whether or not they appear here.
-
abnormal vision
-
aggression
-
anaemia
-
high or low blood pressure
-
confusion
-
fits (seizures)
-
diarrhoea / loose stools
-
dizziness
-
dry mouth
-
quickened heart rate
-
falls
-
fatigue
-
sleepiness
-
fever
-
headache
-
indigestion
-
itching
-
malaise
-
muscle jerks
-
muscle rigidity
-
nightmares/vivid dreams
-
numbness/tingling
-
pain – abdominal
-
pain – joints
-
pain – muscle
-
rash
-
restless legs
-
sexual dysfunction
-
sweating
-
tremor
-
urinary retention
-
vomiting.
Adverse events will be classed as serious if any of the following apply:
-
life-threatening
-
necessitates hospitalisation
-
prolongs hospitalisation
-
causes persistent and / or significant disability or incapacity
-
deliberate self-harm.
A3.3 Bristol Activities of Daily Living Scale
A3.4 Carer Demographic Information
A3.5 Carer Global Impression
A3.6 Carer Registration #1
A3.7 Client Service Receipt Inventory (CSRI)
A3.8 Cornell Scale for Depression in Dementia (CSDD)
Cornell Scale for Depression in Dementia (CSDD) (PDF download)
A3.9 Concomitant medications/DRUGS (Page one)
A3.10 Concomitant treatments/Non Drugs
A3.11 DEMQOL
A3.12 DEMQOL Proxy
A3.13 End of Trial (Routine) Request for Unblinding
End of Trial (Routine) Request for Unblinding (PDF download)
A3.14 EuroQol (EQ-5D) – Carer
A3.15 EuroQol (EQ-5D) – Participant
A3.16 Exclusions from Randomisation
A3.17 General Health Questionnaire (GHQ12)
A3.18 Medical History (Page One)
A3.19 Medication Guess
A3.20 Medication Preference
A3.21 Neuropsychiatric Inventory (NPI)
A3.22 Non-serious Adverse Events Log (Page One)
A3.23 Participant Demographic Information
A3.24 Participant Registration
A3.25 Pill Count
A3.26 Randomisation Request Form: HTA-SADD trial
A3.27 Serious Adverse Event (SAE) Form
A3.28 Short Form 12 (SF-12 Version 2)
A3.29 Mini-Mental State Examination (SMMSE)
A3.30 Trial medication log
A3.31 Withdrawal Form
A3.32 Zarit Caregiver Burden Inventory
22.4 Appendix 4 Policy on Ancillary Studies
This is to be developed.
22.5 Appendix 5 CONSORT Diagram
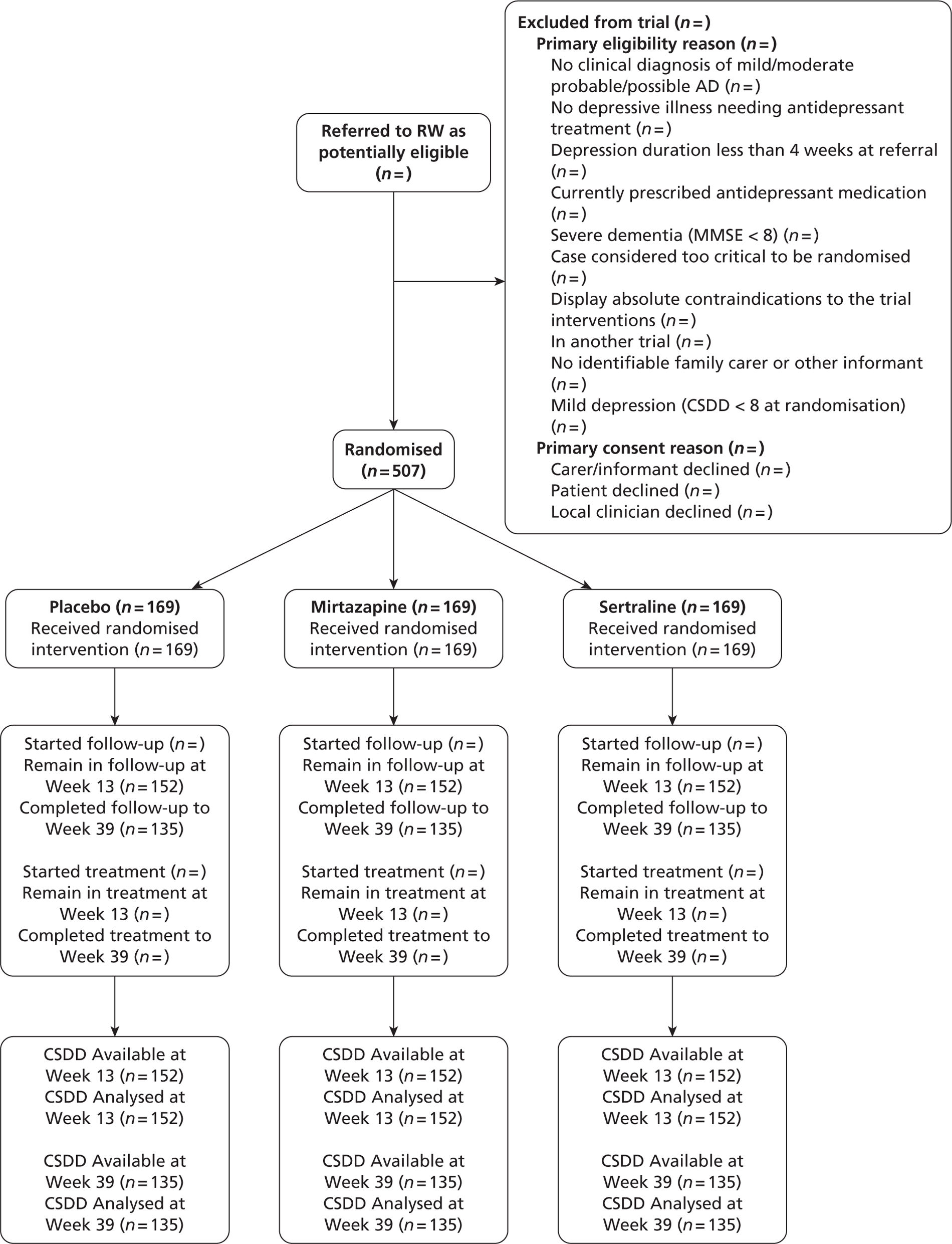
22.6 Appendix 6 Statistical Analysis Strategy
To be agreed by the TMG and the TSC after the start of randomisation.
22.7 Appendix 7 Declaration of Helsinki 1996
Please see: http://www.hku.hk/facmed/research/ec/Declaration_of_Helsinki_1996_version.PDF
List of abbreviations
- AD
- Alzheimer's disease
- AS
- Alzheimer's Society
- BADL
- Bristol Activities of Daily Living
- BPSD
- Behavioural and Psychological Symptoms of Dementia
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CSDD
- Cornell Scale for Depression in Dementia
- CSRI
- Client Service Receipt Inventory
- CTU
- Clinical Trials Unit
- DEMQOL
- Dementia Quality of Life
- DeNDRoN
- Dementia and Neurodegenerative Disease Research Network
- DIADS
- Depression in Alzheimer's Disease Study
- DMEC
- Data Monitoring and Ethics Committee
- DSM
- Diagnostic and Statistical Manual
- ENT
- ear, nose and throat
- EQ-5D
- European Quality of Life-5 Dimensions
- GHQ-12
- General Health Questionnaire version 12
- GP
- general practitioner
- HRQL
- health-related quality of life
- HTA
- Health Technology Assessment
- ICER
- incremental cost effectiveness ratio
- MH&N CTU
- Mental Health & Neurosciences Clinical Trials Unit
- MHRN
- Mental Health Research Network
- MMSE
- Mini Mental State Examination
- NASSA
- noradrenergic and specific serotonergic antidepressant
- NB
- net benefit
- NICE
- National Institute for Health and Clinical Excellence
- NIHR
- National Institute for Health Research
- NINCDS-ADRDA
- National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association
- NPI
- Neuropsychiatric Inventory
- OR
- odds ratio
- QALY
- quality-adjusted life-year
- QRD
- Quality Research in Dementia
- RCT
- randomised controlled trial
- SADD
- Study of Antidepressants for Depression in Dementia
- SCIE
- Social Care Institute for Excellence
- SD
- standard deviation
- SE
- standard error
- SES
- standardised effect size
- SF-12
- Short Form questionnaire-12 items
- SSRI
- selective serotonin reuptake inhibitor
- TCA
- tricyclic antidepressant
- TSC
- Trial Steering Committee
- VAS
- visual analogue scale
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices in which case the abbreviation is defined in the figure legend or at the end of the table.